

Abstract
Functional interactions between glucocorticoids and the endocannabinoid system have been repeatedly documented; yet, to date, no studies have demonstrated in vivo that glucocorticoid hormones regulate endocannabinoid signaling. We demonstrate that systemic administration of the glucocorticoid corticosterone (3 and 10 mg/kg) resulted in an increase in the tissue content of the endocannabinoid N-arachidonylethanolamine (AEA) within several limbic structures (amygdala, hippocampus, hypothalamus), but not the prefrontal cortex, of male rats. Tissue AEA content was increased at 10 min and returned to control 1 h post corticosterone administration. The other primary endocannabinoid, 2-arachidonoylglycerol, was found to be elevated by corticosterone exclusively within the hypothalamus. The rapidity of the change suggests that glucocorticoids act through a non-genomic pathway. Tissue contents of two other N-acylethanolamines, palmitoylethanolamide and oleolyethanolamide, were not affected by corticosterone treatment, suggesting that the mechanism of regulation is neither fatty acid amide nor N-acylphosphatidylethanolamine phospholipase D. These data provide in vivo support for non-genomic steroid effects in mammals and suggest that AEA is a mediator of these effects.
Keywords: FAAH, non-genomic, limbic, cannabinoid, corticosterone, adrenal
Introduction
Glucocorticoid hormones are produced by the adrenal cortex and secreted into the general circulation following exposure to stressful or aversive stimuli; the glucocorticoid hormones promote adaptive physiological responses to the stressor and restoration of homeostasis. The effects of glucocorticoids occur in several phases, although they are typically categorized as those that occur rapidly (seconds to minutes) and those that develop more slowly (minutes to hours; (McEwen, 1994). Within the brain, as well as throughout other systems in the body, glucocorticoids exert effects through activation of cytosolic receptors, which dimerize, enter the nucleus and modify gene transcription through binding to specific response elements in the promoter regions of target genes. However, several studies have demonstrated physiological and behavioral effects of glucocorticoids that occur too rapidly to be mediated by alterations in gene transcription or are unaffected by protein synthesis inhibitors (Haller et al., 2008). Accumulating data indicate that these non-genomic effects of glucocorticoids occur through the activation of either G-protein coupled receptors or membrane-associated cytosolic steroid receptors (Karst et al., 2005; Tasker et al., 2006). These mechanisms produce rapid effects on behavior and physiology contributing to adaptive responses to stress as well as the immediate normalization of homeostasis following stress (Tasker et al., 2006). To date, few in vivo studies have elucidated the biochemical changes that underlie the rapid, nongenomic neurobehavioral effects of the glucocorticoids.
Endocannabinoids, acting through the CB1 cannabinoid receptor, subserve a retrograde signaling system that is widely distributed throughout the brain and functions to modulate axonal release of excitatory and inhibitory neurotransmitters (Freund et al., 2003). There are several lines of evidence which indicate that endocannabinoid signaling is regulated by glucocorticoid hormones. First, exposure of isolated hypothalamic slices to glucocorticoids rapidly induces endocannabinoid mobilization through activation of a G-protein coupled receptor (Di et al., 2003). Second, rapid behavioral responses to glucocorticoid administration in vivo are blocked by administration of CB1 receptor antagonists (Campolongo et al., 2009; Coddington et al., 2007), suggesting that glucocorticoids rapidly recruit endocannabinoid signaling. These data have lead to the hypothesis that endocannabinoids are the synaptic “workhorse” of glucocorticoids (Hill and McEwen, 2009). The purpose of this study was to extend these observations and measure the effects of exogenous glucocorticoid administration on corticolimbic tissue endocannabinoid contents. Our prediction that glucocorticoid administration would rapidly increase endocannabinoid contents was confirmed, lending further support to both the importance of rapid glucocorticoid signaling and the role of endocannabinoids in this mechanism.
Methods
Subjects
Seventy day old male Sprague-Dawley rats (250-325 g; Charles River, Kingston, NY), pair housed in standard maternity bins lined with contact bedding, were used in this study. Colony rooms were maintained at 21 °C, and on a 12 h light/dark cycle, with lights on at 0900 h. All rats were given ad libitum access to Purina Rat Chow and tap water. All protocols were approved by the Institutional Animal Care and Use Committee of Rockefeller University.
