

Abstract
Large chemical and biological systems such as fuel cells, ion channels, molecular motors, and viruses are of great importance to the scientific community and public health. Typically, these complex systems in conjunction with their aquatic environment pose a fabulous challenge to theoretical description, simulation, and prediction. In this work, we propose a differential geometry based multiscale paradigm to model complex macromolecular systems, and to put macroscopic and microscopic descriptions on an equal footing. In our approach, the differential geometry theory of surfaces and geometric measure theory are employed as a natural means to couple the macroscopic continuum mechanical description of the aquatic environment with the microscopic discrete atom-istic description of the macromolecule. Multiscale free energy functionals, or multiscale action functionals are constructed as a unified framework to derive the governing equations for the dynamics of different scales and different descriptions. Two types of aqueous macromolecular complexes, ones that are near equilibrium and others that are far from equilibrium, are considered in our formulations. We show that generalized Navier–Stokes equations for the fluid dynamics, generalized Poisson equations or generalized Poisson–Boltzmann equations for electrostatic interactions, and Newton's equation for the molecular dynamics can be derived by the least action principle. These equations are coupled through the continuum-discrete interface whose dynamics is governed by potential driven geometric flows. Comparison is given to classical descriptions of the fluid and electrostatic interactions without geometric flow based micro-macro interfaces. The detailed balance of forces is emphasized in the present work. We further extend the proposed multiscale paradigm to micro-macro analysis of electrohydrodynamics, electrophoresis, fuel cells, and ion channels. We derive generalized Poisson–Nernst–Planck equations that are coupled to generalized Navier–Stokes equations for fluid dynamics, Newton's equation for molecular dynamics, and potential and surface driving geometric flows for the micro-macro interface. For excessively large aqueous macromolecular complexes in chemistry and biology, we further develop differential geometry based multiscale fluid-electro-elastic models to replace the expensive molecular dynamics description with an alternative elasticity formulation.
Keywords: Variational principle, Multiscale, Geometric flows, Solvation analysis, Electrostatic analysis, Implicit solvent models, Molecular dynamics, Elasticity, Navier–Stokes equation, Poisson–Boltzmann equation, Nernst–Planck equation
1. Introduction
Recently, multiscale modeling and multiscale simulation have emerged as powerful approaches in physical, biological, mathematical, and engineering sciences (Abraham et al., 1998; Knap and Ortiz, 2001;Tadmor et al., 1996;Tang et al., 2006; Engquist et al., 2007; Wang and Shu, 2009). The popularity of these approaches is driven by the human curiosity and desire to understand the behavior of complex systems, such as complex fluids, turbulent flows, micro-fluidics (Zheng et al., 2003; Chen and Conlisk, 2008), solids, interface problems, structure and fluid interactions, wave propagation in random media, stochastic processes, and statistically self-similar problems, to name only a few. In general, multiscale models and methods allow efficient descriptions of key elements in a physical phenomenon such that the subsequent simulations are feasible with the current computational capability, and the simulation results offer insights to the understanding of the phenomenon. A variety of multiscale models and computational methods has been developed. One class of multiscale models addresses multiphysics involved in the problem at hand with multiple governing equations, such as microscopic laws for atoms and molecules at microscopic settings, and transport equations for the conservation of mass, momentum, and energy at macroscopic settings. Computationally, bridging scales and bridging domains may be used to couple macro-micro scales or domains (Tang et al., 2006). Another class of multiscale models is originated from earlier multigrid methods or wavelet multiresolution analysis. These models emphasize the extraction and utilization of information at different time or spatial scales described by one set of governing equations that are of similar nature in mathematics. A typical example is the homogenization problem governed by an elliptic equation. The other class of multiscale models is heterogeneous approaches.
Yet perhaps the oldest and the most elegant multiscale model is the Boltzmann equation, particularly the quantum Boltzmann equation known as the Waldmann–Snider equation (Waldmann, 1957; Snider, 1960) which is derived from the BBGKY hierarchy with appropriate scattering closure and stosszahl ansatz for the two-body density operator. The Boltzmann equation and the Waldmann–Snider equation describe the time evolution of a one-particle statistical distribution or density operator, influenced by the interaction of another particle which represents the effect of all other particles in the system (Alavi et al., 1997). The Boltzmann equation is one of the most important equations of nonequilibrium statistical mechanics. The Boltzmann equation and the Waldmann–Snider equation incorporate three time scales, i.e., the collision time, the mean free time, and the relaxation time (or hydrodynamic time); as well as three spatial scales, i.e., the microscopic description of the dynamics of particles (atoms, electrons, phonons, and molecules), the mesoscopic description of the local density, the local velocity, and the local temperature that are out of equilibrium, and the macroscopic description of the transport quantities, such as the conservation laws of mass, momentum, and energy (Snider et al., 1996a, 1996b). However, the Boltzmann theory is limited to the systems that are essentially homogeneous, such as gases, fluid, and electrons in solids, etc. More general multiscale analysis is required for heterogeneous systems.
The most intriguing and fascinating phenomenon on Earth is life. Amazingly, life encompasses over more than twenty orders of magnitude in time scales from electron transfer, proton dislocation, on the scale of femtoseconds to organism lifetimes on the scale of years; and over ten orders of magnitude in spatial scales from electrons to organisms. Since life has so many scales in space and time, biology is subdivided into molecular biology, cellular biology, development biology, evolutionary biology, organismic biology, population biology, etc., not to mention emerging fields such as systems biology, ecology, and bioinformatics. Biology at each scale and level collects enormous amount information which can easily outrace the theory needed to understand it. Quantitative understanding and theoretical prediction have emerged as a key discipline in the contemporary biology. Therefore, the complexity of life and the need for its understanding present an extraordinary opportunity for multiscale modeling and simulation.
Due to their extraordinary spatiotemporal extension, biological scales are difficult to integrate. An interesting development in theoretical biology is the scale relativity theory approach to integrative systems biology (Auffray and Nottale, 2008; Nottale and Auffray, 2008). The scale relativity theory is an extension of Einstein's theories of relativity, formulated by applying the principle of relativity to both motion and scale transformations of the reference system. This approach has the potential of overcoming the fundamental hurdles of multiscale integration in systems biology. The interested reader is referred to two recent review papers for detail (Auffray and Nottale, 2008; Nottale and Auffray, 2008).
Under physiological conditions, most biological processes, such as ion channel, signal transduction, deoxyribonucleic acid (DNA) specification, transcription, post transcription modification, translation, protein folding, and protein-protein interaction, occur in water, which consists of 65–90% human cell weight. A prerequisite to quantitative descriptions of the above mentioned biological processes is the understanding of solvation—the static and dynamical behavior of macromolecules in the aquatic environment and the synergy of solvent-solute interactions. Solvation models can be roughly divided into two classes: explicit solvent models that treat the solvent in molecular or atomic detail, and implicit solvent models that generally replace the explicit solvent with a dielectric continuum while keeping the atomic detail of the biomolecule (Roux and Simonson, 1999; Warshel and Papazyan, 1998; Simonson, 2001; Sharp and Honig, 1990; Tully-Smith and Reiss, 1970; Fries and Patey, 1985; Beglov and Roux, 1997). Each method has its strengths and weaknesses. While explicit solvent models offer some of the highest levels of detail, they generally require extensive sampling to extract thermodynamic or kinetic properties of interest. This approach becomes intractable for large biomolecules, such as large proteins, molecular motors, and viruses. On the other hand, implicit solvent models focus on the biomolecule of interest, and provide only a mean-field description of the solvent. Because of their fewer degrees of freedom, implicit solvent models have become popular for many applications in molecular simulations (Holst, 1993; Baker, 2004, 2005; Feig and Brooks III, 2004; Dong et al., 2008; Boschitsch and Fenley, 2004, 2007).
Implicit solvent models require a separation of macroscopic and microscopic domains at the solvent-solute boundary. Commonly used boundaries are van der Waals surfaces and molecular surfaces (Richards, 1977). However, these surfaces may admit geometric singularities, such as cusps and self-intersection surfaces, which devastate their computational applications in structural modeling and simulation (Connolly, 1983; Sanner et al., 1996). Image smoothing techniques, including Gaussian kernel and anisotropic diffusion (Xu et al., 2006; Zhang et al., 2006), are applied to improve the regularities of Connolly surfaces. The first partial differential equation (PDE) based molecular surface was constructed in 2005 (Wei et al., 2005). Unlike PDE based surface smoothing techniques which start with a given surface, this approach embeds the atomic information, instead of a surface, in the Eulerian representation or formulation, and generate biomolecular surfaces by gradient driven diffusion and subsequent level-set extraction (Wei et al., 2005). An important question in molecular/structural biology is that “what is the physical boundary of a bio-molecule in solvent.” Therefore, in general, the biomolecular surface morphology should be determined by the optimization of the free energy of the macromolecule in the aquatic environment. Recently, we have addressed this issue by considering a mean curvature flow model of bimolecular surfaces, the minimal molecular surface (MMS) that minimizes the surface free energy functional (Bates et al., 2006, 2008). More recently, we have presented a general procedure for the formation and evolution of biomolecular surfaces by balancing the geometric curvature effects and potential effects (Bates et al., 2009). The mathematical structure of this approach was prototyped in 1999 (Wei, 1999). In our approach, stochastic geometric flows have also been introduced to account for the random fluctuation and dissipation in density and forces near the surface (Bates et al., 2009). Physical properties, such as free energy optimization (area decreasing) and incompressibility (volume preserving), were realized in our potential driven geometric flow equations (Bates et al., 2009). An important issue in implicit solvent models is the lack of sufficient descriptions of ion correlation, polar–nonpolar coupling and solvent-solute interactions (Ashbaugh, 2000; Bostrom et al., 2005; Cerutti et al., 2007; Chorny et al., 2005; Dzubiella et al., 2006a; Fixman, 1979;Forsman, 2004). This problem was considered by Dzubiella et al. (2006a) who proposed an interesting free energy optimization procedure for coupling polar–nonpolar interactions. Their model includes contributions from pressure, Gauss and mean curvatures, short-range repulsion, dispersion and electrostatic effects. Biomolecular surfaces were generated from this model via the level set approach (Cheng et al., 2007) which is similar to our Eulerian geometric flow approaches of biomolecular surfaces (Wei et al., 2005; Bates et al., 2006, 2008, 2009).
Geometric flows (Willmore, 1997) or geometric evolution equations, have been extensively studied in the mathematical context in the past two decades (Evans and Spruck, 1991; Gomes and Faugeras, 2001; Mikula and Sevcovic, 2004). Computational techniques based on level sets were devised by Osher and Sethian (Osher and Sethian, 1988; Rudin and Osher, 1992; Sethian, 2001) and have been developed and applied by many others in geometric flow analysis (Cecil, 2005; Chopp, 1993; Smereka, 2003; Sarti et al., 2002; Sbert and Solé, 2003; Sethian, 2001; Sochen et al., 1998; Du et al., 2004). An alternative approach is to minimize energy functional in the framework of the Mumford–Shah variational functional (Mumford and Shah, 1989) and the Euler–Lagrange formulation of surfaces developed by Chan and coworkers (Blomgren and Chan, 1998), and others (Carstensen et al., 1997; Li and Santosa, 1996; Osher and Rudin, 1990; Rudin and Osher, 1992; Sapiro and Ringach, 1996). We introduced some of the first high-order geometric evolution equations for image analysis (Wei, 1999). Our approach was based on the kinetic theory and generalized conservation principles. Our high-order geometric flow equations have led to many interesting applications (Wei, 1999; Wei and Jia, 2002; Sun et al., 2006a; Lysaker et al., 2003; Gilboa et al., 2004). Recently, Bertozzi and Greer have carried out mathematical analysis of these high order equations in Sobolev space H1 norm (Bertozzi and Greer, 2004; Greer and Bertozzi, 2004a, 2004b), which proved the existence and uniqueness of the solution to a case with H1 initial data and a regularized operator. We also introduced coupled geometric flow equations for image edge detection (Wei and Jia, 2002). Recently, we have proposed an evolution operator based single-step method for image denoising and enhancement (Sun et al., 2006a). Surface gradient flows derived from the optimization of certain functionals defined on the surface are commonly used for applications ranging from surface diffusion (Taubin, 1995), denoising of acquired surface data from 3D scanners (Desbrun et al., 1999), harmonic analysis and structure definition of data (Coifman et al., 2005), shape optimization and surface design (Kobbelt, 2000; Bobenko and Schröder, 2005), minimal surfaces (Pinkall and Polthier, 1993), texture transfer (Dinh et al., 2005), to dynamic evolution of surfaces (Grinspun et al., 2003). Such flows can be either extrinsic, where the embedding of the surface itself changes, as in the mean curvature flow; or intrinsic, where properties such as metric tensor defined on the surface evolve, as in the Ricci flow. The former is the method of choice for the purposes of visualization and most other applications. The latter, however, may provide the necessary supplement for classification, analysis and simplification of large surface datasets.
