

Abstract
Vascular access dysfunction contributes to patient morbidity during maintenance hemodialysis. In this study we determined if knockout of heme oxygenase-1 predisposed to malfunction of arteriovenous fistulas. After three weeks, all fistulas in wild type mice were patent whereas a third of the fistulas in knockout mice were occluded and these exhibited increased neointimal hyperplasia and venous wall thickening. Heme oxygenase-1 mRNA and protein were robustly induced in the fistulas of the wild type mice. In the knockout mice there was increased PAI-1 and MCP-1 expression, marked induction of MMP-2 and MMP-9, but similar expression of PDGFα, IGF-1, TGF-β1, VEGF, and osteopontin compared to wild type mice. We conclude that heme oxygenase-1 deficiency promotes vasculopathic gene expression, accelerates neointimal hyperplasia and impairs the function of arteriovenous fistulas.
Keywords: arteriovenous access, arteriovenous fistula, arteriovenous graft, chronic dialysis, heme oxygenase
Vascular access dysfunction represents the single most important determinant of morbidity and mortality in patients on maintenance hemodialysis.1–11 Of the available vascular accesses, the arteriovenous fistula (AVF) is the favored modality, and yet these accesses commonly fail to subserve the desired function:1–11 AVFs may never attain maturation and thereby never fully acquire the capacity to accommodate and sustain the heightened rate of blood flow required for effective dialysis; additionally, after variable periods of adequate function, blood flow may be increasingly impeded as the patency of the AVF is lost due to venous stenosis and thrombosis. Venous stenosis of the AVF results from neointimal hyperplasia, the latter representing an expansive lesion consisting of proliferating and migrating cells, and matrix accumulation. Neointimal hyperplasia is not only instrumental in the late failure of the AVF, but such lesions, when they occur at juxta-anastomotic sites in the AVF, commonly account for maturational failure of AVFs. The failure of hemodialysis arteriovenous grafts is also largely accounted for by neointimal hyperplasia.12 Understanding the pathogenesis of neointimal hyperplasia is thus a fundamental issue in elucidating the basis for vascular access dysfunction.1–12
This study was prompted by two seminal studies published in this journal in 2006.13,14 The first study, that by Lin et al.,13 demonstrated that impaired patency and functional failure of AVFs were more likely to occur in patients with HO-1 (heme oxygenase-1) gene polymorphisms characterized by relatively long GT repeats; these novel findings suggest that expression of HO-1 may influence the viability of AVFs. HO-1, the inducible HO isoform, can confer protective effects against diverse insults, including those affecting the kidney and vasculature, and involving actions that may be anti-inflammatory, antioxidant, or antiproliferative in nature.15–21
The second study upon which the present observations are based is that by Castier et al.,14 which introduced and characterized a novel murine AVF model; this model recapitulates the salient features of dysfunctional AVFs in humans, in particular, prominent neointimal hyperplasia. Using this model in HO-1−/− mice, this study examined the functional significance of HO-1 as a determinant of the functionality and form of an AVF.
RESULTS
After 3 weeks, the AVFs in all 16 HO-1 +/+ mice were fully patent, whereas no flow occurred in 5 of the 15 AVFs in HO-1−/− mice; patency rates for the AVF were thus reduced from 100% in HO-1+/+ mice to 67% in HO-1−/− mice (P<0. 05, Fischer’s exact test). This impaired patency was attended by more exuberant venous neointimal hyperplasia (Figures 1 and 2). Morphometric studies of the AVF in HO-1−/− mice demonstrated increased thickness of the venous wall and decreased luminal area/venous wall cross-sectional area (Figure 2).
Figure 1. Representative histological sections of the vein of the AVF in HO-1+/+ and HO-1−/− mice at the level of the anastomosis examined 3 weeks after creation of the AVF.
