

Abstract
Tumor-specific variable regions of the clonal immunoglobulin (idiotype, Id) expressed by B cell non-Hodgkin lymphoma (NHL) can be targeted by active immunotherapy. We conducted a phase I/II trial to determine the safety and immunogenicity of a patient-specific, recombinant, mammalian cell-derived Id protein conjugated to keyhole limpet hemocyanin (Id-KLH; MyVax® personalised immunotherapy) in 22 patients with follicular NHL in first remission after chemotherapy. Subjects received five subcutaneous immunisations with MyVax® plus locally administered granulocyte-macrophage colony-stimulating factor (GM-CSF). Among 21 evaluable patients, 62% mounted Id-specific immune responses. Evoked anti-Id antibodies recognised both recombinant Id and native Id, and could specifically stain autologous tumor cells. At median follow-up of more than 6 years, median progression-free survival is 38 months. Immunisation of follicular lymphoma patients with MyVax® Id-KLH is safe and patients often mount tumor-specific immune responses. These results form the basis of a pivotal phase 3 trial of MyVax® in follicular NHL.
Keywords: Immunotherapeutic approaches, neoplasia, immunotherapy, lymphoma and Hodgkin disease, vaccines
Introduction
The tumor-specific immunoglobulin (Ig) expressed by B cell lymphomas can serve as a target for active immunotherapy. This target, referred to as the ‘idiotype’ (Id), is composed of the unique antigenic determinants in the variable regions of the clonal Ig heavy and light chains expressed by the tumor cells [1]. Id proteins contain structures that can be recognised by antibodies [2–6] and by CD4+ [5,7–9] and CD8+ [10–14] T cells, and can be isolated from autologous tumor cells and formulated into a custom-made therapeutic tumor vaccine. The traditional approach for generating patient-specific Id vaccines involves fusion of individual patient's lymphoma cells with myeloma cells, yielding a ‘rescue’ hybridoma secreting large quantities of Id protein [15]. The Id is then chemically conjugated to the highly immunogenic carrier protein keyhole limpet hemocyanin (KLH) rendering it more immunogenic. The resulting Id-KLH conjugate is then injected subcutaneously (s.c.) along with an immunologic adjuvant to evoke tumor-specific antibody and T cell responses.
Results from early clinical trials of Id immunisation for follicular lymphoma using hybridoma-derived Id have included the induction of tumor-specific anti-Id immune responses that correlate with improved disease-free and overall survival [7,8], achievement of molecular complete remissions (bcl-2 negative PCR status) and favorable progression-free survival (PFS) using Id-KLH plus GM-CSF [16,17], and durable tumor regressions following immunisation with Id protein-loaded autologous dendritic cells [18,19]. However, limitations of the rescue hybridoma method include a production failure rate as high as 15%, the need for viable tumor cells for cell fusion, non-uniformity of the Id product (IgG, IgM or other isotype expressed by the tumor) and the instability of Id secretion by tumor hybridomas over time. An alternative technique, ‘molecular rescue’, employs PCR amplification of the tumor-specific variable region Ig sequences from small numbers of tumor cells (107) for cloning into expression vectors carrying the desired isotype backbone [Figure 1(A)]. This molecular approach obviates the need for surgical biopsy, as adequate material can be obtained by fine or core needle biopsy, bone marrow biopsy, involved peripheral blood or fluid collection aspiration. We [20] have developed these newer techniques for the efficient production of Id proteins from lymphoma specimens.
Figure 1.
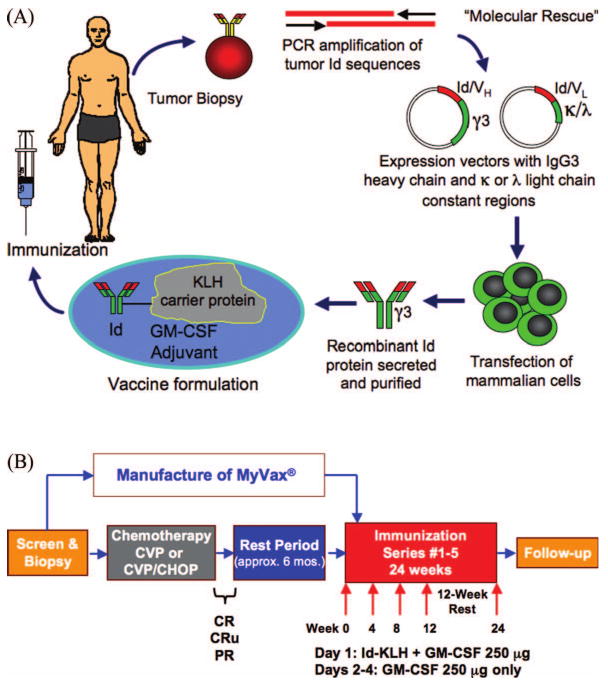
Therapeutic immunisation for lymphoma with recombinant idiotype protein. (A) Schema for production and administration of MyVax®. PCR, polymerase chain reaction; Id, idiotype; VH and VL, heavy and light chain variable regions; KLH, keyhole limpet hemocyanin. (B) Treatment schedule for cytoreductive chemotherapy and immunisations.
We performed a phase I/II clinical trial of a mammalian cell-derived, recombinant patient-specific therapeutic Id vaccine product (MyVax®, Genitope Corporation, Fremont, CA) in patients with follicular NHL in first remission following chemotherapy. The goals of this study were to determine the feasibility, safety and immunogenicity of this novel form of recombinant Id-KLH plus GM-CSF immunisation following standard cytoreductive chemotherapy. We found that recombinant Id-KLH plus GM-CSF was well-tolerated, and that immunogenicity was comparable to that of hybridoma-derived Id. Induced serum anti-Id antibodies recognised native tumor Id structure as well as recombinant Id, and could specifically bind to patient's tumor cells. After more than 6-years of follow-up, a substantial proportion of patients remain progression-free, including many predicted at baseline to have adverse prognosis. These results have provided the basis for a multi-center, randomised phase III trial of this recombinant Id vaccine following initial chemotherapy.