An unsolved problem in implicit solvent models is how the macroscopic description is coupled to the microscopic description. So far, most attention has been paid to the calculation of the electrostatic energy or the solvation energy. To our best knowledge, there is no complete description of both the macroscopic system and the microscopic system on an equal footing in the literature. Without carefully spelling out the detail of macro-micro coupling, implicit solvent models cannot be formally regarded as multiscale models per se. Additionally, an important while essentially neglected issue is how the forces of solvation models generated from microscopic and macroscopic domains are balanced. This issue is particularly important whenever the forces computed from solvation models are either to be utilized for molecular dynamics in any manner (Gilson et al., 1993; Im et al., 1998; Lu et al., 2002; Lu and Luo, 2003; Luo et al., 2002; Prabhu et al., 2004), or to be used in a comparison with those from explicit methods (Swanson et al., 2007) or experimental data. A throughout understanding of force balance and macro-micro coupling requires again a complete description of both the macroscopic subsystem and the microscopic subsystem on an equal footing. Moreover, while the nonequilibrium property of the microscopic subsystem has been studied by implicit-solvent molecular dynamics (Gilson et al., 1993; Im et al., 1998; Lu et al., 2002; Lu and Luo, 2003; Luo et al., 2002; Prabhu et al., 2004), the nonequilibrium property of the macroscopic subsystem has rarely been considered in implicit solvent models. In fact, the nonequilibrium property of the macroscopic subsystem is crucial to the protein hydrophobic collapse and RNA folding transition time in solution observed with microfluidic techniques (Lapidus et al., 2007). It is high time to develop a multiscale paradigm that unifies the macroscopic description of the aquatic environment and the microscopic description of the macromolecule.
Recently, researchers have developed multiscale hybrid simulations of fluid dynamics and molecular dynamics to study a wide range of physical phenomena, including micro/nano-scale nozzle structure of inkjet head or electrospray devices (Budiono et al., 2008) and the structure of normal shock waves in dilute argon (Valentini and Schwartzentruber, 2009). This approach typically resorts to the Lennard–Jones potential for describing molecular interactions. The domain boundary between the fluid dynamics and the molecular dynamics is able to feature the hydrophilic and the hydrophobic surfaces for a jetting system (Budiono et al., 2008). However, similar models for macromolecules in fluid flows are yet to be developed.
Another class of challenging problems concerns complex chemical systems, such as fuel cells (Franco et al., 2006; Gurau and Mann, 2009; Promislow and Wetton, 2009), and complex biological systems, such as ion channels (Cheng et al., 2009; Hwang et al., 2006; Gordon et al., 2009) and adenosine-5′-triphosphate (ATP) synthase motors. One particular challenge of these problems is that they may involve excessively large aqueous macro-molecular complexes. An interesting example is DNA packing and unpacking (Grigoryev et al., 2009). DNA contains the basic genetic information for all modern living things to function, grow, and reproduce. A single human DNA molecule can have billions of atoms. Another challenge is that these systems encompass a wide range of physical phenomena, including electrophoresis, electroosmosis (Zheng et al., 2003; Chen and Conlisk, 2008), electrohydrodynamics, and molecular dynamics. The underlying chemical and biological systems can be far from equilibrium, and thus the Boltzmann distribution used in the Poisson–Boltzmann theory may be no longer valid. The density distributions of charged species have to be accounted by alternative approaches. Usually, these systems are described by the Nernst–Planck equation in conjunction with the Poisson equation and the Navier–Stokes equation (Chen et al., 1995; Chu and Bazant, 2006; Vlassiouk et al., 2008; Abaid et al., 2008; Zhou et al., 2008c). Very often, Brownian dynamics, as well as molecular dynamics is also employed to study these complex chemical/biological systems. A unified multiscale theory that includes Poisson–Nernst–Planck equations, the Navier–Stokes equation and the implicit-solvent molecular dynamics will provide a new approach for the description of these systems. It will also enhance our understanding of the advantage and the limitation of the Navier–Stokes Poisson–Nernst–Planck theory, and the molecular dynamics. Such an understanding is crucial for detailed comparison and the choice of simulation parameters. Additionally, the origin of the drift and the diffusion of individual components in the continuum theory is also needed to be clarified. Therefore, it is imperative to develop a multiscale paradigm that can effectively utilize current computational capabilities to advance our understanding of the complexity of chemical and biological systems, including their equilibrium and nonequilibrium properties.
The objective of this work is to create a fundamental paradigm, i.e., a differential geometry based multiscale variational framework, to address the aforementioned challenges in implicit solvent models and Poisson–Nernst–Planck (PNP) theories. Though these challenges originate from a large number of atoms and a variety of interactions in macromolecular systems including the aquatic environment, the lack of multiscale models to provide an appropriate description of the solvent has led to the current inadequacy in understanding. Utilizing the differential geometry theory of surfaces, we formulate a multiscale paradigm that puts the macroscopic description of the solvent and the microscopic description of solute on an equal footing. We describe the biomolecule in the atomistic detail and describe the transport properties of the solvent with mechanical variables. The interface of the macroscopic and microscopic subsystems is naturally described by the differential geometry theory of surfaces. We set up new free energy functionals for equilibrium analysis and action functionals for non-equilibrium studies of the solvation process. The free energy functionals and action functionals are optimized by the first variation and the least action principle, respectively. Such optimization processes generate the desirable governing equations for both the macroscopic and the microscopic subsystems. While equilibrium properties are determined by the generalized Poisson–Boltzmann equation and potential driven geometric evolution equations, the nonequilibrium properties of the macroscopic and microscopic subsystems are described by generalized Navier–Stokes equations for fluid dynamics and the Newton's equation for molecular dynamics, respectively. Finally, we analyze a class of problems that are far from equilibrium by using our multiscale approach. We formulate generalized geometric, Poisson–Nernst–Planck, Navier–Stokes and Newton's equations for describing chemical reactions, electrohydro-dynamics, electroosmosis, and electrophoresis in ion channels, ATP synthase motors and fuel cells.
The rest of this paper is organized as the following. We first give a brief review of the differential geometry theory of curvatures and surfaces in Section 2. The construction of potential-curvature driven geometric flows is also discussed. Section 3 is devoted to the differential geometry based multiscale models for aqueous macromolecular complexes near equilibrium. Here, aqueous macromolecular complexes mean macromolecules and their aquatic environment, and near equilibrium means the system may be nonequilibrium but the Boltzmann distribution for charged species is still valid to a good approximation. To make it easily understood, we have gradually built up our full differential geometry based multiscale models by considering a family of models, starting from a simple electrostatic system, then adding a nonpolar solvation component, followed by a further inclusion of fluid dynamics, and finally considering the addition of the molecular dynamics. In this manner, the advantages and drawbacks of each model can be clearly demonstrated step by step. In Section 4, existing problems, including some apparent inconsistence, in the current Poisson–Boltzmann based implicit solvent models are analyzed. New multi-scale solvation models that are without the differential geometry based surface description are also provided so that two classes of formulations, either with or without the differential geometry based surface description, can be compared to enhance the understanding. Section 5 provides a differential geometry based multiscale framework to the electrohydrodynamics and eletrophoresis in fuel cells and ion channels. The origin of the drift and diffusion in the PNP theory is analyzed. Two new multiscale models are presented to allow a unified description of self-consistently coupled fluid dynamics, charge drift and diffusion, surface formation and evolution, and the molecular dynamics of macromolecules. Section 6 deals with a class of challenging problems that involve excessively large aqueous macromolecular complexes. For this type of systems, we develop alternative differential geometry based fluid-electro-elastic models in which the expensive molecular dynamics description is replaced with an elastic representation. This paper ends with some concluding remarks.
2. Review of curvatures, surfaces and geometric flows
In this section, the differential geometry theory of surfaces (Willmore, 1997; Wolfgang, 2002) and potential driven geometric flows (Bates et al., 2009; Wei, 1999) are briefly reviewed to establish notations and to facilitate further development.
2.1. Curvature and surface preliminary
We consider a C2 immersion , where is an open set (a chart for an n-manifold). Here, I(u) = (I1(u), I2(u), . . . ,In+1(u)) is a position vector labeled by x = I(u) for a point on a hypersurface, and . We define tangent vectors (or directional vectors) as . The Jacobian matrix of the mapping I is then given by DI = (X1, X2, . . . ,Xn). We denote 〈, 〉 as the Euclidean inner product in . An important quantity is the first fundamental form I of a surface element given by I(Xi, Xj) := 〈Xi, Xj〉 for every two tangent vectors Xi, Xj ∈ TuI, the tangent hyperplane at I(u). In the coordinate I(u) = (I1(u), I2(u), . . . , In+1(u)), the first fundamental form is a symmetric, positive definite matrix (gij) = (I(Xi, Xj)). Let N(u) be the unit normal vector given by the Gauss map ,
| (1) |
where × is the cross product (i.e., wedge product) in and ⊥uI is the normal space of I at point x = I(u). The vector N is perpendicular to the tangent hyperplane TuI at I(u). Note that , the tangent space at x. By means of the normal vector N and tangent vectors Xi, the second fundamental form is given by
| (2) |
With this form, the mean curvature can be calculated from H = hijgji, where we have used the Einstein summation convention of repeated indexes, and (gij) = (gij)−1. The shape operator of I is the Weingarten map defined pointwise by by
| (3) |
where , is the Jacobian of the Gauss map, and the map is a linear isomorphism with well defined inverse mapping (DIu)−1. Note is self-adjoint in the basis and has the property of . With these notations, we can make the connection between the first and second fundamental forms
| (4) |
where II is a symmetric bilinear form on TuI for every .
In general, given an n-dimensional manifold Ξ embedded in and a vector field b defined in, the divergence of b with respect to Ξ is given by , where g = Det(gij) is the Gram determinant of I. The surface is described locally by the mapping to . The gradient of a scalar field b on the maifold is given by . The Laplace–Beltrami operator is given by
| (5) |
We apply the Laplace–Beltrami operator to the position vector x and obtain
| (6) |
where N is the normal vector at x and H is the mean curvature. High-order curvatures have been extensively studied in the context of membrane bending analysis (Canham, 1970; Helfrich, 1973; Ou-Yang and Helfrich, 1989; Iwamoto et al., 2006).
Another important operation is the manifold integration. The integral of a function b on the manifold is given by
| (7) |
This expression gives the surface area when b = 1.
2.2. Lagrangian formulation of geometric flows
The formation of biomolecular surfaces can be described via either the Lagrangian formulation or the Eulerian formulation. In the Lagrangian formalism, surface elements are evolved directly under various driven forces. In the Eulerian formalism, the surface is embedded in a hypersurface and the latter is evolved under prescribed driving forces according to physical and/or biological principles. A sharp surface is obtained from an iso-surface extraction procedure, such as the level set approach. The Lagrangian formalism is straightforward for force prescription and is computationally efficient, but has difficulties in handling geometric singularities, such as surface breaking up or surface merging. The Eulerian formalism can easily handle geometric singularities, but is more time consuming and may have difficulty in prescribing driven forces. However, this difficulty can be overcome by an appropriate construction of the total energy functional or the total action functional, as shown in this work.
In the Lagrangian formulation, the surface deformation or evolution of biomolecules can be postulated as a time-dependent process for a family of smooth surfaces, which are defined as an immersion of two-dimensional manifolds Ξ(t) in . Here, Ξ(t) are parameterized by t, {Ξ(t) : t ≥ 0, Ξ (0) = Ξ0}, where 0 is the initial manifold determined by the input configuration of the molecule with appropriate selections of atomic radii, such as expanded van der Waals radii. Assuming a biomolecule in a solvent, the equation of motion for a position vector x(t) ∈ Ξ(t) on the surface is given by
| (8) |
This is the mean curvature flow and it has been used for the generation of the minimal molecular surface (MMS) in the Eulerian formulation (Bates et al., 2008). Desired MMSs are generated with appropriate geometric constraints originating from molecular boundaries as explained in detail in Bates et al. (2008). Specifically, the van der Waals surface of the molecule is protected during the evolution.
