

Abstract
The receptor for advanced-glycation-end-products (RAGE) has been implicated as a pro-inflammatory factor in chronic inflammatory conditions such as diabetes mellitus and rheumatoid arthritis. The aim of this study was to investigate the inhibitory effect of the soluble-RAGE (sRAGE), the extracellular domain of RAGE, on RAGE expression and NF-κB translocation in human-salivary gland-cell-lines (HSG). Cells were stimulated with agonist S100A4, fusion protein of RAGE encompassing the extracellular domain of RAGE (ex-RAGE), ex-RAGE followed by S100A4, or S100A4 followed by ex-RAGE. Our study indicates that RAGE expression was highest at 150 μg/μl of S100A4 and efficiently down-regulated by 1.8-fold (P < 0.05) when ex-RAGE was incubated prior to agonist S100A4. RAGE protein was also consistently down-regulated by 20–40% with pre-incubation of ex-RAGE. More importantly, nuclear translocation of p65 and p52 of NF-κB by S100A4 was inhibited in the presence of ex-RAGE, confirming anti-inflammatory function of ex-RAGE. In conclusion, ex-RAGE down-regulates RAGE expression and inhibits p65 and p52 activation in HSG, providing evidence that ex-RAGE functions as a “decoy” to RAGE–ligand interaction and thus potentially dampening inflammatory conditions.
A unique receptor of the immunoglobulin superfamily of cell surface molecules known as the receptor for advanced glycation end products (RAGE) has been implicated in chronic inflammatory conditions such as Sjögren's syndrome (SjS), diabetes, and rheumatoid arthritis (Neeper et al., 1992; Katz et al., 2004; Stewart et al., 2008). RAGE is expressed on a variety of cell types such as endothelial and smooth muscle cells and it binds multiple families of ligands, namely advanced glycation end products (AGEs), the S100/calgranulin family, and high-mobility group box 1 (HMGB1; amphoterin) (Schmidt et al., 2001). Unlike other surface receptors, RAGE is unique in that it recognizes three-dimensional structures such as beta-sheets and fibrils rather than specific amino acid structures (Stern et al., 2002).
While a number of different RAGE ligands have been identified, the S100 protein family, in particular, has been implicated in the regulation of inflammatory RAGE signaling (Hofmann et al., 1999). S100 proteins are characterized by their two calcium binding sites of the helix–loop–helix conformation, and most are homodimers linked by a connecting peptide. S100A4 (Mts-1), an 11-kDa member of the S100 family, acts as a potent cytokine via RAGE, stimulating expression of RAGE itself while generating oxidative stress, synthesis and secretion of proinflammatory cytokines, and chemotaxis (Bernhard Moser, 2006).
RAGE–ligand interaction induces sustained activation of the proinflammatory transcription factor NF-κB (Bierhaus et al., 2001), although it is still not clear which subunits of NF-κB are mainly involved in this process. NF-κB resides in the cytoplasm in its inactive form bound to the inhibitor IκBα until activation, upon which IκBα is degraded and NF-κB translocates into the nucleus. Activation of NF-κB results in the transcription of a number of target genes including cytokines, adhesion molecules, and RAGE itself. One unique feature of RAGE-induced NF-κB activation is the positive feedback loop by which the RAGE signal is maintained and amplified. Due to its ability to sustain cellular activation, RAGE can function as a master switch, converting short-lasting pro-inflammatory responses into long-lasting cellular dysfunction (Bierhaus et al., 2001).
One way in which the body regulates RAGE is through a naturally occurring inhibitor known as soluble RAGE (sRAGE), a secreted isoform of RAGE. Blockade of RAGE–ligand interaction using sRAGE, a truncated form of the receptor comprising the extracellular domain of RAGE, functions as an inhibitor and prevents S100 or other ligand binding to the receptor. This interaction has been demonstrated to reduce chronic cellular activation and the outcome of disease in experimental animal models (Hudson et al., 2003). In humans, sRAGE results from alternative splicing of RAGE mRNA. Katz et al. (2004) demonstrated increased expression of RAGE in the minor salivary glands of SjS and, more recently, Stewart et al. (2008) demonstrated that SjS patients exhibit lower sRAGE levels in sera compared to normal patients, suggesting that SjS patients may lack the inhibitory effect of sRAGE on the RAGE-mediated inflammatory process, thus sustaining chronic inflammatory conditions in SjS.