Patients and methods
Study design
The study included previously untreated patients ≥18 years of age with stage III or IV follicular B cell NHL (Grades I and II by WHO classification), with accessible tumor tissue for biopsy expressing monoclonal surface Ig. Subjects were required to have Karnofsky performance status of >70%, adequate hepatic, renal and bone marrow function, and the ability to provide informed consent. Patients with serious concomitant illnesses, CNS involvement, pregnancy, HIV seropositivity or those requiring chronic corticosteroid or immunosuppressive therapy were excluded. The vaccine dose and schedule were chosen to be identical to those safely used in previous trials of hybridoma-derived Id proteins [7,8,16] (see below). For this reason, despite this being a first-in-human trial of the new recombinant Id vaccine, no dose escalation was performed. To be eligible for immunisation, subjects had to achieve and maintain at least a partial response (PR) to chemotherapy for a minimum of 3 months after completion of treatment [21], and until vaccine manufacturing was complete, up to a maximum of 1 year. The study was approved by the Institutional Review Boards of Stanford University and the University of Nebraska Medical Centers and the U.S. Food and Drug Administration, and all patients supplied written informed consent.
Target enrollment was set at 25 subjects, with a goal of completing immunisation in 20 or more subjects. A dropout rate of approximately 20% was estimated from our prior experience in cytoreduction with CVP/CHOP and in laboratory production of recombinant Id vaccines [8,19]. Given that vaccine-associated toxicities were expected to be minimal based on prior experience with similar vaccine preparations, formal stopping rules for vaccine safety were not predetermined. Primary study endpoints included safety, Id-specific immune responses (serum anti-Id antibody titers and Id-specific T cell proliferation) and clinical response status, with secondary endpoints of progression-free and overall survival [21]. The primary criteria for proceeding with a phase III trial were safety and an overall immune response rate (humoral + cellular) of at least 40%, thus approximating the 49% immune response rate observed in our previous trial using hybridoma-derived Id [8].
Patients
Eligible patients were evaluated at baseline for prognosis according to the Follicular Lymphoma International Prognostic Index (FLIPI)[22]. After tissue collection, patients received four to six cycles of CVP (cyclophosphamide 400 mg/m2 PO days 1–5, vincristine 1.4 mg/m2 IV day 1, and prednisone 100 mg PO days 1–5) to best clinical response [Figure 1(B)]. Five patients achieving only PR after CVP were treated with two to six additional cycles of CHOP (cyclophosphamide 750 mg/m2 IV day 1, Adriamycin 50 mg/m2 IV day 1, vincristine 1.4 mg/m2 IV day 1, and prednisone 50 mg/m2 PO days 1–5), in attempt to achieve minimal residual disease burden before vaccination. Clinical responses were classified as PR, complete response (CR), or CR unconfirmed (CRu) according to standardised response criteria [21]. Immunisations were initiated 4–9 months after completion of chemotherapy according to the schedule given below. Repeat staging evaluations including CT scans of the chest, abdomen and pelvis were performed pre-vaccine, post-vaccine (1–3.5 months following completion of vaccinations), every 3 months for the first 2 years, and every 3–6 months thereafter. PFS is measured from the date of last chemotherapy.
Production of recombinant idiotype protein vaccines
The schema for production of MyVax® is shown in Figure 1(A). Total RNA was isolated from tumor biopsies using the RNeasy Midi Kit (Qiagen, Valencia, CA), and first strand cDNA amplified with primers specific for the appropriate heavy chain or light chains as determined by flow cytometry. RT-PCR was performed using family specific Ig variable and constant region primers located 5′ to those used for cDNA synthesis, with a separate reaction for each V region family [23,24]. DNA from individual PCR reactions was directly sequenced, then confirmed by repeat RT-PCR. Id-encoding DNA was subcloned into cDNA plasmid expression vectors containing an IgG3 heavy chain constant region sequence (except patient #1, where IgG1 was used), and the light chain constant region sequence matching that of the tumor. Individual clones were isolated and sequenced, and those matching the original RT-PCR product selected for expression in mammalian cells. Plasmids each expressing the Id, the selectable marker hypoxanthine-guanine phosphoribosyl transferase, and the amplifiable marker dihydrofolate reductase were co-electroporated into BW5147.G.1.4 murine lymphoma cells [25], followed by selection in 2 μg/mL azaserine and 100 μM hypoxanthine. Selected clones were assayed for Ig production by ELISA, and high-expressing clones grown further in methotrexate to amplify the Id genes until the secretion level was adequate for large-scale production.
Production cell lines were incubated in HyQ CCM-1 media (Hyclone, Logan, UT) in 2-L cell culture bags (Medtronic, Chicago, IL) at 37°C for 8–12 days. Recombinant Id was purified from supernatants using HiTrap Protein G columns (GE Healthcare, Newark, NJ), eluting with 100 mM glycine, pH 2.7, then dialysed into normal saline. The Id was then chemically conjugated to clinical grade KLH (Biosyn, Carlsbad, CA) with 0.1% glutaraldehyde (Sigma, St. Louis, MO) at room temperature for 60 min, dialysed into normal saline and aliquoted into vials for storage at −80°C. The final products were tested for sterility and endotoxin content. Rescue hybridoma Id proteins were produced for 12 patients as previously described [15], for use in immune response assays.
Immunisations
Immunisations consisted of 1 mg of Id-KLH (0.5 mg each Id and KLH) plus 250 μg GM-CSF on day 1, divided and injected s.c. into the skin of each anterior thigh or posterior upper arm. On days 2–4, 250 μg GM-CSF was divided equally and injected into these same marked sites. Immunisations were given at weeks 0, 4, 8, 12 and 24. Immune response testing was performed before immunisations 1, 3, 4 and 5, and at weeks 14 and 26. Adverse events (AEs) were recorded according to National Cancer Institute Common Terminology Criteria for AE (CTCAE v3.0).
