

Abstract
The outer leaflet of the outer membranes of Gram-negative bacteria is composed primarily of lipid A, the hydrophobic anchor of lipopolysaccharide. Like Escherichia coli, most Gram-negative bacteria encode one copy of each of the nine genes required for lipid A biosynthesis. An important exception exists in the case of the fourth enzyme, LpxH, a peripheral-membrane protein that hydrolyzes UDP-2,3-diacylglucosamine to form 2,3-diacylglucosamine 1-phosphate and UMP by catalyzing the attack of water at the α-P atom. Many Gram-negative organisms, including all α-proteobacteria and diverse environmental isolates, lack LpxH. Here, we report a distinct UDP-2,3-diacylglucosamine pyrophosphatase, designated LpxI, which has no sequence similarity to LpxH, but generates the same products by a different route. LpxI was identified because its structural gene is located between lpxA and lpxB in Caulobacter crescentus. The lpxI gene rescues the conditional lethality of lpxH-deficient E. coli. Lysates of E. coli in which C. crescentus LpxI (CcLpxI) is over-expressed display high levels of UDP-2,3-diacylglucosamine pyrophosphatase activity. CcLpxI was purified to >90% homogeneity. CcLpxI is stimulated by divalent cations and is inhibited by EDTA. Unlike E. coli LpxH, CcLpxI is not inhibited by increasing the concentration of detergent, and its pH-dependency is different. When the CcLpxI reaction is carried out in the presence of H218O, the 18O is incorporated exclusively into the 2,3-diacylglucosamine 1-phosphate product, as judged by mass spectrometry, demonstrating that CcLpxI catalyzes the attack of water on the β-P atom of UDP-2,3-diacylglucosamine.
Gram-negative bacteria possess inner and outer membranes. The outer membrane is asymmetric, having a periplasm-facing leaflet comprised of phospholipids, and an outer leaflet consisting primarily of lipid A, the hydrophobic anchor of lipopolysaccharide (LPS) (1, 2). The biosynthesis of lipid A is required for growth and viability in most Gram-negative bacteria (1, 3). Therefore, the conserved, constitutive enzymes of lipid A biosynthesis (Scheme 1) are excellent targets against which to develop new antibiotics (4-6). The present study focuses on the catalysis of UDP-2,3-diacylglucosamine hydrolysis (7, 8), the fourth step of the pathway (Scheme 1).
Scheme 1. Role of UDP-diacylglucosamine pyrophosphatases in lipid A biosynthesis.

LpxH is primarily found in γ- and β-proteobacteria, whereas LpxI is present in α-proteobacteria, many δ-proteobacteria, and some environmental isolates, as described in detail in Supporting Table 2. Chlamydia and Cyanobacteria contain neither LpxH nor LpxI, but do possess LpxD and LpxB. Protein sequence data can be found at (http://www.ncbi.nlm.nih.gov/sutils/genom_tree.cgi).
The lpxH gene encodes the essential UDP-2,3-diacylglucosamine hydrolase of Escherichia coli (7, 8). E. coli LpxH (EcLpxH) and its orthologues are distantly related to a large superfamily of metallo-phosphoesterases with diverse functions. EcLpxH is a peripheral membrane enzyme that displays apparent surface dilution kinetics (8), and it requires divalent cations, preferably Mn2+, but not Mg2+, for activity (8). 31P NMR analysis of the EcLpxH products, UMP and 2,3-diacylglucosamine 1-phosphate (lipid X), generated from UDP-2,3-diacylglucosamine in the presence of H218O, revealed that this hydrolase catalyzes the attack of water at the α-P atom of UDP-2,3-diacylglucosamine with incorporation of the 18O isotope into the UMP product (8) (Scheme 2).
Scheme 2. LpxH and LpxI catalyze UDP-diacylglucosamine hydrolysis by different mechanisms.

Oxygen from the water attacking the α-phosphate in the LpxH-catalyzed reaction is shown in red, whereas oxygen from the water attacking the β-phosphate in the LpxI-catalyzed reaction is shown in magenta.
Most Gram-negative bacteria contain one gene coding for each enzyme of the lipid A biosynthetic pathway (Scheme 1) (3). These genes are easily identified by sequence comparisons (9). However, LpxH is a notable exception. Many bacterial species, including all α-proteobacteria, many δ-proteobacteria, all spirochetes, all cyanobacteria, and diverse environmental strains, such as the hyperthermophile Aquifex aeolicus, lack LpxH orthologues. They nevertheless produce lipid A using the same enzymes upstream and downstream of UDP-2,3-diacylglucosamine, as demonstrated for Rhizobium leguminosarum and Rhizobium etli, which lack LpxH (10, 11). These findings suggests that alternative enzyme(s) catalyzing UDP-2,3-diacylglucosamine hydrolysis must exist in these bacteria in order to provide the appropriate substrates for LpxB (Scheme 1).
While lack of sequence homology hampers the search for the genes encoding these alternative hydrolases, genomic context can sometimes assist in the identification of novel gene functions (12–14). In this work, we identify and characterize LpxI, a protein unrelated to LpxH that catalyzes UDP-2,3-diacylglucosamine hydrolysis by attack of water on the β-P atom instead of the α-P atom (Scheme 2). The gene encoding LpxI was identified because of its location between lpxA and lpxB in certain α-proteobacteria, such as Caulobacter crescentus and Mesorhizobium loti, which lack LpxH (Fig. 1) (15, 16). The different hydrolytic mechanism employed by LpxI explains why LpxI and LpxH share no overall sequence similarity or even any conserved active site residues (Fig. 2). LpxI belongs to a unique protein superfamily (DUF1009) (17) for which no biochemical functions have been previously reported.
Figure 1.
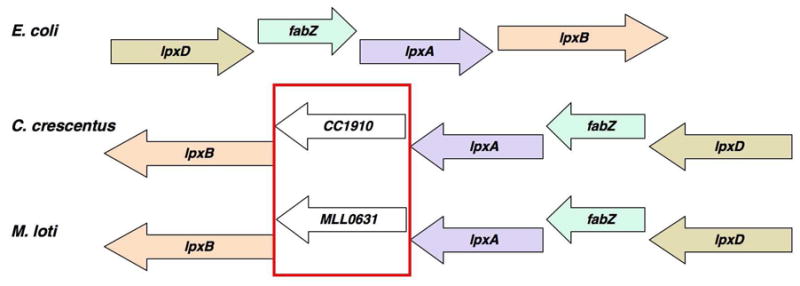
Comparison of the lpxD-fabZ-lpxA-lpxB gene clusters of E. coli, C. crescentus, and M. loti. Arrows represent open reading frames and their directions of transcription. A gene of unknown function encoding a protein designated as DUF2009 is situated between lpxA and lpxB in C. cresentus and M. loti, and also is present in many other bacteria, like Aquifex aeolicus (not shown), that lack LpxH (15, 16, 32). The DUF1009 orthologues, CC1910 and MLL0631, are highlighted with a red box. Gene lengths are not drawn exactly to scale.
Figure 2.

Sequence alignments of LpxH and LpxI orthologues. Panel A. Sequence alignments of five representative LpxH orthologues. Panel B. Alignments of five typical LpxI orthologues. In each panel, the absolutely conserved residues are colored red. There is no conservation of domains or of possible active site motifs between LpxI and LpxH, which are members of distinctly different protein families. The sequences were obtained from (http://www.ncbi.nlm.nih.gov/sutils/genom_tree.cgi).
Materials and methods
Chemicals and reagents
Yeast extract, tryptone and Bacto agar were purchased from Difco (Detroit, MI). Glass-backed silica gel 60 thin layer chromatography (TLC) plates were from EMD Chemicals (Darmstadt, Germany). Chloroform, pyridine, methanol, and acetic acid were from EMD Science (Gibbstown, NJ). The γ-32Pi was purchased from PerkinElmer (Waltham, MA). The 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), phosphate buffered saline components, HCl and salts were purchased from Sigma-Aldrich (St. Louis, MO).
