

Summary
The Hedgehog (Hh) signaling pathway is a developmentally conserved regulator of stem cell function. Several reports suggested that Hh signaling is an important regulator of hematopoietic stem cell (HSC) maintenance and differentiation. Here we test this hypothesis in vivo using both gain- and loss-of-function Hh genetic models. Surprisingly, our studies demonstrate that conditional Smoothened (Smo) deletion or over-activation has no significant effects on adult HSC self-renewal and function. Moreover, they indicate a lack of synergism between the Notch and Hh pathways in HSC function, as compound RBPJ- and Smo-deficiency does not affect hematopoiesis. In agreement with this notion, detailed genome-wide transcriptome analysis reveals that silencing of Hh signaling does not significantly alter the HSC-specific gene expression “signature”. Our studies demonstrate that the Hh signaling pathway is dispensable for adult HSC function and suggest that the Hh pathway can be targeted in future clinical trials addressing the effect of Hh inhibition on leukemia-initiating cell maintenance.
Introduction
Hematopoietic stem cells (HSC) are able to self-renew as well as give rise to all blood lineages. HSCs mainly reside in specialized bone marrow microenvironments, called HSC niches. The niche is thought to provide appropriate signals that maintain the balance between self-renewal and differentiation of HSCs (Adams and Scadden, 2006; Lessard et al., 2004; Moore and Lemischka, 2006; Morrison and Spradling, 2008; Wilson and Trumpp, 2006; Yin and Li, 2006). However, the identity of these signals and the molecular mechanisms governing HSC fate largely remain elusive. Thus, identification of regulators of HSC function is a central issue in stem cell biology.
The roles of developmentally imprinted signaling pathways, more specifically Notch, Wingless (Wnt) and Hedgehog (Hh), in HSC homeostasis have been studied extensively (Maillard et al., 2008; Stier et al., 2002); (Cobas et al., 2004; Reya et al., 2003). Hh is a secreted protein family with 3 members in higher vertebrates (Shh, Ihh, Dhh). In the absence of Hh, the Patched (Ptch) receptor acts as a negative regulator of signaling as it inhibits the action of Smoothened (Smo) (Hammerschmidt et al., 1997). Hh protein binds and inhibits Ptch action, inducing signaling transduction through Smo. This signaling cascade results in the nuclear localization and activation of the Gli family of transcription factors. Although Hh is a major regulator of cell fate decision and body segment polarity (Nusslein-Volhard and Wieschaus, 1980), its role in HSC homeostasis and differentiation remains controversial. Several reports have suggested that Hh signaling is critical for HSC and hematopoietic progenitor differentiation. A study of zebrafish hematopoiesis revealed that embryo mutants of the Hh pathway display defects in HSC formation (Gering and Patient, 2005), indicating that Hh is required for definitive hematopoiesis. Consistent with these data, in vitro studies found that antibodies to Hh inhibited the cytokine-induced proliferation of human primitive hematopoietic stem cells, whereas Shh induced the expansion of human hematopoietic repopulating cells (Bhardwaj et al., 2001). In addition, analysis of Ptch1+/- mice showed that Hh activation expanded primitive bone marrow cells, but continued Hh activation led to HSC exhaustion (Trowbridge et al., 2006). Furthermore, a recent study using an in vivo model of Hh deficiency suggested that HSCs require Smo-mediated signals for their homeostasis (Zhao et al., 2009a). In contrast to these studies, it was proposed that Hh signaling is involved at the level of lymphocyte lineage commitment as a defect in the common lymphoid progenitor (CLP) population was observed upon deletion of Ptch1 (Uhmann et al., 2007). Moreover, Hh signaling has been demonstrated to be important for the differentiation and proliferation of hematopoietic progenitors in the thymus (Crompton et al., 2007; El Andaloussi et al., 2006). Finally, a recent report suggested that Hh signaling is essential for the differentiation of leukemia-initiating cells, introducing Hh inhibitors in clinical trials targeting BCR-ABL+ leukemia (Dierks et al., 2008; Dierks et al., 2007).
As none of these studies directly targeted Hh function specifically in adult HSCs, we decided to address HSC-specific Hh function in vivo. To this end, both gain- and loss-of-function conditional Smo genetic models were used, as the Smo receptor is the only non-redundant element of the Hh pathway. Surprisingly and contrary to the consensus view, Hh signaling appeared to be dispensable for the self-renewal and differentiation of adult bone marrow HSC. Indeed, neither conditional deletion of the Smo signal transducer nor hyper-activation of the Hh pathway had an affect in adult HSC maintenance and function. Interestingly, Hh signaling also appeared to be dispensable for the function of putative leukemia-initiating cells in T-cell leukemia as induction and progression of the disease was unaffected by silencing of the pathway.
Results
Conditional deletion of Smo fails to affect HSC maintenance in vivo
To study the role of Hh signaling in adult HSCs, we generated a Cre-regulated conditional model of Smo deletion (SmoF/FMx1-Cre+, Figure 1A), in which expression of the Cre recombinase is under the control of myxovirus-resistance 1 (Mx1) gene promoter (Mx1-Cre) (Gu et al., 1994) and is induced by interferon-α (via stimulation with polyI:polyC). In these mice, the first exon of the Smo locus is flanked by loxP sites and is deleted upon Cre-mediated recombination (Long et al., 2001). SmoF/FMx1-Cre-(control) and SmoF/FMx1-Cre+ littermate mice were treated with polyI:polyC. This treatment resulted in the efficient deletion of Smo floxed alleles and the generation of a recombined Smo deleted (Δ) alleles (Figure 1B, lane 4). At the mRNA level, Smo was not detectable in SmoΔ/Δ bone marrow cells, and the expression of Ptch1, a key target gene of Hh activation, was significantly reduced compared to the control mice (Figure 1C).
Figure 1. Phenotypically normal HSCs and progenitors in Smo-deficient mice.
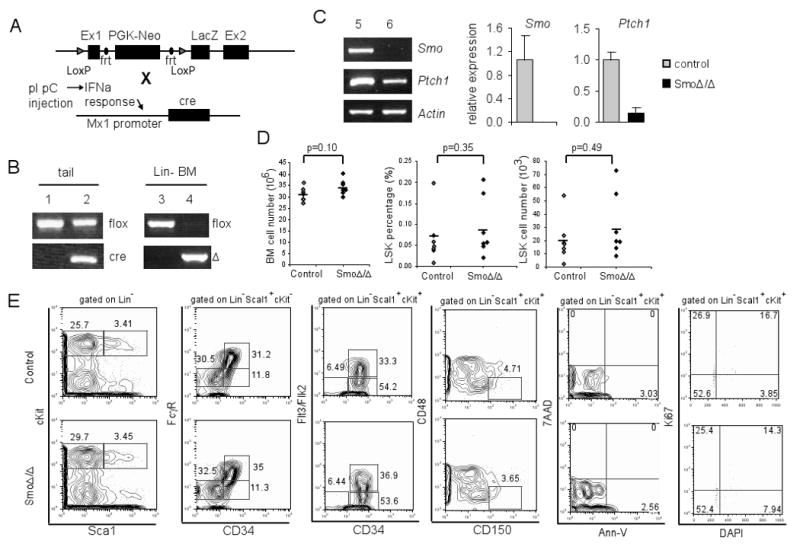
(A) Schematic representation of Smo-floxed allele (upper lane) and Mx1-Cre allele (lower lane). (B) PCR of genomic DNA extracted from mouse tails or lineage negative bone marrow cells to detect the Smo floxed, deleted (Δ), or Cre allele. Lane 1, 3: SmoF/FMxCre-; Lane 2, 4: SmoF/FMxCre+. (C) RT-PCR and quantification of Smo and Ptch1 mRNA in lineage negative bone marrow cells from polyI:polyC injected SmoF/FMxCre- (lane 5) and SmoF/FMxCre+ (lane 6) mice. The expression levels were normalized against β-actin. (D) Total number of bone marrow cells and LSK cells in control and SmoΔ/Δ mice. Each diamond represents a single mouse, and the bar indicates the average numbers. (E) FACS plots of bone marrow from control and SmoΔ/Δ mice. Representative plots (from at least 20 individual experiments) are shown.