Humoral immune response assessments
Anti-Id antibodies were measured in serum by ELISA as previously described [8], with the following modifications. For IgG3 recombinant Id proteins, bound anti-Id antibodies were detected with a cocktail of anti-human IgG1, IgG2 and IgG4 antibodies coupled to horseradish peroxidase (HRP) (Southern Biotech, Birmingham, AL). A response was considered positive when a four-fold increase in anti-Id titer was found compared to both pre-immunisation serum and three isotype-matched irrelevant Id proteins used as specificity controls. Antibody responses to rescue hybrid-derived Id proteins and KLH were measured as previously described [8]. For tumor cell staining experiments, cryopreserved IgM-positive tumor cells were incubated with 1% pre- or post-vaccine serum overnight at 4°C, washed, then incubated with goat anti-human IgG F(ab′)2-phycoerythrin, followed by counter-staining with anti-human IgM F(ab′)2-FITC (Biosource, Camarillo, CA). Stained cells were analysed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA).
Cellular immune response assessment
T cell proliferation assays were performed on patients 1–16 at Stanford University using fresh peripheral blood mononuclear cells (PBMC) as reported previously [8]. Patients 17–21 were treated at the University of Nebraska Medical Center, and because of the difficulties in shipping viable PBMC across the U.S., cellular proliferation assays were not attempted for these cases. This assay was chosen to provide historical comparison with our experience using hybridoma-derived Id proteins [7,8,18,19]. Briefly, PBMC were cultured in quadruplicate in media alone or with tumor Id, irrelevant Id proteins from other patient's tumors, or KLH at 0.1, 1.0, 10 and 100 μg/mL. Incorporation of [3H]-thymidine (Amersham Pharmacia Biotech, Piscataway, NJ, 185 GBq/mmol) was measured after an overnight pulse (1 μCi/well) on day 5. A response was interpreted as positive when incorporation of more than twice background (media alone) with at least one antigen concentration was observed on two or more occasions.
Statistical analysis
Differences in PFS among patient groups were determined using the Kaplan–Meier estimation [26] and log-rank statistic using PRISM software (Graph-Pad Software, San Diego, CA).
Results
Enrollment and cytoreductive chemotherapy
The baseline characteristics of the 21 evaluable patients are summarised in Table I. Detailed descriptions of individual patients and outcomes are presented in Table II. The vast majority of patients had stage IV disease with bone marrow involvement (86%). According to the FLIPI prognostic model [22], nearly all patients had high risk (n = 8, 38%) or intermediate risk (n = 11, 52%) disease; only two subjects (10%) had low-risk disease by FLIPI (Table I).
Table I.
Summary of patient characteristics.
| Characteristic | Patients (n = 21) |
|---|---|
| Age (years) | |
| Median | 49 |
| Range | 32–73 |
| Sex | |
| Male | 11 (52) |
| Female | 10 (48) |
| Type of NHL | |
| Follicular small cleaved cell (Grade 1) | 15 (71) |
| Follicular mixed cell (Grade 2) | 6 (29) |
| Disease stage | |
| Stage III | 3 (14) |
| Stage IV | 18 (86) |
| FcγRIIIa genotype | |
| F/F or V/F | 17 (81) |
| V/V | 4 (19) |
| FLIPI score | |
| 1 (low risk) | 2 (10) |
| 2 (intermediate risk) | 11 (52) |
| 3 or 4 (high risk) | 8 (38) |
Values within parentheses represent percentages.
Table II.
Individual patient characteristics and outcomes summary.
| Patient | Sex/age | Histology/stage | FLIPI | Pre-vaccine chemoRx | Clinical response post-chemoRx | Pre-vaccine clinical status | Post-vaccine clinical status | Progression-free survival (months) (E = expired) | FcRγIII genotype | Anti-Id humoral IR | Anti-Id cellular IR |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M/53 | Gr2/IVA | 1 | 6 CVP | CR | CR | CR | 95.4+ | V/F | Positive | Positive |
| 2 | F/44 | Gr1/IVA | 3 | 8 CVP, 2 CHOP | PR, CR | CR | CR | 91.1+ | F/F | Positive | Negative |
| 3 | F/49 | Gr1/IVA | 2 | 8 CVP | CR | PR | PR | 38 | V/F | Negative | Negative |
| 4 | F/37 | Gr1/IVB | 3 | 6 CVP | PR | CR | PD | 12.5 | V/F | Negative | Negative |
| 5 | M/47 | Gr1/IVA | 2 | 4 CVP, 4 CP | CR | CR | CR | 30.3 | F/F | Positive | Positive |
| 6 | M/32 | Gr1/IVA | 2 | 5 CVP, 6 CHOP | PR, CRu | PR | PD | 12.1 | V/F | Negative | Positive |
| 7 | M/51 | Gr1/IIIB | 2 | 7 CVP | CRu | CRu | CR | 86.2+ | V/V | Negative | Negative |
| 8 | F/51 | Gr1/IIIA | 2 | 8 CVP | PR | CRu | CR | 84.3+ | V/F | Positive | Negative |
| 9 | M/49 | Gr1/IVA | 2 | 6 CVP | CR | CR | CR | 19.5 | F/F | Positive | Negative |
| 10 | F/41 | Gr1/IVA | 3 | 6 CVP | CRu | CRu | CRu | 22.5, E | V/F | Positive | Positive |
| 11 | M/57 | Gr2/IVA | 3 | 8 CVP | CRu | CR | PD | 16.4, E | V/F | Negative | Negative |
| 12 | M/55 | Gr1/IVA | 2 | 8 CVP, 4 CHOP | PR, CR | CRu | PD | 15.2, E | F/F | Positive | Positive |
| 13 | F/52 | Gr1/IVA | 2 | 6 CVP, 2 CHOP | PR, PR | PR | PR | 22.1, E | F/F | Negative | Positive |
| 14 | M/48 | Gr1/IVA | 2 | 6 CVP | PR | PR | CR | 82.7+ | V/V | Negative | Negative |
| 15 | F/67 | Gr1/IVA | 3 | 4 CVP, 2 VP | PR | PR | CR | 77.3+ | V/F | Positive | Negative |
| 16 | M/44 | Gr1/IVA | 3 | 8 CVP | PR | CRu | CR | 67.9+ | V/V | Negative | Positive |
| 17 | F/73 | Gr1/IIIA | 4 | 6 CVP | PR | CRu | CRu | 83.3+ | F/F | Negative | ND |
| 18 | F/39 | Gr2/IVA | 1 | 6 CVP | CR | CR | PD | 15 | V/F | Negative | ND |
| 19 | M/68 | Gr2/IVA | 3 | 6 CVP | CR | CR | CR | 60.9 | F/F | Positive | ND |
| 20 | M/40 | Gr2/IVA | 2 | 6 CVP, 4 CHOP | PR, CRu | CRu | CRu | 75.7+ | V/F | Negative | ND |
| 21 | F/41 | Gr2/IVA | 2 | 6 CVP | CR | CR | PD | 12.7 | V/V | Positive | ND |
Gr, grade; FLIPI, follicular lymphoma international prognostic index; CVP, cyclophosphamide, vincristine and prednisone; CHOP, cyclophosphamide, adriamycin, vincristine and prednisone; CR, complete response; CRu, complete response unconfirmed; PR, partial response; IR, immune response.