Cloning and molecular biology
Amplification of DNA segments, from either genomic or plasmid DNA, was accomplished using KOD Hot Start DNA polymerase, 8 mM dNTP stocks (2 mM each of dATP, dTTP, dGTP, and dCTP), and KOD Hot Start reaction buffer, all obtained from EMD Chemicals (Gibbstown, NJ). Polymerase chain reactions (PCR) conditions were those recommended by the manufacturer, except for the inclusion of 1% (v/v) dimethyl sulfoxide and 1 M betaine in the reaction mixtures. The plasmids described in this study (see Table 1) were stored in and amplified from E. coli strain XL1-Blue (Stratagene, La Jolla, CA). Qiagen Mini-Prep kits and QIAquick Spin kits (Qiagen, Valencia, CA) were used, respectively, to purify plasmids and DNA fragments by protocols described by the manufacturer. Restriction endonucleases, T4 DNA ligase, and calf intestinal alkaline phosphatase were obtained from New England Biolabs (Ipswich, MA). Unless otherwise stated, the following concentrations of antibiotics were used when appropriate: ampicillin, 100 μg/ml; kanamycin, 50 μg/ml; chloramphenicol, 20 μg/ml; tetracycline, 15 μg/ml.
Table 1.
Relevant strains and plasmids.
| Strain | Description | Source |
|---|---|---|
| C41(DE3) | F− ompT hsdSB(rB− mB−) gal dcm (DE3) D(srl-recA)306::Tn10 | (51) |
| W3110A | F− aroA::Tn10 msbA+ , TetR | (28) |
| XL1-Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F'proAB lacIqZDM15 Tn10 (TetR)] | Strategene |
| MN7 | K-12-derived pgsA444 lpxB1; accumulates lipid X | (52) |
| DY330 | W3110 ΔlacU169 gal490λcI857 Δ(cro -bioA) | (19) |
| Ec21XB | XL1-Blue harboring pEcH21b, AmpR | This work |
| Cc21XB | XL1-Blue harboring pCcI21b, AmpR | This work |
| EcH33XB | XL1-Blue harboring pBAD33Ec, Cam R | This work |
| CcI33XB | XL1-Blue harboring pBAD33Cc, Cam R | This work |
| EcH30XB | XL1-Blue harboring pBAD30Ec, Amp R | This work |
| CcI30XB | XL1-Blue harboring pBAD30Cc, Amp R | This work |
| DY330VC | DY330 harboring pBAD30, AmpR | This work |
| DY330Ec | DY330 harboring pBAD30Ec, AmpR | This work |
| DY330Cc | DY330 harboring pBAD30Cc, AmpR | This work |
| DY330ΔHEc | DY330 lpxH::Kan harboring pBAD30Ec, KanR , AmpR | This work |
| DY330ΔHCc | DY330 lpxH::Kan harboring pBAD30Cc, KanR , AmpR | This work |
| W3110AVC | W3110A harboring pBAD33, CamR | This work |
| W3110AEc | W3110A harboring pBAD33Ec, CamR | This work |
| W3110ACc | W3110A harboring pBAD33Cc, CamR | This work |
| W3110AΔHEc | W3110A lpxH::Kan harboring pBAD33Ec, CamR, AmpR | This work |
| W3110AΔHCc | W3110A lpxH::Kan harboring pBAD33Cc, CamR, Amp R | This work |
| VC_21b | C41(DE3) harboring pET21b, AmpR | This work |
| EcH_21b | C41(DE3) harboring pEcH21b, AmpR | This work |
| CcI_21b | C41(DE3) harboring pCcI21b, AmpR | This work |
| Plasmid | Description | Source |
| pET21b | high-copy expression vector containing a T7 promoter, AmpR | Novagen |
| pET28b | high-copy expression vector containing a T7 promoter, KanR | Novagen |
| pBAD30 | arabinose inducible vector, AmpR | (18) |
| pBAD33 | arabinose inducible vector, CamR | (18) |
| pEcH21b | pET21b containing E. coli lpxH, AmpR | This work |
| pCcI21b | pET21b containing C. crescentus lpxI, AmpR | This work |
| pBAD33Ec | pBAD33 containing E. coli lpxH, CamR | This work |
| pBAD33Cc | pBAD33 containing C. crescentus lpxI, CamR | This work |
| pBAD30Ec | pBAD33 containing E. coli lpxH, AmpR | This work |
| pBAD30Cc | pBAD33 containing C. crescentus lpxI, AmpR | This work |
Construction of plasmids
DNA oligomers (Integrated DNA Technologies, Coralville, IA) were used to amplify E. coli K-12 (NP_414724) lpxH and C. crescentus CB15 (NP_420716) LpxI from genomic DNA obtained from ATCC (Rockville, MD). Primers (Supporting Table 1) were designed to confer 5′ NdeI and 3′ BamHI restriction sites upon the ends each of the amplified oligonucleotides. A Mastercycler Gradient Thermocycler (Eppendorf, Hamburg, Germany) was used to amplify linear DNA. These PCR products were digested using NdeI and BamHI, according to manufacturers’ protocols, and subsequently ligated into a similarly digested recipient vector, pET21b (Novage/EMD Chemicals, Gibbstown, NJ). These ligation reactions were then transformed into the XL1-Blue strain of chemically-competent E. coli (Stratagene, La Jolla, CA), according to the manufacturer’s recommended procedure. Plasmids were isolated from transformants, and their sequences confirmed by the Duke Cancer Center DNA Sequencing Facility. The resulting constructs were designated pEcH21b and pCcH21b (see Table 1). The restriction endonucelases XbaI and SalI were used to digest these plasmids, liberating linear E. coli lpxH or C. crescentus lpxI, each flanked by 5′ Xba and 3′ SalI restriction sites, and containing a pET21b-encoded ribosome binding site 7-base-pairs upstream from the start codon. These segments were then ligated, as above, into appropriately digested pBAD30 or pBAD33 plasmids (18). The resulting plasmids are described in Table 1.
Construction of E. coli lpxH deletion strains
A deletion mutant of E. coli LpxH was generated with an in-frame kanamycin (Kan) insertion/substitution. The primers KanFlank_FW and KanFlank_RV (see Supporting Table 1) were used to amplify a Kan cassette from the plasmid pET28b (Novagen/EMD Chemicals, Gibbstown, NJ). These primers were designed with 40 base-pair overhangs complementary to the regions of the E. coli chromosome immediately flanking lpxH. KanFlank_FW also added a ribosome binding site in the optimal position at 7 base-pairs 5′ to the kan cassette’s start codon. The resulting linear oligonucleotide was electroporated into temperature-shifted E. coli strain DY330 (19), harboring either pBAD30, pBAD30Ec, or pBAD30Cc (strains DY330VC, DY330Ec, and DY330Cc, respectively, see Table 1), as previously described (20). The strains were plated on LB-agar supplemented with kanamycin and ampicillin, and grown for 18 hours at 30°C. Transformants were re-purified, and colony PCR was performed using primers Kan_FW and Kan_RV (Supporting Table 1) to amplify the lpxH locus and its 100-base-pair flanking regions. The resulting oligonucleotides were sequenced to confirm their identities. DY330 strains with lpxH::Kan replacements, covered by pBAD30Ec or pBAD30Cc, were designated DY330ΔHEc and DY330ΔHCc, respectively (Table 1).
A P1vir lysate was prepared (21) from DY330ΔHEc, and used to infect E. coli W3110A cells harboring either pBAD33, pBAD33Ec, or pBAD33Cc (strains W3110AVC, W3110AEc, or W3110ACc respectively) (Table 1). Following infection and outgrowth as previously described (21), cells were spread onto LB-agar plates supplemented with chloramphenicol, kanamycin, and 5 mM sodium citrate, and allowed to grow for 20 hours at 30°C. Colonies were selected and re-purified twice to remove traces of contaminating phage. Colony PCR was performed to amplify the chromosomal region ± 100 base-pairs from the lpxH locus, and the resulting oligonucleotides were confirmed by sequencing. The strains thereby created from the transduction of lpxH::Kan into W3110ACc and W3110AEc were designated W3110AΔHEc and W3110AΔHCc (see Table 1).
