

Abstract
Background
Meningococcal epidemics in Africa are generally caused by capsular group A strains, but W-135 or X strains also cause epidemics in this region. Factor H–binding protein (fHbp) is a novel antigen being investigated for use in group B vaccines. Little is known about fHbp in strains from other capsular groups.
Methods
We investigated fHbp in 35 group A, W-135, and X strains from Africa.
Results
The 22 group A isolates, which included each of the sequence types (STs) responsible for epidemics since 1963, and 4 group X and 3 group W-135 isolates from recent epidemics had genes encoding fHbp in antigenic variant group 1. The remaining 6 W-135 isolates had fHbp variant 2. Within each fHbp variant group, there was 92%–100% amino acid identity, and the proteins expressed conserved epitopes recognized by bactericidal monoclonal antibodies. Serum samples obtained from mice vaccinated with native outer membrane vesicle vaccines from mutants engineered to express fHbp variants had broad bactericidal activity against group A, W-135, or X strains.
Conclusions
Despite extensive natural exposure of the African population, fHbp is conserved among African strains. A native outer membrane vesicle vaccine that expresses fHbp variants can potentially elicit protective antibodies against strains from all capsular groups that cause epidemics in the region.
For more than 100 years, devastating epidemics of meningococcal disease have resulted in enormous suffering in sub-Saharan Africa (reviewed in [1]). From 1988 to 1997, >700,000 cases were reported, with >100,000 associated deaths [2]. The public health responses were expensive, only partially effective [3], and deflected scarce resources from efforts to control other diseases. Control of epidemic meningococcal disease in Africa therefore remains an important health priority.
Most meningococcal disease in industrialized countries is caused by strains in capsular groups B, C, or Y, whereas most disease in sub-Saharan Africa is caused by group A strains. A promising group A polysaccharide-protein conjugate vaccine for sub-Saharan Africa is under development by the Meningitis Vaccine Project [4, 5]. However, because strains from other capsular groups, including groups W-135 [6] and X [7, 8], also can cause epidemics in this region, additional vaccine approaches may be needed.
Genome mining [9, 10] and proteomics [11] have identified a large number of novel vaccine targets for the prevention of group B meningococcal disease. One of these targets is factor H–binding protein (fHbp), a surface-exposed lipoprotein that binds human complement factor H [12]. The binding of factor H to the surface of Neisseria meningitidis down-regulated complement activation and enhanced resistance of the organism to bactericidal activity [12–14]. This antigen is part of 2 promising recombinant protein vaccines being developed for the prevention of group B disease [15, 16]. However, it is inaccurate to describe fHbp or other protein antigens, such as NadA [17, 18] or genome-derived neisserial antigen (GNA) 2132 [10, 19], as group B–specific antigens, because they elicit antibodies independent of the capsular polysaccharide group. Therefore, these antigens potentially can protect against disease caused by strains from other capsular groups.
Meningococcal fHbp can be subclassified into different antigenic variant groups on the basis of sequence similarity and antigenic cross-reactivity among fHbp groups [16, 20]. In general, antibodies prepared against fHbp in the variant 1 (v.1) group (also referred to as “subfamily B” [16]) were bactericidal against strains expressing fHbp from the homologous variant or subfamily group, but not against strains expressing fHbp in the variant 2 (v.2) or 3 (v.3) groups (also referred to as “subfamily A” [16]), and vice versa [18, 20]. The purpose of the present study was to investigate antigenic variants of fHbp expressed by epidemic group A, W-135, and X strains from Africa. As proof of concept for the use of an outer membrane vesicle (OMV) vaccine in Africa, we also measured the susceptibility of representative African strains to the bactericidal activity of serum samples obtained from mice vaccinated with prototype native (i.e., non–detergent-treated) OMV vaccines prepared from mutants engineered to express fHbp from different antigenic variant groups [21, 22].
Materials and Methods
Isolate collection
Case isolates recovered from patients in Africa were obtained from the World Health Organization (WHO) Collaborating Centre for Reference and Research on Meningococci, Norwegian Institute of Public Health (Oslo, Norway) (n = 21); the US Centers for Disease Control and Prevention (Atlanta, GA) (n = 15); and J.-M. Alonso, Institute Pasteur (Paris, France) (n = 2). After removal of 3 duplicate isolates, there were 35 unique isolates (table 1). The multilocus sequence typing (MLST) [23] and PorA data were obtained either from the provider of the strain or from the publicly accessible database at the Neisseria and PorA MLST Home Pages [24–25].
Table 1. Characteristics of the meningococcus strains used in the present study.