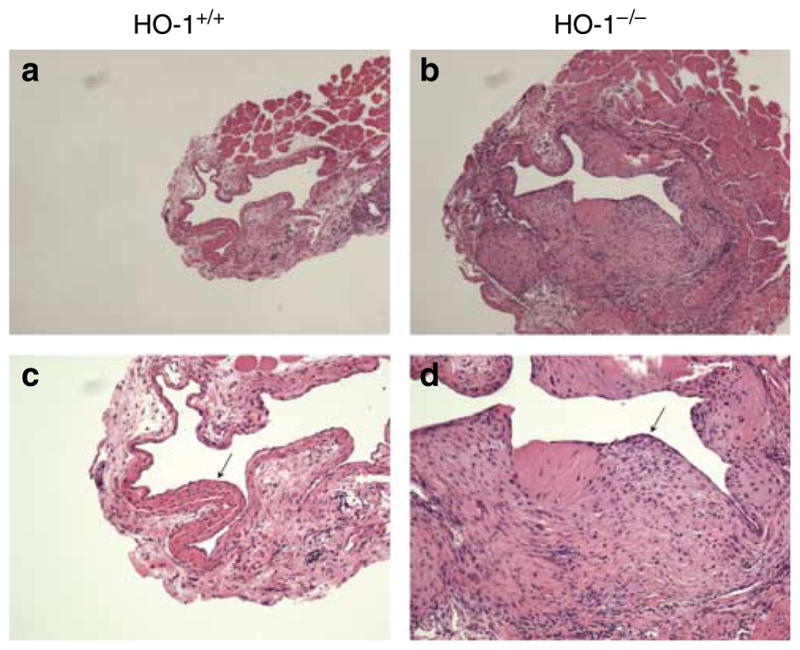
(a) Low-power view of the vein of the AVF in HO-1+/+ mice (original magnification ×50). (b) Low-power view of the vein of the AVF in HO-1−/− mice (original magnification ×50). (c) High-power view of the vein of the AVF in HO-1+/+ mice (original magnification ×100). (d) High-power view of the vein of the AVF in HO-1−/− mice (original magnification ×100). Venous neointimal hyperplasia (marked by arrows) in the AVF was markedly increased in HO-1−/− mice. Foci of calcification were observed in the AVF in HO-1+/+ and HO-1−/− mice, and were more prominent in the latter group (data not shown). The histological appearance of the contralateral, intact (control) jugular vein in HO-1+/+ and HO-1−/− mice was normal (data not shown). All sections are stained by hematoxylin and eosin.
Figure 2. Morphometric analyses of the vein in the AVF in HO-1+/+ and HO-1−/− mice undertaken 3 weeks after creation of the AVF.

(a) Venous wall thickness. In each jugular vein, thickness of the venous wall was measured at 12 sagittal sections evenly spaced around the circumference of the venous wall of the AVF at the level of the anastomosis, and the mean value for each vein was determined; n = 8 and n = 9 for the vein in the AVF in HO-1 +/+ and HO-1−/− mice, respectively; n = 4 for the control vein in HO-1 +/+ and HO-1−/− mice. (b) Luminal area/venous wall cross-sectional area. In sections of the vein at the level of the anastomosis, the circumferential profiles of the lumen and the external elastic lamina of the venous wall were delineated, the areas encompassed by these boundaries were determined, and the ratio of the luminal area to the venous wall cross-sectional area was calculated; n = 8 and n = 9 for the vein in the AVF in HO-1 +/+ and HO-1−/− mice, respectively; n = 4 for the control vein in HO-1 +/+ and HO-1−/− mice. *P<0.05, HO-1−/− mice versus HO-1 +/+ mice for that index for the vein in the AVF; †P<0.05, HO-1−/− mice versus HO-1 +/+ mice for that index for the control vein. The control was provided by the contralateral, intact, jugular vein.
Expression of genes relevant to vascular injury and vascular access dysfunction was assessed 1 week after anastomosis. HO-1 was markedly induced in the AVF in HO-1+/+ mice, whereas no expression of HO-1 was observed in HO-1−/− mice (Figure 3). Expression of the proinflammatory, procoagulant gene, plasminogen activator inhibitor-1 (PAI-1), was markedly increased in the AVF in HO-1−/− mice as compared with the AVF in HO-1+/+ mice; expression of MCP-1 mRNA was increased in HO-1−/− mice as compared with HO-1+/+ mice both in the vein of the AVF and in the contralateral (intact) vein. Platelet-derived growth factor-α (PDGFα) was increased to a comparable degree in the AVF in HO-1 +/+ and HO-1−/− mice. The pattern observed for PDGFα was broadly seen for a number of growth factors/cytokines, including transforming growth factor-β1, insulin-like growth factor-1 (IGF-1), and osteo-pontin; vascular endothelial growth factor mRNA expression in the AVF in HO-1 +/+ and HO-1−/− mice was also comparable, and not increased above the contralateral side (data not shown). Strong HO-1 protein expression in the AVF in HO-1 +/+ mice was exhibited predominantly by cells with phenotypic characteristics of smooth-muscle cells (Figure 3e and f).