More general geometric flows can be cast in the form of
| (9) |
where Vg is the amplitude of the velocity V induced by geometric and potential forces. The surface diffusion flow, Willmore flow (Willmore, 1997) and generalized surface diffusion flows are constructed respectively by Vg = −ΔΞH, Vg = –ΔΞH – 2H(H2 – K), and , where K is the Gauss curvature (Bates et al., 2009. Willmore flow is a special case of the Canham–Helfrich flow (Canham, 1970; Helfrich, 1973) and preserves the sphericity of the geometry. It has been used in cellular membrane modeling. Surfaces with volume preserving and area decreasing properties were constructed in our earlier work (Bates et al., 2009). The volume preserving property is important for computations in microcanonical, canonical, and grand canonical ensembles. Stochastic geometric flows were introduced (Bates et al., 2009). Such a formulation is necessary for the introduction of Brownian dynamics and Langevin dynamics.
2.3. Eulerian formulation of potential driven geometric flows
Similarly, the Eulerian formulation can be obtained via an appropriate choice of the immersion I. Previously, we have chosen I = (x, y, z, S) which maps (Bates et al., 2008). It is quite easy to show that
| (10) |
where is the normal vector in , S = (0, 0, 0, 1), and Ξ is the graph of S. It is noted that N is the normal vector for the surface immersion I, rather than for the hypersurface function S. For a given surface, it is easy to show that its normal vector does not depend on whether its description is based on an implicit function or an explicit one. In our earlier work, potential driven geometric flows (Bates et al., 2009) were constructed as
| (11) |
where , and V is a microscopic interaction potential, such as the Lennard–Jones potential. This approach allows the balance between intrinsic geometric effects and potential interactions. In fact, the basic structure of Eq. (11) was introduced in our earlier high-order geometric evolution equations (Wei, 1999). The computational procedure for the surface formation and evolution of biomolecules was described in detail (Bates et al., 2009). Different numerical methods for biomolecular surface constructions were constructed, analyzed, and compared (Bates et al., 2009). To illustrate the concept, a surface generated by using the potential driven geometric flows is depicted in Fig. 1.
Fig. 1.

A C60 surface generated by geometric and potential driven geometric flows.
Additionally, higher-order geometric flows, including ones similar to our earlier formulations (Wei, 1999), were constructed for the formation and evolution of biomolecular surfaces (Bates et al., 2009). The performance of the higher-order geometric flows was examined (Bates et al., 2009). Typically, higher-order geometric flows produce distinguished molecular morphologies.
In Sect. 3.2, we show that the connection between Eulerian and Lagrangian formulations can be made by the geometric measure theory. From the point of view of computation, it is necessary to express all quantities in either the Eulerian or the Lagrangian formulation.
3. Multiscale models for aqueous macromolecular complexes near equilibrium
The concept of interface often comes along with the separation of different phases of the same matter, such as solid, liquid, and gaseous phases of water molecules. In thermodynamics, phase transitions are associated with a latent heat (first-order) or a divergence in other physical properties, such susceptibility or correlation length (second order). Interface is also used to describe the separation of different materials, such as oil and water, and solvent and solute. These materials may have similar physical properties, such as in the same type of phases, but they differ in chemical and/or other physical properties. In general, the concept of material interface can be used if there is a significant measurable spatial difference in either a chemical property or a physical property. A material interface can be either very sharp or blurred, depending on the nature of the interfacial interactions. In this work, we use the word “boundary” to describe a relatively blurred interface—there is a considerable overlap in the wavefunctions over different spatial regions due to strong interactions. One of our goals is to determine interfacial morphology, i.e., interface or boundary, by means of the fundamental laws of physics. The ability to make the aforementioned spatial discrimination gives us a leverage to select different descriptions for different regions (subdomains) of the spatial domain, according to our needs and computational capability.
In this section, we formulate differential geometry based multiscale models for macromolecules and their aquatic environment that are near equilibrium. As such, the density of charged particles in the continuum domain can be approximated by the Boltzmann distribution and no additional equation is needed to describe the density profile of charged species. We start with a simple model for electrostatic interactions and propose a new generalized Poisson–Boltzmann equation. Then, many modifications to the electrostatic model are made, including the consideration of a nonpolar solvation free energy, and models for hydrodynamics and molecular dynamics.
In our multidomain setting, we take a discrete atomistic description for the macromolecule, and a continuum macroscopic description for the aqueous solvent. We consider the domain , which is essentially divided into two (types of) regions, Ωm and Ωs, i.e., . Here, Ωm and Ωs are domains for macromolecules and aqueous solvents, respectively. An illustration of macromolecular and solvent domains is depicted in Fig. 2. However, at the atomic scale, Ωm and Ωs may overlap each other as the boundary of biomolecules and solvent cannot be perfectly sharp, because of the overlapping distribution of electron densities. Therefore, the solvent-solute boundary is . For the discrete description of macromolecules, we consider a total of Na atoms. We set as the macroscopic variable and as the microscopic variable of Na atoms. We use the hypersurface function to characterize the boundary of the solvent and molecular domains (Bates et al., 2008, 2009). As such, S(x) is essentially a characteristic function of the biomolecular domain, i.e., it is one (S = 1) inside the biomolecule and zero (S = 0) in the aquatic media as indicated in Fig. 2. Consequently, (1 − S) is essentially a characteristic function of the solvent domain. Neither does S nor does (1 − S) behave like a Heaviside function. Instead, it takes a value between zero and one (0 ≤ S ≤ 1) near the boundary of the macromolecule. Such a profile characterizes the boundary between the biomolecule and the aquatic environment. In terms of S and (1 − S), we can also define the molecular subdomain and the aquatic subdomain as Ωm : {x | S(x) ≠ 0} and Ωs : {x | (1 − S(x)) ≠ 0}, respectively.
Fig. 2.
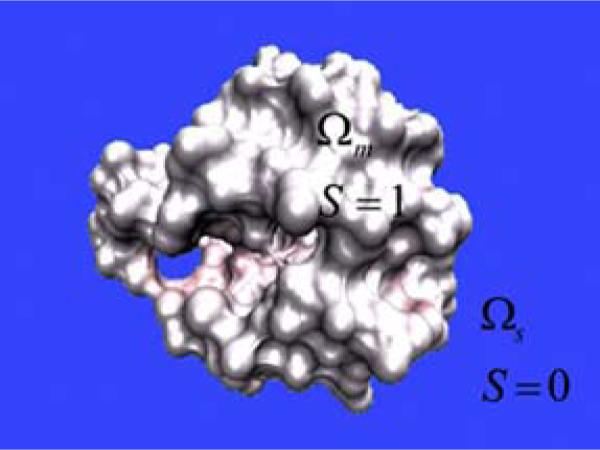
An illustration of the macromolecule and its aquatic environment. The macromolecular domain (Ωm) and its hypersurface function value S = 1 are indicated on protein 451c. Similarly, the aqueous domain (Ωs) and its hypersurface function value S = 0 are indicated on the background.
3.1. Electrostatic modeling
Among various components of molecular interactions, electrostatic interactions are of special importance because of their long range and influence on polar or charged molecules—including water, aqueous ions, and amino or nucleic acids (Warshel et al., 2006; Davis and McCammon, 1990; Dong et al., 2008; Grochowski and Trylska, 2007; Sharp and Honig, 1990; Warshel and Papazyan, 1998; Simonson, 2001; Fogolari et al., 2002; Simonson, 2003; Baker, 2004, 2005; Feig and Brooks III, 2004). Electrostatic interactions are ubiquitous for any system of charged or polar molecules, such as biomolecules (proteins, nucleic acids, lipid bilayers, sugars, etc.) in their aquatic environment. For example, proteins in living cells are made up of 20 types of amino acids, 11 of them are either charged or polar in neutral solution. Moreover, nucleic acids contain long stretches of negatively charged groups in addition to the polar phosphate groups in nucleotides. Electrostatic steering is an essential effect in molecular motors which convert chemical free energy from the hydrolysis of ATP into mechanical movement or work in living organisms. Therefore, electrostatic interactions are some of the most important aspects that determine the physical and chemical properties of biomolecules, such as protein folding, protein-DNA binding, transcription, translation, gene expression, and regulation, etc. As most biological processes occur in aquatic environments, electrostatic solute-solvent interactions are of paramount importance in the exploration of biological mechanism, the analysis of macromolecular behavior, and the modeling of the intramolecular and inter-molecular interactions of biological complexes.
Biomolecular systems are polarizable materials. Therefore, both electric field E and electric displacement D are important quantities. However, in the biophysical community, it is a convention to only deal with the electric field. As such, the polarization effects of neutral biomolecules are treated as explicit partial charges. This approach is simple and reasonable for a system without rapidly changing electric current and magnetic field. In fact, the effects of magnetic field H, magnetic displacement B and magnetic displacement are normally neglected in biomolecular systems.
3.1.1. Free energy functional of the electrostatic discrete-continuum system
The free energy functional of the electrostatic system was given by Sharp and Honig (1990), and Gilson et al. (1993). In this work, we consider a different free energy functional of electrostatic interactions of the form
| (12) |
where ϕ is the electrostatic potential, kB is the Boltzmann constant, T is the temperature, cj is the bulk concentration of jth ionic species, Nc is the number of ionic species, and is the canonical density of molecular free charges, with Qj being charges on (discrete) atoms. Here, ∊m = ∊0εm and ∊s = ∊0εs are the permittivities of the macromolecule and the solvent, respectively, where ∊0 is the permittivity of vacuum and εα (α = m, s) are relative permittivities. We treat εα as constants. Electrostatic potential is of long-range in nature and cannot be simply restricted to subdomains. Note that variables in the bracket [·] of expression GElect[S, ϕ, x] are the ones we wish to keep track of, instead of being the unique set of variables for variations.
The present free energy functional given in Eq. (12) differs much from that given by Sharp and Honig (1990), and by Gilson et al. (1993). It is inherently multidomain and multiscale in nature. The domain is divided into the macromolecular subdomain characterized by S, and the solvent subdomain characterized by 1 –S. Unlike in the previous set-up, these subdomains do not have to be mutually exclusive. Similar to the previous treatment, a discrete description of the macromolecule and a continuum description of the solvent are employed in Eq. (12). It is necessary to demonstrate that the present free energy functional reproduces the classical Poisson–Boltzmann equation and proper electrostatic forces densities at certain limits.
It is important to note that the sign convention used in Eq. (12) is the same as that used by Sharp and Honig (1990), and by Gilson et al. (1993). The square terms in Eq. (12) are always negative and admit the zero upper bound. Consequently, we need to optimize the electrostatic free energy functional, instead of minimizing it. This sign convention will impact all the sign conventions in the rest of this paper. Off course, the physics and mathematics will not be impaired if another convention is chosen.
3.1.2. Generalized Poisson–Boltzmann equation for the electrostatic discrete-continuum system
By optimizing GElect[S, ϕ, x] with respect to ϕ via the Euler–Lagrange equation while keeping S unchanged, we have
| (13) |
By setting Eq. (13) to zero, we have
| (14) |
where
| (15) |
provides in general a smooth dielectric profile near the interface. Equation (14) is a new generalized Poisson–Boltzmann equation for smooth dielectric profiles in overlapping domains. It is important to show that the classic Poisson–Boltzmann equation is a special case of Eq. (14) at the sharp interface limit. For a sharp solvent-solution interface, i.e., , S becomes a Heaviside function and we define
| (16) |
At the sharp interface limit, Eq. (14) reduces to the standard Poisson–Boltzmann equation (Sharp and Honig, 1990; Gilson et al., 1993; Holst, 1993; Yu et al., 2007a; Geng et al., 2007; Zhou et al., 2008a)
| (17) |
and appropriate interface conditions
| (18) |
where Γ is the sharp interface and n is the norm vector of the surface. Recently, we have developed the matched interface and boundary (MIB) method (Zhao and Wei, 2004; Zhou et al., 2006; Zhou and Wei, 2006;Yu et al., 2007b;Yu and Wei, 2007) for solving elliptic equations with discontinuous interfaces. Three generations of MIB based Poisson–Boltzmann (PB) solvers, the MIBPB-I (Zhou et al., 2008a), the MIBPB-II (Yu et al., 2007a) and the MIBPB-III (Geng et al., 2007) have been constructed. The MIBPB has a unique feature that it was the first interface technique based PB solver that rigorously enforces the solution and flux continuity conditions (18) at the dielectric interface in bio-molecular context. It is so far the only known existing second-order PB solver for the electrostatic analysis of macromolecules.
Because the solvent-solute boundary is essentially smooth and free of sharp geometric singularities, it is expected that the numerical solution of the generalized Poisson–Boltzmann equation is easier than that of the standard Poisson–Boltzmann equation (Chen et al., 2010). This will enhance the wider application of the Poisson–Boltzmann approach and particularly, provide a new promise for Poisson–Boltzmann based molecular dynamics simulations.