The aim of our study was to investigate the inhibitory effects of sRAGE on RAGE expression in the human salivary gland cell line (HSG) by utilizing the fusion protein of RAGE encompassing the extracellular domain of RAGE (referred as ex-RAGE herein). The HSG cell line was established from an irradiated human submandibular gland and has been used as an in vitro model for study of the influence of pharmacologic agents on salivary glands, as well as a model system for irradiation-induced damage in salivary glands (Katz et al., 1994, 1995, 1999, 2008; Wu et al., 1994, 1996; Nagler, 1998; Shalita-Chesner et al., 2001; Bulosan et al., 2009). Furthermore, NF-κB, the downstream transcription factor of RAGE, in the presence or absence of ex-RAGE in the HSG cells was investigated to identify NF-κB subunits that regulated by ex-RAGE. Our study provides further molecular insights into ex-RAGE regulation on inflammatory RAGE and NF-κB in HSG cells and will aid us to develop a strategy to dampen the chronic inflammatory process by targeting RAGE–ligand interactions.
Materials and Methods
HSG cell culture
The HSG cells were a kind gift from Dr. Joseph Katz at the University of Florida. Cells were maintained in DMEM supplemented with 10% fetal calf serum, penicillin(100 U/ml), and streptomycin (100 μg/ml) (Life Technologies, Burlington, Ont., Canada). Cells were plated at medium density (2.5 × 104 cells/cm2) in a flask and kept at 37°C with 5.0% CO2 until a confluence of 70–80% was achieved in 4–5 days. Cells were harvested with 0.5% trypsin/0.53 mM EDTA, washed and resuspended in new media.
S100A4 and sRAGE treatments
HSG cells were seeded in 6-well plates (1 × 106 cells per well) and treated with various concentrations (50, 100, 150, 200 ng/ml) of S100A4 for 24 or 48 h. After establishing the optimal concentration for S100A4 treatment (150 ng/ml), HSG cells were treated at varying time points (6, 12, 18, 24, 30 h). The inhibition of S100A4-induced RAGE expression was investigated using a similar 6-well assay plate set-up. The cells were stimulated for 24 h under the following conditions: untreated, S100A4, ex-RAGE, ex-RAGE followed by S100A4, and S100A4 followed by ex-RAGE treatment, with a 30-min interval between S100A4 and ex-RAGE treatments. Recombinant S100A4 was purchased from Abnova (Taipei City, Taiwan) and recombinant human RAGE/Fc chimera, composed of the extracellular domain of human RAGE (Met 1-Ala 344), homologues to sRAGE, fused to the human Fc via a short peptide linker, was utilized as ex-RAGE (R& D systems, Inc., Minneapolis, MN). Lipopolysaccharide, a positive control for NF-κB activation, was used at an 1 μg/ml concentration (Ultrapure E. coli LPS, Invitrogen, San Diego, CA).
RAGE gene expression by RT-PCR
Total RNA was prepared from HSG cells using the RNeasy Mini Kit (Qiagen, Valencia, CA). cDNA was synthesized using 1 μg of total RNA, Superscript II reverse transcriptase and pd(T) 12–18 oligomeric DNA (Amersham Pharmacia, Piscataway, NJ). Following an initial denaturation at 94°C for 4 min, each PCR was carried out for 34 cycles consisting of 94°C for 30 sec, 57°C for 30 sec, and 72°C for 1 min. PCR products were size separated by electrophoresis using 2% agarose gels for further visualization. PCR band intensities were normalized to β-actin using the Flourchem Imaging densitometer system (Alpha Innotech Corporation, San Leandro, CA). The sequences of each primer set for human genes are: β-actin-forward: 5′-CCTGACCCTAAGGCCAACCG-3′, reverse: 5′-GCTCATAGCTCTTCTCCAGGG -3′ (398 bp), h-RAGE-forward: 5′-GTG CCA GGC AAT GAA CAG GAA T-3′, reverse: 5′-TTC ACA GGT CAG GGT TAC GGT T-3′ (570 bp).