| Strain | Alternate designation | Capsular group | STa | PorA VRb | fHbpc gene variant (peptide IDd) | Reactivity of anti-fHbp MAbse | Source country | Year obtained | ||
|---|---|---|---|---|---|---|---|---|---|---|
| JAR 1 | JAR 5 | JAR 31 | ||||||||
| CH1A | Z1275f | A | 1 | 5–2,10 | 1 (4) | + | + | − | Niger | 1963 |
| CH4A | Z5010f | A | 1 | 5–2,10 | 1 (4) | + | + | − | Djibouti | 1966 |
| CH5A | Z5005f | A | 1 | 5–2,10 | 1 (ND) | + | + | − | Morocco | 1967 |
| CH6A | Z1269f | A | 4 | 7,13–1 | 1 (ND) | + | + | − | Burkina Faso | 1963 |
| CH7A | Z1278f | A | 4 | 7,13 | 1 (ND) | + | + | − | Niger | 1963 |
| CH8A | Z1318f | A | 4 | 7–5,13–1 | 1 (5) | + | + | − | Burkina Faso | 1966 |
| CH10A | Z1362f | A | 4 | 7,13 | 1 (ND) | + | + | − | Cameroon | 1966 |
| CH11A | Z1213f | A | 4 | 7,13–1 | 1 (ND) | + | + | − | Ghana | 1973 |
| CH13A | Z2491f | A | 4 | 7,13–1 | 1 (5) | + | + | − | Gambia | 1983 |
| CH14A | Z3667f | A | 4 | 7,13–1 | 1 (ND) | + | + | − | Sudan | 1985 |
| CH15A | Z4421f | A | 4 | 7,13 | 1 (5) | + | + | − | Mali | 1990 |
| CH17A | Z4186f | A | 4 | 7,13–1 | 1 (ND) | + | + | − | Mali | 1990 |
| CH19A | F6124 f | A | 5 | 20,9 | 1 (5) | + | + | − | Chad | 1988 |
| CH20A | Niger 1/95g | A | 5 | 20,9 | 1 (5) | + | + | − | Niger | 1995 |
| CH21A | Senegal 1/99g | A | 5 | 20,9 | 1 (5) | + | + | − | Senegal | 1999 |
| CH22A | E23/03g | A | 7 | 20,9 | 1 (5) | + | + | − | Ethiopia | 2003 |
| CH23A | E2/88g | A | 7 | 20,9 | 1 (ND) | + | + | − | Ethiopia | 2003 |
| CH24A | Niger 12/06g | A | 7 | 20,9 | 1 (5) | + | + | − | Niger | 2006 |
| CH25A | Niga 3/07g | A | 7 | 20,9 | 1 (5) | + | + | − | Nigeria | 2007 |
| CH26A | LNP20868 | A | 2859 | 20,9 | 1 (ND) | + | + | − | Burkina Faso | 2003 |
| CH27A | LNP20790 | A | 2859 | 20,9 | 1 (5) | + | + | − | Burkina Faso | 2003 |
| CH28A | BuFa 6/07g | A | 2859 | 20,9 | 1 (5) | + | + | − | Burkina Faso | 2007 |
| CH30W | BuFa 16/01g | W-135 | 11 | 5,2 | 1 (9) | + | + | − | Burkina Faso | 2001 |
| CH31W | BuFa 6/02g | W-135 | 11 | 5,2 | 2 (23) | − | − | + | Burkina Faso | 2002 |
| CH32W | BuFa 16/02g | W-135 | 11 | 5,2 | 2 (23) | − | − | + | Burkina Faso | 2002 |
| CH33W | M9261 | W-135 | 11 | 5,2 | 2 (ND) | − | − | + | Burkina Faso | 2002 |
| CH34W | M9262 | W-135 | 11 | 5,2 | 2 (23) | − | − | + | Burkina Faso | 2002 |
| CH36W | BuFa 1/03g | W-135 | 11 | 5,2 | 2 (ND) | − | − | + | Burkina Faso | 2003 |
| CH37W | BuFa 2/03g | W-135 | 11 | 5,2 | 2 (23) | − | − | + | Burkina Faso | 2003 |
| CH38W | Su 1/06g | W-135 | 11 | 5,2 | 1 (9) | + | + | − | Sudan | 2006 |
| CH39W | Mali 29/07g | W-135 | 11 | 5,2 | 1 (9) | + | + | − | Mali | 2007 |
| CH43X | BuFa 2/97g | X | 751 | 5–1,10–1 | 1 (73) | + | + | − | Burkina Faso | 1997 |
| CH40X | BuFa 7/07g | X | 181 | 5–1,10–1 | 1 (74) | − | + | − | Burkina Faso | 2007 |
| CH41X | Ug 10/06g | X | 5403 | 19,26 | 1 (74) | − | + | − | Uganda | 2006 |
| CH42X | Ug 13/07g | X | 5403 | 19,26 | 1 (74) | − | + | − | Uganda | 2007 |
NOTE. fHbp, factor H–binding protein; ID, identification no.; MAb, monoclonal antibody; ND, not done; ST, multilocus sequence type; VR, PorA variable region ST.
Determined using the Neisseria MLST Home Page [23], as described elsewhere [24]. ST-1, -4, and -11 are the central STs in the respective ST complexes; ST-5, -7, and -2859 are in the ST-5 complex; ST-181 and -751 are in the same clone complex as each other, which has not yet been designated; ST-5403 is from another clonal complex, also not yet designated, and is unrelated to that of ST-181 and ST-751.
Determined using the Neisseria.org Web site [25].
The fHbp variant was determined by quantitative polymerase chain reaction, as described elsewhere [26].
Assigned using the Neisseria.org fHbp Database [27].
MAbs JAR 1 and 5 are specific for fHbp in the variant 1 (v.1) group [18, 28]. JAR 5 is broadly reactive and JAR 1 reacts with a subset of fHbp in the v.1 group [18, 28]. JAR 31 cross-reacts with fHbp in the variant 2 and variant 3 groups (J.A.W. and D.M.G., unpublished data).
[29].
Strain from the World Health Organization Collaborating Centre for Reference and Research on Meningococci (Oslo, Norway).
Bacterial growth and preparation of heat-killed cells
Meningococci were subcultured on chocolate agar plates (Remel) incubated at 37°C in 5% CO2 for 18 h. Bacteria were suspended in 7 mL of Mueller-Hinton broth (BD Biosciences) containing 0.25% D-glucose (Sigma-Aldrich) at an optical density measured at 620 nm (OD620) of 0.12, and they were grown for 2–3 h to an OD620 of 0.8. The bacteria were killed by heating the suspensions at 56°C for 1 h, collected by centrifugation at 3000 g, and resuspended in PBS to an OD620 of 0.6.
Quantitative polymerase chain reaction (PCR) and DNA sequencing
Quantitative PCR was performed using heat-killed meningococci as the source of the DNA template. The amplification conditions and the primers for fHbp and the 16S rRNA control gene were described elsewhere [26].
For DNA sequencing, genomic DNA was prepared from ∼109 bacterial cells by use of the DNeasy Tissue Kit (Qiagen). The fHbp genes were PCR amplified from genomic DNA by use of A1 and B2 primers described elsewhere [20]. The DNA sequences of the PCR products were determined using the A1 and/or 22 primers [20] (Davis Sequencing). The DNA sequences were analyzed using the DNA Strider software (version 1.4; Comissariat à l'Energie Atomique, France).
Preparation of native OMVs and Western blot analysis
Native OMVs were prepared from blebs released into bacterial culture supernatants, as described elsewhere [30]. Native OMVs (5 μg) or recombinant (r) proteins (1 μg) were separated by SDS-PAGE (4%–12% NuPAGE; Invitrogen). Western blot analysis was performed using heat-killed bacteria (∼108 cfu) or OMV (5 μg), as described elsewhere [31]. The primary antibodies were anti-fHbp monoclonal antibodies (MAbs) JAR 1 (2 μg/mL) [28], JAR 5 (0.1 μg/mL) [28], or JAR 31 (4 μg/mL) diluted in PBS containing 0.1% Tween-20 and 1% (wt/vol) bovine serum albumin (Equitech Bio).