UDP-2,3,-diacylglucosamine hydrolase expression and in vitro TLC assay
The plasmids pET21b and pCcI21b were transformed into E. coli strain C41(DE3) by electroporation and grown for 18 hours at 30°C on LB-agar plates supplemented with ampicillin. Single colonies of the resulting strains VC_21b and CcI_21b (see Table 1) were used to inoculate 5-ml overnight cultures. These, in turn, were used to inoculate 50 ml cultures of appropriately supplemented LB media, at an initial OD600 of ~0.02. Strains VC_21b and CcI_21b were then grown at 30°C with aeration at 220 rpm, until the A600 reached ~0.5 (about 5 hours). Expression was induced by adding isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 250 μM. The cells were grown for an additional 5 hours, until the final A600 of the induced cells was ~3. Cells were collected by centrifugation at 3000 x g, washed with phosphate buffered saline (PBS) (22), and resuspended in 3 ml ice-cold PBS. The washed, re-suspended cells were passed twice through a French Pressure cell at 18,000 psi, and cell debris were removed by centrifugation at 10,000 x g for 30 minutes. Lysates were analyzed by SDS-PAGE and were assayed for UDP-2,3-diacylglucosamine hydrolase activity (8).
For our initial analysis, the addition of 5 μl of diluted lysate from strains VC_21b or CcI_21b was used to start the UDP-2,-3-diacylglucosamine hydrolase reactions in a total volume of 25 μl. Non-radiolabeled UDP-2,3,-diacylglucosamine was prepared as previously described (8). Briefly, the final reactions contained 1 mM UDP-2,3-diacylglucosamine, 20 mM HEPES pH 8.0, and 2 mM MnCl2, and they were equilibrated at 30°C for 15 minutes in 0.5 ml polypropylene Eppendorf tubes. Reactions were started by addition of crude lysates from VC_21b or CcI_21b, diluted as appropriate with PBS. At various time-points, the reactions were quenched by spotting 5 μl portions onto 10 x 10 cm high performance silica TLC plates (Merck, Darmstadt, Germany). These were developed and dried as described (8). Reaction products were visualized by spraying the dried TLC plate with 10% H2SO4 in ethanol, and subsequent charring on a hot plate.
Preparation of [β-32P]-UDP-2,3-diacylglucosamine
[32P]-labeled lipid X was prepared as previously reported (23). Following purification, the lipid X was re-dissolved in 2 ml CHCl3/MeOH (2:1, v/v), transferred to a clean screw-capped glass tube, and thoroughly dried under a nitrogen stream. Next, 10 mg UMP-morpholidate, 50 μl 0.2 M 1-H-tetrazole in acetonitrile, and 1 ml dry pyridine were added. The tube was immediately sealed with a Teflon-coated screw cap and subjected to sonic irradiation for 5 min in a bath sonicator (Avanti Polar Lipids, Alabaster, AL). The mixture was incubated in a rotary water-bath shaker for at least 20 hours at 37°C, shaking at 100 rpm. The reaction mixture was dried under nitrogen until no traces of pyridine remained (~2 hours). The residue at the bottom of the tube was re-dissolved in 100-300 μl of 20 mM HEPES pH 8.0, containing 0.02% (w/v) Triton X-100, and subjected to bath sonic irradiation for 2 min. This material was split into aliquots and stored at −80°C. With this method we could consistently convert over 95% of the [32P]-lipid X to [β-32P]-UDP-2,3-diacylglucosamine.
Expression and purification of C. crescentus LpxI
A colony of E. coli strain CcI_21b (which over-expresses CcLpxI upon induction) was used to inoculate a 25 ml overnight LB medium culture supplemented with ampicillin. Following growth for 16 hours at 30°C, this culture was used to inoculate 1L of LB broth containing ampicillin to an initial A600 of ~0.05. Growth proceeded at 30°C with aeration at 220 rpm, until A600 reached ~0.5. IPTG was then added to a final concentration of 250 μM, and growth was continued for 5 h at 30°C. Cells, which typically grew to A600 of ~3.5, were harvested by centrifugation at 3000 x g for 30 minutes, and subsequently washed with 80 ml 20 mM HEPES, pH 8.0, containing 50 mM NaCl. The washed pellets were stored at −80°C. In a typical purification, ~4 g wet cell pellet was re-suspended in ~300 ml lysis buffer (20 mM HEPES pH 8.0 containing 50 mM NaCl), and cells were lysed by three passages through an ice-cold Cell-Cracker pressure disruption chamber (Microfluidics International Corp., Newton, MA). Cell debris and membranes were removed by ultracentrifugation at 150,000 x g for 1 hour. A Rabbit-Plus peristaltic pump (Rainin Instruments, LLC, Oakland, CA) was used to load the membrane-free lysate at ~2 ml/min onto a 5 ml High-Trap Q Sepharose Fast-Flow anion exchange cartridge (GE Health Sciences, Pittsburgh, PA), previously equilibrated in lysis buffer. The column was washed with 20 volumes (100 ml) of lysis buffer at 3 ml/min and was then attached to an AKTA 600 FPLC system (GE Health Sciences, Pittsburgh, PA). A 30-column-volume (150 ml) continuous gradient from 100% Buffer A (20 mM HEPES pH 8.0 containing 50 mM NaCl) to 100% Buffer B (20 mM HEPES pH 8.0 containing 200 mM NaCl) was applied at a flow rate of 2 ml/min. The protein, which was collected in 5-ml fractions, began to elute at ~100 mM NaCl. SDS-PAGE was used to determine which fractions to pool; typically 6 fractions (30 ml) were combined. The partially-purified CcLpxI was concentrated to ~10 ml using two 15 ml Amicon Ultra 10,000 molecular weight cutoff centrifuge concentrators (Millipore, Billerica, MA), which had been washed in Buffer B. The AKTA 600 FPLC system was used to load the concentrated, pooled Q Sepharose fractions at 1.5 ml/min onto a 330 ml size exclusion column (Superdex 200 XK26/70; GE Healthcare, Waukesha, WI), equilibrated with Buffer B. The peak of LpxI eluted at ~210 ml. Fractions of 5 ml were collected, and their purity determined by SDS-PAGE. Typically, 4 fractions (20 ml) were pooled and subsequently concentrated to ~25 mg/ml as above. Protein aliquots were stored at −80°C, and subjected to fast freeze-thaw cycles upon storage and retrieval.
Autoradiographic in vitro assay of CcLpxI
Unless otherwise noted, 25 μL reactions containing 20 mM HEPES, pH 8.0, 0.5% (w/v) fatty-acid-free bovine serum albumin (BSA), 0.05% (w/v) Triton X-100, 2 mM MgCl2, 100 μM UDP-2,3-diacylglucosamine, 1000 cpm/μl [β-32P]-UDP-2,3-diacylglucosamine, and enzyme were prepared in 0.5 ml polypropylene tubes. Prior to addition of 5 μl enzyme as the last step, the other reaction components were equilibrated at 30 °C for 15 minutes. Unless otherwise noted, enzyme samples were diluted in a buffer identical to the assay mixture, but lacking UDP-2,3-diacylglucosamine. At various time-points, 3 μl reaction portions were removed and spotted onto 20 × 20 cm silica gel TLC plates (EMD Chemicals, Inc., Darmstadt, Germany). These were then developed, dried, scanned, and quantified as previously described (8).
Kinetic parameters, pH optimum, and detergent dependence of CcLpxI
To determine the effect of pH upon the apparent specific activity of CcLxpI, partially purified enzyme was assayed as described above, but with the usual 20 mM HEPES, pH 8.0, replaced with a triple-buffer system consisting of 100 mM sodium acetate, 50 mM bis(2-hydroxyethyl)iminotris(hydroxymethyl)hexane, and 50 mM Tris (24). CcLpxI activity was measured from pH 4.0-9.0. A single-limb pKa curve was fit to the data using KaleidaGraph. Typically, enzyme was present at 5–50 nM.