Figure 3. mRNA expression of HO-1 and other genes in the vein in the AVF in HO-1 +/+ and HO-1−/− mice, and immunohistochemical localization of HO-1 protein in the AVF in HO-1 +/+ mice, undertaken 1 week after creation of the AVF.

mRNA expression for HO-1 (a), PAI-1 (b), MCP-1 (c), and PDGFα (d) was determined by quantitative real-time reverse transcriptase-polymerase chain reaction. For each group n = 6–10. *P<0.05, HO-1−/− mice versus HO-1 +/+mice for the AVF. †P<0.05, HO-1−/− mice versus HO-1 +/+ mice for the control vein. The control was provided by the contralateral, intact, jugular vein. Immunohistochemical localization of HO-1 protein in the AVF in HO-1 +/+ mice is shown in (e, f) (original magnification ×400, AVFs from 2 HO-1 +/+ mice) and in the contralateral, intact (control) jugular vein in HO-1 +/+ mice in (g) (original magnification ×400). As shown in panels (e) and (f), strong expression of HO-1 in the AVF was exhibited predominantly by cells with phenotypic characteristics of smooth-muscle cells, some of which are labeled by closed arrows; expression of HO-1 was also exhibited by cells, considerably fewer in number in the AVF, and which had phenotypic characteristics of leukocytes, some of which are labeled by open arrows. No such expression of HO-1 was observed in the contralateral, intact (control) jugular vein in HO-1 +/+ mice (g), the latter showing staining for HO-1 only in some of the endothelial cells (g); staining for HO-1 was also observed in some of the endothelial cells in the AVF (e, f). Immunohistochemical localization was performed using a polyclonal antibody (SPA-895; Stressgen, Ann Arbor, MI, USA).
As interest is focused on metalloproteinases in vascular injury and access dysfunction,5,6,8,22–24 we assessed two relevant metalloproteinases. Matrix metalloproteinase-9 (MMP-9) mRNA was dramatically increased in the AVF in HO-1−/− mice as compared to HO-1 +/+ mice, and was attended by increased enzyme activity and protein expression (Figure 4). Increased activity and protein expression were also observed for MMP-2 (data not shown).
Figure 4. Assessment of MMP-9 in the vein in the AVF in HO-1+/+ and HO-1−/− mice undertaken 1 week after creation of the AVF.

(a) Studies of MMP-9 mRNA by quantitative real-time reverse transcriptase- polymerase chain reaction. For each group, n = 6. *P<0.05, HO-1−/− mice versus HO-1 +/+ mice for the AVF. (b) Assessment of pro-MMP-9 enzyme activity (by zymography) and protein expression (by western analysis). Each lane represents protein extract pooled from venous homogenates prepared from two mice for that condition. The numbers below each lane represent the individual densitometric reading, and the means of the three values for each experimental group are provided below this. Densitometry for proMMP-9 was performed with a GS-800 calibrated densitometer (Bio Rad, Hercules, CA, USA).
DISCUSSION
That certain polymorphisms in the HO-1 gene increase the risk for dysfunction of human AVFs suggests that HO-1 expression may influence the viability of human AVFs.13 However, as analyses of these HO-1 gene polymorphisms were not accompanied by assessment of HO-1 expression or HO activity,13 it is uncertain whether decreased or increased HO-1 expression/HO activity confers an increased risk for AVF dysfunction observed with these polymorphisms. In other diseases, lesser inducibility of HO-1 may be associated with HO-1 polymorphisms characterized by long GT repeats,25–27 thus raising the possibility that diminished HO-1 expression/HO activity may contribute to the increased risk for AVF dysfunction as observed by Lin et al. To the best of our knowledge, our findings are the first to demonstrate that the failure to express HO-1 adversely affects an AVF in a murine model.