Accessibility of fHbp on the bacterial surface
We used flow cytometry to measure the binding of serum anti-fHbp antibodies to the surface of live N. meningitidis bacteria. Serum samples were obtained from mice vaccinated with recombinant fHbp in the v.1 group (gene from strain MC58) or v.2 group (gene from strain RM1090). Details about the method used have been described elsewhere [14, 28].
Mouse immunogenicity
We tested bactericidal activity against epidemic group A, X, and W-135 strains in serum samples obtained from mice vaccinated in a previous study with a mixture of 2 native OMV vaccines prepared from mutants of group B strains [32]. One of the OMV vaccines was obtained from a previously described LpxL1 knockout mutant of strain H44/76, which was engineered to overexpress fHbp in the v.1 group [22]. The second vaccine was from a new LpxL1 knockout mutant of strain NZ98/254, which was engineered to express both endogenous fHbp v.1 and a heterologous fHbp in the v.2 group [32]. The respective PorA variable region types of the 2 strains were P1.7,16 and P1.7–2,4. Mice were vaccinated with 3 injections, each separated by 3 weeks. The dose used for each injection was 2.5 μg of OMV protein (1.25 μg of each OMV vaccine) and 600 μg of aluminum hydroxide. Blood samples were collected 3 weeks after the third injection was administered. The control vaccine consisted of a mixture of 2 detergent-extracted OMV vaccines prepared from the corresponding wild-type strains of H44/76 and NZ98/254, in accordance with the procedure described by the Norwegian Institute of Public Health [33], and adsorbed with aluminum hydroxide.
Complement-mediated bactericidal activity
Serum bactericidal activity was measured using washed, log-phase bacteria grown to an OD620 of 0.6 in Mueller-Hinton broth supplemented with 0.25% glucose and 0.02 mmol/L cytidine monophosphate N-acetyl neuraminic acid [34]. The buffer was Dulbecco's PBS containing 0.9 mmol/L CaCl2 and 0.5 mmol/L MgCl2 × 6H20 (Mediatech) with 1% (wt/vol) bovine serum albumin (Equitech-Bio). The complement source was human serum samples lacking endogenous bactericidal activity. For the group X and W-135 strains, we depleted IgG from the complement by applying 1 mL of serum to a protein G column (HiTrap Protein G HP [1 mL]; GE Healthcare), which was displaced with 1 mL of PBS (Roche Applied Science). Adsorption of IgG was monitored by ELISA on the flow-through and elution fractions. Preservation of hemolytic complement activity was confirmed using the EZ Complement CH50 Test (Diamedix). The serum bactericidal titer was defined as the reciprocal dilution that provided a 50% decrease in the number of colony-forming units noted after incubation at 37°C for 1 h, compared with the number of colony-forming units noted at time 0 in the negative control reactions.
Results
Strains and genetic lineages
The 35 meningococcal isolates were recovered from patients residing in 14 countries (figure 1). Twenty-two of the isolates were capsular group A strains, 9 were group W-135 strains, and 4 were group X strains (table 1). With one exception (strain CH19A from Chad, ST-5, 1988) (table 1), the group A isolates recovered from 1963 through 1990 belonged to the ST-1 or ST-4 clonal complexes (ST-1 or -4, respectively). After 1990, the group A isolates that were recovered belonged to the ST-5 complex (ST-5, -7, or -2859). The 9 recently recovered W-135 isolates all were ST-11. The 2 group X isolates recovered from cases in Burkina Faso 10 years apart were not assigned to a designated clonal complex but were from the same clonal complex, based on sharing 5 of the 7 MLST alleles. The 2 group X isolates recovered in Uganda in 2006–2007 had an identical ST (ST-5403), which was unrelated to that of the group X strains from Burkina Faso.
Figure 1.
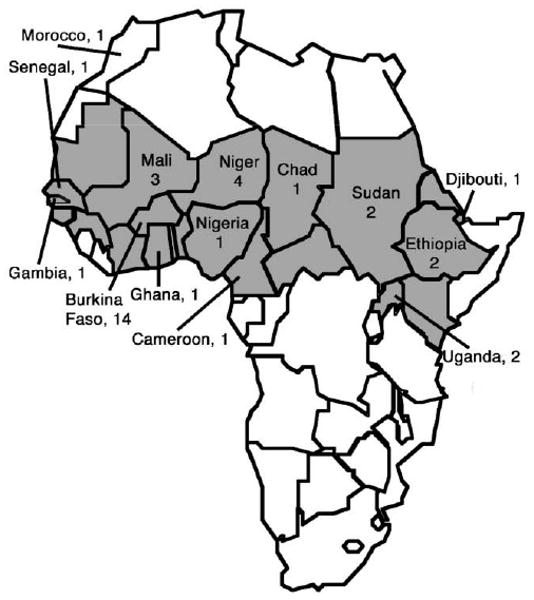
Map of Africa showing the source countries and nos. of isolates that were examined in this study. Sub-Saharan Africa is depicted in gray.
Genes encoding fHbp
On the basis of quantitative PCR assays, all of the group A and X isolates had genes encoding fHbp in the v.1 group, whereas the group W-135 isolates had genes encoding fHbp in the v.1 or v.2 group (table 1).
We determined the DNA sequences of genes encoding fHbp from 24 of the 35 isolates (13 group A, 7 group W-135, and all 4 group X isolates). The inferred peptide identification numbers are shown in table 1, and the relative amino acid conservations of each of the proteins, compared with different reference antigenic variants of fHbp, are summarized in table 2, which appears only in the electronic version of the Journal. The group A isolates, which were selected to include each of the ST clones obtained since 1963, had genes encoding a highly conserved fHbp (≥99% amino acid identity). The fHbp genes of the group X isolates encoded proteins with ≥93% amino acid identity to each other and ≥92% amino acid identity to fHbp of the group A strains. The amino acid sequences deduced from the fHbp v.1 gene of the group W-135 isolates were identical to each other and ≥93% identical to fHbp of the group A isolates, whereas the amino acid sequences of the W-135 strains with fHbp v.2 genes had 99% identity to the v.2 fHbp of group C strain FAM18 [35]. The relative amino acid sequence similarities of fHbp from strains representative of each of the STs are shown in figure 2.
Table 2. Amino acid sequence identity of factor H–binding protein (fHbp) from African meningococcus isolates.
| The table is available in its entirety in the online edition of the Journal of Infectious Diseases. |
Figure 2.