Procedure
Rats were administered a subcutaneous injection of vehicle (1% ethanol in sesame oil; Sigma-Aldrich, St. Louis MO) or corticosterone (3 or 10 mg/kg in vehicle; Sigma-Aldrich) and killed by decapitation 10 min or 1 h following injection. Brains were removed and the prefrontal cortex (a tissue block composed of medial prefrontal cortex and anterior cingulate, which was anatomically defined as the area dorsal to the anterior olfactory nucleus and medial to the corpus callosum and claustrum formation), amygdala (composed of central, basolateral, medial and cortical nuclei), hippocampus (containing all subregions of the hippocampus, as well as both dorsal and ventral regions) and hypothalamus (a tissue block that was anatomically defined by a dorsal barrier of the top of the third ventricle and laterally by the striatum and fornix) were rapidly dissected and frozen on dry ice within 5 min of decapitation. Tissue was stored at -80 °C until analysis. Trunk blood was also collected for measurement of circulating corticosterone.
Endocannabinoid Extraction and Analysis
The contents of the two primary endocannabinoids AEA and 2-AG, as well the non-cannabinoid NAEs palmitoylethanolamide (PEA) and oleoylethanolamide (OEA), were measured in lipid extracts of brain tissue using isotope-dilution liquid chromatography–mass spectrometry as described previously (Patel et al., 2005).
Corticosterone Radioimmunoassay (RIA)
Blood samples were centrifuged at 3000 × g for 20 min after which plasma was extracted and stored at -80 °C until analysis. Plasma corticosterone (50 μl) was measured in duplicate using a commercial RIA kit (Coat-A-Count; Siemens Medical Solutions Diagnostics, Los Angeles, CA). The quality control standard of this assay in our hands was 5.3%.
Statistics
Analyses of the effects of glucocorticoid administration on the tissue contents of AEA, 2-AG, PEA and OEA were performed using a one way analysis of variance (ANOVA); post-hoc analysis was performed using a Dunnett's t-test to compare the effects of the two doses of corticosterone to the same control. All analyses used p < 0.05 as an indication of significance.
Results
Within the amygdala, AEA content was found to be elevated 10 min following corticosterone administration [F (2, 16) = 11.39, p < 0.005; Fig. 1]. Post hoc analysis demonstrated that AEA content was increased in response to both the 3 mg/kg (p < 0.05) and the 10 mg/kg (p < 0.005) doses of corticosterone compared to vehicle treated controls. At 1 h following corticosterone administration, AEA content within the amygdala was also increased [F (2, 16) = 4.43, p < 0.05; Fig. 1]; however, at this time point, AEA content was elevated only in the amygdala of rats receiving the 10 mg/kg dose of corticosterone (p < 0.05). There was no effect of corticosterone on amygdalar 2-AG content at either the 10 min [F (2, 16) = 2.14, p > 0.05] or 1 h [F (2, 16) = 2.56, p > 0.05] following administration.
Figure 1. Corticosterone administration rapidly mobilizes anandamide in limbic structures and 2-AG within the hypothalamus.
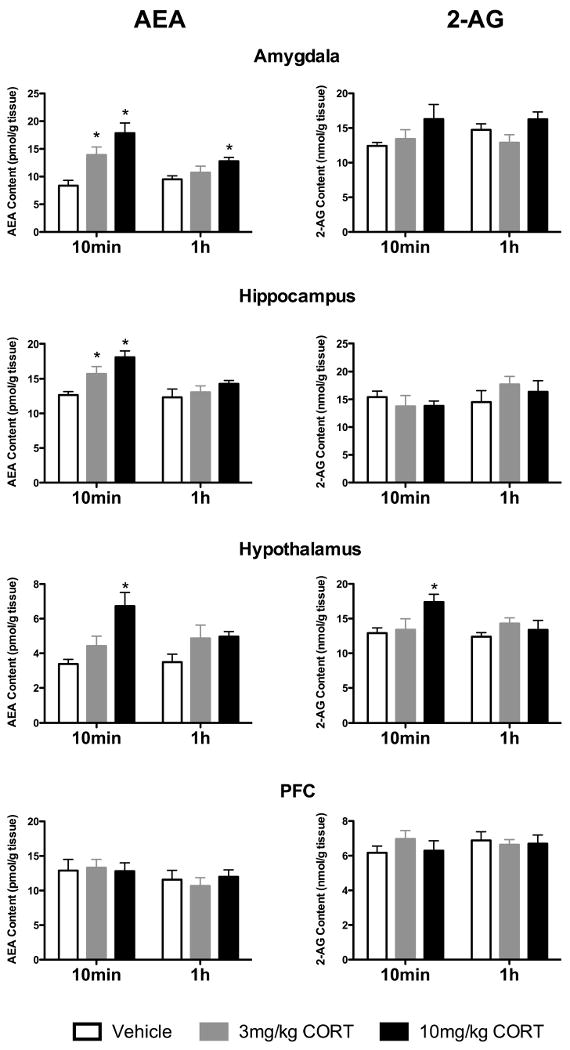
Systemic administration of corticosterone (CORT), at doses of 3 mg/kg and 10 mg/kg, resulted in a significant increase in the tissue content of anandamide (AEA) within the amygdala, hippocampus and hypothalamus, but not the prefrontal cortex. Similarly, a significant increase in the tissue content of 2-arachidonoylglycerol (2-AG) within the hypothalamus, but not the prefrontal cortex, amygdala or hippocampus was seen following administration of the 10 mg/kg dose of corticosterone exclusively. These effects were all seen 10 min following corticosterone administration and had largely dissipated at 1 h following administration. Values denoted are means ± SEM; n = 5-6 for each treatment condition. * denotes significant differences (p < .05).