Initial analysis of control and Smo-deficient mice at 4 weeks post polyI:polyC injection demonstrated no significant alteration in the overall bone marrow cellularity (p=0.10). Further analysis showed that Smo deletion had no effect on the relative frequency (p=0.35) or the absolute number of Lin-Sca1+cKit+ (LSK), a cell population enriched for HSCs (p=0.49) (Figure 1D). HSCs differentiate and give rise to myeloid progenitors (MP, Lin-Sca1-cKit+), which can be subdivided into common myeloid progenitors (CMP, Lin-Sca1-cKit+CD34+FcγRlow), granulocyte-monocyte progenitors (GMP, Lin-Sca1-cKit+ CD34+FcγRhi), and megakaryocyte/erythrocyte progenitors (MEP, Lin-Sca1-cKit+CD34-FcγRlow). Our analysis showed that CMP, GMP and MEP compartments were comparable between Smo-deficient and control mice (Figure 1E). Moreover, the percentages of terminally differentiated B- and T-lymphocytes appear normal in Smo-deficient spleen and bone marrow (Figure S1). In the thymus, the distribution of mature (CD4+, CD8+) and immature (CD4+8+, CD4-8-) compartments appeared similar to controls. Further subdivision of the CD4-8- compartment using the CD25 and CD44 surface antigen expression also revealed a normal distribution (Figure S1). The lack of a perturbation of the hematopoietic compartment was not due to early time-point analysis as Smo-deficient mice at 16 weeks post polyI:polyC injection also displayed normal populations of LSK, progenitors and lymphocytes despite the complete absence of Smo mRNA (Figure S2).
We further examined the LSK population, which can be subdivided into long term (LT)-HSC (LSKCD34-Flt3/Flk2-), short term (ST)-HSC (LSKCD34+Flt3/Flk2-) and multipotent progenitors (MPP, LSKCD34+Flt3/Flk2+). We observed comparable numbers of LT-, ST-HSC and MPPs between control and Smo-deficient mice (Figure 1E). Also similar analysis using CD150 and CD48 as markers of LT-HSC (LSKCD48-CD150+) did not reveal any abnormalities in Smo-deficient mice (Figure 1E). Additionally, we investigated the pro-survival and pro-proliferative functions of the Hh pathway. Staining for the pro-apoptotic marker AnnexinV did not reveal abnormal induction of cell death in Smo-deficient LSKs. Furthermore cell cycle analysis of the marker for proliferation Ki67 in conjunction with DAPI to measure DNA content did not reveal any differences in the cell cycle profiles between control and Smo-deficient LSKs (Figure 1E). These findings strongly suggested that Smo-mediated Hh signaling is dispensable for adult HSC and progenitor homeostasis and differentiation. To further test this hypothesis, we analyzed Gli1lacZ/lacZ mice (Bai et al., 2002), in which Gli1, a key transcription activator of the Hh pathway, is deleted and replaced by a lacZ allele. We did not detect any defects in the HSC compartment or in T and B lymphopoiesis in the bone marrow and the thymus of Gli1lacZ/lacZ mice (Figure S3), suggesting that Gli1 function is dispensable for hematopoiesis.
Smo-deletion does not alter differentiation ability of progenitor cells
To test functionality of Smo-deficient stem cells and progenitors, LSKs were flow-purified from either control or Smo-deficient bone marrows and methylcellulose assays were performed in presence of the appropriate cytokines. Both types of LSK cells generated similar numbers of colony-forming units (CFU) in both primary and secondary platings (Figures 2A and 2B). The deletion of Smo was confirmed by colony-specific PCR. The results showed that 14 out of the 15 studied colonies derived from Smo-deficient LSKs deleted the Smo allele. The expression of Smo mRNA was not detectable by quantitative RT-PCR; moreover, the expression of Ptch1 mRNA was significantly reduced in Smo-deficient LSK-derived colonies (Figures 2C and 2D). These results suggest that Smo is dispensable for short-term differentiation ability of hematopoietic progenitor cells.
Figure 2. Physiological differentiation of Smo-deficient progenitors.
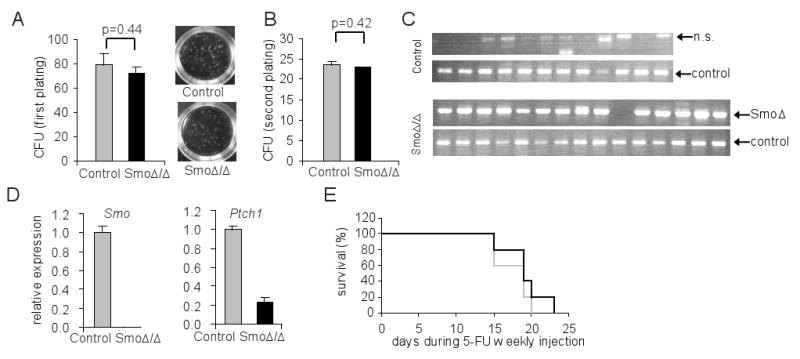
(A) Number of colonies was scored on day 7 of methylcellulose assay, and images of plates are shown on right panel. (B) Number of colonies was scored on day 7 after re-plating cells from A. (C) PCR on genomic DNA of colonies formed from A. The Smo Δ allele and a loading control genomic allele are shown. (D) Quantitative RT-PCR of Smo and Ptch1 on colonies formed from A. The expression levels were normalized against β-actin. (E) Survival curve of control (grey) or SmoΔ/Δ (black) mice after weekly 5-FU injection (n=5).
ST-HSC cells are able to give rise rapidly to colonies in the spleen when transplanted into lethally irradiated hosts. To study the effect of Smo deletion in this process, CFU-spleen units (CFU-S) were scored after transplanting either control or Smo-deficient bone marrow cells. We obtained identical CFU-S scores for the two groups (Figure S4), again indicating that Smo function is dispensable for rapid progenitor differentiation. To test the capability of Smo-deficient progenitors to expand and replenish the immune system, control and Smo-deficient mice were challenged weekly with a dose of 5-fluorouracil (5-FU) to eradicate cycling cells (Berardi et al., 1995) and the survival of these mice was observed. Similar survival percentage in the two groups (Figure 2E) suggested that the Smo-deficient progenitor cells were able to enter into cell cycle at a comparable level as wild-type cells. Collectively these results demonstrate that Smo is dispensable for short-term differentiation of adult progenitor cells both in vitro and in vivo.