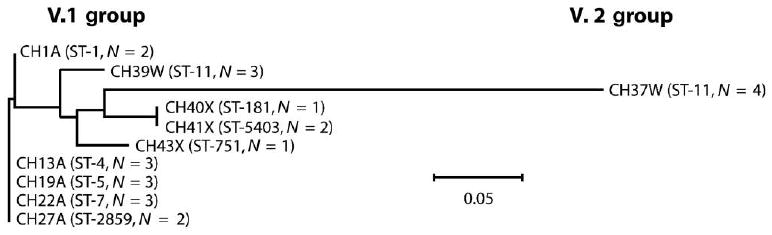
Phylogram of the factor H–binding protein (fHbp) polypeptide sequences from African strains. The relative distance between respective peptides is shown on the horizontal line; the scale bar denotes 5 changes/100 amino acids. One fHbp amino acid sequence from each sequence type (ST) is represented, except for ST-11 (group W-135), for which data from strains with fHbp variant 1 (v.1) or 2 (v.2) are shown. N, the no. of isolates with identical capsular group, ST, and fHbp polypeptide sequence. Peptide identifiers for each strain are shown in table 1.
fHbp epitope expression
We determined the ability of 3 anti-fHbp MAbs (JAR 1, 5, and 31) to bind with each of the 35 strains, by use of Western blot analysis. Binding to OMV preparations from representative group A, W-135, and X strains is shown in figure 3, and the results for all 35 strains are summarized in table 1. All of the group A, X, and W-135 isolates with genes encoding fHbp v.1 were positive for JAR 5, which is broadly reactive with fHbp in the v.1 group [28, 31]. The group A strains, the W-135 strains with fHbp v.1, and 1 of the 4 group X strains also reacted with JAR 1, which is specific for a subset of fHbps in the v.1 group [18, 28]. The W-135 strains with fHbp v.2 genes were negative for binding to JAR 1 or JAR 5, but they reacted with JAR 31, which is specific for fHbp in the v.2 and v.3 groups.
Figure 3.
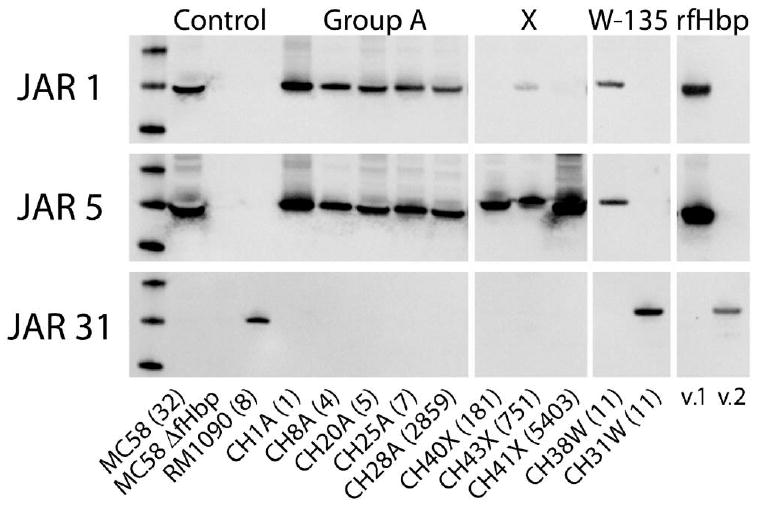
Expression of factor H–binding protein (fHbp) in outer membrane vesicle (OMV) preparations of representative isolates, as measured by Western blot analysis. Monoclonal antibodies (MAbs) JAR 1 and JAR 5 were raised against fHbp variant 1 (v.1) (gene from strain MC58); MAb JAR 31 was raised against fHbp variant 3 (v.3) (gene from strain M1239), but it cross-reacts with fHbp variant 2 (v.2) and v.3 proteins. OMV from control strains included MC58 wt (fHbp v.1), MC58 fHbp knockout (ΔfHbp) mutant, and RM1090 WT (fHbp v.2). The sequence type (ST) of each isolate is shown in parentheses. rfHbp, recombinant fHbp.
To quantify surface-accessible fHbp, we measured the binding of serum anti-fHbp antibody to live bacterial cells of representative group A, W-135, and X isolates by use of flow cytometry (figure 4, white histograms). For use as a negative control, we measured either binding of the anti-fHbp antiserum to the respective fHbp knockout strain (figure 4, black histograms) or binding of a negative control mouse antiserum to the respective wild-type strain (figure 4, gray histograms). Among isolates with fHbp in the v.1 group, fHbp was most abundant in the group X strains, and it was at its lowest level in the group W-135 strains. The 2 group A isolates tested from different ST clones had fHbp levels that were intermediate between those noted in the group X and group W-135 isolates. The 2 group W-135 isolates with fHbp v.2 had more surface-accessible fHbp than did a group C isolate known to have low expression of ST-11, which served as a positive control.
Figure 4.
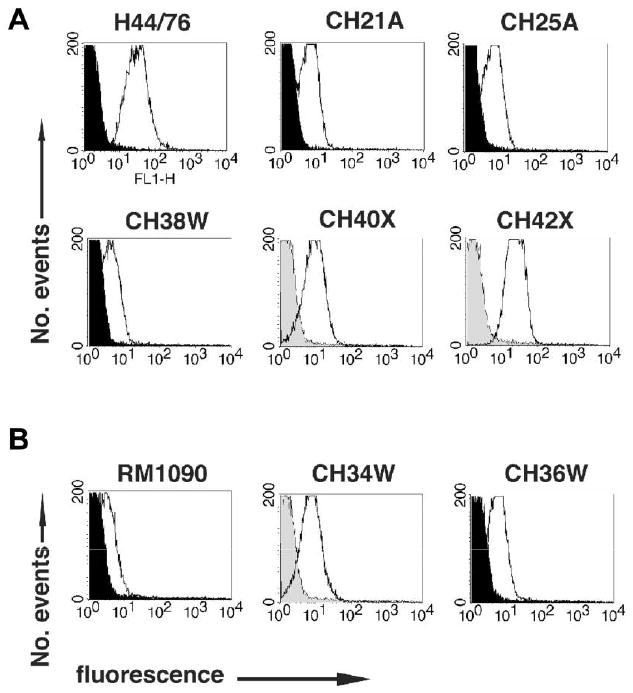
Surface-accessibility of factor H–binding protein (fHbp) on live bacterial cells, as measured by flow cytometry with mouse anti-fHbp variant 1 (v.1) (A) or 2 (v.2) (B) antisera. The H44/76 positive control is known to express high levels of fHbp v.1 [14]. The RM1090 positive control is known to express low levels of fHbp v.2 [21] with 99% amino acid identity to fHbp v.2 of the group W-135 test strains. White and black histograms denote the respective binding of anti-fHbp antisera (1:250) to wild-type and fHbp knockout strains. For strains for which fHbp knockout mutants were not available, the gray histograms denote binding of a 1:50 dilution of a negative control antiserum with the wild-type strain. Strain designations are those shown in table 1.