Boldface type indicates subjects in ongoing first complete remission.
All patients achieved at least a PR to CVP chemotherapy. Because the rate of detectable immune responses to Id immunisation is improved in patients with minimal residual tumor [8], we attempted further cytoreduction in five PR patients using two to six cycles of CHOP. In four cases, response was improved to CR or CRu (Table II). One patient received rituximab (three 4-week courses) before, during and after Id-KLH immunisations by an outside oncologist in violation of the protocol, and was thus not evaluable for immune and clinical responses, but was included in safety evaluations. In this study, rituximab was not permitted because of its resulting depletion of normal B cells and severe impairment of humoral responses to vaccines [27–30].
Production and administration of recombinant idiotype vaccines
Recombinant Id proteins were successfully produced for all 22 patients (100%) eligible for immunisation during chemotherapy and the subsequent ‘rest’ period for immunologic recovery after chemotherapy [Figure 1(B)]. This rest period ranged from 4.6 to 8.8 months following chemotherapy (median 6.5 months), depending on the time required to complete vaccine production. All patients remained in remission during the rest period (Table II). The total time required to produce vaccines varied from 3 to 13 months, but most vaccines could be manufactured within 3–6 months. The clinical status before immunisation was PR in 5 patients (24%), CRu in 7 patients (33%), and CR in 9 patients (43%). Patient 6 had early radiographic evidence of tumor growth pre-immunisation, though not meeting criteria for progressive disease, and thus proceeded to complete immunisations. Four additional patients had been enrolled in the study, but were not eligible for immunisations because of inadequate response to chemotherapy (n = 3, including one subject whose progressive disease had transformed to diffuse large B cell lymphoma), or lack of surface Ig expression by collected tumor cells (n = 1).
Safety of immunisations
All 22 patients completing the five scheduled immunisations were evaluable for safety. Immunisations were well-tolerated, with AEs primarily limited to mild-to-moderate local injection site reactions and transient flu-like symptoms (Table III). Injection site reactions were characterised by erythema (100%) and less often induration, local discomfort and itching. The vast majority of AEs were Grade 1, self-limiting and largely attributable to GM-CSF.
Table III.
Adverse events in 22 patients evaluable for safety.
| Adverse events | Patients (n = 22) (%) |
|---|---|
| Injection site reactions | Any grade* |
| Erythema | 100 |
| Induration | 86 |
| Pain | 86 |
| Pruritus | 64 |
| Bruising | 32 |
| Inflammation | 27 |
| Headache | 55 |
| Fatigue | 45 |
| Back pain | 36 |
| Pain NOS | 32 |
| Nasopharyngitis | 32 |
| Arthralgia | 27 |
| Influenza-like illness | 23 |
| Myalgia | 23 |
| Fever | 18 |
| Nausea | 18 |
| Dyspepsia | 14 |
| Rigors | 14 |
Grade 1 except for one patient each (5%) with Grade 2 myalgia, Grade 2 headache, Grade 2 dyspepsia, two patients (9%) with Grade 2 pain NOS, and one patient (5%) with Grade 3 muscle spasms.
Immune responses to recombinant idiotype vaccine
Among 21 subjects evaluable for humoral anti-Id response and 16 subjects evaluable for T cell response, the overall Id-specific immune response rate was 62% (13/21). This included 10 patients with antibody responses (48%), 7 patients with T cell responses (44%), and 4 patients mounting both antibody and T cell responses (25%). All patients mounted both humoral and T cell proliferative responses to KLH. The frequency of Id-specific immune responses did not differ substantially between patients treated with CVP (9/16, 56%) versus CVP plus CHOP (4/5, 80%). An example of a tumor-specific anti-Id antibody response is shown in Figure 2(A), with patient's post-immunisation serum reacting with the recombinant Id. Figure 2(B) is an example of a tumor-specific of humoral response, in which significant binding is detected only against autologous Id, but not to Id derived from three other patient's tumors.
Figure 2.