Smo deletion does not affect HSC self-renewal and reconstitution ability
One explanation for the lack of an overt effect of Smo loss on HSC maintenance or function is that the potential Hh function is masked due to the nature of the analysis utilized, and that it can be revealed only in a competitive setting. To test the reconstitution capacity of Smo-deficient HSCs, competitive bone marrow transplantations (BMT) were performed. Bone marrow cells from either control or Smo-deficient mice (CD45.2+/Ly5.2+) were competed with an equal number of bone marrow cells from isogenic CD45.1+/Ly5.1+ mice and transplanted into lethally irradiated Ly5.1+ recipients (Figure 3A). Peripheral blood analysis of chimerism of the recipients showed that Smo-deficient HSCs were able to compete with wild-type HSCs, in a manner similar to control HSCs (Figure 3B). Similar assays were performed by mixing flow-purified LSKs from either control or Smo-deficient mice with competing Ly5.1 bone marrow cells. Once more, no significant differences were observed between control and Smo-deficient LSKs 14 weeks after BMT (Figure 3C). This lack of phenotype was not due to partial or inefficient deletion of the Smo, as quantitative RT-PCR in flow-purified Ly5.2+Lin- bone marrow cells of recipient mice 16 weeks after BMT showed a complete loss of Smo mRNA expression (Figure 3D). Indeed, at week 16 after BMT, Ly5.2+ Smo-deficient donor cell-derived LSK cells were present in the bone marrow, B220+ B and CD3+ T cells in the spleen of recipients (Figure 3E), demonstrating the repopulation ability of Smo-deficient HSCs.
Figure 3. Physiological competitive ability of Smo-deficient hematopoietic progenitors.
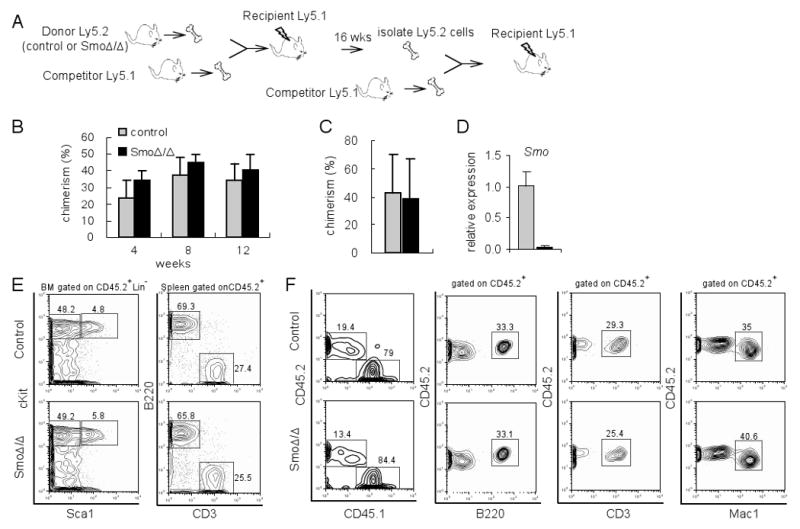
(A) Scheme of primary and secondary bone marrow transplantation (BMT). (B) Percentage of chimerism in peripheral blood of recipient mice at different time points after primary competition BMT. Donor cells were total bone marrow cells. Mean ± S.D. are shown (n=8). (C) Chimerism of peripheral blood of recipient mice 14 weeks after primary competition BMT. Donor cells were flow-purified LSK cells. Mean ± S.D. are shown (n=4). (D) Quantitative RT-PCR of Smo in flow-purified Ly5.2+Lin- bone marrow of recipients 16 weeks after BMT. Grey: control; Black: SmoΔ/Δ. The expression levels were normalized against β-actin. (E) Representative FACS plot of bone marrow and spleen of primary recipient mice 16 weeks after BMT. (F) Representative FACS plot of peripheral blood in the recipient (n=4 for each genotype) 12 weeks after secondary competition BMT.
To more rigorously test the repopulation ability Smo-deficient HSCs, a secondary competitive BMT was performed using donor-derived Ly5.2+ bone marrow cells isolated from the recipients of the primary transplant. We observed that the reconstitution ability of Smo-deficient HSCs was identical to that of control HSCs even in this sensitive serial transplantation setting. As shown in Figure 3F, at 12 weeks post secondary BMT, the chimerism in peripheral blood was comparable between recipients that had received control or Smo-deficient cells, and donor-derived B220+, CD3+ and Mac1+ cells were present at similar percentages. Taken together these data indicate that deletion of Smo has no significant effect on HSC repopulation ability.
Smo deletion does not alter HSC-specific gene expression signature
The absence of a phenotypic defect in HSCs that lack Smo led us to search for a putative role for Hh signaling in stem cell and progenitor gene expression patterns. To examine whether the loss of Smo results in changes at the molecular level in HSCs, microarray analysis was performed using flow-purified LSK and MP populations from either control or Smo-deficient mice. The array analysis (Figure S5C) and qRT-PCR studies (data not shown) showed a complete loss of Smo expression in both LSK and MP compartments. When control LSKs were compared with control MPs in duplicate experiments, 739 genes changed expression levels by 2-fold or greater (Figure S5A). For the purpose of this analysis, these 739 genes were regarded as an HSC-enriched gene expression “signature”. As a proof of principal, it was shown that this specific gene “signature” was lost upon deletion of Fbw7, a ubiquitin ligase that is essential for the maintenance of HSC quiescence (Thompson et al., 2008). Indeed, 43% (315 out of 739) of these selected genes were significantly down-regulated in Fbw7-deficient LSKs. In contrast, less than 10% (70 out 739) of these genes changed (up- or down-regulated) in response to Smo deletion (Figure S5B), suggesting that the HSC gene signature is largely preserved in Smo-deficient LSKs.
Previous reports (Forsberg et al., 2005; Jankovic et al., 2007; Mansson et al., 2007; Terskikh et al., 2003; Thompson et al., 2008) have defined a list of genes closely associated with LT-HSC activity. These genes are highly expressed in LT-HSCs but are down-regulated as HSCs lose their self-renewal abilities. These genes include transcription factors/cofactors important for HSC self-renewal and differentiation (Meis1, Egr1, Eya1/2), surface receptors (Mpl, Thy1, Agpt) as well as regulators of HSC survival (Mcl1). Our array analyses showed that the expression of these genes was not altered by the inducible deletion of Smo (Figure S5C). These data demonstrate that Smo is not required for the maintenance of adult HSC properties at the molecular level and support our findings that HSCs are phenotypically normal in the absence of Smo.