NF-κB protein expression by Western blot
Using the same cell culture set-up as described above, the Protein and RNA Isolation System (PARIS, Ambion Inc, Austin, TX) was used to separate the cytoplasmic and nuclear proteins for each treatment. Each fraction was loaded onto a 4–20% Tris-HCl Bio-Rad ready gel (Bio-Rad Inc., Hercules, CA). The resolved proteins were transferred to PVDF membranes and incubated with 5% non-fat dry milk in TBS with 0.1% Tween-20, 10 mM Tris (pH 7.5) and 100 mM NaCl. The membrane was incubated with five separate primary antibodies corresponding to the NF-κB subunits: p65, p50, p52, c-Rel, RelB (1:100 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at room temperature, followed by goat anti-rabbit IgG secondary antibody incubation for an hour (1:1,000 dilution; Sigma, St. Louis, MO). After washing, the immunoreactive bands were detected using ECL chemiluminescence reagents and X-ray films. ImageJ (NIH) was used to measure the density of the bands.
RAGE protein expression by In-Cell Western (whole cell immunofluoresent staining)
Using the same stimulation protocol as described above, In-Cell Western assays were performed using a In-Cell Western Kit (LI-COR Biosciences, Lincoln, NE). The cells were fixed using 3.7% formaldehyde in 1 × PBS, washed using 1 × PBS containing 0.1% Triton X-100, blocked using 150 μl of Odyssey Blocking Buffer, and incubated with rabbit anti-human RAGE antibody at a dilution of 1: 100 overnight at 4°C The secondary antibody for rabbit IgG, which were labeled with IRDye 680 and IRDye 800CW were subsequently added at a dilution of 1:1,000 in the dark. The cells were imaged using a LI-COR Scanner and analyzed by the Odyssey System (Li-COR Bioscienes).
Detection of p65 NF-κB activation
The spatial translocation of NF-κB from the cell cytoplasm to the nucleus was optimally quantified used a NF-κB Activation HitKit™ (Cellomics, Pittsburgh, PA). In brief, HSG cells were stimulated for 24 h as described previously. A primary antibody for p65 (1:100) and a secondary antibody (1:1,000) conjugated with the fluorophore Alexa Fluor 488 and DNA-specific Hoeschst Dye were used. Microscopic images were taken at 20× and 40× magnifications using a Zeiss Axiovert 200 M microscope and a Zeiss AxioCam MRm camera. Image J (NIH) was also used to measure the green fluorescence intensity of nucleus.
Statistical analyses
Student's t-test was used for statistical analysis and P < 0.05 was considered to be significant.
Results
Regulation of RAGE expression by S100A4
To test the potential inhibitory effect of ex-RAGE on RAGE expression in HSG cells in vitro, we first determined the optimal concentration and incubation time for S100A4 treatment. HSG cells were incubated for 24 or 48 h with varying concentrations of S100A4 (50, 100, 150, 200 ng/ml). Our semiquantitative RT-PCR data indicate that the expression of RAGE was dramatically increased with 150 ng/ml of S100A4 after a 24 h incubation period (Fig. 1A). With a longer incubation time of 48 h, the expression level began to increase at 50 ng/ml of S100A4, and then decreased slightly at 200 ng/ml. Subsequently, HSG cells were treated with S100A4 (150 ng/ml) for 6, 12, 18, 24, and 30 h (Fig. 1B). While cells stimulated for 12 or 24 h showed increased RAGE gene expression (1.9-fold increase, P < 0.05), cells treated for 6, 18, or 30 h showed no change or reduced RAGE expression as compared to non-stimulated cells (Fig. 1B). Based on these data, subsequent experiments were performed using 150 ng/ml of S100A4 treatment for 24 h.