AEA content was increased in the hippocampus 10 min following corticosterone administration [F (2, 17) = 9.90, p < 0.005; Fig. 1], with significant increases in hippocampal AEA content in rats injected with both 3 mg/kg (p < 0.05) and 10 mg/kg (p < 0.005) corticosterone compared to vehicle treated rats. There was no significant difference in hippocampal 2-AG content between vehicle treated or corticosterone treated rats at 10 min following injection [F (2, 17) = 0.45, p > 0.05; Fig. 2]. There were no differences in AEA [F (2, 17) = 1.14, p > 0.05; Fig. 1] or 2-AG [F (2, 17) = 0.75, p > 0.05; Fig. 2] contents among the three treatment groups in hippocampal tissue harvested at 1 hour after injection.
Within the hypothalamus, both AEA [F (2, 17) = 8.74, p < 0.01; Fig. 1] and 2-AG [F (2, 17) = 3.92, p < 0.05; Fig. 2] were elevated following administration of the 10 mg/kg dose (for both p < 0.01) but not the 3 mg/kg dose (p > 0.05) of corticosterone 10 min following injection. At 1 h following corticosterone, there were no significant differences in the hypothalamic contents of AEA [F (2, 16) = 2.07, p > 0.05; Fig. 1] or 2-AG [F (2, 16) = 0.95, p > 0.05; Fig. 2] among the three treatment groups.
There were no significant differences in AEA content within the prefrontal cortex at either 10 min [F (2, 17) = 0.04, p > 0.05; Fig. 1] or 1 h [F (2, 17) = 0.33, p > 0.05; Fig. 1] after injection among the three treatment groups. Similarly, there were no significant differences in 2-AG content within the prefrontal cortex at either 10 min [F (2, 17) = 0.73, p > 0.05; Fig. 2] or 1 h [F (2, 17) = 0.08, p > 0.05; Fig. 2] after injection among the three treatment groups.
We also examined the effect of corticosterone administration on two other NAEs, PEA and OEA in the same brain regions (Table 1). In brain tissues harvested 10 min following injection, there were no differences among vehicle, 3 or 10 mg/kg corticosterone treatment groups in PEA content in the amygdala [F (2, 16) = 0.20, p > 0.05], hippocampus [F (2, 17) = 2.62, p > 0.05], hypothalamus [F (2, 17) = 1.24, p > 0.05] or prefrontal cortex [F (2, 16) = 1.64, p > 0.05]. Similarly, there was no effect of corticosterone administration on OEA content at 10 min post injection within the amygdala [F (2, 16) = 0.54, p > 0.05], hippocampus [F (2, 17) = 0.93, p > 0.05], hypothalamus [F (2, 17) = 0.51, p > 0.05] or the prefrontal cortex [F (2, 16) = 0.81, p > 0.05] compared to vehicle treated rats. There were no differences in PEA content in the amygdala [F (2, 16) = 0.02, p > 0.05], hippocampus [F (2, 17) = 0.92, p > 0.05], hypothalamus [F (2, 17) = 2.95, p > 0.05] or the prefrontal cortex [F (2, 16) = 1.06, p > 0.05] among the treatment groups in tissues harvested 1 h following injection. Similarly, there were no differences in OEA content in the amygdala [F (2, 16) = 0.13, p > 0.05], hippocampus [F (2, 17) = 0.16, p > 0.05], hypothalamus [F (2, 17) = 0.54, p > 0.05] or the prefrontal cortex [F (2, 16) = 0.21, p > 0.05] among the treatment groups in tissues harvested 1 h following injection.