Absence of functional redundancy between the Notch and Hh pathways in hematopoiesis
The possibility remains that redundancy between signaling pathways masked a potential function for Hh in the early stages of hematopoiesis. We have shown previously that the Hh and Notch pathways share similar expression patterns and putative functions. Also, Smo mRNA expression appears to be significantly induced in response to Notch activation in Linneg bone marrow progenitors (El Andaloussi et al., 2006; Vilimas et al., 2007). These observations suggest a functional redundancy between the two pathways in adult HSC function. To test this hypothesis, we generated mice deficient for both Smo and RBPJ, a DNA binding factor required for canonical Notch signaling and performed competitive BMT. We injected polyI:polyC into RBPJF/FSmoF/FMx1-Cre+ mice, and confirmed the excision of Smo- and RBPJ-floxed alleles as well as the recombination of both alleles in the bone marrow (Figure 4A). Next we performed competitive BMT and found that RBPJ/Smo-deficient (DKO) cells were able to efficiently reconstitute irradiated hosts. Analysis of chimerism in the peripheral blood 6 to 10 weeks after BMT did not reveal any defects for DKO cells (Figure 4B). In fact, donor cells derived from DKO bone marrow were able to give rise to LSKs in bone marrow (Figure 4C) and B220+ cells in the spleen (Figure 4F). There were no significant differences in the number of donor-derived LSKs between the two groups 12 weeks post BMT (Figure 4D). As expected, donor-derived T cells (CD4+CD8+ T cells in thymus, and TCRβ+ cells in spleen) (Figures 4E and 4F) and marginal zone B cells (B220+CD21highCD23low/- cells in spleen) (Figure 4F) were reduced in recipients transplanted with DKO bone marrow, since Notch signaling is required for the development of these two lineages. Collectively these results show that neither Notch nor Hh signaling are necessary for adult HSC maintenance and differentiation. Furthermore these data suggest that these two pathways are not redundant in governing HSC fate.
Figure 4. Physiological competitive ability of RBPJ- and Smo- double deficient progenitors.
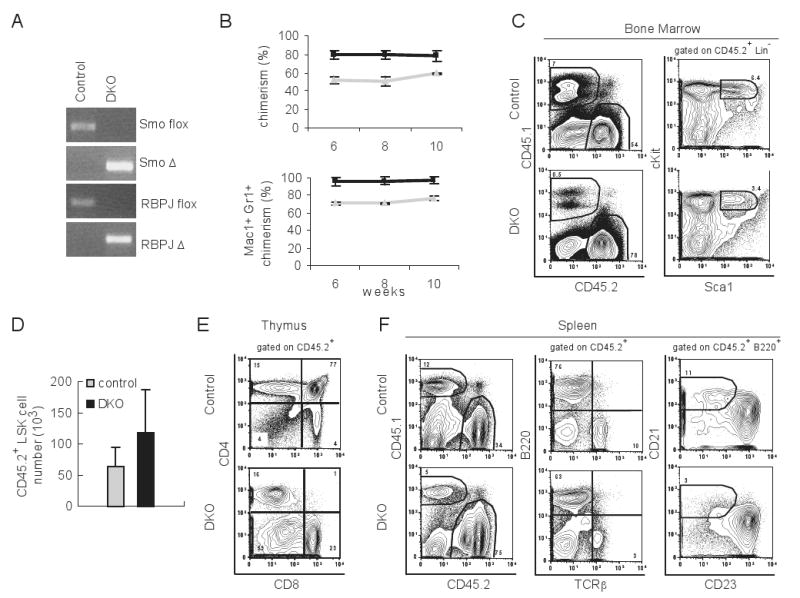
(A) PCR of genomic DNA extracted from bone marrow cells of polyI:polyC injected RBPJF/FSmoF/FMx1-Cre- mice (control) and RBPJF/FSmoF/FMx1-Cre+ mice (DKO). Smo and RBPJ floxed and deleted (Δ) alleles were detected. (B) Chimerism of total peripheral blood (upper panel) or Mac1+Gr1+ cells in peripheral blood (lower panel) of recipient mice at different time points after primary competition BMT. Donor cells were mixed of 1:2 ratios of Ly5.1+ cells and Ly5.2+ cells. Ly5.2+ cells were either from control (grey) or DKO mice (black). Mean ± S.D. are shown (n=2 for control, n=6 for DKO). (C) Representative FACS plot of bone marrow in the recipient mice 12 weeks after competitive BMT. (D) Number of donor derived LSK cells in bone marrow 12 weeks after BMT. Mean ± S.D. are shown (n=10 for control, n=4 for DKO). (E) Representative FACS plot of thymus in the recipient at 12 weeks after competition BMT. (F) Representative FACS plot of spleen in the recipient 12 weeks after competitive BMT.
Mapping of Hh signaling component expression in HSC and their niche
The absence of a phenotype in Smo-deficient HSCs suggested that the Hh signaling might not be active or of low activity in these cells. To determine whether elements of the Hh signaling network can be detected in either HSCs or the HSC “niche”, the expression of the components of this pathway was examined in flow-purified LSKs and differentiated MPs. We found that both the Smo transducer and Ptch1 receptor mRNAs (which is also a target gene of Hh signaling) were expressed in LSKs and MPs, (Figure S6A). In contrast, the members of Hh ligand family, Ihh and Dhh but not Shh mRNA was detected in primary preparations of calvarial osteoblasts, cells that comprise the osteoblastic HSC niche (Figure S6B), demonstrating that Hh ligands are available to HSCs. Several mouse and human osteoblastic lines showed similar Hh expression profiles (not shown). However, the expression of the downstream transcription factors Gli1, Gli2 and Gli3 was not detectable in either LSKs or MPs by quantitative PCR (Figure S6C) and microarray analysis (data not shown), a result that suggested low levels of Hh activity in both LSK and MP populations. Therefore, these data indicate that HSCs and progenitors have the ability to receive Hh signaling since they express both the Smo and Ptch1 receptors and Hh ligands are present in the niche. Nevertheless, there is little ongoing Hh activity as the transcription factors are not expressed, which is consistent with the described lack of HSC phenotype in Smo-deficient mice.
Hh pathway activation does not expand HSC or enhance their engrafting ability
It has been proposed previously that Hh morphogens could be used for in vitro expansion of primitive stem cell and progenitor populations and thus could be beneficial in transplantation protocols (Bhardwaj et al., 2001). Our results have shown an incomplete Hh activation in HSCs suggesting that Hh pathway activation could either expand HSC or provide them with competitive advantage in transplantation settings. To directly test this hypothesis, we used a Hh gain-of-function model (R26SmoM2), in which enhanced yellow fluorescent protein (EYFP) was fused with the constitutively active W539L point mutation of the mouse smoothened homolog gene (SmoW539L), and “knocked” into the ubiquitously-expressed ROSA26 locus (Jeong et al., 2004). The expression of SmoM2/EYFP fusion gene is blocked by a loxP-flanked STOP fragment inserted between the ROSA26 promoter and the SmoW539L/EYFP sequence (Figure 5A). We crossed these mice to the Mx1-Cre stain and generated R26SmoM2/SmoM2Mx1-Cre+ (referred to hereafter as Cre+) or R26SmoM2/SmoM2 Mx1-Cre- (referred to as Cre-), and induced SmoW539L/EYFP expression by injecting polyI:polyC. As shown in Figure 5B, YFP expression was detected by flow cytometry in LSKs of Cre+ mice after polyI:polyC administration. At the mRNA level, both Smo and Ptch1 expression were significantly increased in the LSKs of Cre+ compared to Cre- mice (Figure 5C), demonstrating the over-expression of Smo and activation of the Hh pathway. Also, it was found that elements of the Hh pathway (Ptch, Gli1) were aberrantly expressed in differentiated hematopoietic cells (thymic CD4+8+ cells) in which the pathway is normally silent (not shown). An additional indication of non physiological Hh activation was that the majority of Cre+ mice died later in life due to the development of tumors (primarily medulloblastomas and skin tumors, data not shown). However, Cre+ mice did not show any increase in absolute numbers of LSKs (Figure 5D), illustrating that the hyper-activation of Hh signaling was unable to result in expansion of the LSK compartment. Moreover, LSKs of Cre+ mice did not show any signs of enhanced (or suppressed) apoptosis or aberrant cell cycle profiles as shown by AnnexinV or Ki67 staining. Finally, no major defects in lymphopoiesis were detected (Figure 5E).