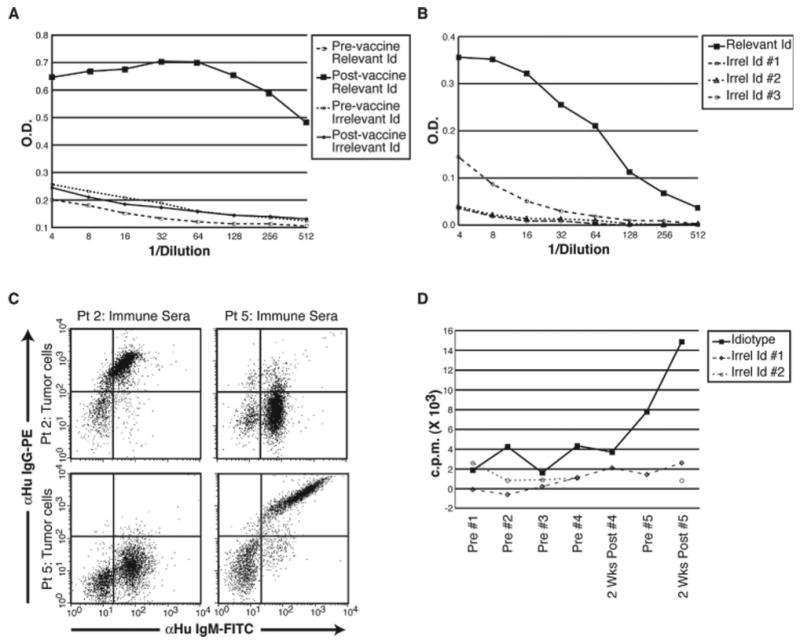
Idiotype-specific immune responses following Id-KLH immunisation. (A) Patient 5 humoral response measured by ELISA. Pre- or post-vaccination serum was serially diluted and tested for binding (indicated by optical density, O.D.) to the patient's own tumor Id vs. another patient's (irrelevant) Id protein as a control. Only post-vaccine serum shows a high degree of tumor Id-specific binding. (B) Specificity of an anti-Id antibody response after immunisation. Post-immunisation sera (patient 1) was serially diluted and tested by ELISA for binding (indicated by optical density, O.D.) to the patient's own tumor (relevant) Id versus three other patient's tumor Id proteins as controls. The solid black line indicates a highly tumor-specific antibody response, with minimal binding to other patient's tumor Ids (irrelevant; dashed lines). (C) Flow cytometric demonstration that anti-Id antibodies induced by recombinant Id-KLH vaccine specifically recognise autologous tumor cells. Tumor cells from patients 2 and 5 (upper and lower sets of panels, respectively) were incubated with either autologous post-vaccine serum or that of the other patient as a control. Bound anti-Id IgG antibodies were detected by anti-IgG-PE (Y-axis), and tumor B cells (IgM+) were counterstained by anti-IgM-FITC (X-axis). Each patient's serum specifically recognises only the autologous tumor cells, but not the control, irrelevant tumor cells. (D) Tumor Id-specific T cell proliferative response post-vaccination. Proliferation of PBMC cultured with autologous tumor Id or irrelevant control Id proteins (100 μg/mL) from other patient's tumors were measured by 3H-thymidine incorporation (counts per minute, c.p.m.). The kinetics of the T cell proliferative response to tumor versus control Id proteins during the course of the five immunisations is depicted. Maximal T cell response is achieved after the 5th injection.
To confirm that immune responses generated against recombinant Id were relevant to native Id structure, tumor-specific rescue hybridomas were produced for 12 patients. The resulting rescue Id proteins were tested alongside recombinant Id proteins as targets for binding to anti-Id antibodies in post-immunisation serum. Among cases available for comparison, in each case of positive humoral reactivity to recombinant Id (n = 5), comparable reactivity to rescue hybridoma-derived Id was also demonstrated (data not shown). Conversely, when there was no humoral response to recombinant Id (n = 7), reactivity to rescue hybridoma Id was also negative. To further verify the ability of induced serum anti-Id antibodies to bind to the Id in its native conformation, we performed tumor cell staining experiments with post-immunisation serum from two patients having high anti-Id antibody titers using flow cytometry. As shown in Figure 2(C), each patient's serum antibodies binds only to autologous tumor cells, thus demonstrating again the tumor-specific nature of the evoked anti-Id humoral immune response. Id-specific T cell proliferation was also measured as in previous trials, and a representative response is shown in Figure 2(D). Notably, the response becomes positive only after the fourth immunisation, and reaches a high level 2 weeks after the fifth Id-KLH injection. As predicted, the single patient in this study who received rituximab failed to mount a humoral anti-Id response, and the humoral response to KLH was delayed and of low titer (data not shown).
Clinical outcomes following immunisation
At a median follow-up of 77 months since the end of chemotherapy, 9 of 21 patients (43%) have remained in continuous first remission, remaining progression-free for 67.9+ to 95.4+ months (Table II). Twelve of 21 (57%) patients have progressed, with a median TTP of 38 months (Figure 3). Seventeen of 21 patients (81%) remain alive, with four deaths due to lymphoma. Of the 9 patients experiencing long-term disease-free survival, 7 are in CR and 2 are in CRu. Prolonged PFS was noted in patients with both intermediate (n = 4) and high-risk disease (n = 4) according to the FLIPI. Thus, adverse prognosis did not appear to preclude a favorable outcome in this cohort treated with Id immunisation after chemotherapy. The proportion of long-term CR/CRu patients was similar in patients receiving CVP alone (7/16, 44%) versus CVP plus CHOP (2/5, 40%). As in earlier studies, we attempted to correlate immunologic responses to clinical outcomes [8]. In this study, positive results in the humoral and/or T cell proliferation immune response assays used did not correlate significantly with TTP (Table II).
Figure 3.
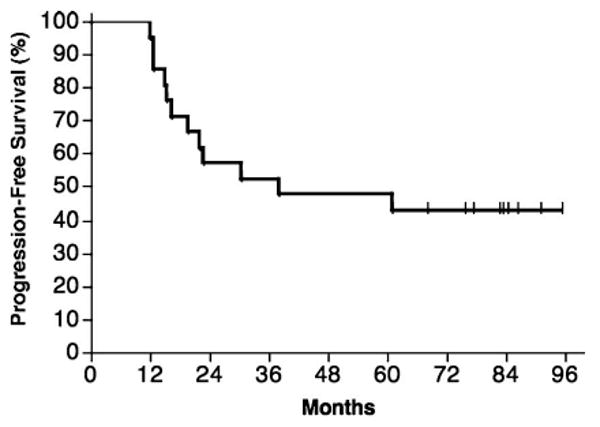
Progression-free survival (PFS) of all 21 evaluable subjects, measured from the date of last chemotherapy. Nine of 21 subjects (43%) remain in remission after median of follow-up of 77 months. Median PFS is 38 months.
Discussion
Therapeutic immunisation with patient-specific tumor Id offers the opportunity to specifically target lymphoma cells for immune-mediated destruction [1]. Numerous early phase clinical trials have provided strong rationale for this approach [2,6,7,16–19]. Until recently, however, the rapid and efficient production of Id for immunisation of large numbers of NHL patients was not feasible because of the cumbersome hybridoma procedures involved. We report here the first method for large-scale production of patient-specific recombinant Id proteins in mammalian cells. This technology reduces the time required for Id protein production by half (to 3–4 months in some cases), while improving the success rate to nearly 100%. Among more than 500 NHL biopsies collected for the ongoing MyVax® phase 3 trial, Id protein has been produced in more than 99% of cases in which tumor cells express surface Ig (D. Denney, unpublished data). Although all patients in the current study had excisional biopsies, surgical biopsy is not required for the production of recombinant Id, as adequate tumor cells can be obtained by FNA, core needle biopsy, bone marrow aspiration or sampling of involved peripheral blood.