Fig. 1.

Enhanced RAGE expression in HSG by S100A4. A: Using RT-PCR, HSG cells were incubated at two different time points (24 and 48 h) at varying concentrations of S100A4. B: HSG cells were subsequently treated at a single concentration of S100A4 (150 ng/ml) over five different time points to determine the proper incubation time for this study.
Inhibition of S100A4-induced RAGE gene expression by ex-RAGE
To test the inhibitory effect of ex-RAGE on S100A4-induced RAGE gene expression in HSG cells, HSG cells were treated with either S100A4 (150 ng/ml), ex-RAGE (50 ng/ml), ex-RAGE followed by S100A4, or S100A4 followed by ex-RAGE for 24 h. RAGE genes expression was then analyzed using RT-PCR (Fig. 2A). Decreased RAGE expression was observed when cells were stimulated with ex-RAGE followed by S100A4 as compared to cells treated with S100A4 alone (1.85-fold decrease, P < 0.05). The effect of ex-RAGE inhibition on RAGE expression was slightly less when ex-RAGE was added before S100A4 stimulation than when S1004 was added prior to ex-RAGE (1.26-fold difference, not statistically significant).
Fig. 2.
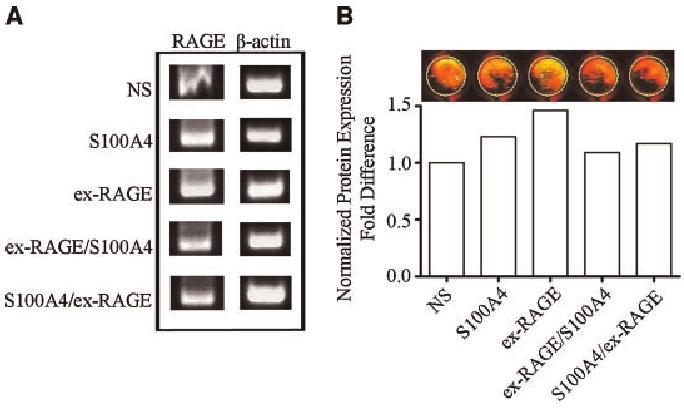
Inhibition of S100A4-induced RAGE gene expression by ex-RAGE. A: The greatest gene expression was demonstrated by RT-PCR for HSG cells treated with ex-RAGE alone (50 ng/ml). A decreased gene expression was observed when ex-RAGE was added prior to or after S100A4 treatments (separated by a 30 min interval). B: Protein expression of RAGE was examined by In-Cell Western analysis using the same experimental design for RT-PCR analysis. Following a rabbit anti-human RAGE primary antibody incubation (1: 100 dilution), the cells were treated with an anti-rabbit IgG secondary antibody (1: 1,000 dilution) conjugated with IRDye800CW. The absorbance values were normalized for analysis.
Inhibition of S100A4-induced RAGE protein expression by ex-RAGE
To determine if down-regulated RAGE gene expression is correlated with RAGE protein expression, HSG cells were treated with S100A4 and ex-RAGE using the same experimental design as the RT-PCR analysis and detected by In-Cell Western (Fig. 2B). The signal intensity was first normalized based on the cell numbers and the normalized value of non-stimulated cells was then converted to 1 for comparisons (Fig. 2B, bar graph). S100A4 and ex-RAGE stimulated cells showed increased RAGE protein expression (25% and 40%, respectively) shown as more yellow image than the ones with ex-RAGE and S100A4 showing red signal. Interestingly, RAGE protein expression was decreased by 20% when ex-RAGE was added prior to S100A4 stimulation compared to S100A4 alone.