3.1.3. Electrostatic forces
It remains to investigate whether the proposed new free energy functional reproduces appropriate electrostatic forces. To this end, we carry out the variation of the electrostatic free energy functional, Eq. (12)
| (19) |
where the terms associated with δϕ vanished by the generalized Poisson–Boltzmann equation (14). It is important to note that because two terms are associated with domains. By using integration by parts, the last term in Eq. (19) can be written as
| (20) |
By further writing δS = ∇S · δx, we have
| (21) |
where fDB, fID, and fRF are force densities of dielectric boundary (DB), ionic boundary (IB) and reaction field (RF), respectively,
| (22) |
| (23) |
| (24) |
These are force density expressions for smooth solvent-solute interfaces. Except for a minor difference in the DB force, there expressions are consistent with those in the literature (Gilson et al., 1993).
3.2. Solvation modeling
3.2.1. Free energy functional of the discrete-continuum solvation system
The solvation process of macromolecules involves a number of interactions. Typically, the free energy of solvation models includes polar and nonpolar contributions (Levy et al., 2003; Dzubiella et al., 2006b; Gallicchio and Levy, 2004; Wagoner and Baker, 2006; Huang et al., 2001). For a system near equilibrium, the polar part is standardly represented by the free energy functional of Sharp and Honig (1990). In this work, we use Eq. (12) for describing the polar solvation energy. For the nonpolar solvation energy, we first consider the interface energies of surface curvature and the mechanical work
| (25) |
where γ is the surface tension and p(x) is the hydrodynamic pressure. The first term is the surface energy and the second team is the mechanical work of creating the vacuum of a biomolecular size in the fluid (Sitkoff et al., 1996). The surface energy measures the disruption of intermolecular and/or intramolecular bonds that occurs when a surface is created. Energetically, the surface creation must be intrinsically unfavorable otherwise the macromolecule would be unstable and fail to exist in the folded form in the solvent. Therefore, the interface energy is hydrophobic in nature for most biomolecules.
Note that the first term in Eq. (25) is given in terms of a surface integration over the manifold as discussed in Sect. 2.1 (see Eq. (7)). To optimize the energy, it is necessary to change this integral into a volume one. To this end, we make use of the coarea formula (Federer, 1959) in geometric measure theory. When reduced to the case of 3D (Euclidean) space, this formula states that for a scalar field B with C1 continuity conditions, integrating a function b over each of its isolevels c in a region can be done directly by a volume integral over through (Federer, 1959)
| (26) |
where c denotes an isovalue of B, B−1(c) represents the c-isosurface (i.e., the set of 3D points {xc} such that B(xc) = c), and B(·) is an arbitrary function of space. In other words, the term ∥∇ B∥ measures a local “density of isosurface area.” Consequently, if we think of the foliation consisting of all B−1(c) as the representation of a “blurred interface,” the coarea formula elucidates the relationship between the sum of area integrals and a global volume integral. In our case, the hypersurface function S is essentially a characteristic function of the biomolecule and takes all the isosurface values from 0 to 1. Therefore, the volume integral gives the mean surface area of a family of isosurfaces. Consequently, we approximate the interface mechanical energy by
| (27) |
Wagoner and Baker (2006) have shown that the attractive dispersion effects near the solvent-solute interface described by the Weeks–Chandler–Andersen (WCA) potential (Weeks et al., 1971) play a crucial role in the solvation analysis. A similar solvent-solute interaction term was considered by Dzubiella et al. (2006b) with the Lennard–Jones potential. In this work, we use a general potential term in the nonpolar contribution to obtain the following nonpolar solvation free energy functional:
| (28) |
where ρs is the solvent density and u(x, z) is a general solvent-solute interaction (SSI) potential which involves those atoms in the molecule that are near the solvent-solute interface. The integration of the potential term is over the solvent domain and the contribution from atoms is given by a summation.
The total free energy functional of solvation is given by
| (29) |
This total free energy functional can be used to derive governing equations for the solvation system.
3.2.2. Surface evolution equation of the discrete-continuum solvation system
First, it is easy to show that by optimizing Gtotal[S, ϕ, x] with respect to ϕ, we obtain the desirable generalized Poisson–Boltzmann equation (14) again. In this work, we are interested in the derivation of potential driven eometric flows (Bates et al., 2009) from the discrete-continuum solvation system. To this end, we carry out the variation of Gtotal[S, ϕ, x] with respect to the hypersurface function, S
| (30) |
where is a generalized Laplace–Beltrami operator. It is related to the mean curvature and minimizes the surface area of the macromolecule.
An efficient approach for the optimization of the total action and the generation of the solvent-solute boundary profile is to construct potential driven geometric flows by using the steepest descent scheme as proposed in our earlier work for solvation analysis (Bates et al., 2009). By examining the structure of Eq. (11), we have
| (31) |
The steady-state solution to Eq. (31) balances the hydrophobic surface energy, hydrostatic pressures, and electrostatic energies. From the point of the view of differential geometry, Eq. (31) is a potential driven geometric flow in a Riemannian space with a new conformal metric. Clearly, if we rewrite Eq. (31) as
| (32) |
with V being appropriate non-curvature terms, we essentially recover the potential driven geometric flow introduced in our previous studies (Bates et al., 2009). An important feature of these potential driven geometric flows is that they are multiscale in nature, and mix macroscopic and microscopic descriptions. We can solve Eq. (31) and extract a sharp solvent-solute interface using the same procedures and similar initial/boundary conditions as discussed in our previous work (Bates et al., 2006, 2008, 2009). However, in the present work, we do not assume a sharp solvent-solute boundary.
3.2.3. Solvation forces
Solvation forces are often evaluated to check the consistence of different solvation theories (Wagoner and Baker, 2006). The derivation of solvation forces is the same as that of electrostatic forces
| (33) |
Here, terms associated with the variation of the electrostatic potential, δϕ, vanish because of the enforcement of the generalized Poisson–Boltzmann equation (14). Similarly, terms associated with the variation of the hypersurface function, δS, vanish because of the enforcement of the potential driven geometric flow equation (31). Integrating by parts, we have
Finally, terms associated with the variation of the spatial variable, δx, can be written as
| (34) |
| (35) |
where fP, fSSI, and fRF are force densities of pressure gradient, solvent-solute interaction and reaction field (RF), respectively,
| (36) |
| (37) |
| (38) |
A few remarks are in order. First, these force expressions differ much from those of the electrostatic system given in Eq. (22) because of the present consideration of nonpolar solvation energies. Additionally, a very interesting point is that the dielectric boundary force and ionic boundary force do not appear now because of the enforcement of the potential driven geometric flow equation (31). This means that forces can be balanced in different manners according to settings. If the molecular surface is used instead of the surface generated by using the potential driven geometric flow, the dielectric boundary force and the ionic boundary force will present as shown in Section 3.1.3. Moreover, the energy functional considered in this section can be regarded as a system in equilibrium. As such, the equilibrium pressure is homogeneous in space and the pressure gradient force vanishes fP = 0. Furthermore, the force due to solvent-solute interaction is important only near the interface. The solvent density, ρs, may be regarded as a constant at equilibrium for a dilute solution. Of course, the densities of charged components of the solvent satisfy the Boltzmann distribution. Note that the effect of solvent orientations (Fries and Patey, 1985; Cerutti et al., 2007) and the density distortion near the solvent-solute boundary as that described in the integral equation theory (Tully-Smith and Reiss, 1970; Beglov and Roux, 1997) are not concerned in the present work. Finally, as atomic centers are away from the interface, the hypersurface function is near unity, i.e., S ~ 1, at atomic centers. One has fRF = ρm ∇ϕ, which is the same as the reaction field force derived in the last section.
3.3. Fluid and static solute interactions
A major problem in the preceding two subsections is that there is no mechanism to eliminate all forces. Stated differently, the forces in the system are unbalanced. This is an indication that something may have gone wrong in the model because the action functional cannot be minimized (or optimized). In this subsection, we consider a system of fluid and static solute interaction. We keep in mind that the macromolecule is placed in the solvent in this system. The atoms in the biomolecule are partially charged. Ions in the solvent are not described as discrete particles but as a continuous distribution. Additionally, there are interactions between the macromolecule and the solvent that lead to forces. We assume that these forces are balanced by fluid motion, which is also subject to appropriate initial and boundary conditions. We also assume that the atoms in the bio-molecule are essentially at their equilibrium positions, and densities of ionic species can be approximated by their equilibrium Boltzmann distributions.
3.3.1. Action functional for fluid and static solute interactions
For inviscid flows, the fluid energy can be accurately classified as kinetic energy and potential energy (Φ + p), where ρs is the density of the fluid, v is the fluid velocity vector, Φ is the potential energy mostly due to the gravitation, and p is the hydrodynamic pressure. Such a total energy is constant everywhere in the fluid
| (39) |
according to Bernoulli's principle. However, for viscous flows, the sum of the kinetic energy and the potential energy is no longer a conservative quantity. The fluid may lose energy to the shear stress and/or extensional stress. From the kinetic theory point of view (Snider et al., 1996a, 1996b), the nature of the stress tensor involves microscopic many-body interactions in the fluid, which are absent from macroscopic models. Such unaccounted interactions normally lead to irreversible dissipation. Therefore, the exact form of the stress tensor is generally difficult to determine. In practical, the form of the stress tensor depends on the level of the approximation. Phenomenologically, many expressions, including those under the assumptions of Newtonian fluid and non-Newtonian fluid, have been used for the stress tensor. In an earlier work, we simplified stress tensors by their symmetry properties and analyzed their connection to the quantum kinetic theory (Wei, 2002). The selection of the stress tensor for biological systems is an active area of research.
In the present work, we consider a simple fluid model, a Newtonian fluid, and its stress tensor is given by
| (40) |
where μf is the viscosity of the fluid, symbol T denotes the transpose, is the identity tensor and the Einstein summation convention is used. The stress tensor is a symmetric and traceless second rank tensor. In the present work, we restrict our discussion to incompressible fluid flows
| (41) |
which is a good approximation for most aquatic chemical and biological systems at ordinary (or physiological) conditions. Therefore, we have
| (42) |
Due to its dissipative nature, the stress energy depends on the fluid history. Therefore, we denote the stress energy density as a semidefinite integral
| (43) |
where the velocity v is considered as a function of time t′. Here, the unspecified lower time limit refers to a time when the fluid is at rest, and thus the stress energy is zero. The Lagrangian of an incompressible viscous flow is given by
| (44) |
where u is the potential interaction between the fluid and the atoms in the biomolecule, i.e., the solvent-solute interaction (SSI). The stress term takes a negative sign because it costs the dissipation of the fluid energy.
The total action functional of the system includes contributions from the solvation energy discussed in the preceding subsection, and the Lagrangian of the fluid
| (45) |
where the Einstein summation convention is used only for tensorial quantities. Similarly, we make use of the Einstein notation only for stress and strain tensors in the rest of this paper. Here, we have chosen a negative sign for the fluid Lagrangian (LMacro) so that the signs for potential energies are consistent with their signs used in the preceding analysis. Since the solvent-solute interaction has been considered as a part of the interface energy, it is omitted from the fluid Lagrangian to avoid double counting. The gravitation potential energy (Φ) has also been omitted due to the small length scale of the problem under consideration. Nonetheless, keeping this term does affect much of the following discussion.
3.3.2. Surface evolution equation for fluid and static solute interactions
We are interested in the governing equations for fluid-electrostatic interactions. First, the generalized Poisson–Boltzmann equation (14) can be derived by optimizing the total action functional () in Eq. (45) with respect to ϕ. As the generalized Poisson–equation depends in the hypersurface function S, to solve the generalized Poisson–Boltzmann equation, one needs to determine S. To derive an appropriate equation for S, we use the same procedure discussed earlier, i.e., optimizing the total action functional () in Eq. (45) with respect to S, following by the use of the steepest descent scheme as proposed in our earlier work (Bates et al., 2008, 2009). We have the following governing equation for the time evolution of the hypersurface function:
| (46) |
Note that two pressure terms in Eq. (46) do not cancel each other because they are in different domains. Equation (46) can be solved by using a similar procedure developed in our earlier work (Bates et al., 2008, 2009).