Accentuated neointimal hyperplasia occurred in the AVF of HO-1−/− mice, and in one-third of these, blood flow ceased; characterizing the cellular composition of the AVF in HO-1−/− mice is the focus of future studies. Such accentuated neointimal hyperplasia may reflect enhanced expression of PAI-1 and MCP-1, both of which are inhibitable by HO-1 and its products;28–31 additionally, the heightened PAI-1 expression in the AVF of HO-1−/− mice may exert thrombotic effects, the latter likely contributing to AVF failure in these mice. The AVF in HO-1+/+ and HO-1−/− mice exhibited comparable mRNA expression for assorted cytokines – PDGFα, IGF-1, transforming growth factor-β1, osteopontin – incriminated in neointimal hyperplasia; thus, if the adverse effects of an HO-1−/− state involve these cytokines, such effects would be ‘distal’ to cytokine gene expression.
A notable finding was obtained with MMP-9, a basement membrane-disrupting molecule of interest in access dysfunction. Although required in physiological processes, MMP-9, when induced aberrantly or inordinately, can injure tissues, and is incriminated in the pathogenesis of autoimmune diseases, access dysfunction, and atherosclerosis.5,6,8,22–24 By disrupting basement membranes, MMP-9 facilitates proliferation and migration of smooth-muscle cells, and the influx of inflammatory cells, processes fundamental to the pathogenesis of neointimal hyperplasia and atherosclerosis, and destabilization of atherosclerotic plaques; MMP-9 may promote outward as well as inward remodeling.5,6,8,22–24 A nuclear factor-κB-dependent gene, MMP-9 is inducible by oxidants and proinflammatory cytokines, and in prior studies, MMP-9 was induced by increased production of peroxynitrite. We speculate that the exaggerated induction of MMP-9 in HO-1−/− mice adversely affects the AVF in this mutant strain, and such induction reflects the prooxidant effects, proinflammatory effects, or increased nuclear factor-κB activation recognized in an HO-1 deficiency state.
That HO-1 deficiency adversely affects the function of an AVF raises the possibility that overexpression of HO-1 or the provision of its products may safeguard the functionality of an AVF. Easily accessible because of its superficial position, the AVF can be readily targeted by locally applied therapies, the efficacy of such therapies founded on HO-1 and its products would thus be of interest.
MATERIALS AND METHODS
All studies were approved by the IACUC of the Mayo Clinic and performed in accordance with NIH guidelines. An AVF was constructed, as previously described, by an end-to-side anastomosis between the right common carotid artery and jugular vein in mice weighing 25–35 g.14,22 The HO-1 +/+ and HO-1−/− murine colony was maintained as previously described, and mice were age-matched (in the range of 18 to 53 weeks) and sex-matched in all studies.30,32 mRNA gene expression was assessed by quantitative real-time reverse transcriptase-polymerase chain reaction in the AVF at 1 week, whereas at 3 weeks, histological and morphometric studies were evaluated in perfusion-fixed AVFs.30,32,33 Gel zymography and western analysis were performed on venous protein extract fractionated by electrophoresis; immunoblotting was performed with polyclonal anti-MMP-9 and anti-MMP-2 antibodies (R&D Systems, Minneapolis, MN, USA) overnight at 4 °C.34,35
STATISTICS
Data are expressed as mean±s.e. Comparisons between HO-1−/− and HO-1 +/+ mice for that condition were performed with the Student’s t-test and the Mann-Whitney test for parametric and nonparametric data respectively. Results are considered significant for P<0.05.
Acknowledgments
Source of support: DK-70124 (KAN, ZSK, JPG).
Footnotes
DISCLOSURE
None.