Bactericidal activity of serum samples obtained from mice vaccinated with native OMV vaccines
We measured the bactericidal activity of serum samples obtained from mice vaccinated in a previous study with a mixture of 2 native OMV vaccines prepared from mutants of H44/76 and NZ98/254 with attenuated endotoxin activity and engineered to express heterologous fHbp in the v.1 and v.2 groups [32] (for description of vaccines, see Methods above). The test isolates included 3 group A, 3 W-135, and 2 X case isolates from Africa, as well as 2 Hajj-related ST-11, W-135 case isolates from France (LNP17592 with fHbp v.1 and LNP19995 with fHbp v.2). All 10 isolates had heterologous PorA from the strains used to prepare the OMV vaccines. The serum samples obtained from the mice vaccinated with the mixture of the 2 native OMV vaccines from the mutants had broad bactericidal activity against all 10 strains (figure 5). In contrast, the serum samples obtained from control mice vaccinated with a mixture of 2 detergent-extracted OMV vaccines prepared from the respective wild-type strains had much lower titers.
Figure 5.
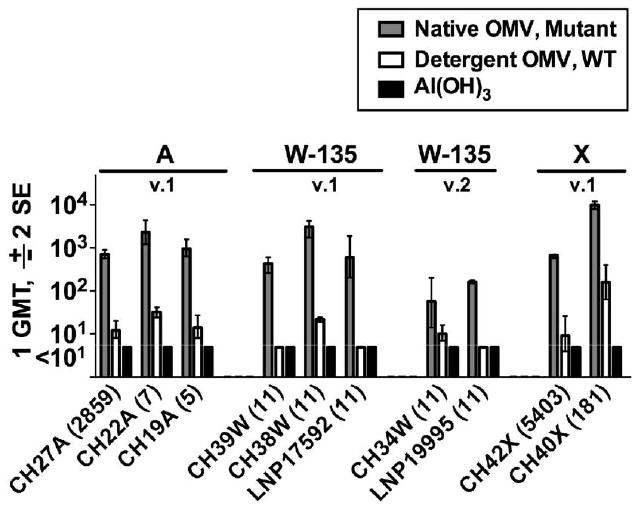
Serum bactericidal titers against epidemic group A, W-135 and X strains. Eight strains were from Africa and 2 (LNP17592 and LNP19995) were Hajj-related sequence type (ST)–11, W-135 case isolates from France. All of the strains had heterologous PorA from the vaccine strains. The multilocus STs are shown in parentheses. The vaccine groups were as follows: Native outer membrane vesicle (OMV), Mutant (with “Mutant” denoting LpxL1 knockout strains of H44/76 and NZ98/254 expressing fHbp variant 1 and variant 2); Detergent OMV, WT (detergent-treated OMV from respective wild-type strain); and Al(OH)3 (aluminum hydroxide only).
Discussion
Group A strains account for most epidemics of meningococcal disease in sub-Saharan Africa. However, in 2001–2002, an epidemic caused by a capsular group W-135 strain resulted in >30,000 cases in Burkina Faso [36–38]. Recent outbreaks of meningococcal disease caused by capsular group X strains also were reported in Ghana [8, 39], Niger [7], and Kenya and Uganda [40]. These observations underscore the complex epidemiologic profile of meningococcal disease in Africa as well as the potential for strains from capsular groups other than group A to emerge once group A epidemics are controlled by introduction of the group A polysaccharide-protein conjugate vaccine.
The current strategy to provide coverage against strains from capsular groups other than group A is based on the use of multivalent polysaccharide or polysaccharide-protein conjugate vaccines. However, nonconjugated polysaccharide vaccines do not confer protection in young children or prime for immunologic memory at any age. Quadrivalent A, C, W-135, and Y polysaccharide-protein conjugate vaccines are licensed and/or in development in industrialized countries and have the potential to be effective at all ages [41]. However, these vaccines do not elicit coverage against epidemic group X strains or against group Z or 29E strains, which have not yet caused epidemic disease in Africa but could emerge. The quadrivalent conjugate vaccines also are expensive (∼$80/dose) and are unlikely to be affordable in sub-Saharan Africa, one of the poorest regions of the world. Development of a new multivalent capsular polysaccharide-protein conjugate vaccine for Africa, one that would likely need to include group X strains, would also be technically challenging, expensive, and time consuming.
The purpose of the present study was to investigate fHbp sequence variants and antigenic expression among epidemic meningococcal isolates from Africa and determine the feasibility of using a noncapsular-based OMV vaccine for the prevention of meningococcal disease in Africa. The data indicated that fHbp may be a promising vaccine candidate for Africa, because all of the group A isolates tested from strains with STs responsible for epidemics involving millions of cases over 45 years expressed a highly conserved fHbp in the v.1 group (table 1 and table 2, the latter of which appears only in the electronic version of the Journal). Thus, there was little if any natural immune selection for fHbp antigenic variants induced by the extensive exposure to group A meningococci during this period. Similarly, the group X isolates investigated in the present study expressed conserved fHbps in the antigenic v.1 group, as did a subset of the ST-11, group W-135 isolates (table 1 and table 2, the latter of which appears only in the electronic version of the Journal). The remaining ST-11, W-135 isolates expressed a conserved fHbp in the v.2 group, which was nearly identical to fHbp in the v.2 group in the ST-11, group C strain FAM18 [35].
The ST-11, W-135 isolates from Africa that had fHbp in the v.1 group were recovered from patients in Burkina Faso during 2001 and from patients in Sudan and Mali during 2006 and 2007, whereas the ST-11, W-135 isolates that had fHbp in the v.2 group were recovered from cases occurring at the height of a large epidemic in Burkina Faso during 2002–2003 [36, 37, 42, 43]. On the basis of fHbp variant groups, there were at least 2 ST-11, W-135 subclones circulating in Africa during 2001–2007, and the subclone with fHbp in the v.2 group was responsible, at least in part, for the epidemic in Burkina Faso in 2002–2003.