3.3.3. Generalized Navier–Stokes equation of fluid and static solute interactions
It is well known that the fluid velocity in a simple setting is governed by the Navier–Stokes equation
| (47) |
| (48) |
where is the (deviatoric) stress tensor, and F represents body forces per unit volume. The divergence condition ∇ · v = 0 is for the incompressibility of the fluid. The force term, F may have many different origins and is balanced with forces on the macromolecular boundary (Geng and Wei, 2009). In real fluid, the exact form of the stress tensor is unknown. In fluid mechanical modeling, the selection of the stress tensor usually depends on the level of approximation, such as, Newtonian fluid or non-Newtonian fluid, etc. The Navier–Stokes equation has been applied to an extremely wide variety of situations, ranging from ocean flows, weather prediction, aerodynamics, chemical reactors to blood circulations. When biomolecular systems are subject to unsteady solvent environments, the consideration of the Navier–Stokes equation for the solvent motion becomes a reasonable alternative to more expensive full atom description.
As the governing equation for the momentum conservation of the fluid, usually, the Navier–Stokes equation is derived either from a kinetic equation, e.g., the Boltzmann equation (Snider et al., 1996a, 1996b), or from a formal analysis of conservation laws. An explicit variational derivation of the Navier–Stokes equation was given by Sciubba (2004) with a brief discussion of historical perspective. In general, the derivation of the Navier–Stokes from the Euler–Lagrange equation or from the least action principle has not been well accepted, despite that the derivation of the ideal fluid is well known (Sciubba, 2004). An important goal of the present work is to derive the Navier–Stokes equation (47) by the least action principle in our multiscale framework. To this end, we apply the principle of the least action to the total action functional (45)
| (49) |
where the variation of the fluid part has not been carried out yet. Clearly, Eq. (49) consists of coupled fluid dynamics and hypersurface dynamics. First, terms associated with δϕ vanish because of the generalized Poisson–Boltzmann equation (14). Additionally, terms associated with δS vanish because of the hypersurface evolution equation (46).
Before we discuss terms associated with δx, we further carry out the variation of the fluid Lagrangian. Since the fluid is assumed to be incompressible, the variation is restricted by the condition that the mass should not be varied, i.e.,
| (50) |
In the Lagrangian formulation, we have
| (51) |
where we have used the relation . We could have distinguished the general macroscopic position variable x and the position of the fluid particle by denoting the latter with a different symbol. However, since the Jacobian connecting these two vectors is an identity matrix, this more rigorous treatment does not lead to anything new. Here, S is considered as time independent. Note that the time dependence of S is created by the steepest decant procedure, which is devised to efficiently minimize the term associated with dS. Similar to the geometric flows discussed in Sect. 2, the fluid equations can be expressed in either the Eulerian formulation or the Lagrangian formulation. In the Lagrangian formulation, one tracks a fluid particle as it moves from some initial point to a neighboring point. The motion and acceleration of the fluid particle are described directly by hydrodynamic equations. In the Eulerian formulation, we consider a rectangular reference system which is at rest. All field quantities, such as density, velocity, pressure, etc. are considered as at a fixed point in space. We use operator dv/dt to describe two consecutive positions of the same fluid particle in the Lagrangian formulation. However, unlike geometric flows, the fluid flow is moving with the stream velocity v. Consequently, the Lagrangian description of the rate of change (dv/dt) is related to the Eulerian description by
| (52) |
This equation is used to switch the Lagrangian formulation in Eq. (51) into the Eulerian formulation.
The variation over the pressure term gives . For the stress term, we have
where is the stress at an unspecified time. It is reasonable to assume that the fluid at some unspecified time is static, then vanishes. We therefore drop the for simplicity. Alternatively, one may just keep it and define the difference as a new stress tensor.
Finally, we collect all terms associated with δx
| (53) |
where force f is defined in Eqs. (34)–(38). Equation (53) describes the coupling of the fluid dynamics to the hypersurface dynamics and electrostatic interactions. To understand the relative separation of these two parts, one needs to recall that the hypersurface function S is essentially a characteristic function of the biomolecular subsystem, and similarly, (1 − S) is essentially a characteristic function of the fluid subsystem. The S and the (1 − S) regions are essentially independent to each other except that the force associated with the biomolecule in the S region is balanced by the pressure gradient and fluid acceleration in the (1 − S) region.
It is easy to show that for (1 − S) ≠ 0, the quantity in the integrand of Eq. (53) is the desirable generalized Navier–Stokes equation
| (54) |
where the force is given by
| (55) |
Note that the molecular force F impacts the fluid motion only at the interface region. For the fluid region away from the macromolecule, i.e., S = 0, the form of our generalized Navier–Stokes equation (54) becomes identical to the original Navier–Stokes equation (47). Additionally, when S → 0, the stress term reduces to μf∇2v and we arrive at the celebrated Navier–Stokes equation
| (56) |
where f̄ = limS→0 F.
In the present multiscale framework, we have derived three governing equations. The electrostatic interactions are described by the generalized Poisson–Boltzmann equation (14). While the surface formation and evolution are determined by the hypersur-face equation (46). Then the fluid dynamics is governed by the generalized Navier–Stokes equation (54). These equation are coupled via the discrete-continuum interface and by three variables (ϕ, S, v). Their solutions are to be constructed by appropriate iterations that take care of the nature of strong nonlinear couplings. Computational aspects of Eq. (56), including appropriate initial and boundary conditions, are extensively studied (Liu and Shu, 2000; Wan et al., 2002; Zhou and Wei, 2003; Sun et al., 2006b).
In many biological systems, fluid velocities are very slow, the length-scales of the flow are very small, and/or the viscosities are very large. Consequently, the Reynolds number becomes low and is much smaller than 1. As a result, the advective inertial forces of the flow are very small compared with viscous stress forces. The fluid flow for this class of problems is the generalized Stokes flow
| (57) |
Obviously, this flow is a special case in our multiscale framework.
3.4. Molecular dynamics in static solvent
A fundamental issue in biological modeling and computation is how to deal with a tremendously large number of degrees of freedom resulting from various interactions. Under physiological condition, a biomolecular complex, such as a virus or an ion channel, and its interacting environment may involve millions of atoms and water molecules. In principle, the solvent-solute complex can be described entirely at the microscopic scale, i.e., atomistic description or a more detailed description of electrons and nuclei. However, such an approach cannot be productive and does not provide proper theoretical predictions of physical properties of interest. It is impossible at present, and formidably expensive in the near future, to describe in full-atomic detail of the biomolecular complex. On the other hand, a simple-minded macroscopic description of the complex system might be incapable of revealing the molecular and atomic information of the macromolecule and its dynamics. We therefore reduce the number of degrees of freedom of this problem by a multiscale variational framework. In our multiscale models, we describe the aquatic environment by a hydro-continuum, i.e., a macroscopic description. As such, we are able to dramatically reduce the number of degrees of freedom associated with millions of surrounding water molecules. However, since the biomolecule is the objective of interest, we describe the macromolecule in atomic detail, i.e., a microscopic, discrete description. We assume that the aquatic environment is essentially static, and ionic densities are near equilibrium.
3.4.1. Energy functional for molecular dynamics in static solvent
The microscopic energy contributions include the kinetic energy of each individual atom and the potential energy due to various atomic interactions. The Lagrangian of the microscopic part is given by
| (58) |
where ρj = mj(δzj − xj) is the mass density of the j th atom, mj and xj are the mass and the macroscopic position of the j th atom, respectively. Here, and U(z) are respectively the kinetic and the potential energies of the j th atom with . Most important potential interactions among atoms include the van der Waals type described by the Lennard–Jones potential and the charge-charge interactions described by the Coulomb potential. Other interactions, such as dispersion, dipolar, quadrupolar, and Pauli's exclusion principle effects can be included for more accurate description in the present formulation.
The total action functional consists of the total energy of the solvation (29) and the Lagrangian of the microscopic molecular dynamics. We therefore have the total action functional of the form
| (59) |
Note that there is an issue of sign convention in the expression of the total action. We choose a negative sign for the atomic part LMicro so that the forces from the present calculations will be consistent with those in the last section.
Here, the choice of the atomic interaction potential, U , is quite subtle because the electrostatic interactions could be doubly counted, i.e., once in U and the other in electro-static energy, . Therefore, there are two alternative ways to select U . way is to exclude the electrostatic interactions in U and leave the interaction to be accounted by the generalized Poisson–Boltzmann system as discussed above. The other way is to include the atomic electrostatic interactions in U . As such, one has to compute the reaction field force twice, once with appropriate dielectric constants in both S and (1 − S) regions, and the other with a uniform dielectric constant. The difference of these two calculations is used to account for the effect of the solvent that is not explicitly included in the molecular dynamics (Gilson et al., 1993).
3.4.2. Surface evolution equation of molecular dynamics in static solvent
As discussed earlier, the optimization of Stotal[S, ϕ, x, z] with respect to ϕ leads to the desirable generalized Poisson–Boltzmann equation (14) again. In solving the generalized Poisson–Boltzmann equation, we also need to update the equation for the evolution of the hypersurface function S. In a procedure similar to that used in the derivation of Eqs. (30) and (31), we arrive at the following governing equation for the hypersurface function
| (60) |
Equation (60) describes the balances of the hydrophobic surface energy, static pressures, electrostatic energy, and atomic energies. Similarly, Eq. (60) can also be cast into the form of Eq. (32), i.e., the form of potential driven geometric flows proposed in our earlier work (Bates et al., 2009). Note that the evolution of the hypersurface function S depends not only on macroscopic variable x, but also the macroscopic positions xj due to the microscopic variables zj and the role of the mass density ρj.
3.4.3. Newton's equations of molecular dynamics in static solvent
An interesting issue is how to balance forces derived from the action functional. To understand this aspect, we carry out the variation of the action functional (59)
| (61) |
where . As in the earlier treatment, the variation is restricted by the condition that the mass should not be varied, i.e., δ(ρjdzj) = 0. Terms associated with δϕ vanish because of the generalized Poisson–Boltzmann equation (14). Similarly, terms associated with δS vanish because of the hypersurface evolution equation (60). Therefore, we still have two terms that are associated with δzj and δx, respectively,
| (62) |
where the macroscopic force f has been defined in Eq. (35). Here, the microscopic force associated with the jth atom is with the components defined as
| (63) |
| (64) |
| (65) |
where , and are respectively, solvent-solute interaction force, reaction field force, and potential interaction force. Note that since S ~ 0 in the molecular domain, the force contribution from the interaction between the solvent and solute () is very small except that the j th atom is very close to the interface. Whereas the reaction field force () is of the same scale as those from the other potential interaction effects ().
The least action in Eq. (62) requires the vanishing of the integration, which means the vanishing of the integrand. As δx and δzj vary independently and are nonzero, the only way for the integrand to vanish is for terms associated with δx and δzj to be zero, respectively. Therefore, for S ≠ 0, we arrive at the desirable Newton's equation for the molecular dynamics (MD)
| (66) |
It is seen from Eq. (63) that the driven forces of this molecular dynamics have a term associated with solvent-solute interaction near the interface (), in addition to the normal electrostatic potential effect () and other potential effects ().
It is pointed out that we do not have a complete force balance in the present model. This problem will be resolved in the next model. The system considered in the present subsection can be regarded as macroscopically at equilibrium, while microscopic subsystem is out of equilibrium and its motion is governed by the Newton's equation. This may be reasonable because the microscopic system has much smaller time and spatial scales.
3.5. Coupled fluid dynamics and molecular dynamics
Similar to the solvation system, the system described in the last subsection has a problem of lacking a mechanism to balance macroscopic forces f. This problem is addressed in the present subsection. Here, we consider a force-balanced multiscale system that includes not only the electrostatic interaction, surface dynamics, and fluid dynamics, but also molecular dynamics. As such, both the macroscopic subsystem and the microscopic subsystem are out of equilibrium. Nevertheless, we assume that densities of ionic species are near equilibrium and can be approximated by their equilibrium Boltzmann distributions. To describe this multiscale system, we need a Lagrangian for the macroscopic subsystem of the hydrodynamic continuum. Furthermore, the total action functional also includes the free energies due to the surface tension or curvature effect and the mechanical work of immersing the macromolecule into the solvent. This system is suitable for the description of the dynamics of macromolecules in microchannel and nano-channel flows.
3.5.1. Action functional for coupled fluid dynamics and molecular dynamics
The electrostatic free energy functional is given in Eq. (12). The nonpolar energy is prescribed in Eq. (28). The Lagrangian of the fluid subsystem is presented in Eq. (44), and finally, the Lagrangian of the molecular subsystem is provided in (58). Therefore, we have the total action functional for a system of fluid-molecular interactions
| (67) |
As discussed in an earlier case, we have chosen negative signs for the atomic Lagrangian (LMicro) and the fluid Lagrangian (LMacro) so that the potential energies have positive signs. The solvent-solute interaction u is considered as a part of the nonpolar energy.