To determine the KM and VMAX of CcLpxI with respect to UDP-2,3-diacylglucosamine, the purified enzyme was assayed as above, but with the concentration of UDP-2,3-diacylglucosamine varied from 5 to 1000 μM. KaleidaGraph was used to fit velocities to the Michaelis-Menten equation (25). The concentration of CcLpxI in the assay was varied from 2 and 200 nM in order to maintain linear conversion to product with time at different UDP-2,3-diacylglucosamine concentrations.
To probe whether CcLpxI activity is affected by detergent, the typical assay conditions were employed, except that the concentration of Triton X-100 was varied from 0 to 1.0% w/v. In order to measure LpxI activity in the absence of detergent, the [β-32P]UDP-2,3-diacylglucosamine was suspended in buffer lacking Triton X-100. An enzyme concentration of 10 nM was used in these assays.
Metal dependence of CcLpxI
To determine whether divalent metal ions stimulate CcLpxI activity in vitro, the standard assay condition was employed, except that the 2 mM MgCl2 was replaced by either no additive, or by 2.0, 0.2, or 0.02 mM of the chloride salts of each of the following: Mg2+, Ca2+, Co2+, Ni2+, Cu2+, Zn2+, and Mn2+; a similar EDTA control was also included. The concentration of CcLpxI in the assay was varied from 2 and 200 nM in order to follow linear conversion to product with time in the presence of the different additives. A separate assay, in which MgCl2 was titrated from 0 to 20 mM, was performed in the presence of 20 nM CcLpxI.
Preparation of the CcLpxI reaction products in the presence of H218O
Reaction mixtures (50 μl each) consisting of 100 μM UDP-2,3-diacylglucosamine, 2 mM MgCl2, 20 mM HEPES pH 8.0, and 66 nM of purified CcLpxI were set up in the presence of either 100% H216O or H218O/ H216O (77:23, v/v) (Cambridge Isotopes, Andover, MA). The reactions were allowed to proceed at 30°C for two hours and were quenched by conversion to a 1.9 ml single-phase, acidic Bligh-Dyer system (26). A 20 μl portion of this material, containing 37 ng of lipid X, was analyzed by normal-phase liquid chromatography (LC) and mass spectrometry. Briefly, the sample was loaded onto a 25 cm x 2.1 mm Ascentis silica HPLC column (Sigma-Aldrich, St. Louis, MO) using an Agilent 1200 Quaternary LC system (Santa Clara, CA), coupled to a QSTAR XL quadrupole time-of-flight mass spectrometer (Applied Biosystems, Foster City, CA), operating in the ESI negative ion mode. Following injection onto the column, which was equilibrated in mobile phase A consisting of CHCl3/methanol/saturated aqueous NH4OH (160/39/1, v/v/v), the sample was subjected to chromatography at a constant flow-rate of 300 μl/min. First, an isocratic gradient of 100% mobile phase A was applied for 2 minutes, followed by a linear gradient from 100% mobile phase A to 100% mobile phase B consisting of CHCl3/methanol/water/saturated aqueous NH4OH (120/68/10/1, v/v/v/v) over 14 minutes. Next, 100% mobile phase B was applied for 11 minutes, followed by a linear gradient from 100% mobile phase B to 100% mobile phase C consisting of CHCl3/methanol/water/saturated aqueous NH4OH (90/90/19/1, v/v/v/v) over 3 minutes. Finally, mobile phase C was applied for 3 minutes, followed by a linear transition to mobile phase A over 30 seconds; the latter was passed over the column for an additional 5 minutes. The parameters used for negative ion electrospray mass spectrometry (ESI-MS) and MS/MS analysis were: ionization source energy = −4500 V, curtain gas pressure = 20 psi, GSI=20 psi, DP=−55 V, FP=−150 V, with nitrogen used as the collision gas. The Analyst QS software suite (Applied Biosystems, Foster City, CA) was employed for data analysis.
Results
Identification of a novel UDP-2,3-diacylglucosamine hydrolase in bacteria lacking lpxH
The Clusters of Orthologous Genes database (17) was used to compare the genomes of Gram-negative bacteria lacking lpxH orthologues. Given that the gene(s) encoding a novel UDP-2,3-diacylglucosamine hydrolase analogue(s) might be located near other lipid A biosynthetic genes and that in many species lpxD, fabZ, lpxA, and lpxB cluster in an operon, we searched for species that lacked lpxH but possessed additional open reading frames of unknown function in the lpxD-fabZ-lpxA-lpxB locus. Several bacteria, including C. crescentus (16), S. meliloti (27) and Mesorhizobium loti (15), met this criterion in that a ~1000 base-pair open reading frame of unknown function was inserted between lpxA and lpxB (Fig. 1). The corresponding gene product, annotated as DUF1009 in each of these three strains (17), is also present in many (though not all) of the other organisms that produce lipid A but lack lpxH. LpxH orthologues display no sequence similarity to the DUF1009 sequences (Fig. 2), which have a distinctly different set of conserved amino acid residues.
The gene encoding the C. crescentus DUF1009 orthologue, CC1910 (16), was amplified from genomic DNA, cloned into the high copy pET21b plasmid, and expressed in E. coli C41(DE3), yielding strain CcI_21b. This construct was grown in parallel with its empty-vector control (VC_21b) and induced to over-express the CC1910 protein. Cells from each strain were harvested and lysed. Over-expression of the expected 29 kDa CC1910 protein was confirmed by SDS-PAGE (Supporting Fig. 1, panel A). Equal concentrations of lysates from VC_21b versus CcI_21b were used to detect UDP-2,3-diacylglucosamine hydrolase activity by TLC, similar to the method described for E. coli LpxH (8). This analysis revealed that the CcI_21b lysate contained over 1000-fold higher UDP-2,3-diacylglucosamine hydrolase activity than its matched vector control (Supporting Fig. 1, panel B, lane 2 versus lane 4), suggesting that CC1910, hereafter referred to as CcLpxI, is functionally similar to LpxH. Upon incubation of these lysates with CDP-diacylglycerol, under otherwise similar assay conditions, no difference in the rate of CDP-diacylglycerol hydrolysis was observed between extracts of VC_21b and CcI_21b, showing that LpxI, in contrast to CDP-diacylglucosamine hydrolase (7, 8), is selective for UDP-2,3-diacylglucosamine (data not shown).
C. crescentus lpxI can replace lpxH in E. coli
To determine whether C. crescentus lpxI possesses UDP-2,3-diacylglucosamine hydrolase activity in vivo, the essential lpxH gene was replaced in E. coli DY330 (19) with a kanR cassette, and covering plasmids expressing either E. coli lpxH or C. crescentus lpxI were used to test for complementation of lpxH deficiency. Whereas the empty vector could not complement the deletion of the chromosomal lpxH gene, plasmids expressing either E. coli lpxH or C. crescentus lpxI allowed this deletion mutant to form colonies (Supporting Fig. 2). The lpxH::kan deletion could also subsequently be transduced into the wild-type strain E. coli W3110A (28) harboring either E. coli lpxH or C. crescentus lpxI on a pBAD plasmid. The replacement of lpxH with kan in W3110A was confirmed by PCR amplification of the lpxH locus and its 100 base-pair flanking regions. The identities of these PCR products were confirmed by sequencing.
Expression and purification of CcLpxI
Induced cells of E. coli strain CcI_21b massively over-produced a protein of the molecular weight expected for LpxI and its associated UDP-2,3-diacylglucosamine hydrolase activity. To characterize CcLpxI, it was purified to greater than 90% homogeneity using ion-exchange and size-exclusion chromatography (Fig. 3). The pellet from 500 ml of CcI_21b cells grown to A600 ~3.5 yielded ~30 mg of CcLpxI. The specific activity of the protein increased only 1.2 fold during the purification, consistent with the efficient over-expression, and the total activity yield was ~20% (Table 2). CcLpxI elutes from a gel filtration column in a symmetric peak (Fig. 3, panel A), consistent with either a dimeric or monomeric solution structure. Unlike EcLpxH, which is a peripheral membrane protein, recombinant CcLpxI did not sediment with membranes, but remained in the supernatant following ultracentrifugation.