References
- 1.Hakim R, Himmelfarb J. Hemodialysis access failure: a call to action. Kidney Int. 1998;54:1029–1040. doi: 10.1046/j.1523-1755.1998.00122.x. [DOI] [PubMed] [Google Scholar]
- 2.Schwab SJ. Improving access patency: pre-end-stage renal disease strategies. J Am Soc Nephrol. 1998;9:S124–S129. [PubMed] [Google Scholar]
- 3.Roy-Chaudhury P, Kelly BS, Zhang J, et al. Hemodialysis vascular access dysfunction: from pathophysiology to novel therapies. Blood Purif. 2003;21:99–110. doi: 10.1159/000067863. [DOI] [PubMed] [Google Scholar]
- 4.Konner K, Nonnast-Daniel B, Ritz E. The arteriovenous fistula. J Am Soc Nephrol. 2003;14:1669–1680. doi: 10.1097/01.asn.0000069219.88168.39. [DOI] [PubMed] [Google Scholar]
- 5.Roy-Chaudhury P, Kelly BS, Melhem M, et al. Vascular access in hemodialysis: issues, management, and emerging concepts. Cardiol Clin. 2005;23:249–273. doi: 10.1016/j.ccl.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Dixon BS. Why don’t fistulas mature? Kidney Int. 2006;70:1413–1422. doi: 10.1038/sj.ki.5001747. [DOI] [PubMed] [Google Scholar]
- 7.Dixon BS. Weighing in on fistula failure. Kidney Int. 2007;71:12–14. doi: 10.1038/sj.ki.5002021. [DOI] [PubMed] [Google Scholar]
- 8.Roy-Chaudhury P, Spergel LM, Besarab A, et al. Biology of arteriovenous fistula failure. J Nephrol. 2007;20:150–163. [PubMed] [Google Scholar]
- 9.Kanwar YS. Functional duality of progenitor cells influxing into arteriovenous fistula during its neoangiogenesis. Am J Physiol Renal Physiol. 2007;293:F468–F469. doi: 10.1152/ajprenal.00237.2007. [DOI] [PubMed] [Google Scholar]
- 10.Ravani P, Spergel LM, Asif A, et al. Clinical epidemiology of arteriovenous fistula in 2007. J Nephrol. 2007;20:141–149. [PubMed] [Google Scholar]
- 11.Schwab SJ. Hemodialysis vascular access: the Achilles’ heel remains. Kidney Int. 2007;72:665–666. doi: 10.1038/sj.ki.5002470. [DOI] [PubMed] [Google Scholar]
- 12.Roy-Chaudhury P, Kelly BS, Miller MA, et al. Venous neointimal hyperplasia in polytetrafluoroethylene dialysis grafts. Kidney Int. 2001;59:2325–2334. doi: 10.1046/j.1523-1755.2001.00750.x. [DOI] [PubMed] [Google Scholar]
- 13.Lin CC, Yang WC, Lin SJ, et al. Length polymorphism in heme oxygenase-1 is associated with arteriovenous fistula patency in hemodialysis patients. Kidney Int. 2006;69:165–172. doi: 10.1038/sj.ki.5000019. [DOI] [PubMed] [Google Scholar]
- 14.Castier Y, Lehoux S, Hu Y, et al. Characterization of neointima lesions associated with arteriovenous fistulas in a mouse model. Kidney Int. 2006;70:315–320. doi: 10.1038/sj.ki.5001569. [DOI] [PubMed] [Google Scholar]
- 15.Nath KA, Balla G, Vercellotti GM, et al. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest. 1992;90:267–270. doi: 10.1172/JCI115847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase-1 GeneAblation and expression. J Am Soc Nephrol. 2000;11:965–973. doi: 10.1681/ASN.V115965. [DOI] [PubMed] [Google Scholar]
- 17.Abraham NG, Kappas A. Heme oxygenase and the cardiovascular-renal system. Free Radic Biol Med. 2005;39:1–25. doi: 10.1016/j.freeradbiomed.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 18.Chen S, Kapturczak MH, Wasserfall C, et al. Interleukin 10 attenuates neointimal proliferation and inflammation in aortic allografts by a heme oxygenase-dependent pathway. Proc Natl Acad Sci USA. 2005;102:7251–7256. doi: 10.1073/pnas.0502407102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70:432–443. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 20.Hill-Kapturczak N, Agarwal A. Haem oxygenase-1: a culprit in vascular and renal damage? Nephrol Dial Transplant. 2007;22:1495–1499. doi: 10.1093/ndt/gfm093. [DOI] [PubMed] [Google Scholar]