Table 1. The effects of corticosterone administration on the tissue content of palmitoylethanolamine and oleoylethanolamine.
There was no effect of corticosterone (CORT) administration (either 3 mg/kg or 10 mg/kg dose) on the tissue content of either palmitoylethanolamide (PEA) or oleoylethanolamine (OEA) at both 10 minutes and 1 hour following administration in any brain structure examined. Values denoted are means ± SEM. For all treatment conditions, n = 5-6.
| Vehicle | 3 mg/kg CORT | 10 mg/kg CORT | |
|---|---|---|---|
| 10 min post administration | |||
| Prefrontal Cortex | |||
| PEA (pmol/g tissue) | 58.4 +/- 5.2 | 74 +/- 7.1 | 72.8 +/- 8.5 |
| OEA (pmol/g tissue) | 27.0 +/- 3.2 | 34.6 +/- 3.9 | 31.2 +/- 4.7 |
| Amygdala | |||
| PEA (pmol/g tissue) | 72.8 +/- 11.1 | 78.5 +/- 7.5 | 81.5 +/- 10.7 |
| OEA (pmol/g tissue) | 54.0 +/- 4.5 | 60.5 +/- 5.7 | 62.4 +/- 6.9 |
| Hippocampus | |||
| PEA (pmol/g tissue) | 95.4 +/- 5.0 | 83.9 +/- 3.2 | 99.6 +/- 6.8 |
| OEA (pmol/g tissue) | 50.8 +/- 2.8 | 52.4 +/- 4.8 | 59.0 +/- 5.6 |
| Hypothalamus | |||
| PEA (pmol/g tissue) | 170.8 +/- 10.9 | 185.6 +/- 19.6 | 193.7 +/- 16.7 |
| OEA (pmol/g tissue) | 86.5 +/- 8.2 | 94.4 +/- 10.7 | 108.2 +/- 8.2 |
| 1 h post administration | |||
| Prefrontal Cortex | |||
| PEA (pmol/g tissue) | 67.7 +/- 11.7 | 65.3 +/- 5.1 | 82.7 +/- 9.4 |
| OEA (pmol/g tissue) | 29.2 +/- 4.2 | 26.3 +/- 2.5 | 28.9 +/- 2.9 |
| Amygdala | |||
| PEA (pmol/g tissue) | 71.1 +/- 10.7 | 70.6 +/- 9.4 | 72.9 +/- 10.6 |
| OEA (pmol/g tissue) | 55.5 +/- 7.7 | 53.5 +/- 5.7 | 50.8 +/- 6.3 |
| Hippocampus | |||
| PEA (pmol/g tissue) | 89.4 +/- 8.1 | 82.9 +/- 3.3 | 98.1 +/- 10.6 |
| OEA (pmol/g tissue) | 52.7 +/- 8.5 | 55.6 +/- 3.9 | 53.1 +/- 3.6 |
| Hypothalamus | |||
| PEA (pmol/g tissue) | 152.2 +/- 10.6 | 144.4 +/- 14.2 | 174.2 +/- 7.9 |
| OEA (pmol/g tissue) | 83.9 +/- 8.9 | 81.1 +/- 7.1 | 91.8 +/- 6.6 |
Plasma concentrations of corticosterone were significantly increased in rats treated with both doses of corticosterone compared to vehicle injected rats at 10 minute [vehicle: 51.9 +/- 13.3 ng/ml; 3 mg/kg: 765.8 +/- 55.5 ng/ml; 10 mg/kg: 1470.0 +/- 236.5 ng/ml; F (2, 17) = 25.49, p < 0.0001] and 1 h [vehicle: 20.3 +/- 4.3 ng/ml vs. 3 mg/kg: 717.1 +/- 73.3 ng/ml vs. 10 mg/kg: 1414.0 +/- 166.4 ng/m F (2, 17) = 44.03, p < 0.0001] following injections.
Discussion
In the present study, we demonstrated that systemic administration of corticosterone resulted in a rapid, transient increase in AEA contents in the amygdala, hippocampus and hypothalamus, but not the PFC. Specifically, tissue AEA concentrations were increased at 10 minutes following corticosterone administration, but had largely returned to baseline concentrations within 1 hour, despite the fact that plasma corticosterone remained elevated. These data suggest that AEA-mediated signaling is a transient response to increased circulating glucocorticoids. The rapidity of this effect suggests that corticosterone affects AEA content through a non-genomic mechanism, findings consistent with previous in vitro and behavioral studies demonstrating that glucocorticoids rapidly mobilize endocannabinoids (Campolongo et al., 2009; Coddington et al., 2007; Di et al., 2003). These data provide support for the hypothesis that glucocorticoids can influence brain function in mammals via non-genomic signaling cascades.