Figure 3.
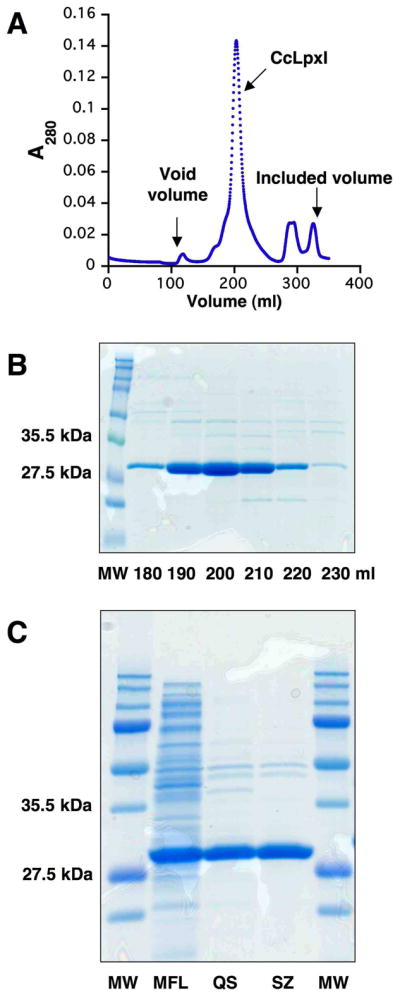
Purification of untagged recombinant CcLpxI. Panel A. Elution of CcLpxI monitored at A280 from a size exclusion column. Panel B. A 12% SDS-PAGE analysis of selected fractions from the gel filtration column. The lanes correspond to the elution volume of the trace shown in Panel A. Equal volumes were loaded. Panel C. SDS-PAGE analysis of protein from each step of the CcLpxI purification. MFL, QS, and SZ denote membrane-free lysate, pooled fractions from the Q-Sepharose anion exchange column, and pooled fraction from the Superdex S200 sizing column, respectively. Approximately 20 μg of protein is loaded in each lane.
Table 2.
Purification of CcLpxI from E. coli strain CcI_21b
| Step | Protein (mg) | Volume (ml) | Units (mmol/min) | Specific Activity (μmol/min/mg) | Yield (%) | Fold Purification |
|---|---|---|---|---|---|---|
| Membane-free lysate | 168 | 280 | 4.1 | 25 | 100 | 1.0 |
| Q-Sepharose anion exchange | 34 | 30 | 0.9 | 26 | 22 | 1.1 |
| S200 size exclusion | 27 | 20 | 0.8 | 30 | 20 | 1.2 |
An autoradiographic assay of CcLpxI using [ββ β-32P]-UDP-2,3-diacylglucosamine
Using [β-32P]-UDP-2,3-diacylglucosamine as the substrate, we developed a quantitative TLC-based assay for CcLpxI. Enzymatic activity is linear with time and with protein concentration (Fig. 4, panels A and B). Moreover, CcLpxI quantitatively converts [β-32P]-UDP-2,3-diacylglucosamine to [32P]lipid X (Fig. 4, panel C).
Figure 4.

Linearity of CcLpxI activity with time and protein concentration. Panel A. Fraction of 100 μM [β-32P]UDP-2,3-diacylglucosamine converted to the product lipid X at various times under standard assay conditions in the presence of 12.5 nM CcLpxI. Panel B. Fraction of 100 μM [β-32P]UDP-2,3-diacylglucosamine converted to lipid X per minute as a function of enzyme concentration. Panel C. An image of a silica TLC plate of an in vitro CcLpxI reaction run at an enzyme concentration of 40 nM.
Apparent kinetic parameters and detergent dependence of CcLpxI
The apparent KM of purified CcLpxI with respect to UDP-2,3-diacylglucosamine was 105 ± 25 μM, while the apparent Vmax was 69 ± 5 μmols/min/mg (Fig. 5, panel A). The pH rate profile was determined for CcLpxI, and a single pKa curve was fit to the data using Kaleidograph. The pKa was estimated to be 6.15 ± 0.86 (Fig. 5, panel B), although the data deviates from a single ionization model at low pH. The detergent dependence of CcLpxI was determined by varying the concentration of Triton X-100 in the assay from 0 to 1% w/v. While stimulated ~3-fold in the presence of 0.05% w/v Triton X-100, the apparent activity did not decrease in high concentrations of Triton X-100 (Fig. 5, panel C) showing that CCLpxI activity is not apparently decreased by surface dilution of the substrate (29).
Figure 5.
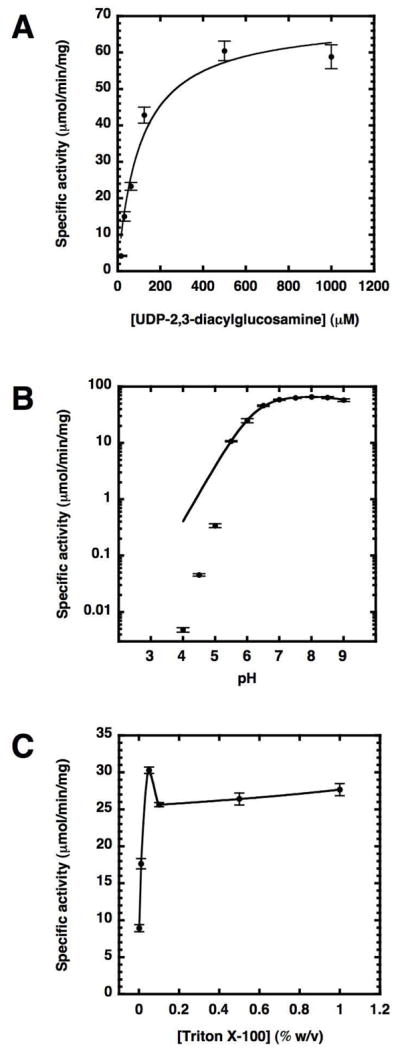
Substrate, pH, and detergent dependence of CcLpxI activity. Panel A. UDP-2,3-diacylglucosamine concentration dependence of CcLpxI activity under standard assay conditions. The apparent KM, fit using Kaleidograph, was 105 ± 25 μM; the apparent Vmax was 69 ± 5 μmols/min/mg. Panel B. CcLpxI activity as a function of pH. The activity is relatively constant at pH values between 6.5 and 9, but declines sharply below 6. Panel C. Effect of Triton X-100 concentration on the apparent specific activity of CcLpxI at 100 μM UDP-2,3-diacylglucosamine. Surface dilution kinetics are not apparent, and there is measurable activity in the absence of Triton.
Metal dependence of CcLpxI activity in vitro
The purified CcLpxI was assayed under the standard conditions, except that 0.02, 0.2, or 2 mM of the chloride salts of each of the following were added: Mg2+, Ca2+, Co2+, Ni2+, Cu2+, Zn2+, and Mn2+, as explained in the legend to Fig. 6. For each concentration, a control assay was included in which divalent cations were not added (Fig. 6A, no add); in another set 0.02, 0.2, or 2 mM EDTA was included in place of the divalent cations. The apparent specific activity of purified CcLpxI increased 10-fold in the presence of Mg+2 versus no added metal, or 6-fold in the presence of added Mn2+ or Co2+ (Fig. 6, panel A). The presence of EDTA at either 2.0 or 0.2 mM completely inhibited CcLpxI. This inhibition was reversed by diluting the EDTA-treated enzyme into reactions containing 2 mM Mg2+ or Mn2+ (data not shown). Inhibition was also seen at the higher concentrations of Cu2+ and Zn2+.