- 21.Orozco LD, Kapturczak MH, Barajas B, et al. Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ Res. 2007;100:1703–1711. doi: 10.1161/CIRCRESAHA.107.151720. [DOI] [PubMed] [Google Scholar]
- 22.Castier Y, Brandes RP, Leseche G, et al. p47phox-dependent NADPH oxidase regulates flow-induced vascular remodeling. Circ Res. 2005;97:533–540. doi: 10.1161/01.RES.0000181759.63239.21. [DOI] [PubMed] [Google Scholar]
- 23.Ram M, Sherer Y, Shoenfeld Y. Matrix metalloproteinase-9 and autoimmune diseases. J Clin Immunol. 2006;26:299–307. doi: 10.1007/s10875-006-9022-6. [DOI] [PubMed] [Google Scholar]
- 24.Newby AC. Do metalloproteinases destabilize vulnerable atherosclerotic plaques? Curr Opin Lipidol. 2006;17:556–561. doi: 10.1097/01.mol.0000245262.48258.b4. [DOI] [PubMed] [Google Scholar]
- 25.Chen YH, Lin SJ, Lin MW, et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Hum Genet. 2002;111:1–8. doi: 10.1007/s00439-002-0769-4. [DOI] [PubMed] [Google Scholar]
- 26.Exner M, Minar E, Wagner O, et al. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic Biol Med. 2004;37:1097–1104. doi: 10.1016/j.freeradbiomed.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 27.Brydun A, Watari Y, Yamamoto Y, et al. Reduced expression of heme oxygenase-1 in patients with coronary atherosclerosis. Hypertens Res. 2007;30:341–348. doi: 10.1291/hypres.30.341. [DOI] [PubMed] [Google Scholar]
- 28.Fujita T, Toda K, Karimova A, et al. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med. 2001;7:598–604. doi: 10.1038/87929. [DOI] [PubMed] [Google Scholar]
- 29.Nath KA, Vercellotti GM, Grande JP, et al. Heme protein-induced chronic renal inflammation: suppressive effect of induced heme oxygenase-1. Kidney Int. 2001;59:106–117. doi: 10.1046/j.1523-1755.2001.00471.x. [DOI] [PubMed] [Google Scholar]
- 30.Pittock ST, Norby SM, Grande JP, et al. MCP-1 is upregulated in unstressed and stressed HO-1 knockout mice: pathophysiologic correlates. Kidney Int. 2005;68:611–622. doi: 10.1111/j.1523-1755.2005.00439.x. [DOI] [PubMed] [Google Scholar]
- 31.Murali NS, Ackerman AW, Croatt AJ, et al. Renal upregulation of HO-1 reduces albumin-driven MCP-1 production: implications for chronic kidney disease. Am J Physiol Renal Physiol. 2007;292:F837–F844. doi: 10.1152/ajprenal.00254.2006. [DOI] [PubMed] [Google Scholar]
- 32.Tracz MJ, Juncos JP, Grande JP, et al. Renal hemodynamic, inflammatory, and apoptotic responses to lipopolysaccharide in HO-1−/− mice. Am J Pathol. 2007;170:1820–1830. doi: 10.2353/ajpath.2007.061093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diaz Encarnacion MM, Griffin MD, Slezak JM, et al. Correlation of quantitative digital image analysis with the glomerular filtration rate in chronic allograft nephropathy. Am J Transplant. 2004;4:248–256. doi: 10.1046/j.1600-6143.2003.00311.x. [DOI] [PubMed] [Google Scholar]
- 34.Zhao H, Ito A, Sakai N, et al. RECS1 is a negative regulator of matrix metalloproteinase-9 production and aged RECS1 knockout mice are prone to aortic dilation. Circ J. 2006;70:615–624. doi: 10.1253/circj.70.615. [DOI] [PubMed] [Google Scholar]
- 35.Gerlach RF, Demacq C, Jung K, et al. Rapid separation of serum does not avoid artificially higher matrix metalloproteinase (MMP)-9 levels in serum versus plasma. Clin Biochem. 2007;40:119–123. doi: 10.1016/j.clinbiochem.2006.10.007. [DOI] [PubMed] [Google Scholar]