Figure 5. Hyper-activation of the Hh pathway does not expand HSC compartment.

(A) Schematic representation of R26SmoM2 locus (upper panel) and Mx1-Cre locus (lower panel). (B) Histogram of YFP gated on LSKs. Grey line: Rosa26SmoM2/SmoM2Mx1-Cre-. Black line: R26SmoM2/SmoM2Mx1-Cre+. (C) Quantative RT-PCR of Smo and Ptch1 in LSKs. The expression levels were normalized against β-actin. (D) Frequency of LSK cells. Each diamond represents a single mouse, and the bar indicates the average number. (E) Representative FACS plots of bone marrow from R26SmoM2/SmoM2Mx1-Cre- and R26SmoM2/SmoM2Mx1-Cre+ mice. (F) Number of colonies was scored on first and secondary plating of methylcellulose assay. (G) Histogram of YFP gated on lineage negative cells of bone marrow, which were used for BMT in H. Grey line: Rosa26SmoM2/SmoM2Mx1-Cre-. Black line: R26SmoM2/SmoM2Mx1-Cre+. (H) Percentage of peripheral blood chimerism in recipient mice after competitive BMT of R26SmoM2/SmoM2Mx1-Cre- (grey) and R26SmoM2/SmoM2Mx1-Cre+ (black) at different time points. One line represents one mouse (n=4).
To further examine whether hyper-activation of Hh influences the ability of LSKs to differentiate, CFU methylcellulose-based assays were performed. We observed that Smo-mutant LSKs gave rise to similar number of colonies as controls. Moreover, replating of the colonies that originated from the Hh hyper-active LSKs also generated identical number of colonies as controls (Figure 5F).
To directly test the reconstitution ability of Hh hyper-active HSCs, we transplanted bone marrow cells from polyI:polyC-treated Cre+ mice (Ly5.2+) mixed with Ly5.1+ competing bone marrow into lethally irradiated Ly5.1+ hosts. Bone marrow cells used for competitive BMT were confirmed to express YFP as shown in Figure 5G. The chimerism in the peripheral blood 4-12 weeks after transplant was similar between Cre- and Cre+ groups (Figure 5H), indicating that over-expression of an activated Smo does not provide a competitive advantage to HSCs. These observations argued against the suggestion that Hh hyper-activation affects HSC expansion and in vivo fitness.
Hedgehog signaling is dispensable for the induction or maintenance of lymphoblastic leukemia
Our studies so far do not support a role for Hh signaling in physiological adult HSC function. Recent reports (Dierks et al., 2008; Zhao et al., 2009b) suggested that Hh could affect BCR-ABL+ leukemia stem cell function and disease progression. These conclusions led us to study the potential role of Hh in the induction and maintenance of a different leukemia type, acute lymphoblastic leukemia (ALL). It has been shown that the majority of primary cases of T cell ALL (T-ALL) carry activating NOTCH1 mutated alleles (Weng et al., 2004). The study of these tumors have revealed that the Hh pathway was active in T-ALL, since both GLI1 and PTCH1 were highly expressed in many Notch1 mutant T-ALL cell lines (unpublished data) and PTCH1 were expressed in majority (38 out of 48) of primary T-ALL cases (Figure 6A). Thus, to test whether Hh signaling is required for the transformation of hematopoietic progenitors in T-ALL, a well characterized transplantation model (Vilimas et al., 2007) was used. Lineage-depleted bone marrow from either polyI:polyC injected SmoF/FMx1-Cre- or SmoF/FMx1-Cre+ mice was isolated, and infected with a bicistronic retroviral vector expressing the intracellular domain of Notch1 (Notch-IC), and green fluorescent protein (GFP). As expected, peripheral blood analysis of recipients of Notch-IC infected control cells revealed that the majority of cells were GFP+, a marker of Notch-IC expression, and most of which were CD4+8+, a manifestation of T-ALL (Figure 6B). Examination of recipients that had received Notch-IC infected Smo-deficient cells showed similar percentages of GFP+CD4+CD8+ cells in the peripheral blood as well as kinetics of leukemogenesis (Figures 6B and 6C). We further confirmed that T-ALLs developed from Smo-deficient cells deleted Smo-floxed and harbored SmoΔ allele (Figure 6D), which demonstrated that Hh signaling is dispensable for T-ALL generation.
Figure 6. Leukemia (T-ALL) induction and maintenance is not altered by Smo deficiency.
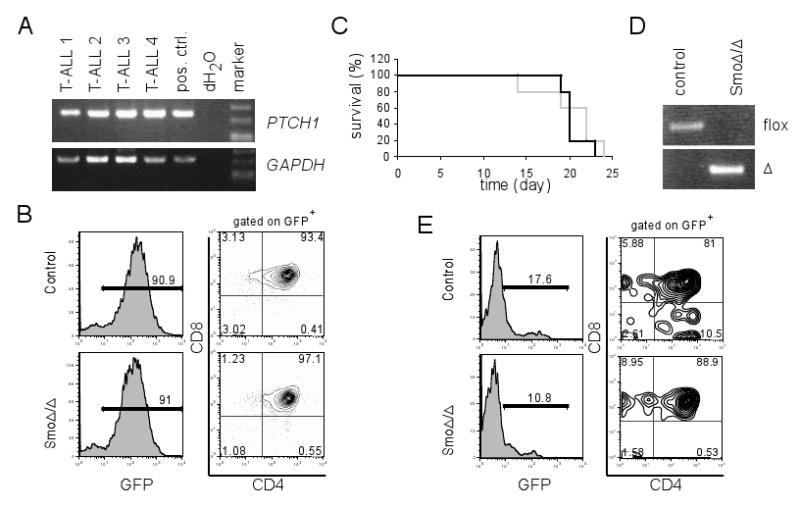
(A) RT-PCR of PTCH1 in primary T-ALL samples. GAPDH served as a loading control. (B) Representative FACS plots for CD4 and CD8 staining of peripheral blood from the recipients 2 weeks following transplantation with Notch-IC infected control or SmoΔ/Δ lineage-negative bone marrow cells. Notch-IC infected cells were identified by gating on GFP+ cells. (C) Survival curve of host mice transplanted with Notch-IC infected control (black) or SmoΔ/Δ lineage-negative bone marrow cells (grey) (n=5). (D) PCR of Smo-floxed and Δ alleles on genomic DNA purified from GFP+CD4+CD8+ peripheral blood of host mice in B. (E) Representative FACS plots for CD4 and CD8 staining of peripheral blood from the recipients (n=5) 3 weeks after secondary BMT. 5×106 GFP+ bone marrow cells from primary recipients were used for secondary BMT.