All of the group A, X, and W-135 strains expressed proteins with conserved epitopes recognized by bactericidal anti-fHbp MAbs (table 1). On the basis of flow cytometric studies, the group X isolates had relatively abundant surface-accessible fHbp, whereas the antigen was sparse on the surface of a group W-135 isolate with a v.1 protein and was present in intermediate amounts on the group A isolates with fHbp v.1 and group W-135 isolates with fHbp v.2. In previous studies, strains with low expression of fHbp were resistant to the bactericidal activity of antibodies elicited by recombinant fHbp vaccines [21, 22]. However, as described below, most of these strains with low expression of fHbp were susceptible to the bactericidal activity of anti-fHbp antibodies elicited by native OMV vaccines with over-expressed fHbp [22, 44].
To decrease endotoxin activity, conventional OMV vaccines are prepared by treatment with detergents to extract lipooligosaccharide [45]. This treatment also extracts potentially desirable vaccine antigens, such as fHbp [22] and GNA 2132 [19]. To circumvent the need for treatment with detergents, we prepared native OMV vaccines from mutants in which we inactivated the genes encoding LpxL1 [22, 32], which resulted in a lipooligosaccharide with penta-acylated lipid A. The lipooligosaccharide from the mutant was known to have substantially less endotoxin activity than lipooligosaccharide with hexa-acylated lipid A from wild-type strains [46]. We also engineered the mutants to express heterologous fHbps in the v.1 and v.2 groups. Similar to control detergent-treated OMV vaccines from the wild-type strains, the native OMV vaccines from the mutants stimulated low proinflammatory cytokine responses by human PBMCs. Thus, native OMV vaccines prepared from LpxL1 knockout mutants may be safe to administer to humans. In mice, the mixture of the 2 native OMV vaccines elicited broad serum bactericidal antibody responses against genetically diverse N. meningitidis group B strains with heterologous PorA and with fHbp in each of the antigenic variant groups. In the present study, we found that the serum samples obtained from these mice also had complement-mediated bactericidal activity against epidemic group A, W-135, and X strains from Africa, including some strains with relatively low natural expression of fHbp. These data were for mice vaccinated with vaccines designed to prevent group B disease. Native OMV vaccines prepared from mutants of African strains expressing PorA and fHbp antigenic variants that match those of strains from Africa may provide even better protection.
In the present study, the control mice that were vaccinated with a mixture of 2 detergent-treated OMV vaccines from the wild-type strains had serum titers with low bactericidal activity against most of the African strains (figure 5). The control vaccines were selected to bridge experience with similarly prepared OMV vaccines from strains H44/76 and NZ98/254 that had been tested extensively in humans and had been shown to be safe and to confer protection against group B disease [47–51]. Adults who are naturally primed and vaccinated with detergent-treated OMV vaccines had higher bactericidal responses [52] with broader serum bactericidal activity than did vaccinated infants or young children, whose responses were largely specific to strains with PorA antigens that matched the vaccine strain [53]. The serum bactericidal responses of immunologically naive mice vaccinated with the control detergent-treated OMV vaccines in the present study also were largely limited to the strains used to prepare the vaccines [32]. Thus, the data for the mice vaccinated with detergent-treated OMV paralleled the PorA-specific bactericidal responses observed in vaccinated human infants. In contrast, all 10 group A, W-135, and X strains tested with heterologous PorA antigens from the vaccine strains were killed by serum samples from mice vaccinated with the mixture of the 2 native OMV vaccines from mutant strains with genetically attenuated endotoxin and expression of heterologous fHbp variants. Collectively, the results support further studies of the mutant native OMV vaccine approach to prevent meningococcal disease caused by strains from all capsular groups responsible for epidemics in Africa.
Acknowledgments
We thank Ray Chen, Serena Giuntini, Rachel Lown-Hecht, Esther Mun-gai, Ryan Palapaz, and Tracy Wong for expert technical assistance.
Financial support: National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH; Public Health Service grants R01 AI 046464 [to D.M.G.] and AI 070955 [to P.T.B.]). The Children's Hospital Oakland Research Institute facility was funded by the National Center for Research Resources, NIH (Research Facilities Improvement Program grant C06 RR 16226).
Footnotes
Potential conflicts of interest: D.M.G. is principal investigator of laboratory research conducted on behalf of Children's Hospital Oakland Research Institute, which is funded by grants from Novartis Vaccines and Diagnostics and Sanofi Pasteur; holds a paid consultancy from Novartis; and is an inventor on patents or patent applications associated with meningococcal B vaccines. O.K. is currently an employee of Novartis Vaccines, and D.A.C. has received research funding from Wyeth Vaccines and Sanofi Pasteur. P.T.B. and J.A.W. report no relevant conflicts of interest.
Presented in part: 16th International Pathogenic Neisseria Conference, September 2008, Rotterdam, The Netherlands (abstract P125).