Inhibition of NF-κB p65 activation in the presence of ex-RAGE in S100A4-treated HSG cells
To examine if ex-RAGE inhibits the NF-κB pathway in order to suppress RAGE expression in S100A4 stimulated cells, the activation status of NF-κB p65 subunit was examined by immunocytochemistry. HSG cells were treated with S100A4 and ex-RAGE as described above with the addition of a positive control of LPS for NF-κB activation. The cells were then stained for p65 (green) and counterstained with Hoeschst Dye (blue) (Fig. 3A). Quantitative results were obtained by measuring the density of the green fluorescence in the nucleus using ImageJ (Fig. 3B). As indicated in Figure 3B, the highest level of NF-κB activation, which leads to the translocation of subunit p65 into the nucleus, was demonstrated by LPS treated cells and S100A4 treated cells. The lowest level of activation was demonstrated in the ex-RAGE/S100A4 treated cells, suggesting that ex-RAGE does inhibit NF-κB p65 translocation.
Fig. 3.
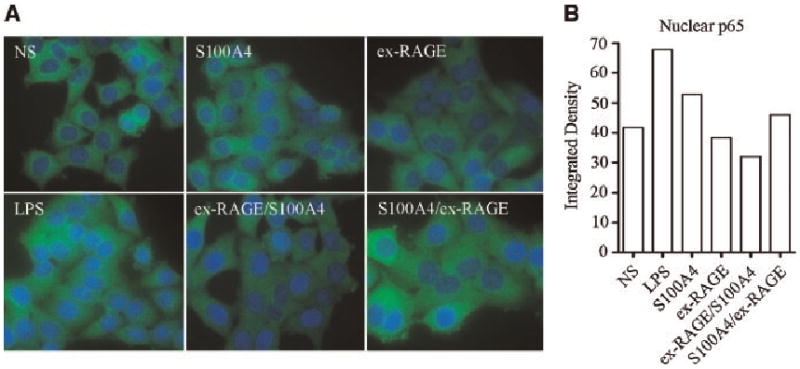
Inhibition of NF-κB p65 translocation by ex-RAGE. A: The activation status of NF-κB p65 subunit was examined by immunocytochemistry. HSG cells were stained for p65 (green) following treatments and counterstained with Hoeschst Dye (blue). B: Quantitative analysis using ImageJ, which measures green fluorescence intensity in the nucleus, indicated an inhibitory response of NF-κB p65 translocation in ex-RAGE/SI00A4 treated HSG cells.
Inhibition of p65 and p52 subunits of NF-κB translocation to the nucleus in the presence of ex-RAGE
In order to identify which NF-κB subunits are involved in S100A4-induced RAGE expression and inhibition of nuclear translocation by ex-RAGE, we examined all five subunits of NF-κB (p65, p52, p50, c-Rel, RelB) by Western blot analysis. HSG cells were treated with S100A4 and ex-RAGE as previously described. As shown in Figure 4, the subunits p65 and p52 both demonstrated single banding patterns at 65 and 52 kDa, respectively, as expected, while p50, c-Rel and RelB showed multiple bands, potentially forming hetero-or homodimers (Figs. 4A). Integrated density generated by Image J revealed that p65, p52 and p50 demonstrated increased cytoplasmic expression with LPS and S100A4 stimulation (Fig. 4B) whereas other subunits were not altered with LPS and S100A4 stimulation (graphs not shown). Nuclear expression of p65, p52 and p50 was also increased with LPS and S100A4, suggesting nuclear translocation. Interestingly, the translocation of these three subunits was decreased by in the presence of ex-RAGE (Fig. 4A,B).
Fig. 4.