3.5.2. Governing equations for coupled fluid dynamics and molecular dynamics
In this system, we derive four governing equations by employing the principle of the least action to the total action functional () in Eq. (67)
| (68) |
Here, δS, δϕ, δx, and δz are four infinitesimally small but nonzero perturbations. In order for the first variation to vanish, the terms associated δS, δϕ, δx, and δz have to vanish independently. First, terms associated with δϕ give rise to the generalized Poisson–Boltzmann equation (14).
Additionally, the surface evolution equation can be constructed by requiring terms associated with δS in Eq. (68) to vanish, following by the use of the steepest descent scheme
| (69) |
This is very similar to the surface evolution equation for the fluid-electrostatic interactions, except there is an extra term for the molecular energy.
Moreover, it is easy to see that the vanishing of terms associated with δx gives rise to the generalized Navier–Stokes equation of continuum fluid dynamics presented in Eq. (54), with the stress tensor (42) and interaction force (55).
Finally, the Newton's equation for molecular dynamics (66) is derived from terms associated with δzj, with appropriate force expressions given in Eqs. (63)–(65).
In this system, all forces are balanced. The fluid dynamics, the molecular dynamics, the electrostatic subsystem, and the hypersurface function are all coupled.
4. Multiscale models without the differential geometry based solvent-solute interface
The differential geometry based multiscale approach proposed in this work provides an efficient paradigm to minimize or optimize the surface free energy, together with other energies. The present approach utilizes the hypersurface function in the Eulerian formulation. A somewhat equivalent differential geometry based multiscale approach is to make use of the Lagrangian formulation, which will be presented elsewhere. Certainly, one can also carry out multiscale variational modelings without the use of differential geometry based surfaces. As such, the surface free energy and mechanical work will not be minimized and their effects in the model depend on the choice of the surface model, such as solvent excluded surfaces or solvent accessible surfaces. In this section, we demonstrate one of such multiscale approaches. A comparison of two different multiscale formulations, i.e., multiscale models with and without the differential geometry based solvent-solute boundary, is given.
The foundation of electrostatic forces associated with the generalized Poisson–Boltzmann equation was due to Gilson et al. (1993) based on the free energy expression given by Sharp and Honig (1990). In this section, we also analyze the force balance in the earlier electrostatic formulation. We show that although the classical electrostatic force theory is incomplete, there are some consistencies between the theory of Gilson et al. and the present differential geometry based multiscale formalism.
4.1. Force paradox in the classical Poisson–Boltzmann system
Gilson et al. formulated the theory of electrostatic forces associated with the Poisson–Boltzmann equation (Gilson et al., 1993) and their force expressions have been implemented in implicit solvent based molecular dynamics (Gilson et al., 1993;Im et al., 1998; Lu et al., 2002; Lu and Luo, 2003; Luo et al., 2002; Prabhu et al., 2004). We are interested in the understanding of how well electrostatic forces couple to the molecular dynamics in the implicit molecular dynamics theory of Gilson et al. (1993). To this end, we add a molecular dynamic term to the free energy functional of the electrostatic system proposed by Sharp and Honig (1990), and by Gilson et al. (1993)
| (70) |
where ∊(x) = ∊0ε(x) is the permittivities, λ(x) is characteristic function for the solvent, and other quantities have been defined in preceding sections.
By taking the least action for the above total action functional, Eq. (70), one has
| (71) |
where the terms associated with δϕ vanish by the standard Poisson–Boltzmann equation
| (72) |
In Eq. (71), fDB, fID, fRF, and are force densities of dielectric boundary (DB), ionic boundary (IB), reaction field (RF), and potential interaction (PI), respectively,
| (73) |
| (74) |
| (75) |
| (76) |
The first three terms are exactly the force density expressions given in the literature (Gilson et al., 1993). Since Eq. (71) vanishes by the least action principle, terms associated with δx and those associated with δzj have to vanish independently. As such, we have
and
Since δx and δzj are nonzero infinitesimal perturbations, we have and the Newton's law for molecular dynamics . Consequently, the macroscopic forces associated with δx cannot be balanced by atomic motions governed by the Newton's equation associated with δzj. An implication of this result is that the dielectric boundary force and ionic boundary force derived in the theory of Gilson et al. (1993) may not be applied to atoms. On the other hand, Im et al. (1998) have shown that the total force computed with the theory of Gilson et al. is consistent with that computed from the force definition, i.e., the negative gradient of the free energy functional. Apparently, there is an interesting force paradox associated with the classical Poisson–Boltzmann system.
4.2. Direct coupling between fluid dynamics and molecular dynamics
In this work, we resolve the above apparent force paradox by a multiscale description of the electrostatic and molecular dynamic system. We show that if the fluid dynamics is included in the description, all the forces in the system can be balanced. An important observation which is crucial to the understanding is that the solvent-solute interface, such as the van der Waals surface or the molecular surface, is determined directly by atomic positions of the biomolecule described by the microscopic variable z; and is indirectly affected by the electrostatic interaction. However, the solvent-solute interface is referenced in the Poisson–Boltzmann equation described in the macroscopic variable, x. As such, we assume that the permittivity is a dependent function of x and z, i.e., ∊ = ∊(x, z). Similar, the characteristic function depends also on x and z, i.e., λ = λ(x, z). We will discuss the consequence of an alternative assumption that ∊ = ∊(z) and λ = λ(z) after the derivation of the full set of governing equations.
We therefore consider the following total action functional
| (77) |
Here, the electrostatic interaction is balanced by the fluid action functional and molecular action functional.
Note that surface free energy and the mechanical work of immersing a macromolecule into the solvent are not presented in the above action functional because this model admits a priorly fixed surface model, such as a van der Waals surface, or a molecular surface. However, in the evaluation of the total solvation energy, one should still include the contribution from the surface area and mechanical work.
The governing equations are obtained by the principle of the least action
| (78) |
According to the principle of least action, terms associated with each independent variation have to vanish. Here, terms associated with δϕ give rise to the classical the Poisson–Boltzmann equation (72). Terms associated with δx lead to the desirable form of the Navier–Stokes equation
| (79) |
where the force is given by
Similarly, from terms in Eq. (78) associated with δzj, we have
| (80) |
where the potential interaction force is defined in Eq. (76), and
| (81) |
| (82) |
| (83) |
A few comments are in order. First, it is clear that the dielectric boundary force and the ionic boundary force indeed couple to atoms and molecular dynamics, along with forces from other potential interactions. The above mentioned apparent force paradox is due to the incomplete description of the complex multiscale system of discrete and continuum interactions. A sufficient description of the system must use two sets of independent variables, the macroscopic variable x and the microscopic variable z. This is physically sound because interface indeed depends on the atomic position and motion, the solvent and solvent-solute interaction.
Additionally, the Poisson–Boltzmann equation requires ∊ and λ to be functions of macroscopic variables x. Therefore, the present treatment ∊ = ∊(x, z) and λ = λ(x, z) appears to be a unique option to couple macroscopic with microscopic without resorting to an additional interaction potential describing the solvent-solute interaction as discussed in preceding sections.
Moreover, since we treat ∊ and λ as functions of x, the interface induced forces have to be balanced by the fluid motion governed by the Navier–Stokes equation. The inclusion of a term describing the solvent-solute interaction in the fluid action functional can be easily done, i.e.
Such an extra term (ρsu) will generate forces near the interface that impact both the atomic and fluid motions.
Finally, the reaction field force in Eq. (81) may look strange, as the electrostatic potential ϕ does not depend on the microscopic variable zj . However, this term is fine because the charge density ρm is a set of Dirac delta functions.
4.3. Comparison between multiscale models with and without differential geometry based interfaces
An interesting issue is that what happens if we set the hypersurface as a function of x and z, i.e., S = S(x, z). This arrangement will slightly change the formulation in some earlier sections where microscopic variable z was not included. However, it has no impact to Sections 3.4 and 3.5. Note that in Eqs. (60) and (69), the surface evolution is already under the influence of atomic kinetic energies described in terms of the microscopic variable z, although such variables are forced into macroscopic positions by the associated mass densities ρj.
In practical computations, atomic coordinates are used as a set of boundary conditions for our hypersurface evolution equations (Bates et al., 2008, 2009). Additionally, the potential driven evolution operator of the hypersurface function in our recent work (Bates et al., 2009) depends on atomic interactions. Therefore, the hypersurface function has always been a function of the microscopic variable.
A main difference of the multiscale models of Sections 4.2 and 3.5 is that without appropriate interface terms, the total energy of the system is incomplete and energy optimization does not lead to a physical minimum. Therefore, the current description of the interface energy is an important element of the solvation system.
Another main difference of the multiscale models of Sections 4.2 and 3.5 is that in differential geometry based multiscale models, one does not need to compute dielectric boundary force and ionic boundary force. However, we have to solve a surface evolution equation as shown in Eqs. (60) and (69). The computational efficiency of these two approaches is to be examined in the future. The result of comparing these two approaches may also depend on computational algorithms employed.
There is yet another way to see the consistency of the approaches in Sections 4.2 and 3.5. In Eq. (68), we have a hypersurface related term
If we assume that S = S(x, z) we have . We therefore recover the dielectric boundary force and ionic boundary force, in∇addition to many other force terms. However, we will not explicitly list these expressions in the present work.
It is clear that there is a flexibility in the selection of a set of independent variables in the total variation. One can make use of this flexibility to choose an appropriate set of equations to be solved. Sometimes, different selections may lead to different physical interpretations and the use of different computational techniques. We will not elaborate these interesting aspects any further in the present work.
Finally, we comment on the similarities in multiscale models with and without differential geometry based interfaces. These similarities are some of the most interesting aspects in our multiscale models that do not make use of differential geometry based surfaces. First, we still set up a total energy functional or a total action functional for a given system of interest. Therefore, energies from differential scales are put in an equal footing. Although the surface energy is fixed a priori. Additionally, as an energy approach, it is convenient to utilize the variational principle to derive governing equations for the system. These governing equations are normally coupled to each other. Moreover, we also seek the balance of the forces in the governing equations. In our view, this balance is not only necessary to make the multiscale description complete, but also offers the technical means for the system to reach its optimal state.
5. Multiscale models for aqueous macromolecular complexes far from equilibrium
Electrophoresis and charge diffusion and migration in solution or solid play an important role in a wide range of science and technology, from the transport of electrons and holes in nano electronic transistors and integrated circuits, chemical and physical processes of fuel cells, to ion channels in the plasma membrane of living cells. These systems are far from equilibrium and often involve many physical phenomena, such as electro-osmotic flows, electrophoresis, and polarization in electrolytes (Zheng et al., 2003; Chen and Conlisk, 2008). These phenomena are usually described by the Poisson–Nernst–Planck (PNP) equations, or the coupled Poisson–Nernst–Planck (PNP) and Navier–Stokes equations (Chen et al., 1995; Chu and Bazant, 2006; Hwang et al., 2006; Vlassiouk et al., 2008; Abaid et al., 2008; Zhou et al., 2008c). In this work, we present differential geometry based multiscale models to describe these phenomena. We invoke the differential geometry description so that the formation and evolution of the discrete-continuum boundary are included in the total energy optimization. The interface description is important in polymer electrolyte membrane (PEM) fuel cells, where a hydrophobic polymer membrane, functionalized by acidic side chains, is used as the electrode separator (Franco et al., 2006; Gurau and Mann, 2009; Promislow and Wetton, 2009). The working conditions of PEM fuel cells are far from equilibrium and demand detailed description of chemical reactions, charge transport, and the atomic level understanding of the polymer activity.
In this section, we consider a kinematics which is similar to that described in Section 3.1. The macromolecule is described in atomic detail and its domain is characterized by hypersurface function S. The atomic positions are obtained either from experimental data or from theoretical calculations, such as molecular dynamics or in principle, quantum dynamics, also. Molecular motion is coupled to the aquatic environment which is described by continuum mechanics, i.e., hydrodynamics. The aquatic environment may have multiple species, which may undergo chemical reactions. The macroscopic domain is characterized by (1 − S). The boundary between microscopic and macroscopic domains is coupled with the static and/or dynamical interactions between the macromolecule and the aquatic environment. The electrostatic interactions are described by the Poisson type of equations, with both discrete charges in the macromolecular domain and continuum charge distributions in the aquatic environment. As the continuum system is far from equilibrium due to the fluid and charge transport, we must abandon the Boltzmann distribution of charged particles and allow the density of each species to be determined by the mass conservation law. The atomistic interactions among the atoms, and interactions between atoms near the micro-macro interface and the solvent are also a part of our description. The state and dynamics of the whole system are determined by the optimization of the total free energy functional or the total action functional. We call it an optimization when we deal with a “minimum-maximum” problem.