To address the ability of Smo-deficient tumors to regenerate, we performed secondary BMTs using Notch1-transformed (GFP+) leukemic cells. No significant differences in the induction of secondary leukemia were noted, in opposition to a role for Hh signaling in the regulation of putative “leukemia-initiating” cells. (Figure 6E). To further demonstrate that Hh signaling is not essential for the maintenance of the transformed cells, several T-ALL lines (PTCH1 and GLI1 positive) were incubated with the potent and specific Smo inhibitor, cyclopamine. In agreement with our in vivo data, the presence of cyclopamine did not affect the leukemic cell line survival or the rate of proliferation (not shown). Taken together, these results indicate that Hh activation is dispensable for the transformation of hematopoietic progenitors and the progression of Notch-induced T-ALL.
Discussion
In this study, we demonstrate that inducible genetic deletion of the only non-redundant element of the Hh cascade, Smo, was unable to affect adult hematopoiesis, specifically at the level of the HSC. Smo-deficient HSCs display normal abilities to differentiate, self-renew and regenerate the immune system. In agreement with these phenotypic and functional studies, gene expression profiling analysis demonstrated that HSC-specific gene expression “signature” was preserved in Smo-deficient HSCs. Interestingly, the simultaneous ablation of both the Hh and Notch pathways was also unable to affect HSC differentiation and function. Moreover, using a gain-of-function model, we found that Hh hyper-activation did not lead to expansion of the HSC compartment. Finally, Smo deletion had no effect on the ability of the Notch1 oncogene to transform early hematopoietic stem cells and progenitors and to induce T-ALL. All of these findings are of unique importance as they directly question the current consensus on the role of Hh signaling in adult hematopoiesis.
Our studies are in contrast to a recent report by Zhao et al. that also used a conditional Smo allele deletion (Zhao et al., 2009). One possible explanation for this discrepancy is the utilization of a distinct mode of deletion. Zhao et al. use the Vav-cre deleter strain that appears to be hematopoietic specific; however it is able to delete the Smo alleles in both adult and fetal hematopoiesis. Indeed it was previously shown that the Vav promoter can efficiently drive Cre-recombinase expression in e.d. 13.5 fetal liver HSC (Stadtfeld and Graf, 2005). It is possible that the reported HSC defects in the Vav-creSmoF/F model reflect Hh signaling functioning not in adult but in fetal HSC function and hematopoiesis. Although future work is required to identify putative Hh roles in fetal hematopoiesis, our data clearly demonstrate that Hh signaling is dispensable for adult HSC function.
Our observations suggest that Hh hypeactivation is unable to expand bone marrow stem cells and progenitors, a conclusion that is inconsistent with a report by Trowbridge and colleagues (Trowbridge et al., 2006). A potential reason for this discrepancy could be the utilization of different animal models. In the germline Ptch+/- model both HSC and/or the HSC “niche” could contribute to phenotype, whereas in the inducible SmoM2 model expression of the activated allele is largely restricted to the hematopoietic compartment. Moreover, putative differences on the effect of Hh hyper-activation on HSC/LSK cell cycle progression could be explained by the differential analysis performed. Indeed, Trownbridge et al. study the cell cycle status of Ptch+/- LSKs after transplantation while we study steady- state LSKs shortly after SmoM2 activation. Finally, it is important to note that the gain-of-function of a pathway effector (SmoM2) may well engender a different hematopoietic phenotype than the loss-of-function of a negative regulator (Ptch) that may have effects on other signaling pathways that could influence hematopoiesis.
Our analyses also failed to demonstrate a significant effect of Smo deletion on T cell differentiation, as proposed previously by several studies including one from our own laboratory, in which Smo was deleted in early T cell progenitors using the Lck-cre strain (Crompton et al., 2007; El Andaloussi et al., 2006). This discrepancy could be due to the differential mode of Cre-recombinase activation and pathway deletion. Indeed, Lck-cre is only active in early thymocytes and it ensures deletion in both fetal and adult thymus, suggesting again that fetal and adult hematopoiesis has unique and distinct Hh signaling requirements. Another reason for the phenotypic discrepancy could be that the Lck-cre-driven thymic effect was only partial. It is thus possible that our current studies, aimed mainly at HSC function, were not quantitative enough to reveal slight alterations of early T cell differentiation. It is more difficult to explain the differential effects on thymic size and progression of T cell maturation of the Mx1-cre-mediated Smo deletion reported by El Andaloussi et al. The timing of the analysis could provide a potential explanation. In this study thymi were analyzed at week 4 and 16 post-deletion, while El Andaloussi et al analyzed mice only one week after the last polyI:polyC injection. It is thus possible that the outcome of these studies was dictated by the timing of the analysis, especially as the thymus is a tissue with enormous regenerative capacity. Additional explanations could also include background differences as the mice studied here (and by Hoffman et al.) are C57Bl/6 SmoF/F while El Andaloussi et al. utilized 129X1/SvJ SmoF/null animals. It is thus possible that the effects on T cell development were influenced by the genetic background of the analyzed mice. Future studies that directly compare T cell development in the different Hh deficient strains are necessary to address the extent of Hh function in T cell development.
Is there any role for Hh in hematopoiesis? The strongest evidence supporting a pivotal role of Hh signaling in hematopoiesis came from the study of zebrafish embryo Hh mutants. (Gering and Patient, 2005). As zebrafish hematopoiesis shares striking similarities to the mammalian fetal blood development, it is possible that the Hh pathway, as previously suggested, plays a more prominent role during fetal blood development. Moreover, it is possible that Hh function is masked by the synergistic function of other signaling pathways. Indeed, pathways such as Notch and Wnt, which have been previously shown to be capable of interacting with Hh (Hallahan et al., 2004; Mak et al., 2006; Yang and Niswander, 1995; Yokota et al., 2004), could collaborate with each other to ensure self-renewal and specify differentiation (Duncan et al., 2005). In this report, we showed that deletion of both RBPJ and Smo did not affect HSC function, suggesting Notch and Hh signaling do not play synergistic roles. However, we cannot exclude potential redundancy with other signaling cues.
Two recent reports (Dierks et al., 2008; Zhao et al., 2009) have identified Smo as a drug target for the targeting of BCR-ABL+ human leukemic stem cells, introducing the notion that the Hedgehog pathway could be important for malignant hematopoiesis and the maintenance of leukemia. In the light of these seminal findings, our results are of further importance as they prove that pharmacological targeting of Hedgehog in leukemia is feasible as physiological HSC function and progression of hematopoiesis remains unaffected. They also suggest that not all blood malignancies can be treated using similar therapeutic protocols as the progression of T-ALL is not affected by the silencing of Hedgehog function.