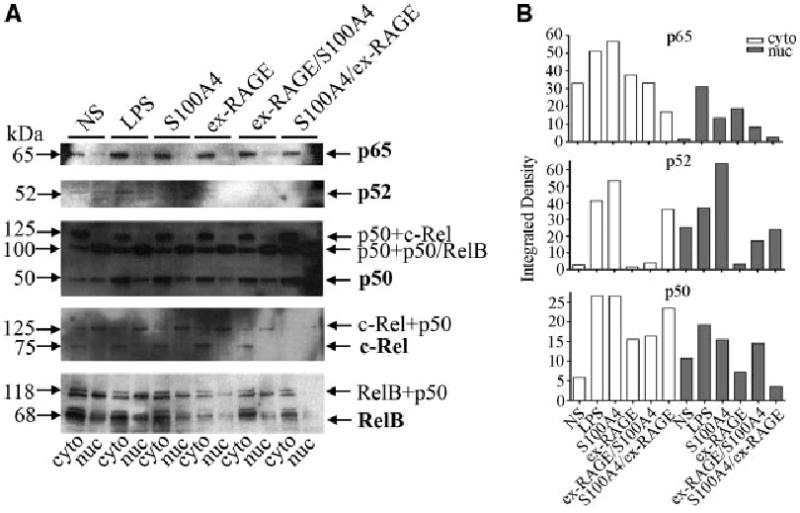
Inhibition of NF-κB subunit translocation by ex-RAGE in S100A4 treated HSG cells. A: Western blot analysis of five subunits (p65, p52, p50, c-Rel, RelB) indicates elevated p65, p52, and p50 subunit expression both in the cytoplasm and nucleus by S100A4 compared to non-stimulated HSG cells. B: Integrated density of NF-κB subunits generated by Image J indicates that increased cytoplasmic expression of p65, p52, and p50 with LPS and S100A4 stimulation, but down-regulated by ex-RAGE incubation.
Discussion
In the present study, we demonstrated that ex-RAGE containing extracelluar domain of RAGE inhibits RAGE gene/protein expression as well as NF-κB activation in the HSG cell line. As a ligand of RAGE, S100A4 showed the ability to upregulate RAGE gene expression at certain concentrations and ex-RAGE functions as a potential inhibitor of the enhanced RAGE gene expression caused by S100A4. Interestingly, p65 translocation was inhibited in the presence of ex-RAGE in S100A4 treated HSG cells, indicating that ex-RAGE may influence the expression or translocation of this subunit of NF-κB. The translocation of NF-κB subunit p52 and p50 were also induced by S100A4 treatment, but inhibited in the presence ex-RAGE.
It is worthwhile to note that S100A4 up-regulates RAGE expression in a dose-dependent manner. In addition, inhibitory effect of ex-RAGE was also tested at different concentrations similar to testing the concentration of S100A4 to determine the ex-RAGE concentration used in our study. Because this inhibitory effect was demonstrated only when optimal concentrations of the ligand S100A4 and the decoy ex-RAGE were applied, circulating endogenous sRAGE levels or ration of RAGE ligands and sRAGE could be critical to block the interaction between RAGE–ligand interaction in vivo. Therefore, it may be logical to assume that reduced sRAGE levels in the sera of SjS patients (Stewart et al., 2008) could implicate persistence or amplification of inflammation in the absence of the proper inhibitory roles of sRAGE in chronic inflammation of SjS.
An inverse relationship between sRAGE and tissue RAGE expression in SjS was supported by the following observations: (a) RAGE belongs to the same immunoglobulin superfamily as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) and serum levels of soluble forms of ICAM-1 and VCAM-1 are elevated in patients with SjS, reflecting upregulation of these adhesion molecules in endothelial cells (Ferri et al., 1998; Rosei et al., 2005), (b) RAGE ligands are positive regulators of cell expression of RAGE, and serum sRAGE levels are positively associated with circulating ligand levels in humans in non-inflammatory conditions (Yamagishi et al., 2006), and (c) sRAGE is downregulated in inflamed salivary glands and in sera of SjS (Katz et al., 2004; Stewart et al., 2008). These studies suggest that circulating endogenous sRAGE expression may negatively reflect salivary gland RAGE expression and reduced level of sRAGE implicates its involvement in chronic inflammatory disease conditions.