5.1. The origin of drift and diffusion in the Nernst–Planck equation
We consider an aqueous phase of multiple components, including the water. The mass density of the αth component is denoted as ρα. The mass conservation gives rise to
| (84) |
where vα is the velocity of component α and ναJj is the production of α per unit volume in the jth chemical reaction. The inclusion of chemical reactions is pertinent to the PEM fuel cells where most chemical reactions take place at the solid-liquid interface. Since mass is conserved in each chemical reaction, we have
Assuming the absence of chemical reactions, all the production terms vanish, i.e., Jj = 0. Clearly, the mass of each species is conserved, and there is no drift or diffusion in the mass conservation equation (84) for component α. Then an interesting question is where does the drift and diffusion in the Nernst–Planck equation come from.
To understand the origin of the drift and the diffusion in the Poisson–Nernst–Planck theory, we first note that Eq. (84) describes the fluid from the individual component point of view. To solve Eq. (84), we need to know vα, which may be obtained either from an equation or from experimental measurements. However, we do not have an equation for vα, and usually, the velocity of an individual component is not measured in a homogeneous fluid. Instead, the fluid barycenter velocity v is mostly observed in fluid mechanical experiments. Therefore, we need an alternative description of the fluid that is consistent with experiment observations. To this end, we consider a collective description of the fluid. The law of mass conservation requires
| (85) |
where the total solvent density ρs and barycenter velocity v are defined as
| (86) |
| (87) |
Equation (85) means that in a fluid, the rate of change of the total density is due to the motion along the barycentric velocity, in addition to a possible volume compression or extension
| (88) |
| (89) |
For incompressible fluid (∇ · v = 0), we have , i.e., the total mass is conserved. However, it is important to note that the rate of change of the density of an individual component is much more complicated. To illustrate this point, we rewrite Eq. (84), the mass conservation equation of component α, in terms of the collective variable, i.e., the fluid barycenter velocity v
| (90) |
where the second term on the left-hand side describes the motion of the mass density ρα moving along with the fluid barycentric velocity. Here, the first term on the right-hand side vanishes for incompressible fluids. The second term (∇ · (vα – v)ρα = ∇ · Jα) is commonly regarded the “diffusion flow” component α defined with respect to the barycentric motion. In a homogeneous fluid, the component velocity vα is not directly described by a governing equation and thus has to be eliminated.
First, we analyze the drift-flux contribution to the diffusion flow due to the electrophoresis effect. For a particle of charge qα, its velocity (vα) is proportional to the local electric field E, i.e., vα ~ E. The electric field can be expressed in terms of the gradient of the electrostatic potential as E = −∇ϕ. This expression is also valid when there is a static external electric field, which can be incorporated in the boundary conditions of the Poisson equation or the Poisson–Boltzmann equation. We therefore make use of the Nernst–Einstein equation for the electrophoresis flux
| (91) |
where qα = −|qα| for negative ions and Dα is the diffusion coefficient of component α. Next, it is convenient to assume that the diffusion flux (vα − v)ρα also has a contribution from the gradient of ρα
| (92) |
This gives rise to the desirable diffusion flux due to the material gradient. Thus, the total diffusion flux for component α is set to
| (93) |
Consequently, the mass conservation equation for each component can be written as
| (94) |
where we have assumed that the fluid is incompressible (∇ · v = 0). Equation (94) is the desired form of the Nernst–Planck equation for the mass conservation of component α. Here, an important feature is that the component velocity (vα) is not a variable. Clearly, the origin of both the drift and the diffusion of component α in a homogeneous fluid is due to the relative motion of such an individual component with respect to the barycentric motion of the fluid.
Equation (94) describes two possible steady states that have often been used in practical simulations. First, if we set , we have the stationary motion of component α along with the stream velocity v. Additionally, if we set , we have the local balance between the drift-diffusion and the reactive production
| (95) |
This form of the Nernst–Planck equation is often solved in practical applications, in particular, in the ion channel problems for ion density distributions and current-voltage curves.
The total charge current density Ic can be expressed as
| (96) |
where ρsqv is the charge current due to convection and is charge conduction current. Here, the total charge per unit mass is defined as . For ion channels, it is important to observe and analyze the current density map, which should be consistent with the electrostatic potential map. Both the current density map and the electrostatic potential map reflect the channel molecular structure and gating mechanism. Finally, the conservation law for total charge is
| (97) |
5.2. Coupled geometric, Navier–Stokes, Poisson–Nernst–Planck equations
In ion channels, the ionic selectivity and gating effect are partially due to the electrostatic interaction of the ion and the channel in a close vicinity. However, in many practical simulations, the “close vicinity” depends on the surface model used for the channel. The solvent accessible surface and the van der Waals surface of a given ion channel exhibit very different profiles of the vicinity. In this work, we develop differential geometry based approaches so that the surface formation is coupled to the Navier–Stokes equation and Poisson–Nernst–Planck equations. We slightly modify the formulation developed in Section 3.3. Here, we consider the following total action functional:
| (98) |
where qα takes a positive sign if component α is positively charged, e.g., a cation, and takes a negative sign if component α is negatively charged, e.g., an anion. Here, we assume that the fluid encompasses multiple ionic species and water molecules. It is important to note that since the system may be far from equilibrium, the Boltzmann distribution of the ionic density is not assumed. Instead, the mass density of each component satisfies the drift-diffusion equation (94). This is a crucial aspect in systems far from equilibrium, such as those in PEM fuel cells, which are described by the drift-diffusion equation (94), the Navier–Stokes equation and the Poisson equation. Note that ρm is the charge density, while ρα is the mass density.
By optimizing the total action functional () in Eq. (98) with respect to ϕ, we have a generalized equation
| (99) |
where ε(S) is defined in Section 3.1.2. This new Poisson equation includes free discrete charge contributions (ρm) from atoms in the macromolecular domain S and free continuum charge contributions () from the solvent domain (1 – S). This equation is coupled to the mass density equation (94) and the equation for the hypersurface function.
In order to derive an appropriate governing equation for the hypersurface (S), we minimize the total action functional () in Eq. (98) with respect to S, following by the use of the steepest descent scheme to construct
| (100) |
This equation has a similar structure as the curvature and potential driven geometric flows (Bates et al., 2009). It couples to the mass density equation (94), the generalized Poisson equation (99) and the generalized Navier–Stokes equation describing the velocity field v.
The derivation procedure for the generalized Navier–Stokes equation from the multi-scale total action functional (98) is the same as that described in preceding sections. To this end, we make use the least action principle
| (101) |
where δS, δϕ and δx are infinitesimally small but non-zero perturbations. Because each independent variation vanishes, for (1 − S) ≠ 0, we have the following generalized Navier–Stokes equation:
| (102) |
The form of the new generalized Navier–Stokes equation (102) is identical to the original Navier–Stokes equation (47) when S = 0. However, in Eq. (102), the force is given by
| (103) |
where f is defined in Eqs. (34)–(38) and
| (104) |
is a generalized reaction field force. Note that without the present hypersurface description, we would result in a normal electric force term of the form
where E =−∇ϕ is the electric field vector. This force term is exactly the same as the force in the usual Navier–Stokes equation for electrohydrodynamics. It is to point out that the generalized electric field force fE vanishes if all fluid components are uniformly charged, i.e., where q is the uniform charge. To see this, we need to realize that this force term is originated from the variation . This result may appear to be inconsistent with electroosmotic flows. However, note that the uniform charge effect has already been accounted for in the hypersurface motion (100) and the generalized Poisson equation (99) in the present multiscale framework. On the other hand, the foundation of the present theory needs to be reexamined if fluid components are uniformly charged. Some assumptions may no longer be valid and the full set of the Maxwell's equations should be used for such a highly charged fluid. We will not further elaborate on this aspect as it does not usually occur in our aqueous chemical and biological systems.
In the present multiscale framework, we have derived three governing equations for the discrete-continuum electrostatic-fluid system. The generalized Poisson equation (99), the hypersurface evolution equation (100), and the generalized Navier–Stokes equation (102) are coupled among themselves and to the mass conservation equation (94). These equations form a new set of governing equations for describing ion channels and other fluidelectrostatic systems.
For many applications in biology, such as many ion channels, the barycentric motion of the fluid may be absent, i.e., v ~ 0. As such, the present description simplifies accordingly. One just needs to solve the mass conservation equation (94), the generalized Poisson equation (99), the hypersurface evolution equation (100), and a generalized Stokes equation
| (105) |
5.3. Coupled geometric, Newton, Navier–Stokes, and Poisson–Nernst–Planck equations
It is important to understand that for complex systems at nonequilibrium, the Boltzmann distribution of the electrostatic energy for the ion concentration is not valid. The alternative description presented above is useful for such cases. However, a full description of these systems should include the molecular dynamics for the macromolecule. Therefore, we consider the total action functional for a system of fluid-molecular interactions
| (106) |
Here, on the right-hand side of the first row is the nonpolar solvation free energy density, the second row is electrostatic solvation free energy density, and the third row contains Lagrangians of both the fluid part and the molecular dynamics part. This system requires the input of ρα from Eq. (94) or Eq. (95).
To derive other governing equations, we apply the principle of the least action to the total action functional () in Eq. (106)
| (107) |
where, δS, δϕ, δx, and δz are nonzero perturbations. Therefore, the terms associated δS, δϕ, δx,and δz vanish independently.
As such, we recover the generalized Poisson equation (99) from the δϕ term. Additionally, from the δS term, we construct a geometric flow equation for surface formation and evolution by using the steepest descent scheme
| (108) |
This is very similar to the geometric flow equation for the fluid-electrostatic interactions, except for a change in the electrostatic energy.
Moreover, the vanishing of terms associated with δx gives rise to the generalized Navier–Stokes equation presented in Eq. (102), with the stress tensor given in Eq. (42), and interaction force prescribed in Eqs. (103)–(104).
Likewise, from terms associated with δzj , we recover the Newton's equation for molecular dynamics given in Eq. (66), with appropriate force expressions given in Eqs. (63)–(65). It is easy to understand that a change in the macroscopic description of the continuum charge distribution has no direct effect in the microscopic forces.
Furthermore, it is emphasized that these four equations are coupled to the mass conservation equation for each component (94). We therefore have to solve multiple governing equations in our multiscale models. Simplifications are possible for given situations. However, a detailed discussion of the simplifications is beyond the scope of the present work.
Finally, the inclusion of the molecular dynamics is crucial for the situation where biomolecules may undergo significant configuration changes during a nonequilibrium process. Gated ion channels are typical examples. A channel can open or close by chemical, or electrical, or photonic, or temperature signals, depending on the variety of circumstances. The molecular dynamics description provides a unique means to capture the configuration change during the channel transport process.
6. Multiscale models for excessively large aqueous macromolecular complexes
There are chemical and biological systems that are excessively large. Their atomic description, particularly the detailed molecular dynamics, involves excessively large numbers of degrees of freedom and is intractable with current computer capability. Therefore, these systems pose a fabulous challenge to theoretical description and prediction. Deoxyribonucleic acid (DNA) and the membrane of PEM fuel cells are examples of excessively large aqueous macromolecular complexes. The challenges from these systems demand different tactics in modeling and simulation.
DNA is a long polymer made from nucleotides and contains the genetic information used in the development and functioning of all known living organisms and some viruses. The largest human DNA polymer has about 220 million base pairs (bp) and is about 67,000 µm in length. However, human DNA polymers are highly packed in chromosomes which are only about a few µm long. Each 146 DNA pb is wrapped around an octamer of small basic proteins called histones. Wrapped histones, together with about 54 DNA bp to form a nucleosome. Nucleosomes are further wrapped around to form chromatins, which are organized in loops, scaffolds, and domains in the chromosomes (Grigoryev et al., 2009). A quantitative description of this DNA packing process is crucial to the in-depth understanding of the protein specification, gene regulation, and gene expression.
Another interesting system is the PEM fuel cell, which is a prime candidate for compact power devices in mobile and other applications. The PEM serves as a conductor for protons and an insulator for electrons, anions, and gases. The membrane is usually made of synthetic polymers, such as Nafion, which relies on liquid water humidification to transport protons. The hydrophobic polymer backbone provides good mechanical stability, while the sulfonic acid functional groups self-organize into hydrophilic water channels of about a few nano-meter diameter to allow the transport of protons. The mechanical properties of the membrane depend on the water management and are crucial to the membrane performance. The membrane is often manufactured at a wide variety of sizes, from nano-scale to macroscopic dimensions (Gurau and Mann, 2009; Promislow and Wetton, 2009).