Elevations in anandamide levels were seen at 10 minutes following corticosterone administration, but had largely returned to baseline levels within 1 hour, despite the fact that corticosterone was still elevated at this time point. This suggests that the biochemical pathway subserving this induction exhausts rapidly, and the ability of glucocorticoids to promote endocannabinoid signaling is a phenomenon of limited duration. However, an alternate possibility is that an interaction between the arousal of the injection stress and the corticosterone occurred to maximally promote endocannabinoid mobilization. This hypothesis is in accord with previous work demonstrating that the rapid effects of glucocorticoids are amplified by concurrent emotional arousal and noradrenergic transmission (Roozendaal et al., 2008).
Tissue content of the other endocannabinoid, 2-AG, was elevated within the hypothalamus exclusively following corticosterone administration, indicating some degree of specificity in this phenomenon. The mechanism for this divergent regulation is not understood, but is not surprising given the distinct biochemical pathways mediating AEA and 2-AG synthesis and metabolism (Ahn et al., 2008).
The biochemical substrate through which glucocorticoids exert rapid changes in cellular function is not well understood. In amphibian tissue, a glucocorticoid sensitive G-protein coupled receptor (GPCR) has been characterized (Orchinik et al., 1991), and similarly, in vitro studies also suggest that mobilization of endocannabinoids by glucocorticoids requires activation of a GPCR and subsequent increase in cAMP-PKA activity (Malcher-Lopes et al., 2006). Reports that cytosolic steroid receptors possess the ability to insert into the cell membrane to modulate intracellular function suggests that cytosolic steroid receptors can have non-canonical signaling roles, including perhaps the initiation of rapid intracellular signaling (Karst et al., 2005). The current data does not eliminate this mechanism; however, other studies have found that recruitment of endocannabinoid signaling by glucocorticoids in the hypothalamus is insensitive to antagonists at the classical steroid receptors (Di et al., 2003).
The finding that glucocorticoid administration increased tissue contents of AEA, but not PEA or OEA, sheds light on the biochemical pathway involved in the increase in AEA contents. With respect to AEA synthesis, three processes have been identified that convert the AEA precursor N-arachidonylphosphatidylethanolamine (NAPE) to AEA (see (Ahn et al., 2008) for review). Of these pathways, one involving a NAPE-selective phospholipase D (NAPE-PLD) is involved in the generation of all of AEA, PEA and OEA, while a recently identified pathway involving the sequential actions of α/β-hydrolase-4 and glycerophosphodiesterase (GDE1), increases selectively synthesis of polyunsaturated NAE's, such as AEA, but not saturated and monounsaturated NAE's, such as PEA and OEA (Simon and Cravatt, 2006; Simon and Cravatt, 2008). The present data that AEA, but not OEA or PEA, are increased in response to exogenous glucocorticoids, is most consistent with increased activity of the α/β-hydrolase-4 synthetic pathway. This pattern of effects on the three NAEs also indicates that the mechanism does not involve changes in the kinetics of fatty acid amide hydrolase (FAAH) since AEA, PEA and OEA are all FAAH substrates for this enzyme (Cravatt et al., 2001; Patel et al., 2005).
The doses used in the present study are typically used for both behavioural and neurobiological studies (Campolongo et al., 2009; Kavushansky and Richter-Levin, 2006), though it should be noted that the circulating corticosterone levels that were achieved are surprisingly higher than expected. Following administration of the 3 mg/kg dose of corticosterone, plasma levels reached are at the upper limit of those which would be achieved following stress exposure, while the levels seen following administration of the 10 mg/kg dose of corticosterone were supra-physiological. Given that the mobilization of AEA within the hippocampus and amygdala was seen following administration of the 3 mg/kg dose of corticosterone, this phenomenon likely represents a biologically relevant change to fluctuations in corticosterone within the physiological range. However, the induction of both AEA and 2-AG within the hypothalamus only occurred at the higher dose of corticosterone, which limits the generalizability of this response to a biological process which would occur under physiological levels of corticosterone. Interestingly, the in vitro studies from Tasker and colleagues demonstrates that 1 µM of corticosterone is required to induce endocannabinoid-mediated suppression of neuronal transmission within the hypothalamus (Di et al., 2003), which approximately equates to the supra-physiological level of corticosterone achieved by the 10 mg/kg dose of corticosterone. Given the heterogeneity of the hypothalamus, it is possible that specific nuclei within the hypothalamus, such as the PVN, exhibit an endocannabinoid response to much lower, and physiologically relevant, levels of corticosterone, but that this effect is diluted when the entire hypothalamus is examined as other nuclei may not respond similarly.