While the activation of NF-κB by LPS is well established, the exact subunits involved in S100A4 and ex-RAGE inhibition had yet to be elucidated. To address this issue, we have demonstrated by immunocytochemistry that the p65 subunit of NF-κB was translocated to the nucleus in the presence of S100A4 or LPS and was inhibited in the presence of ex-RAGE, suggesting the involvement of p65 in this pathway. Our further analyses on all five subunits of NF-κB by Western blot analyses indicate that LPS and S100A4 induces p65, p52, and p50 and their translocation into the nucleus. In addition, S100A4 added prior to ex-RAGE incubation leads to decreased expression of those three subunits in the nucleus compared to LPS (positive control) and S100A4 (agonist) alone, underscoring the inhibitory effect of ex-RAGE on RAGE expression and NF-κB subunit translocation to the nucleus.
In conclusion, we identified that extracellular domain of RAGE inhibits RAGE expression and NF-κB translocation induced by S100A4, a RAGE ligand associated with chronic inflammation. As a result, this study provides important evidence for future utilization of recombinant sRAGE for chronic inflammatory diseases such as SjS. A basic understanding of the inflammatory pathway and potential inhibitors to this process in vitro utilizing HSG cells will allow us to apply our knowledge and observations to in vivo study models in the future. Additional endeavors to optimize dose and delivery strategies and development of recombinant sRAGE for in vivo studies certainly need to be further investigated. Analyses of cytokine regulation by sRAGE, for example, would enhance our understanding of the inflammatory responses sustained by RAGE–ligand interactions.
Acknowledgments
We would like to acknowledge Miss. Kyumee Yo and Mr. Huy Huynh for their technical assistance.
Contract grant sponsor: NIH/NIDCR;
Contract grant numbers: DE016509, DE01670, DE007200.
Literature Cited
- Bernhard Moser KCHAMS. Receptor for advanced glycation end products and its igands: Initiators or amplifiers of joint inflammation—A bit of both? Arthritis Rheum. 2006;54:14–18. doi: 10.1002/art.21522. [DOI] [PubMed] [Google Scholar]
- Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Haring HU, Schleicher E, Nawroth PP. Diabetes-associated sustained activation of the transcription factor nuclear factor-{kappa}B. Diabetes. 2001;50:27920–2808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- Bulosan M, Pauley KM, Yo K, Chan EK, Katz J, Peck AB, Cha S. Inflammatory caspases are critical for enhanced cell death in the target tissue of Sjogren's syndrome before disease onset. Immunol Cell Biol. 2009;87:81–90. doi: 10.1038/icb.2008.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri C, Desideri G, Baldoncini R, Bellini C, De Angelis C, Mazzocchi C, Santucci A. Early activation of vascular endothelium in nonobese, nondiabetic essential hypertensive patients with multiple metabolic abnormalities. Diabetes. 1998;47:660–667. doi: 10.2337/diabetes.47.4.660. [DOI] [PubMed] [Google Scholar]
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- Hudson Bl, Bucciarelli LG, Wendt T, Sakaguchi T, Lalla E, Qu W, Lu Y, Lee L, Stern DM, Naka Y, Ramasamy R, Yan SD, Yan SF, D'Agati V, Schmidt AM. Blockade of receptor for advanced glycation endproducts: A new target for therapeutic intervention in diabetic complications and inflammatory disorders. Arch Biochem Biophys. 2003;419:80–88. doi: 10.1016/j.abb.2003.08.030. [DOI] [PubMed] [Google Scholar]