Figure 6.

Metal dependence of CcLpxI. Panel A. Specific activity of CcLpxI in the presence of various divalent cations or EDTA. Three sets of assays were performed, each of which included a no additive (No add) control, as shown at the left. Each bar in the series of three bars represents a different concentration of each additive (e.g. 0.02, 0.2, or 2 mM). Standard assay conditions were used with enzyme at 10 nM. Panel B. The standard CcLpxI assay was used with 10 nM enzyme, but with increasing MgCl2 from 0 to 20 mM.
Next, LpxI activity was measured under standard assay conditions, but with the concentration of Mg2+ varied from 0 to 20 mM. CcLpxI activity was maximal at 0.2 mM Mg2+ and remained about the same up to 20 mM Mg2+ (Fig. 6, panel B). To determine whether divalent cations are associated with purified CcLpxI, inductively coupled plasma mass spectrometry was performed. No significant amounts of stoichiometric, tightly-bound metal ions were detected in the purified protein in two separate determinations.
CcLpxI catalyzes the attack of H2O on the β ββ-phosphorus atom of UDP-2,3-diacylglucosamine
In order to determine which phosphorous atom of UDP-2,3-diacylglucosamine is attacked during hydrolysis catalyzed by CcLpxI, excess purified enzyme was used to convert UDP-2,3-diacylglucosamine to products quantitatively in the presence H216O or of H218O/H216O (77:23, v/v). The lipid X and UMP products were separated by normal phase LC and analyzed by ESI-MS in the negative ion mode (Figs. 7A and 7B). The lipid X (predicted [M-H]− at m/z 710.424) eluted between minutes 24.4 and 24.6; the UMP emerged during column regeneration between minutes 38.3 and 39.0. For the H218O labeled reaction, the proportional amount (about 77%) of the lipid X product contained 18O, as shown by the presence of a much more intense peak at m/z 712.441 versus m/z 710.440 (Fig. 7B). This result is expected for the [M-H]− ion of a lipid X molecule containing a single 18O atom (predicted [M-H]− at m/z 712.429) versus lipid X containing only 16O (predicted [M-H]− at m/z 710.424). No 18O was incorporated into the UMP product isolated from the CcLpxI 18O reaction mixture (Fig. 7B inset). We conclude that ccLpxI catalyzes the attack of water exclusively on the β-phosphorus atom of UDP-2,3-diacylglucosamine (Scheme 2).
Figure 7.

Incorporation of 18O into lipid X during CcLpxI-catalyzed hydrolysis of UDP-2,3-diacylglucosamine in 77% H218O. CcLpxI-catalyzed hydrolysis of UDP-2,3-diacylglucosamine was carried out in the absence or presence of 77% H218O. Panel A. LC/ESI-MS analysis of lipid X generated by CCLpxI in the presence of 100% H216O. Panel B. LC/ESI-MS analysis of lipid X generated by CCLpxI in the presence of H218O/ H216O (77:23, v/v). The insets show LC/ESI-MS analyses of the UMP generated in the reactions.
Discussion
Although essential for growth in E. coli and present in most γ- and β-proteobacteria (8, 30), LpxH is missing in α-proteobacteria and many other groups (Fig. 8 and Supporting Table 1). Here, we describe the identification of a different UDP-2,3-diacylglucosamine hydrolase present in many of the species that lack LpxH, including almost all α-proteobacteria, most strains of Leptospira (31), and many diverse organisms, like Aquifex aeolicus (32). The relevant gene, which encodes a protein of unknown function annotated as DUF1009, was recognized because of its location between lpxA and lpxB in C. crescentus and M. loti (Fig. 1) (15, 16). Members of the DUF1009 family possess no sequence similarity to EcLpxH or to any other type of protein (Fig. 2, panels A and B).
Figure 8.

Distribution of LpxH and LpxI orthologues among diverse bacterial groups. This figure, modified from the NCBI bacterial genomes web site (http://www.ncbi.nlm.nih.gov/sutils/genom_tree.cgi), shows the approximate distribution of putative LpxH (cyan) and LpxI orthologues (yellow) in diverse bacterial groups for which complete genome sequences are currently available. Light green shading indicates approximately equal proportions of organisms having putative LpxH and LpxI orthologues. Important Gram-negatives, such as the Chlamydiae and the Cyanobacteria, contain neither protein, even though both possess LpxC (Supporting Table 2) and LpxB (Scheme 1). Many Spirochaetales, such as Treponema pallidum and Borrelia Burgdorferi, do not synthesize lipid A. However, species of Leptospira do make lipid A and contain LpxI orthologues.
We cloned the gene encoding DUF1009 from C. cresentus (CC1910) into a high-copy vector and expressed it in E. coli, resulting in massive over-production of the expected 29 kDa protein (Supporting Fig. 1, panel A). E. coli lysates of strain CcI_21b displayed >1000-fold increased UDP-2,3-diacylglucosamine hydrolase activity relative to vector controls (Supporting Fig. 1, panel B). The presence of CC1910, renamed lpxI, permitted the deletion and replacement of the chromosomal lpxH gene with a kanamycin-resistance cassette in E. coli (Supporting Fig. 2). These results demonstrate that LpxI functions as an alternative UDP-2,3-diacylglucosamine hydrolase in vivo.
Taken together with the dissimilarity of their conserved motifs (Fig. 2), we propose that EcLpxH and CcLpxI evolved separately to generate the same products by different hydrolytic mechanisms. The presence of lpxI, but not lpxH, in more ancient species like Aquifex aeolicus, suggests that lpxI may have evolved earlier than lpxH. Moreover, some groups of organisms that make lipid A, such as the Cyanobacteria (33) and the Chlamydiae (34, 35), lack both lpxH and lpxI, suggesting that at least one additional class of UDP-2,3-diacylglucosamine hydrolases may exist.
To determine which P atom of UDP-2,3-diacylglucosamine is attacked by water during CcLpxI-catalyzed hydrolysis, reactions were run in the presence or absence of H218O. Analysis of the products by ESI-MS (Fig. 7) revealed that the 18O was incorporated exclusively into lipid X. CcLpxI therefore catalyzes the attack of water on the β-P atom, whereas EcLpxH specifically attacks the α-P atom (Scheme 2) (8). This observation, together with the lack of homology between EcLpxH and CcLpxI, strongly suggests that these enzymes perform catalysis by different mechanisms.
The incorporation of H218O into the β-P atom of UDP-2,3-diacylglucosamine by LpxI is formally analogous to the chemistry performed by phosphatidylserine synthase (PssA) of E. coli, which catalyzes the transfer of the phosphatidyl moiety of CDP-diacylglycerol to the hydroxyl group of serine (36). PssA catalysis is thought to proceed via a phosphatidyl-enzyme intermediate (37), but PssA can also cleave CDP-diacylglycerol to generate phosphatidic acid and CMP in the absence of serine, with concomitant incorporation of H218O into the phosphatidic acid product by attack of water on the β-P atom of CDP-diacylglycerol (36). This consideration raises the intriguing converse possibility that LpxI (or other enzymes like it) might be able to transfer the 2,3-diacylglucosamine 1-phosphate moiety from UDP-2,3-diacylglucosamine to an acceptor molecule other than water, analogous to the process of phosphatidyl transfer in glycerophospholipid metabolism.
The significance of two mechanisms for cleaving UDP-2,3-diacylglucosamine remains unclear. Phosphohydrolases are ubiquitous in biology, and have probably evolved many times on different protein scaffolds (38). Phosphohydrolases are often metal dependent, and much is known about their mechanisms (39). They play diverse roles in the modification and processing of proteins (40, 41), nucleic acids (42, 43), lipids (44–47) and diverse small molecules (39, 48–50). The mechanisms of these enzymes generally involve the positioning of a deprotonated water molecule for attack on a P atom of a coordinated phosphate moiety of the substrate (39). The roles played by the metal ions may vary. In some cases, the metal coordinates the oxygen atoms of the phosphate group, while in others, it positions the water for attack on the P atom (39, 48–50). In still other instances, the metal appears to coordinate a network of ordered water molecules, which in turn position a “catalytic water” for attack on the substrate (48). Although purified CcLpxI does not contain any tightly bound divalent cations, it is dependent upon them for activity (Fig. 6, panel A). Structural and mechanistic studies will be required to elucidate the roles of metals in CcLpxI catalysis and the basis for the selectivity of CcLpxI for UDP-2,3-diacylglucosamine.