Materials and Methods
Animals
SmoF/F mice (Long et al., 2001) were a gift of Dr. A. McMahon (Harvard University, Boston). Genotyping of SmoF/F (Long et al., 2001; Zhang et al., 2001) and RBPJF/F mice (Han et al., 2002; Tanigaki et al., 2004) was performed as previously reported. SmoF/FMx1-Cre animals were injected with 20μg polyI:polyC per gram of body weight for a total of 3 injections. The injections were initiated 14 days after birth and done every two days. Animals were analyzed 4-6 weeks after the last injection unless indicated otherwise. All animal experiments were done in accordance to the guidelines of the NYU School of Medicine. Gli1lacZ mice were a gift of Dr. A. Joyner (Memorial Sloan Kettering Cancer Center, New York). ROSA26SmoM2 mice (Jeong et al., 2004) were purchased from Jackson laboratory. For 5-FU experiments, 150 μg of 5-FU per gram of body weight were intra-peritoneally injected every week.
Antibodies and FACS analysis
Antibody staining and FACS analysis was performed as previously described (Aifantis et al., 1999). All antibodies were purchased from BD-Pharmingen or e-Bioscience. We used the following antibodies: c-kit (2B8), Sca-1 (D7), Mac-1 (M1/70), Gr-1 (RB6-8C5), NK1.1 (PK136), TER-119, CD3 (145-2C11), CD19 (1D3), IL7Rα(A7R34), CD34 (RAM34), FcγII/III (2.4G2), Flk-2/Flt-3 (A2F10.1), CD4 (RM4-5), CD4 (H129.19), CD8 (53-6.7), CD25 (PC61), CD44 (IM7), CD45.1 (A20), CD45.2 (104), CD150 (9D1), CD48 (HM481), Ki67, AnnexinV, 7-AAD. Bone marrow lineage antibody cocktail includes: Mac-1, Gr-1, NK1.1, TER-119, CD3, CD19. For Ki67 and DAPI staining, briefly, the cells were first treated with Fix and Perm reagents according to manufacturer's instruction (Invitrogen), stained with Ki67 for 20 minutes at room temperature, then washed and resuspended in PBS with 5μg/ml RNaseA and 2μg/ml DAPI.
RT-PCR
Total RNA was isolated using the RNeasy Plus Mini Kit (Qiagen) and cDNA was synthesized using the SuperScript First-Strand Kit (Invitrogen). Quantitative PCR was performed using iQ SYBR Green Supermix and an iCycler (Bio-Rad) using the primer sequences (Tm=60°C used for all primers) provided in supplemental table 1. T-ALL patient samples were provided by collaborating institutions in the United States (St. Jude Children's Research Hospital, Memphis, TN) and Canada (Hospital for Sick Children, Toronto, Canada) (Thompson et al., 2007).
Methylcellulose assay
LSK cells were flow-purified from polyI:polyC injected mice. LSK cells were plated in duplicate (500 LSK/35mm dish) into cytokine-supplemented methylcellulose medium (MethoCult 3434, Stem Cell Technologies), and the number and morphology of colonies were scored 7 days later. For secondary plating, cell colonies were pooled from the first plating, and 4,000 cells were plated in duplicate.
Bone marrow transplantation
5×105 bone marrow cells (Ly5.2+) or 500 LSKs (Ly5.2+) were transplanted by retro-orbital i.v. injections into lethally irradiated (960 cGy) BL6SJL (Ly5.1+) recipient mice in competition with 5×105 B6SJL (Ly5.1+) bone marrow cells. Peripheral blood of recipient mice was collected at 4, 8, and 12 weeks after transplant. For secondary transplanst, recipient mice were sacrificed 16 weeks after primary transplant. Ly5.2+ bone marrow cells were flow-purified and 5×105 cells were transplanted by retro-orbital i.v. injections into lethally irradiated (960 cGy) BL6SJL (Ly5.1+) recipient mice in competition with 5×105 B6SJL (Ly5.1+) bone marrow cells.
Microarray analysis
A group of four mice was pooled for each condition. Microarray analysis was performed as previously described (Thompson et al., 2008). Briefly, freshly isolated cells were sorted by surface marker expression, and total RNA was extracted using the RNeasy kit (QIAGEN, CA). In order to generate sufficient sample quantities for oligonucleotide gene chip hybridization experiments, we used the GeneChip Two-Cycle cDNA Synthesis Kit (Affymetrix, San Jose, CA) for cRNA amplification and labeling. The amplified cRNA was labeled and hybridized to the MOE430 Plus 2 oligonucleotide arrays (Affymetrix). The Affymetrix gene expression profiling data was normalized using the previously published Robust Multi-array Average (RMA) algorithm using the GeneSpring 7 software (Agilent, Palo Alto, CA). The gene expression intensity presentation was generated with MeV software (http://www.tm4.org). Microarray data were deposited under GEO database with the accession number GSE15194.
Retroviral infection of lineage-negative bone marrow cells
Bone marrow cells were enriched for lineage-negative cells using EasySep kit (StemCell Technology), and cultured in OPTI-MEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 ng/ml of SCF and Flt3l, 10 ng/ml of IL6 and IL7. For retroviral production, phoenix cells were transfected with pMigNotch-IC by calcium phosphate method. Virus supernatant was collected 48 hr post transfection and used directly for spin infection of lineage-depleted bone marrow cells at 2500 rpm for 90 minutes. Forty-eight hours after infection, 1×105 lineage-negative GFP-positive cells were i.v. injected into one lethally irradiated (960 cGy) C57BL/6J host mouse.
Statistical analysis
The means of each data set were analyzed using the Student's t test, with a two-tailed distribution and assuming equal sample variance.