- Katz J, Nagler R, Barak S, Livneh A, Baum B, Atkinson J, Shemer J. Cytokines modulate interleukin-6 production by human salivary gland cell line. Cell Immunol. 1994;159:211–219. doi: 10.1006/cimm.1994.1308. [DOI] [PubMed] [Google Scholar]
- Katz J, Weiss H, Goldman B, Kanety H, Stannard B, LeRoith D, Shemer J. Cytokines and growth factors modulate cell growth and insulin-like growth factor binding protein secretion by the human salivary cell line (HSG) J Cell Physiol. 1995;165:223–227. doi: 10.1002/jcp.1041650202. [DOI] [PubMed] [Google Scholar]
- Katz J, Nasatzky E, Werner H, Le Roith D, Shemer J. Tumor necrosis factor alpha and interferon gamma–induced cell growth arrest is mediated via insulin-like growth factor binding protein-3. Growth Horm IGF Res. 1999;9:174–178. doi: 10.1054/ghir.1999.0101. [DOI] [PubMed] [Google Scholar]
- Katz J, Stavropoulos F, Bhattacharyya I, Stewart C, Perez FM, Caudle RM. Receptor of advanced glycation end product (RAGE) expression in the minor salivary glands of patients with Sjogren's syndrome: A preliminary study. Scand J Rheumatol. 2004;33:174–178. doi: 10.1080/03009740310004775. [DOI] [PubMed] [Google Scholar]
- Katz J, Blake E, Medrano TA, Sun Y, Shiverick KT. Isoflavones and gamma irradiation inhibit cell growth in human salivary gland cells. Cancer Lett. 2008;270:87–94. doi: 10.1016/j.canlet.2008.04.051. [DOI] [PubMed] [Google Scholar]
- Nagler RM. Effects of radiotherapy and chemotherapeutic cytokines on a human salivary cell line. Anticancer Res. 1998;18:309–314. [PubMed] [Google Scholar]
- Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- Rosei EA, Rizzoni D, Muiesan ML, Sleiman I, Salvetti M, Monteduro C, Porteri E. Effects of candesartan cilexetil and enalapril on inflammatory markers of atherosclerosis in hypertensive patients with non-insulin-dependent diabetes mellitus. J Hypertens. 2005;23:435–444. doi: 10.1097/00004872-200502000-00027. [DOI] [PubMed] [Google Scholar]
- Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalita-Chesner M, Katz J, Shemer J, Werner H. Regulation of insulin-like growth factor-I receptor gene expression by tumor necrosis factor-alpha and interferon-gamma. Mol Cell Endocrinol. 2001;176:1–12. doi: 10.1016/s0303-7207(01)00484-1. [DOI] [PubMed] [Google Scholar]
- Stern D, Du Yan S, Fang Yan S, Marie Schmidt A. Receptor for advanced glycation endproducts: A multiligand receptor magnifying cell stress in diverse pathologic settings. Adv Drug Del Rev. 2002;54:1615–1625. doi: 10.1016/s0169-409x(02)00160-6. [DOI] [PubMed] [Google Scholar]
- Stewart C, Cha S, Caudle RM, Berg K, Katz J. Decreased levels of soluble receptor for advanced glycation end products in patients with primary Sjogren's syndrome. Rheumatol Int. 2008;28:771–776. doi: 10.1007/s00296-008-0529-4. [DOI] [PubMed] [Google Scholar]
- Wu AJ, Kurrasch RH, Katz J, Fox PC, Baum BJ, Atkinson JC. Effect of tumor necrosis factor-alpha and interferon-gamma on the growth of a human salivary gland cell line. J Cell Physiol. 1994;161:217–226. doi: 10.1002/jcp.1041610205. [DOI] [PubMed] [Google Scholar]
- Wu AJ, Chen ZJ, Baum BJ, Ambudkar IS. Interferon-gamma induces persistent depletion of internal Ca2+ stores in a human salivary gland cell line. Am J Physiol. 1996;270:C514–C521. doi: 10.1152/ajpcell.1996.270.2.C514. [DOI] [PubMed] [Google Scholar]
- Yamagishi S, Adachi H, Nakamura K, Matsui T, Jinnouchi Y, Takenaka K, Takeuchi M, Enomoto M, Furuki K, Hino A, Shigeto Y, Imaizumi T. Positive association between serum levels of advanced glycation end products and the soluble form of receptor for advanced glycation end products in nondiabetic subjects. Metabolism. 2006;55:1227–1231. doi: 10.1016/j.metabol.2006.05.007. [DOI] [PubMed] [Google Scholar]