Supplementary Material
Acknowledgments
We thank Drs. Hak Suk Chung, Jinshi Zhao, and Ziqiang Guan for their careful reading of the manuscript, and other members of our laboratory for their helpful discussion and criticism. The metal analysis of LpxI was carried out by Dr. Aaron Atkinson in the Division of Hematology at the University of Utah, Salt Lake City, UT.
This research was supported by NIH Grant GM-51310 to C. R. H. Raetz and the LIPID MAPS Large Scale Collaborative Grant GM-069338. Louis Metzger was supported by National Science Foundation Graduate Research Fellowship 2005029353.
The abbreviations are
- BCA
bicinchoninic acid
- DTT
dithiothreitol
- dUTPase
dUTP diphosphatase
- ESI-MS
electrospray ionization mass spectrometry
- FPLC
fast protein liquid chromatography
- GlcN
glucosamine
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- LC
liquid chromatography
- Lipid X
2,3-diacylglucosamine 1-phosphate
- LPS
lipopolysaccharide
- PAGE
polyacrylamide gel electrophoresis
- PAP
purple acid phosphatase
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- TLC
thin layer chromatography
Footnotes
Supporting information available
The primers used to construct various LpxI over-expressing strains and additional information regarding the distributions of LpxI and LpxI among diverse bacteria are shown in Supporting Tables 1 and 2 respectively. Additional information on the purification of LpxI, and of the ability to lpxI to complement the E. coli lpxH mutation, are provided in Supporting Figs. 1 and 2, respectively. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, Raetz CRH. Antibacterial agents that inhibit lipid A biosynthesis. Science. 1996;274:980–982. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 5.Barb AW, McClerren AL, Snehelatha K, Reynolds CM, Zhou P, Raetz CRH. Inhibition of lipid A biosynthesis as the primary mechanism of CHIR-090 antibiotic activity in Escherichia coli. Biochemistry. 2007;46:3793–3802. doi: 10.1021/bi6025165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McClerren AL, Endsley S, Bowman JL, Andersen NH, Guan Z, Rudolph J, Raetz CRH. A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid A biosynthesis with antibiotic activity comparable to ciprofloxacin. Biochemistry. 2005;44:16574–16583. doi: 10.1021/bi0518186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Babinski KJ, Kanjilal SJ, Raetz CRH. Accumulation of the lipid A precursor UDP-2,3-diacylglucosamine in an Escherichia coli mutant lacking the lpxH gene. J Biol Chem. 2002;277:25947–25956. doi: 10.1074/jbc.M204068200. [DOI] [PubMed] [Google Scholar]
- 8.Babinski KJ, Ribeiro AA, Raetz CRH. The Escherichia coli gene encoding the UDP-2,3-diacylglucosamine pyrophosphatase of lipid A biosynthesis. J Biol Chem. 2002;277:25937–25946. doi: 10.1074/jbc.M204067200. [DOI] [PubMed] [Google Scholar]
- 9.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Price NPJ, Kelly TM, Raetz CRH, Carlson RW. Biosynthesis of a structurally novel lipid A in Rhizobium leguminosarum: identification and characterization of six metabolic steps leading from UDP-GlcNAc to (Kdo)2-lipid IVA. J Bacteriol. 1994;176:4646–4655. doi: 10.1128/jb.176.15.4646-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez V, Santamaria RI, Bustos P, Hernandez-Gonzalez I, Medrano-Soto A, Moreno-Hagelsieb G, Janga SC, Ramirez MA, Jimenez-Jacinto V, Collado-Vides J, Davila G. The partitioned Rhizobium etli genome: genetic and metabolic redundancy in seven interacting replicons. Proc Natl Acad Sci U S A. 2006;103:3834–3839. doi: 10.1073/pnas.0508502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crowell DN, Anderson MS, Raetz CRH. Molecular cloning of the genes for lipid A disaccharide synthase and UDP-N-acetylglucosamine acyltransferase in Escherichia coli. J Bacteriol. 1986;168:152–159. doi: 10.1128/jb.168.1.152-159.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sweet CR, Ribeiro AA, Raetz CRH. Oxidation and transamination of the 3”-position of UDP-N-acetylglucosamine by enzymes from Acidithiobacillus ferrooxidans. Role in the formation of lipid A molecules with four amide-linked acyl chains. J Biol Chem. 2004;279:25400–25410. doi: 10.1074/jbc.M400596200. [DOI] [PubMed] [Google Scholar]
- 14.Sweet CR, Williams AH, Karbarz MJ, Werts C, Kalb SR, Cotter RJ, Raetz CRH. Enzymatic synthesis of lipid A molecules with four amide-linked acyl chains. LpxA acyltransferases selective for a new analogue of UDP-N-acetylglucosamine in which an amine replaces the 3”-hydroxyl group. J Biol Chem. 2004;279:25411–25419. doi: 10.1074/jbc.M400597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaneko T, Nakamura Y, Sato S, Asamizu E, Kato T, Sasamoto S, Watanabe A, Idesawa K, Ishikawa A, Kawashima K, Kimura T, Kishida Y, Kiyokawa C, Kohara M, Matsumoto M, Matsuno A, Mochizuki Y, Nakayama S, Nakazaki N, Shimpo S, Sugimoto M, Takeuchi C, Yamada M, Tabata S. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 2000;7:331–338. doi: 10.1093/dnares/7.6.331. [DOI] [PubMed] [Google Scholar]
- 16.Nierman WC, Feldblyum TV, Laub MT, Paulsen IT, Nelson KE, Eisen J, Heidelberg JF, Alley MR, Ohta N, Maddock JR, Potocka I, Nelson WC, Newton A, Stephens C, Phadke ND, Ely B, DeBoy RT, Dodson RJ, Durkin AS, Gwinn ML, Haft DH, Kolonay JF, Smit J, Craven MB, Khouri H, Shetty J, Berry K, Utterback T, Tran K, Wolf A, Vamathevan J, Ermolaeva M, White O, Salzberg SL, Venter JC, Shapiro L, Fraser CM. Complete genome sequence of Caulobacter crescentus. Proc Natl Acad Sci U S A. 2001;98:4136–4141. doi: 10.1073/pnas.061029298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, Tatusova TA, Wagner L. Database resources of the National Center for Biotechnology. Nucleic Acids Research. 2003;31:28–33. doi: 10.1093/nar/gkg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma B, Reynolds CM, Raetz CR. Periplasmic orientation of nascent lipid A in the inner membrane of an Escherichia coli LptA mutant. Proc Natl Acad Sci U S A. 2008;105:13823–13828. doi: 10.1073/pnas.0807028105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller JR. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 22.Dulbecco R, Vogt M. Plaque formation and isolation of pure lines with poliomyelitis viruses. J Exp Med. 1954;99:167–182. doi: 10.1084/jem.99.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metzger LEI, Raetz CRH. Purification and characterization of the lipid A disaccharide synthase (LpxB) from Escherichia coli, a peripheral membrane protein. Biochemistry. 2009;48:11559–11571. doi: 10.1021/bi901750f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McClerren AL, Zhou P, Guan Z, Raetz CRH, Rudolph J. Kinetic analysis of the zinc-dependent deacetylase in the lipid A biosynthetic pathway. Biochemistry. 2005;44:1106–1113. doi: 10.1021/bi048001h. [DOI] [PubMed] [Google Scholar]