Supplementary Material
Acknowledgments
We would like to thank Dr. A. McMahon for the SmoF/F animals, Dr. A. Joyner for Gli1lacZ animals, Jiri Zavadil for advice on microarray analysis and Peter Lopez for excellent cell sorting support. Drs. T. Reya, G. Gilliland for sharing unpublished observations and C.W. Brains for constructive discussions. The Aifantis laboratory is supported by a generous donation from the Helen L. and Martin S. Kimmel Stem Cell Center, the National Institutes of Health (R56AI070310, RO1CA105129, RO1CA133379 to I.A., RO1 DK52208 to S.D.N. and CA099978 to B.K.), the American Cancer Society (RSG0806801 to I.A.), the Leukemia and Lymphoma Society (Scholar Award to I.A. and a SCOR Award to S.D.N.), the NY State Department of Health (CO23058), the Irma T. Hirchl Trust and the E. Mallinckrodt Foundation (to I.A.). F.R. is supported by the Swiss National Science Foundation (F3100A0-119725) and the Swiss Cancer League (KLS-01840-02-2006). S.G. is supported by the MSTP program of the University of Chicago.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams GB, Scadden DT. The hematopoietic stem cell in its place. Nat Immunol. 2006;7:333–337. doi: 10.1038/ni1331. [DOI] [PubMed] [Google Scholar]
- Aifantis I, Feinberg J, Fehling HJ, Di Santo JP, von Boehmer H. Early T cell receptor beta gene expression is regulated by the pre-T cell receptor-CD3 complex. J Exp Med. 1999;190:141–144. doi: 10.1084/jem.190.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- Berardi AC, Wang A, Levine JD, Lopez P, Scadden DT. Functional isolation and characterization of human hematopoietic stem cells. Science. 1995;267:104–108. doi: 10.1126/science.7528940. [DOI] [PubMed] [Google Scholar]
- Bhardwaj G, Murdoch B, Wu D, Baker DP, Williams KP, Chadwick K, Ling LE, Karanu FN, Bhatia M. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol. 2001;2:172–180. doi: 10.1038/84282. [DOI] [PubMed] [Google Scholar]
- Cobas M, Wilson A, Ernst B, Mancini SJ, MacDonald HR, Kemler R, Radtke F. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med. 2004;199:221–229. doi: 10.1084/jem.20031615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton T, Outram SV, Hager-Theodorides AL. Sonic hedgehog signalling in T-cell development and activation. Nat Rev Immunol. 2007;7:726–735. doi: 10.1038/nri2151. [DOI] [PubMed] [Google Scholar]
- Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt-Graeff A, Landwerlin K, Veelken H, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14:238–249. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, Veelken H, Engelhardt M, Mertelsmann R, Kelleher JF, et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007;13:944–951. doi: 10.1038/nm1614. [DOI] [PubMed] [Google Scholar]
- Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C, Yoon K, Cook JM, Willert K, Gaiano N, et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol. 2005;6:314–322. doi: 10.1038/ni1164. [DOI] [PubMed] [Google Scholar]
- El Andaloussi A, Graves S, Meng F, Mandal M, Mashayekhi M, Aifantis I. Hedgehog signaling controls thymocyte progenitor homeostasis and differentiation in the thymus. Nat Immunol. 2006;7:418–426. doi: 10.1038/ni1313. [DOI] [PubMed] [Google Scholar]
- Forsberg EC, Prohaska SS, Katzman S, Heffner GC, Stuart JM, Weissman IL. Differential expression of novel potential regulators in hematopoietic stem cells. PLoS Genet. 2005;1:e28. doi: 10.1371/journal.pgen.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gering M, Patient R. Hedgehog signaling is required for adult blood stem cell formation in zebrafish embryos. Dev Cell. 2005;8:389–400. doi: 10.1016/j.devcel.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794–7800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M, Brook A, McMahon AP. The world according to hedgehog. Trends Genet. 1997;13:14–21. doi: 10.1016/s0168-9525(96)10051-2. [DOI] [PubMed] [Google Scholar]
- Han H, Tanigaki K, Yamamoto N, Kuroda K, Yoshimoto M, Nakahata T, Ikuta K, Honjo T. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. International immunology. 2002;14:637–645. doi: 10.1093/intimm/dxf030. [DOI] [PubMed] [Google Scholar]
- Jankovic V, Ciarrocchi A, Boccuni P, DeBlasio T, Benezra R, Nimer SD. Id1 restrains myeloid commitment, maintaining the self-renewal capacity of hematopoietic stem cells. Proc Natl Acad Sci U S A. 2007;104:1260–1265. doi: 10.1073/pnas.0607894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004;18:937–951. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J, Faubert A, Sauvageau G. Genetic programs regulating HSC specification, maintenance and expansion. Oncogene. 2004;23:7199–7209. doi: 10.1038/sj.onc.1207940. [DOI] [PubMed] [Google Scholar]
- Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128:5099–5108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- Maillard I, Koch U, Dumortier A, Shestova O, Xu L, Sai H, Pross SE, Aster JC, Bhandoola A, Radtke F, et al. Canonical notch signaling is dispensable for the maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2008;2:356–366. doi: 10.1016/j.stem.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak KK, Chen MH, Day TF, Chuang PT, Yang Y. Wnt/beta-catenin signaling interacts differentially with Ihh signaling in controlling endochondral bone and synovial joint formation. Development. 2006;133:3695–3707. doi: 10.1242/dev.02546. [DOI] [PubMed] [Google Scholar]
- Mansson R, Hultquist A, Luc S, Yang L, Anderson K, Kharazi S, Al-Hashmi S, Liuba K, Thoren L, Adolfsson J, et al. Molecular evidence for hierarchical transcriptional lineage priming in fetal and adult stem cells and multipotent progenitors. Immunity. 2007;26:407–419. doi: 10.1016/j.immuni.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311:1880–1885. doi: 10.1126/science.1110542. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- Stadtfeld M, Graf T. Assessing the role of hematopoietic plasticity for endothelial and hepatocyte development by non-invasive lineage tracing. Development. 2005;132:203–213. doi: 10.1242/dev.01558. [DOI] [PubMed] [Google Scholar]
- Stier S, Cheng T, Dombkowski D, Carlesso N, Scadden DT. Notch1 activation increases hematopoietic stem cell self-renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood. 2002;99:2369–2378. doi: 10.1182/blood.v99.7.2369. [DOI] [PubMed] [Google Scholar]
- Tanigaki K, Tsuji M, Yamamoto N, Han H, Tsukada J, Inoue H, Kubo M, Honjo T. Regulation of alphabeta/gammadelta T cell lineage commitment and peripheral T cell responses by Notch/RBP-J signaling. Immunity. 2004;20:611–622. doi: 10.1016/s1074-7613(04)00109-8. [DOI] [PubMed] [Google Scholar]
- Terskikh AV, Miyamoto T, Chang C, Diatchenko L, Weissman IL. Gene expression analysis of purified hematopoietic stem cells and committed progenitors. Blood. 2003;102:94–101. doi: 10.1182/blood-2002-08-2509. [DOI] [PubMed] [Google Scholar]
- Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007;204:1825–1835. doi: 10.1084/jem.20070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, Zavadil J, Nimer SD, Aifantis I. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med. 2008;205:1395–1408. doi: 10.1084/jem.20080277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge JJ, Scott MP, Bhatia M. Hedgehog modulates cell cycle regulators in stem cells to control hematopoietic regeneration. Proc Natl Acad Sci U S A. 2006;103:14134–14139. doi: 10.1073/pnas.0604568103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhmann A, Dittmann K, Nitzki F, Dressel R, Koleva M, Frommhold A, Zibat A, Binder C, Adham I, Nitsche M, et al. The Hedgehog receptor Patched controls lymphoid lineage commitment. Blood. 2007;110:1814–1823. doi: 10.1182/blood-2007-02-075648. [DOI] [PubMed] [Google Scholar]
- Vilimas T, Mascarenhas J, Palomero T, Mandal M, Buonamici S, Meng F, Thompson B, Spaulding C, Macaroun S, Alegre ML, et al. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med. 2007;13:70–77. doi: 10.1038/nm1524. [DOI] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- Yang Y, Niswander L. Interaction between the signaling molecules WNT7a and SHH during vertebrate limb development: dorsal signals regulate anteroposterior patterning. Cell. 1995;80:939–947. doi: 10.1016/0092-8674(95)90297-x. [DOI] [PubMed] [Google Scholar]
- Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116:1195–1201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota N, Mainprize TG, Taylor MD, Kohata T, Loreto M, Ueda S, Dura W, Grajkowska W, Kuo JS, Rutka JT. Identification of differentially expressed and developmentally regulated genes in medulloblastoma using suppression subtraction hybridization. Oncogene. 2004;23:3444–3453. doi: 10.1038/sj.onc.1207475. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Ramalho-Santos M, McMahon AP. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell. 2001;105:781–792. [PubMed] [Google Scholar]
- Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009 doi: 10.1038/nature07737. Advanced online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.