These data provide in vivo evidence of a rapid increase in limbic AEA concentrations by glucocorticoids, a finding that suggests rapid recruitment of endocannabinoid signaling by glucocorticoids. The finding that glucocorticoids can rapidly elevate AEA concentrations in vivo adds to the short list of the non-genomic physiological effects of glucocorticoids (Haller et al., 2008) and provides evidence for the existence of such processes in mammals. Given that endocannabinoids are important regulators of synaptic transmission, these data support the hypothesis that endocannabinoids are the functional “workhorse” of glucocorticoids within the synapse (Hill and McEwen, 2009). In this model, glucocorticoids bind to a membrane associated receptor and rapidly mobilize AEA, and perhaps 2-AG, which in turn modulates the balance of excitation and inhibition within a given circuit. In agreement with this model, endocannabinoids have previously been shown to mediate glucocorticoid-induced inhibition of the HPA axis (Di et al., 2003), inhibition of sexual behavior (Coddington et al., 2007) and facilitation of aversive emotional memory consolidation (Campolongo et al., 2009). Enhanced understanding of the tight relationship between glucocorticoids and endocannabinoid signaling in the brain could aid in the development of novel therapeutic targets for diseases which involve dysregulation of one, or both, of these systems; such as affective illnesses, autoimmune disorders and metabolic conditions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn K, et al. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108:1687–707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campolongo P, et al. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc Natl Acad Sci U S A. 2009;106:4888–93. doi: 10.1073/pnas.0900835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coddington E, et al. Endocannabinoids mediate the effects of acute stress and corticosterone on sex behavior. Endocrinology. 2007;148:493–500. doi: 10.1210/en.2006-0740. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98:9371–6. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, et al. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–7. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, et al. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–66. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Haller J, et al. The effects of non-genomic glucocorticoid mechanisms on bodily functions and the central neural system. A critical evaluation of findings. Front Neuroendocrinol. 2008;29:273–91. doi: 10.1016/j.yfrne.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Hill MN, McEwen BS. Endocannabinoids: The silent partner of glucocorticoids in the synapse. Proc Natl Acad Sci U S A. 2009;106:4579–80. doi: 10.1073/pnas.0901519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst H, et al. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci U S A. 2005;102:19204–7. doi: 10.1073/pnas.0507572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavushansky A, Richter-Levin G. Effects of stress and corticosterone on activity and plasticity in the amygdala. J Neurosci Res. 2006;84:1580–7. doi: 10.1002/jnr.21058. [DOI] [PubMed] [Google Scholar]
- Malcher-Lopes R, et al. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci. 2006;26:6643–50. doi: 10.1523/JNEUROSCI.5126-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS. Steroid hormone actions on the brain: when is the genome involved? Horm Behav. 1994;28:396–405. doi: 10.1006/hbeh.1994.1036. [DOI] [PubMed] [Google Scholar]
- Orchinik M, et al. A corticosteroid receptor in neuronal membranes. Science. 1991;252:1848–51. doi: 10.1126/science.2063198. [DOI] [PubMed] [Google Scholar]
- Patel S, et al. The postmortal accumulation of brain N-arachidonylethanolamine (anandamide) is dependent upon fatty acid amide hydrolase activity. J Lipid Res. 2005;46:342–9. doi: 10.1194/jlr.M400377-JLR200. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, et al. Adrenal stress hormones, amygdala activation, and memory for emotionally arousing experiences. Prog Brain Res. 2008;167:79–97. doi: 10.1016/S0079-6123(07)67006-X. [DOI] [PubMed] [Google Scholar]
- Simon GM, Cravatt BF. Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for alpha/beta-hydrolase 4 in this pathway. J Biol Chem. 2006;281:26465–72. doi: 10.1074/jbc.M604660200. [DOI] [PubMed] [Google Scholar]
- Simon GM, Cravatt BF. Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J Biol Chem. 2008;283:9341–9. doi: 10.1074/jbc.M707807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker JG, et al. Minireview: rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology. 2006;147:5549–56. doi: 10.1210/en.2006-0981. [DOI] [PMC free article] [PubMed] [Google Scholar]