- 25.Bartling CM, Raetz CRH. Steady-state kinetics and mechanism of LpxD, the N-acyltransferase of lipid A biosynthesis. Biochemistry. 2008;47:5290–5302. doi: 10.1021/bi800240r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bligh EG, Dyer JJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 27.Galibert F, Finan TM, Long SR, Puhler A, Abola P, Ampe F, Barloy-Hubler F, Barnett MJ, Becker A, Boistard P, Bothe G, Boutry M, Bowser L, Buhrmester J, Cadieu E, Capela D, Chain P, Cowie A, Davis RW, Dreano S, Federspiel NA, Fisher RF, Gloux S, Godrie T, Goffeau A, Golding B, Gouzy J, Gurjal M, Hernandez-Lucas I, Hong A, Huizar L, Hyman RW, Jones T, Kahn D, Kahn ML, Kalman S, Keating DH, Kiss E, Komp C, Lelaure V, Masuy D, Palm C, Peck MC, Pohl TM, Portetelle D, Purnelle B, Ramsperger U, Surzycki R, Thebault P, Vandenbol M, Vorholter FJ, Weidner S, Wells DH, Wong K, Yeh KC, Batut J. The composite genome of the legume symbiont Sinorhizobium meliloti. Science. 2001;293:668–672. doi: 10.1126/science.1060966. [DOI] [PubMed] [Google Scholar]
- 28.Doerrler WT, Reedy MC, Raetz CRH. An Escherichia coli mutant defective in lipid export. J Biol Chem. 2001;276:11461–11464. doi: 10.1074/jbc.C100091200. [DOI] [PubMed] [Google Scholar]
- 29.Carman GM, Deems RA, Dennis EA. Lipid signaling enzymes and surface dilution kinetics. J Biol Chem. 1995;270:18711–18714. doi: 10.1074/jbc.270.32.18711. [DOI] [PubMed] [Google Scholar]
- 30.Babinski KJ, Raetz CRH. Identification of a gene encoding a novel Escherichia coli UDP-2,3-diacylglucosamine hydrolase. FASEB J. 1998;12:A1288. [Google Scholar]
- 31.Ren SX, Fu G, Jiang XG, Zeng R, Miao YG, Xu H, Zhang YX, Xiong H, Lu G, Lu LF, Jiang HQ, Jia J, Tu YF, Jiang JX, Gu WY, Zhang YQ, Cai Z, Sheng HH, Yin HF, Zhang Y, Zhu GF, Wan M, Huang HL, Qian Z, Wang SY, Ma W, Yao ZJ, Shen Y, Qiang BQ, Xia QC, Guo XK, Danchin A, Saint Girons I, Somerville RL, Wen YM, Shi MH, Chen Z, Xu JG, Zhao GP. Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature. 2003;422:888–893. doi: 10.1038/nature01597. [DOI] [PubMed] [Google Scholar]
- 32.Deckert G, Warren PV, Gaasterland T, Young WG, Lenox AL, Graham DE, Overbeek R, Snead MA, Keller M, Aujay M, Huber R, Feldman RA, Short GM, Olsen GJ, Swanson RV. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- 33.Snyder DS, Brahamsha B, Azadi P, Palenik B. Structure of compositionally simple lipopolysaccharide from marine Synechococcus. J Bacteriol. 2009;191:5499–5509. doi: 10.1128/JB.00121-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qureshi N, Kaltashov I, Walker K, Doroshenko V, Cotter RJ, Takayama K, Sievert TR, Rice PA, Lin JS, Golenbock DT. Structure of the monophosphoryl lipid A moiety obtained from the lipopolysaccharide of Chlamydia trachomatis. J Biol Chem. 1997;272:10594–10600. doi: 10.1074/jbc.272.16.10594. [DOI] [PubMed] [Google Scholar]
- 35.Rund S, Lindner B, Brade H, Holst O. Structural analysis of the lipopolysaccharide from Chlamydia trachomatis serotype L2. J Biol Chem. 1999;274:16819–16824. doi: 10.1074/jbc.274.24.16819. [DOI] [PubMed] [Google Scholar]
- 36.Bulawa CE, Hermes JD, Raetz CRH. Chloroform-soluble nucleotides in Escherichia coli. Role of CDP-diglyceride in the enzymatic cytidylylation of phosphomonoester acceptors. J Biol Chem. 1983;258:14974–14980. [PubMed] [Google Scholar]
- 37.Larson TJ, Dowhan W. Ribosomal-associated phosphatidylserine synthetase from Escherichia coli: purification by substrate-specific elution from phosphocellulose using cytidine 5'-diphospho-1,2-diacyl-sn-glycerol. Biochemistry. 1976;15:5212–5218. doi: 10.1021/bi00669a003. [DOI] [PubMed] [Google Scholar]
- 38.Gerlt JA, Babbitt PC. Divergent evolution of enzymatic function: mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu Rev Biochem. 2001;70:209–246. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- 39.Cleland WW, Hengge AC. Enzymatic mechanisms of phosphate and sulfate transfer. Chem Rev. 2006;106:3252–3278. doi: 10.1021/cr050287o. [DOI] [PubMed] [Google Scholar]
- 40.Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 41.Rudolph J. Cdc25 phosphatases: structure, specificity, and mechanism. Biochemistry. 2007;46:3595–3604. doi: 10.1021/bi700026j. [DOI] [PubMed] [Google Scholar]
- 42.Dominski Z. Nucleases of the metallo-beta-lactamase family and their role in DNA and RNA metabolism. Crit Rev Biochem Mol Biol. 2007;42:67–93. doi: 10.1080/10409230701279118. [DOI] [PubMed] [Google Scholar]
- 43.Li WM, Barnes T, Lee CH. Endoribonucleases--enzymes gaining spotlight in mRNA metabolism. FEBS J. 2010;277:627–641. doi: 10.1111/j.1742-4658.2009.07488.x. [DOI] [PubMed] [Google Scholar]
- 44.Stukey J, Carman GM. Identification of a novel phosphatase sequence motif. Protein Sci. 1997;6:469–472. doi: 10.1002/pro.5560060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maehama T, Taylor GS, Dixon JE. PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem. 2001;70:247–279. doi: 10.1146/annurev.biochem.70.1.247. [DOI] [PubMed] [Google Scholar]
- 46.Wang X, Karbarz MJ, McGrath SC, Cotter RJ, Raetz CRH. MsbA transporter-dependent lipid A 1-dephosphorylation on the periplasmic surface of the inner membrane: topography of Francisella novicida LpxE expressed in Escherichia coli. J Biol Chem. 2004;279:49470–49478. doi: 10.1074/jbc.M409078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, McGrath SC, Cotter RJ, Raetz CRH. Expression cloning and periplasmic orientation of the Francisella novicida lipid A 4'-phosphatase LpxF. J Biol Chem. 2006;281:9321–9330. doi: 10.1074/jbc.M600435200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mildvan AS, Xia Z, Azurmendi HF, Saraswat V, Legler PM, Massiah MA, Gabelli SB, Bianchet MA, Kang LW, Amzel LM. Structures and mechanisms of Nudix hydrolases. Arch Biochem Biophys. 2005;433:129–143. doi: 10.1016/j.abb.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 49.Persson R, Cedergren-Zeppezauer ES, Wilson KS. Homotrimeric dUTPases; structural solutions for specific recognition and hydrolysis of dUTP. Curr Protein Pept Sci. 2001;2:287–300. doi: 10.2174/1389203013381035. [DOI] [PubMed] [Google Scholar]
- 50.Holtz KM, Kantrowitz ER. The mechanism of the alkaline phosphatase reaction: insights from NMR, crystallography and site-specific mutagenesis. FEBS Lett. 1999;462:7–11. doi: 10.1016/s0014-5793(99)01448-9. [DOI] [PubMed] [Google Scholar]
- 51.Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 52.Nishijima M, Bulawa CE, Raetz CRH. Two interacting mutations causing temperature-sensitive phosphatidylglycerol synthesis in Escherichia coli membranes. J Bacteriol. 1981;145:113–121. doi: 10.1128/jb.145.1.113-121.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.