References
- 1.Greenwood B. Manson Lecture. Meningococcal meningitis in Africa. Trans R Soc Trop Med Hyg. 1999;93:341–53. doi: 10.1016/s0035-9203(99)90106-2. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. WHO report on global surveillance of epidemic-prone diseases. [16 March 2009];2001 Available at: http://www.who.int/csr/resources/publications/surveillance/WHO_CDS_CSR_ISR_2000_1/en/
- 3.Miller MA, Wenger J, Rosenstein N, Perkins B. Evaluation of meningococcal meningitis vaccination strategies for the meningitis belt in Africa. Pediatr Infect Dis J. 1999;18:1051–9. doi: 10.1097/00006454-199912000-00005. [DOI] [PubMed] [Google Scholar]
- 4.LaForce FM, Konde K, Viviani S, Préziosi MP. The Meningitis Vaccine Project. Vaccine. 2007;25(Suppl 1):A97–100. doi: 10.1016/j.vaccine.2007.04.049. [DOI] [PubMed] [Google Scholar]
- 5.Jodar L, LaForce MF, Ceccarini C, Aguado MT, Granoff DM. Meningococcal conjugate vaccine for Africa: a model for developing new vaccines for the poorest countries. Lancet. 2003;361:4997–9. doi: 10.1016/S0140-6736(03)13494-0. [DOI] [PubMed] [Google Scholar]
- 6.Mueller JE, Borrow R, Gessner BD. Meningococcal serogroup W135 in the African meningitis belt: epidemiology, immunity and vaccines. Expert Rev Vaccines. 2006;5:319–36. doi: 10.1586/14760584.5.3.319. [DOI] [PubMed] [Google Scholar]
- 7.Boisier P, Nicolas P, Djibo S, et al. Meningococcal meningitis: unprecedented incidence of serogroup X-related cases in 2006 in Niger. Clin Infect Dis. 2007;44:657–63. doi: 10.1086/511646. [DOI] [PubMed] [Google Scholar]
- 8.Gagneux SP, Hodgson A, Smith TA, et al. Prospective study of a serogroup X Neisseria meningitidis outbreak in northern Ghana. J Infect Dis. 2002;185:618–26. doi: 10.1086/339010. [DOI] [PubMed] [Google Scholar]
- 9.De Groot AS, Rappuoli R. Genome-derived vaccines. Expert Rev Vaccines. 2004;3:59–76. doi: 10.1586/14760584.3.1.59. [DOI] [PubMed] [Google Scholar]
- 10.Pizza M, Scarlato V, Masignani V, et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science. 2000;287:1816–20. doi: 10.1126/science.287.5459.1816. [DOI] [PubMed] [Google Scholar]
- 11.Williams JN, Skipp PJ, Humphries HE, Christodoulides M, O'Connor CD, Heckels JE. Proteomic analysis of outer membranes and vesicles from wild-type serogroup B Neisseria meningitidis and a lipopolysaccharide-deficient mutant. Infect Immun. 2007;75:1364–72. doi: 10.1128/IAI.01424-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madico G, Welsch JA, Lewis LA, et al. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J Immunol. 2006;177:501–10. doi: 10.4049/jimmunol.177.1.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneider MC, Exley RM, Chan H, et al. Functional significance of factor H binding to Neisseria meningitidis. J Immunol. 2006;176:7566–75. doi: 10.4049/jimmunol.176.12.7566. [DOI] [PubMed] [Google Scholar]
- 14.Welsch JA, Ram S, Koeberling O, Granoff DM. Complement-dependent synergistic bactericidal activity of antibodies against factor H–binding protein, a sparsely distributed meningococcal vaccine antigen. J Infect Dis. 2008;197:1053–61. doi: 10.1086/528994. [DOI] [PubMed] [Google Scholar]
- 15.Giuliani MM, Adu-Bobie J, Comanducci M, et al. A universal vaccine for serogroup B meningococcus. Proc Natl Acad Sci U S A. 2006;103:10834–9. doi: 10.1073/pnas.0603940103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fletcher LD, Bernfield L, Barniak V, et al. Vaccine potential of the Neisseria meningitidis 2086 lipoprotein. Infect Immun. 2004;72:2088–100. doi: 10.1128/IAI.72.4.2088-2100.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Comanducci M, Bambini S, Brunelli B, et al. NadA, a novel vaccine candidate of Neisseria meningitidis. J Exp Med. 2002;195:1445–54. doi: 10.1084/jem.20020407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beernink PT, Welsch JA, Harrison LH, Leipus A, Kaplan SL, Granoff DM. Prevalence of factor H–binding protein variants and NadA among meningococcal group B isolates from the United States: implications for the development of a multicomponent group B vaccine. J Infect Dis. 2007;195:1472–9. doi: 10.1086/514821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welsch JA, Moe GR, Rossi R, Adu-Bobie J, Rappuoli R, Granoff DM. Antibody to genome-derived neisserial antigen 2132, a Neisseria meningitidis candidate vaccine, confers protection against bacteremia in the absence of complement-mediated bactericidal activity. J Infect Dis. 2003;188:1730–40. doi: 10.1086/379375. [DOI] [PubMed] [Google Scholar]
- 20.Masignani V, Comanducci M, Giuliani MM, et al. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J Exp Med. 2003;197:789–99. doi: 10.1084/jem.20021911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou VC, Koeberling O, Welsch JA, Granoff DM. Protective antibody responses elicited by a meningococcal outer membrane vesicle vaccine with overexpressed genome-derived neisserial antigen 1870. J Infect Dis. 2005;192:580–90. doi: 10.1086/432102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koeberling O, Seubert A, Granoff DM. Bactericidal antibody responses elicited by a meningococcal outer membrane vesicle vaccine with overexpressed factor H–binding protein and genetically attenuated endotoxin. J Infect Dis. 2008;198:262–70. doi: 10.1086/589308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maiden MC, Bygraves JA, Feil E, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–5. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neisseria MLST Home Page. [23 February 2009]; Available at: http://pubmlst.org/neisseria.
- 25.Neisseria.org Home Page. [23 February 2009]; Available at: http://neisseria.org.
- 26.Beernink PT, Leipus A, Granoff DM. Rapid genetic grouping of factor H–binding protein (genome-derived neisserial antigen 1870), a promising group B meningococcal vaccine candidate. Clin Vaccine Immunol. 2006;13:758–63. doi: 10.1128/CVI.00097-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neisseria.org fHbp Database. [23 February 2009]; Available at: http://neisseria.org/perl/agdbnet/agdbnet.pl?file=nm_fhbp.xml&page=browse&locus=public_FHBP_p.