The mammalian cell Id expression system used here also offers potential advantages over non-mammalian sources of recombinant Id proteins such as those produced in insect cells or bacteria [20,31]. Production in mammalian lymphoid cells ensures that Id proteins will maintain their natural glycosylation patterns and promotes proper three-dimensional protein folding. Tumor-specific Igs expressed by follicular NHL frequently harbor N-linked glycosylation sites in their variable region domains, and the associated carbohydrate chains may affect recognition of tumor Id by anti-Id antibodies [32].
Several lines of evidence suggest that the induction of a strong anti-Id antibody response is an important goal of Id immunisation. First, passively administered patient-specific anti-Id antibodies have been shown to have striking clinical activity in follicular lymphoma patients, with some apparently cured [2,6]. More importantly, improved long-term outcome of follicular lymphoma patients has been correlated with induction of humoral anti-Id immunity. In our recent retrospective analysis of 136 follicular NHL patients treated with Id-KLH vaccines (including the 21 patients described here), those mounting detectable anti-Id antibody responses experienced significantly longer PFS than those who did not [33]. Moreover, patients harboring the FcγRIIIa V/V polymorphism, previously shown to predict superior response to rituximab, also had prolonged PFS. The V allele corresponds to an FcγR with higher affinity for IgG and enhanced ADCC against antibody-coated tumor cells. This suggests that some subjects may have a cytotoxic anti-tumor antibody response that is undetectable using our current methods. It is reasoned that at a threshold humoral anti-Id response, FcγR-bearing natural killer cells and monocytes carry out efficient ADCC in patients with the V/V genotype.
Given the potential importance of anti-Id antibodies, it was important to ensure that our recombinant Id vaccine could induce humoral anti-Id immunity that was relevant to the native Id conformation as expressed by tumor cells. Anti-Id antibody responses were detectable in 48% of patients in this study, comparable to the rate of 35–42% seen in our earlier series using rescue hybrid-derived Id protein [8,33]. Importantly, there was complete concordance between the measured immune response to the recombinant Id protein used for immunisation and hybridoma-derived proteins, and sera from two patients could specifically stain their own tumor cells. The two patients exhibiting this tumor-specific cell staining both had favorable outcomes; one remains in complete remission at 91.1+ months following initial chemotherapy (#2), and another whose remission lasted more than 30 months (#5).
Positive results in the T cell proliferation immune response assay used did not correlate significantly with TTP in this study (Table II). This particular assay was chosen to allow comparison to our earlier experience with hybridoma-derived Id proteins [7,8,18,19]. It is acknowledged that this assay measures only cell proliferation in response to Id, whereas newer T cell assays such as ELISPOT [30,34,35], cytokine release [16] or intracellular cytokine staining [17,20,36] are more sensitive, particularly in the detection of CD8+ T cell responses. The design of future Id vaccine trials should incorporate such T cell assays to increase sensitivity and to identify relevant immunologic correlates of tumor regression or improved survival.
After a median follow-up of more than 6 years (77 months), 43% of our patients remain in continuous first complete remission (CR or CRu) for periods ranging from 67.9+ to 95.4+ months. The FLIPI has been reported to strongly predict both overall survival [22] and PFS following initial therapy [37–39] in follicular lymphoma. Ninety percent of our subjects had high-risk (38%) or intermediate-risk (52%) disease, which contrasts with the frequency of these risk groups in other larger follicular lymphoma cohorts (high-risk 21–27%, intermediate risk 31–37%) [22,38]. However, in this non-randomised trial, we cannot rule out that these outcomes are due to chemotherapy alone. The lack of correlation between adverse FLIPI score and outcome in this trial may be secondary to the small sample size in this trial.
Despite the demonstrated immunogenicity of the recombinant Id vaccine used in this trial, there remains considerable room for improvement in immune and clinical outcomes after Id immunisation. Id-KLH vaccines used this and other trials to date fail to elicit anti-Id immune responses in up to half of patients [8,19,20,31], and many first remission patients continue to experience early disease recurrence [30,33,40]. Fortunately, recent advances in vaccine technology may offer substantial improvements in Id-KLH vaccine potency. For example, an alternative chemical process for linking Id to KLH has recently been described that markedly augments anti-Id antibody and T cell responses and permits eradication of established lymphoma [41]. This process could easily be incorporated into existing Id-KLH immunotherapies, as could additional new immunologic adjuvants and cytokines [42]. Second generation Id-KLH vaccines incorporating one or more of these refinements may thus have substantially improved efficacy.
In conclusion, we have demonstrated that immunisation of NHL patients with recombinant mammalian cell-derived Id protein (MyVax®) is feasible, safe and leads to frequent Id-specific immune responses. These results have served as the basis for the phase III trial of MyVax® in follicular NHL patients in first remission after standard CVP chemotherapy [43]. As further trials are expected to introduce second-generation Id vaccines with even greater immunologic potency, it is hoped that Id-KLH vaccines will offer important clinical efficacy to patients with NHL.
Acknowledgments
J.M.T. is the recipient of a Clinical Associate Physician award from the NIH (RR-00070-CAP). R.L. is an American Cancer Society Clinical Research Professor.