- 28.Welsch JA, Rossi R, Comanducci M, Granoff DM. Protective activity of monoclonal antibodies to genome-derived neisserial antigen 1870, a Neisseria meningitidis candidate vaccine. J Immunol. 2004;172:5606–15. doi: 10.4049/jimmunol.172.9.5606. [DOI] [PubMed] [Google Scholar]
- 29.Wang JF, Caugant DA, Li X, et al. Clonal and antigenic analysis of serogroup A Neisseria meningitidis with particular reference to epidemiological features of epidemic meningitis in the People's Republic of China. Infect Immun. 1992;60:5267–82. doi: 10.1128/iai.60.12.5267-5282.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moe GR, Zuno-Mitchell P, Hammond SN, Granoff DM. Sequential immunization with vesicles prepared from heterologous Neisseria meningitidis strains elicits broadly protective serum antibodies to group B strains. Infect Immun. 2002;70:6021–31. doi: 10.1128/IAI.70.11.6021-6031.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beernink PT, Welsch JA, Bar-Lev M, Koeberling O, Comanducci M, Granoff DM. Fine antigenic specificity and cooperative bactericidal activity of monoclonal antibodies directed at the meningococcal vaccine candidate factor H–binding protein. Infect Immun. 2008;76:4232–40. doi: 10.1128/IAI.00367-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koeberling O, Giuntini S, Seubert A, Granoff DM. Meningococcal outer membrane vesicle vaccines derived from mutant strains engineered to express factor H–binding proteins from antigenic variant groups 1 and 2. Clin Vaccine Immunol. 2009;16:156–62. doi: 10.1128/CVI.00403-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fredriksen JH, Rosenqvist E, Wedege E, et al. Production, characterization and control of MenB-vaccine “Folkehelsa”: an outer membrane vesicle vaccine against group B meningococcal disease. NIPH Ann. 1991;14:67–79. discussion, 79–80. [PubMed] [Google Scholar]
- 34.Welsch JA, Granoff D. Immunity to Neisseria meningitidis group B in adults despite lack of serum bactericidal activity. Clin Vaccine Immunol. 2007;14:1596–602. doi: 10.1128/CVI.00341-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bentley SD, Vernikos GS, Snyder LA, et al. Meningococcal genetic variation mechanisms viewed through comparative analysis of serogroup C strain FAM18. PLoS Genet. 2007;3:e23. doi: 10.1371/journal.pgen.0030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicolas P, Norheim G, Garnotel E, Djibo S, Caugant DA. Molecular epidemiology of Neisseria meningitidis isolated in the African Meningitis Belt between 1988 and 2003 shows dominance of sequence type 5 (ST-5) and ST-11 complexes. J Clin Microbiol. 2005;43:5129–35. doi: 10.1128/JCM.43.10.5129-5135.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koumare B, Ouedraogo-Traoré R, Sanou I, et al. The first large epidemic of meningococcal disease caused by serogroup W135, Burkina Faso, 2002. Vaccine. 2007;25(Suppl 1):A37–41. doi: 10.1016/j.vaccine.2007.04.038. [DOI] [PubMed] [Google Scholar]
- 38.Mueller JE, Sangaré L, Njanpop-Lafourcade BM, et al. Molecular characteristics and epidemiology of meningococcal carriage, Burkina Faso, 2003. Emerg Infect Dis. 2007;13:847–54. doi: 10.3201/eid1306.061395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gagneux S, Wirth T, Hodgson A, et al. Clonal groupings in serogroup X Neisseria meningitidis. Emerg Infect Dis. 2002;8:462–6. doi: 10.3201/eid0805.010227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Materu S, Cox HS, Isaakidis P, Baruani B, Ogaro T, Caugant DA. Serogroup X in meningococcal disease, Western Kenya. Emerg Infect Dis. 2007;13:944–5. doi: 10.3201/eid1306.070042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Snape MD, Perrett KP, Ford KJ, et al. Immunogenicity of a tetravalent meningococcal glycoconjugate vaccine in infants: a randomized controlled trial. JAMA. 2008;299:173–84. doi: 10.1001/jama.2007.29-c. [DOI] [PubMed] [Google Scholar]
- 42.Nathan N, Rose AM, Legros D, et al. Meningitis serogroup W135 outbreak, Burkina Faso, 2002. Emerg Infect Dis. 2007;13:920–3. doi: 10.3201/eid1306.060940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mueller JE, Yaro S, Traore Y, et al. Neisseria meningitidis serogroups A and W-135: carriage and immunity in Burkina Faso, 2003. J Infect Dis. 2006;193:812–20. doi: 10.1086/500511. [DOI] [PubMed] [Google Scholar]
- 44.Koeberling O, Welsch JA, Granoff DM. Improved immunogenicity of a H44/76 group B outer membrane vesicle vaccine with over-expressed genome-derived Neisserial antigen 1870. Vaccine. 2007;25:1912–20. doi: 10.1016/j.vaccine.2006.03.092. [DOI] [PubMed] [Google Scholar]
- 45.Frasch C, Van Alphen L, Holst J, Poolman J, Rosenqvist E. Outer membrane protein vesicle vaccines for meningococcal disease. In: Pollard AJ, Maiden MC, editors. Meningococcal vaccines: methods and protocols. Totowa, New Jersey: Humana Press; 2001. pp. 81–107. [DOI] [PubMed] [Google Scholar]
- 46.van der Ley P, Steeghs L, Hamstra HJ, ten Hove J, Zomer B, van Alphen L. Modification of lipid A biosynthesis in Neisseria meningitidis lpxL mutants: influence on lipopolysaccharide structure, toxicity, and adjuvant activity. Infect Immun. 2001;69:5981–90. doi: 10.1128/IAI.69.10.5981-5990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelly C, Arnold R, Galloway Y, O'Hallahan J. A prospective study of the effectiveness of the New Zealand meningococcal B vaccine. Am J Epidemiol. 2007;166:817–23. doi: 10.1093/aje/kwm147. [DOI] [PubMed] [Google Scholar]
- 48.Bjune G, Hoiby EA, Gronnesby JK, et al. Effect of outer membrane vesicle vaccine against group B meningococcal disease in Norway. Lancet. 1991;338:1093–6. doi: 10.1016/0140-6736(91)91961-s. [DOI] [PubMed] [Google Scholar]
- 49.Holst J, Feiring B, Naess LM, et al. The concept of “tailor-made,” protein-based, outer membrane vesicle vaccines against meningococcal disease. Vaccine. 2005;23:2202–5. doi: 10.1016/j.vaccine.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 50.Taha MK, Zarantonelli ML, Alonso JM, et al. Use of available outer membrane vesicle vaccines to control serogroup B meningococcal outbreaks. Vaccine. 2007;25:2537–8. doi: 10.1016/j.vaccine.2005.12.059. [DOI] [PubMed] [Google Scholar]
- 51.Holst J. Strategies for development of universal vaccines against meningococcal serogroup B disease: the most promising options and the challenges evaluating them. Hum Vaccin. 2007;3:290–4. doi: 10.4161/hv.4513. [DOI] [PubMed] [Google Scholar]
- 52.Oster P, O'Hallahan J, Aaberge I, Tilman S, Ypma E, Martin D. Immunogenicity and safety of a strain-specific MenB OMV vaccine delivered to under 5 year olds in New Zealand. Vaccine. 2007;25:3075–9. doi: 10.1016/j.vaccine.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 53.Tappero JW, Lagos R, Ballesteros AM, et al. Immunogenicity of 2 serogroup B outer-membrane protein meningococcal vaccines: a randomized controlled trial in Chile. JAMA. 1999;281:1520–7. doi: 10.1001/jama.281.16.1520. [DOI] [PubMed] [Google Scholar]