References
- 1.Timmerman JM. Therapeutic idiotype vaccines for non-Hodgkin's lymphoma. Adv Pharmacol. 2004;51:271–293. doi: 10.1016/S1054-3589(04)51012-8. [DOI] [PubMed] [Google Scholar]
- 2.Miller RA, Maloney DG, Warnke R, Levy R. Treatment of B-cell lymphoma with monoclonal anti-idiotype antibody. N Engl J Med. 1982;306:517–522. doi: 10.1056/NEJM198203043060906. [DOI] [PubMed] [Google Scholar]
- 3.Miller RA, Hart S, Samoszuk M, Coulter C, Brown S, Czerwinski D, et al. Shared idiotypes expressed by human B-cell lymphomas. N Engl J Med. 1989;321:851–857. doi: 10.1056/NEJM198909283211302. [DOI] [PubMed] [Google Scholar]
- 4.George AJ, Tutt AL, Stevenson FK. Anti-idiotypic mechanisms involved in suppression of a mouse B cell lymphoma, BCL1. J Immunol. 1987;138:628–634. [PubMed] [Google Scholar]
- 5.Campbell MJ, Carroll W, Kon S, Thielemans K, Rothbard JB, Levy S, et al. Idiotype vaccination against murine B cell lymphoma. Humoral and cellular responses elicited by tumor-derived immunoglobulin M and its molecular subunits. J Immunol. 1987;139:2825–2833. [PubMed] [Google Scholar]
- 6.Davis TA, Maloney DG, Czerwinski DK, Liles TM, Levy R. Anti-idiotype antibodies can induce long-term complete remissions in non-Hodgkin's lymphoma without eradicating the malignant clone. Blood. 1998;92:1184–1190. [PubMed] [Google Scholar]
- 7.Kwak LW, Campbell MJ, Czerwinski DK, Hart S, Miller RA, Levy R. Induction of immune responses in patients with B-cell lymphoma against the surface-immunoglobulin idiotype expressed by their tumors. N Engl J Med. 1992;327:1209–1215. doi: 10.1056/NEJM199210223271705. [DOI] [PubMed] [Google Scholar]
- 8.Hsu FJ, Caspar CB, Czerwinski D, Kwak LW, Liles TM, Syrengelas A, et al. Tumor-specific idiotype vaccines in the treatment of patients with B- cell lymphoma – long-term results of a clinical trial. Blood. 1997;89:3129–3135. [PubMed] [Google Scholar]
- 9.Lauritzsen GF, Weiss S, Dembic Z, Bogen B. Naive idiotype-specific CD4+ T cells and immunosurveillance of B-cell tumors. Proc Natl Acad Sci USA. 1994;91:5700–5704. doi: 10.1073/pnas.91.12.5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao W, Myers-Powell BA, Braciale TJ. Recognition of an immunoglobulin VH epitope by influenza virus-specific class I major histocompatibility complex-restricted cytolytic T lymphocytes. J Exp Med. 1994;179:195–202. doi: 10.1084/jem.179.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chakrabarti D, Ghosh SK. Induction of syngeneic cytotoxic T lymphocytes against a B cell tumor. II. Characterization of anti-idiotypic CTL lines and clones. Cell Immunol. 1992;144:443–454. doi: 10.1016/0008-8749(92)90258-q. [DOI] [PubMed] [Google Scholar]
- 12.Abe A, Emi N, Taji H, Kasai M, Kohno A, Saito H. Induction of humoral and cellular anti-idiotypic immunity by intradermal injection of naked DNA encoding a human variable region gene sequence of an immunoglobulin heavy chain in a B cell malignancy. Gene Ther. 1996;3:988–993. [PubMed] [Google Scholar]
- 13.Osterroth F, Garbe A, Fisch P, Veelken H. Stimulation of cytotoxic T cells against idiotype immunoglobulin of malignant lymphoma with protein-pulsed or idiotype-transduced dendritic cells. Blood. 2000;95:1342–1349. [PubMed] [Google Scholar]
- 14.Trojan A, Schultze JL, Witzens M, Vonderheide RH, Ladetto M, Donovan JW, et al. Immunoglobulin framework-derived peptides function as cytotoxic T-cell epitopes commonly expressed in B-cell malignancies. Nat Med. 2000;6:667–672. doi: 10.1038/76243. [DOI] [PubMed] [Google Scholar]
- 15.Carroll WL, Thielemans K, Dilley J, Levy R. Mouse × human heterohybridomas as fusion partners with human B cell tumors. J Immunol Methods. 1986;89:61–72. doi: 10.1016/0022-1759(86)90032-3. [DOI] [PubMed] [Google Scholar]
- 16.Bendandi M, Gocke CD, Kobrin CB, Benko FA, Sternas LA, Pennington R, et al. Complete molecular remissions induced by patient-specific vaccination plus granulocyte-monocyte colony-stimulating factor against lymphoma. Nat Med. 1999;5:1171–1177. doi: 10.1038/13928. see comments. [DOI] [PubMed] [Google Scholar]
- 17.Inoges S, Rodriguez-Calvillo M, Zabalegui N, Lopez de Cerio A, Villanueva H, Soria E, et al. Clinical benefit associated with idiotypic vaccination in patients with follicular lymphoma. J Natl Cancer Inst. 2006;98:1292–1301. doi: 10.1093/jnci/djj358. [DOI] [PubMed] [Google Scholar]
- 18.Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 19.Timmerman JM, Czerwinski DK, Davis TA, Hsu FJ, Benike C, Hao ZM, et al. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood. 2002;99:1517–1526. doi: 10.1182/blood.v99.5.1517. [DOI] [PubMed] [Google Scholar]
- 20.Redfern CH, Guthrie TH, Bessudo A, Densmore JJ, Holman PR, Janakiraman N, et al. Phase II trial of idiotype vaccination in previously treated patients with indolent non-Hodgkin's lymphoma resulting in durable clinical responses. J Clin Oncol. 2006;24:3107–3112. doi: 10.1200/JCO.2005.04.4289. [DOI] [PubMed] [Google Scholar]
- 21.Cheson BD, Horning SJ, Coiffier B, Shipp MA, Fisher RI, Connors JM, et al. Report of an international workshop to standardize response criteria for non-Hodgkin's lymphomas. NCI sponsored international working group. J Clin Oncol. 1999;17:1244. doi: 10.1200/JCO.1999.17.4.1244. [DOI] [PubMed] [Google Scholar]; Erratum. J Clin Oncol. 2000;18:2351. [Google Scholar]
- 22.Solal-Celigny P, Roy P, Colombat P, White J, Armitage JO, Arranz-Saez R, et al. Follicular lymphoma international prognostic index. Blood. 2004;104:1258–1265. doi: 10.1182/blood-2003-12-4434. [DOI] [PubMed] [Google Scholar]
- 23.Denney D. Vaccines for treatment of lymphoma and leukemia. United States Patent 5,972,334. Genitope Corporation; 1999 Available at http://www.uspto.gov/patft/index.html.
- 24.Campbell MJ, Zelenetz AD, Levy S, Levy R. Use of family specific leader region primers for PCR amplification of the human heavy chain variable region gene repertoire. Mol Immunol. 1992;29:193–203. doi: 10.1016/0161-5890(92)90100-c. [DOI] [PubMed] [Google Scholar]
- 25.Okada CY, Wong CP, Denney DW, Levy R. TCR vaccines for active immunotherapy of T cell malignancies. J Immunol. 1997;159:5516–5527. [PubMed] [Google Scholar]
- 26.Kaplan EL, Meier P. Non-parametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 27.McLaughlin P, Grillo-Lopez AJ, Link BK, Levy R, Czuczman MS, Williams ME, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol. 1998;16:2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez-Stawinski GV, Yu PB, Love SD, Parker W, Davis RD., Jr Hapten-induced primary and memory humoral responses are inhibited by the infusion of anti-CD20 monoclonal antibody (IDEC-C2B8, Rituximab) Clin Immunol. 2001;98:175–179. doi: 10.1006/clim.2000.4980. [DOI] [PubMed] [Google Scholar]
- 29.van der Kolk LE, Baars JW, Prins MH, van Oers MH. Rituximab treatment results in impaired secondary humoral immune responsiveness. Blood. 2002;100:2257–2259. [PubMed] [Google Scholar]
- 30.Neelapu SS, Kwak LW, Kobrin CB, Reynolds CW, Janik JE, Dunleavy K, et al. Vaccine-induced tumor-specific immunity despite severe B-cell depletion in mantle cell lymphoma. Nat Med. 2005;11:986–991. doi: 10.1038/nm1290. [DOI] [PubMed] [Google Scholar]
- 31.Bertinetti C, Zirlik K, Heining-Mikesch K, Ihorst G, Dierbach H, Waller CF, Veelken H. Phase I trial of a novel intradermal idiotype vaccine in patients with advanced B-cell lymphoma: specific immune responses despite profound immunosuppression. Cancer Res. 2006;66:4496–4502. doi: 10.1158/0008-5472.CAN-05-4233. [DOI] [PubMed] [Google Scholar]
- 32.Zhu D, McCarthy H, Ottensmeier CH, Johnson P, Hamblin TJ, Stevenson FK. Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood. 2002;99:2562–2568. doi: 10.1182/blood.v99.7.2562. [DOI] [PubMed] [Google Scholar]
- 33.Weng WK, Czerwinski D, Timmerman J, Hsu FJ, Levy R. Clinical outcome of lymphoma patients after idiotype vaccination is correlated with humoral immune response and immunoglobulin G Fc receptor genotype. J Clin Oncol. 2004;22:4717–4724. doi: 10.1200/JCO.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Neelapu SS, Baskar S, Gause BL, Kobrin CB, Watson TM, Frye AR, et al. Human autologous tumor-specific T-cell responses induced by liposomal delivery of a lymphoma antigen. Clin Cancer Res. 2004;10:8309–8317. doi: 10.1158/1078-0432.CCR-04-1071. [DOI] [PubMed] [Google Scholar]
- 35.Malyguine A, Strobl SL, Shafer-Weaver KA, Ulderich T, Troke A, Baseler M, et al. A modified human ELISPOT assay to detect specific responses to primary tumor cell targets. J Transl Med. 2004;2:9. doi: 10.1186/1479-5876-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maecker HT, Auffermann-Gretzinger S, Nomura LE, Liso A, Czerwinski DK, Levy R. Detection of CD4 T-cell responses to a tumor vaccine by cytokine flow cytometry. Clin Cancer Res. 2001;7:902s–908s. [PubMed] [Google Scholar]
- 37.Leonard JP, Coleman M, Kostakoglu L, Chadburn A, Cesarman E, Furman RR, et al. Abbreviated chemotherapy with fludarabine followed by tositumomab and iodine I 131 tositumomab for untreated follicular lymphoma. J Clin Oncol. 2005;23:5696–5704. doi: 10.1200/JCO.2005.14.803. [DOI] [PubMed] [Google Scholar]
- 38.Arcaini L, Colombo N, Passamonti F, Burcheri S, Orlandi E, Brusamolino E, et al. Correlation of the FLIPI score for follicular lymphoma with period of diagnosis and type of treatment. Leuk Res. 2006;30:277–282. doi: 10.1016/j.leukres.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 39.Buske C, Hoster E, Dreyling M, Hasford J, Unterhalt M, Hiddemann W. The Follicular Lymphoma International Prognostic Index (FLIPI) separates high-risk from intermediate- or low-risk patients with advanced-stage follicular lymphoma treated front-line with rituximab and the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) with respect to treatment outcome. Blood. 2006;108:1504–1508. doi: 10.1182/blood-2006-01-013367. [DOI] [PubMed] [Google Scholar]
- 40.Koc O, Redfern C, Wiernik PH, Rosenfelt F, Winter J, Guthrie TH, et al. Id/KLH vaccine (FavId TM) following treatment with rituximab: An analysis of response rate improvement (RRI) and time-to-progression (TTP) in follicular lymphoma (FL) Blood. 2004;104:170a. abstract #587. [Google Scholar]
- 41.Betting DJ, Kafi K, Abdollahi-Fard A, Hurvitz SA, Timmerman JM. Sulfhydryl-based tumor antigen-carrier protein conjugates stimulate superior anti-tumor immunity against B cell lymphomas. J Immunol. 2008;181:4131–4140. doi: 10.4049/jimmunol.181.6.4131. [DOI] [PubMed] [Google Scholar]
- 42.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hurvitz SA, Timmerman JM. Current status of therapeutic vaccines for non-Hodgkin's lymphoma. Curr Opin Oncol. 2005;17:432–440. doi: 10.1097/01.cco.0000174040.52427.83. [DOI] [PubMed] [Google Scholar]