One common feature of the above two systems is that electrostatic interactions play an essential role in their static and dynamic properties. DNA is highly charged. The interaction of DNA and histone is electrostatic in nature. DNA packing is also driven by electrostatic interactions (Grigoryev et al., 2009). The membrane in the PEM fuel cells is highly charged too. The sulfonate ion clusters of Nafion determine the function of the membrane. Another common feature is that both systems are so excessively large that their description in full atomic detail is beyond the current capability of computers. To construct quantitative models for DNA polymers and PEM fuel cells, we must change our strategies. Coarse-grained models may be utilized and are useful. These models often involve an appropriate reparameterization of the force field. However, the underlying mathematical and theoretical formulations are the same as those given in the preceding sections. In this work, we introduce alternative multiscale models for the fluid, electro-static, and elastic interactions of the DNA polymers and PEM fuel cells. We refer this model as to a fluid-electro-elastic model because the expensive molecular dynamics of the macromolecule is replaced by a simplified elastic dynamics. A similar treatment was considered by Zhou et al. in a model without the fluid and hypersurface aspects (Zhou et al., 2008b). Some pioneer work on fluid-structure interactions was due to Peskin (1977). A phase field approach of the elastic bending energy for vesicle membranes was also considered by Du et al. (2004). In our approach, the system may still maintain a microscopic description of charges when it is necessary. Appropriate coarse-grained charge assignment or distribution may be implemented in our models, also. These aspects can be both involving and empirical. A more detailed elaboration of the coarse-grained charge distribution is beyond the scope of the present work.
6.1. Lagrangian of elasticity
We assume that the macromolecule behaves as a deformable body under the stress, which is a measure of the internal distribution or intensity (per unit area of a surface) of the total internal forces. For a given stress, the deformation, i.e., the relative displacement between particles in the macromolecule, is geometrically measured by strain. We assume that strain is linearly proportional to the stress and the coefficient of the proportion is the Young's modulus. It is convenient to assume the macromolecule behaves elastically so that it returns to its undeformed state when an applied stress is removed.
Consider a point x in that is deformed to x̄ due to a displacement w, i.e.,
| (109) |
The infinitesimal distance between x̄ and x is
| (110) |
where the Einstein notation is used for tensorial quantities. The strain tensor describes the distance of the points between before and after the elastic deformation
| (111) |
For relatively small deformations, the term that is nonlinear in w is usually small and can be omitted for simplicity,
| (112) |
It is convenient to define the stress tensor of the elastic macromolecule as
| (113) |
where λe is the elastic modulus or stress/strain ratio, and μe is the shear modulus. This linear stress-strain relation has been widely used in elasticity analysis. Obviously, the stress tensor is symmetric
| (114) |
For isotropic system, the elastic potential energy density in the Einstein notation takes the form
| (115) |
Additionally, the Lagrangian of the elastic system is defined as the combination of kinetic and potential energies
| (116) |
where ρe is the mass density of the elastic macromolecule and ẇ is the velocity of the displacement. Note the sign convention for the elastic potential energy in contrast to the kinetic energy.
Generalizations to nonlinear elasticity and systems of particular symmetries can be obtained. It is also possible to include viscoelasticity, and viscoplasticity in our formulation. Due to complexity of biological systems, we expect intensive research on these generalizations in the future. However, these aspects are beyond the scope of the present work.
There are two types of fluid-electro-elastic systems, i.e., near equilibrium and far from equilibrium systems. Formulations for these two types of systems differ very much. For near equilibrium systems, we can make use of the Boltzmann distribution approximation for the densities of ionic species in the solvent. The main advantage of this approach is that one does not need to solve conservation law equations, or the Nernst–Planck equations for ionic species. This is a reasonable approach for many applications that are near equilibrium. An example is the DNA packing or unpacking in salt solutions where we do not expect a dramatic variation of the ion density distribution over space and time. On the other hand, for systems far from equilibrium, we cannot assume the Boltzmann distribution approximation and have to solve the conservation law equations, such as the Nernst–Planck equation, for the densities of ionic species. Typical examples of this type of systems include PEM fuel cells, ATP synthase motors, etc. Very often in these systems, both chemical reaction and charge transfer occur. In the following two subsections, we develop multiscale models for the aforementioned two types of electro-fluid-elastic systems.
6.2. Fluid-electro-elastic systems near equilibrium
The action functional can be constructed by making use of the system developed in Section 3.3. We incorporate the elastic Lagrangian into the fluid and electrostatic action functional
| (117) |
Here, the term (1 − S)ρsu now represents the interaction between the fluid and elastic macromolecule. The charge density ρm in the macromolecule can be either a set of discrete charges or a continuous function over the molecule domain. These two terms are functions of the position and the displacement of the macromolecule. As in the earlier treatment, the gravitation potential energy has been omitted due to the small length scale of the problem under consideration.
We make use of the least action principle to derive governing equations for the fluid and for the elastic macromolecule from the action functional . To this end, we consider the variation of Eq. (117)
| (118) |
where , ρe is treated as a constant, ρsu and ρmϕ are functions of the macromolecular deformation. Here, we assume the condition that the mass should not be varied, i.e., δ(ρedx) = 0.
Governing equations for the system can be deduced. First, we obtain the generalized Poisson–Boltzmann equation (14) by requiring terms associated with δϕ to vanish in Eq. (118). There is no change in the form of the Poisson–Boltzmann equation except a possible replacement of the discrete change density ρm with a continuous one. The selection of this continuum treatment depends on the system under study.
Additionally, the terms associated with δS in Eq. (118) are minimized by the following hypersurface equation constructed by using the steepest descent scheme
| (119) |
As discussed earlier, two pressure terms in Eq. (119) cannot cancel each other because of their different domains.
Moreover, the terms associated with δx in Eq. (118) lead to the generalized Navier–Stokes equation (54). However, the interaction environment in the generalized Navier–Stokes equation is quite different now. It describes the fluid-structure interaction (FSI) in the present setting.
Finally, the variation of the elastic energy term is discussed in the following:
| (120) |
where some surface integrals have been discharged.
Therefore, all terms associated with δw can be written
| (121) |
where are the forces acting on the elastic macromolecule. The fluid-structure interaction (FSI) force and reaction field (RF) force are given by
| (122) |
| (123) |
Unlike in previous cases, the force due to the atomic potential interactions does not appear. This force contributes to elastic properties, i.e., the elastic modulus and the shear modulus.
We therefore have the governing equation for the dynamics of the elastic macromolecule when S ≠ 0
| (124) |
By using the stress tensor defined in Eq. (113), we can rewritten the first term in Eq. (124) as
| (125) |
Additionally, because the symmetry of the stress tensor (114), we can write Eq. (124) in the desirable form
| (126) |
where is the elastic stress tensor. Equation (126) is the generalized elastic equation of motion.
The solution of Eq. (124) is subject to appropriate initial and boundary conditions. Typically, the steady state solution
| (127) |
is pursued in most applications. The Dirichlet boundary condition can be directly applied to the displacement vector
However, it is convenient to impose the free boundary condition on the stress tensor
| (128) |
If a surface force fe is applied on the boundary, we have
| (129) |
where nj is the jth component of the norm vector n and is the jth component of the force f e at the boundary. Computational aspects for structural analysis are available in the literature (Wei, 2001a, 2001b; Wei et al., 2002).
6.3. Fluid-electro-elastic systems far from equilibrium
In this section, we formulate a multiscale model for systems that are far from equilibrium. The main feature is that we cannot assume the convenient Boltzmann distribution for ionic densities. Additional governing equations have to be solved to attain the density of each species. We therefore utilize the generalized Nernst–Planck equation (94) for this purpose.
The action functional of the present fluid-electro-elastic model can be constructed by modifying Eq. (98) with the additional elastic Lagrangian
| (130) |
Here, various components have been explained in the previous section.
As usual, we derive all the governing equations by applying the least action principle to the multiscale total action functional (130)
| (131) |
Details of this variation have been discussed in the preceding sections. Here, δS, δϕ, δw, and δx are infinitesimally small but nonzero perturbations. By variational principle, each independent variation vanishes.
It is easy to see that the generalized Poisson equation (99) can be deduced from terms associated with δϕ. This equation requires the input from the generalized Nernst–Planck equation (94) and information about the hypersurface function S.
From the terms associated with δS, we construct the following evolution equation for the hypersurface function by the steepest descent scheme:
| (132) |
This equation is coupled to all other quantities, such as ϕ, w,and v.
From the terms associated with δx, we obtain again the generalized Navier–Stokes equation (102). This equation is solved together with the generalized Poisson equation (99), the generalized Nernst–Planck equation (94), the hypersurface equation (132), and an equation for the deformation of the macromolecule, which can derived from the terms associated δw in Eq. (131). The final form of the governing equation for the dynamics of the elastic macromolecule is the same as that given in Eq. (124). The detailed derivation has been given in the last subsection.
The multiscale formulations presented in this section can be easily converted into ones without differential geometry based surfaces. The procedure for the conversion is the same as that developed in Section 4. The consequences of such a conversion is similar to those discussed and compared in Section 4. We will restrained from a further elaboration on this aspect.
In addition to applications discussed in Section 6, this multiscale model can also be very useful to voltage-gated ion channels, which open and close depending on the voltage gradient across the membrane. Most important voltage-gated ion channels include sodium channels, calcium channels, potassium channels, and some proton channels. Typically, voltage-gated ion channels often involve two or more configurations during the ion transport circle. The present coupled fluid-electro-elastic model may provide a viable description of complex voltage-gated ion channels, such as voltage-gated sodium channels. The core of a voltage-gated sodium channel consists of four homologous repeat domains, and involves about 4,000 amino acids. Each of the four domains comprises six trans-membrane segments (S1–S6). The real time molecular dynamics simulation of sodium channels is intractable at present.
7. Concluding remarks
We propose a differential geometry based multiscale paradigm for the description and analysis of aqueous chemical, biological systems, such as protein complex, molecular motors, ion channels, and PEM fuel cells. Our multiscale paradigm provides a macroscopic continuum description of the fluid or solvent, a microscopic discrete description of the macromolecule, a differential geometric formulation of the micro-macro interface, and a mixed micro-macro description of the electrostatic interaction. In the proposed framework, we have derived four types of governing equations for different parts of complex systems: fluid dynamics, molecular dynamics, electrostatic interactions, and surface dynamics. These four types of governing equations are generalized Navier–Stokes equations, Newton's equations, generalized Poisson or Poisson–Boltzmann equations, and hypersurface evolution equations. For systems far from equilibrium, coupled geometric evolution equations, generalized Navier–Stokes equations, Newton's equations, and Poisson–Nernst–Planck (PNP) equations are formulated. For excessively large chemical and biological systems, we replace the expensive molecular dynamics with a macroscopic elastic description and develop alternative differential geometry based fluid-electro-elastic models.
A series of multiscale models ranging from simple to complex are introduced in the present work. Two different metric settings, and are used in our derivations. As such, the definitions of various densities (ρs,ρm,ρj ), surface tension γ, pressure p, interaction potential energy densities (u and U ), etc. should have changed appropriately according to two different metric settings, and . In fact, they differ by a volume factor. For simplicity and to avoid confusion, the same set of symbols has been used in the two settings. A more formal treatment would have kept all the quantities defined in the full spatial setting . However, our informal treatment does not affect the form of the governing equations derived in the present work.
The multiscale approaches provided in the present work can be easily generalized to other chemical, physical, and biological systems. For example, it is straightforward to extend the present fluid models to compressible flows, and to the equation of change of the thermal energy, including the energy production due to the chemical reactions. The inclusion of the thermal energy is of particular importance to the modeling of PEM fuel cells where reactive energy production, temperature control, and water management are crucial aspects for the performance of the energy devices. However, these aspects are beyond the scope of the present work and will be addressed elsewhere. In fact, thermal energy is not a conservative quantity as shown in our earlier formal kinetic theory for a system involving kinetic energy and potential energy into conversion (Snider et al., 1996a, 1996b).
Additionally, the proposed differential geometry based multiscale models can be easily generalized to complex systems with multiple interfaces. An interesting topic is the modeling of fluid-electro-elastic and macromolecule interactions. One can maintain macroscopic continuum descriptions for the fluid and the solid while employing a discrete description of the macromolecule of interest. A typical example is those ion channels whose open channel configurations differ significantly from their close channel configurations. The dramatical changes in configurations may involve receptor binding and rearrangement of membrane lipid bilayers. As such, a fluid-electro-elastic and molecular dynamics model will provide a fluid dynamics description of the solvent, an elastic description of the membrane lipid bilayers, and a molecular dynamics description of channel ions, membrane proteins, and their possible binding.
Likewise, the introduction of random effects in the molecular dynamics is another important aspect (Nottale and Auffray, 2008). This can be done either in the manner of the Brownian dynamics (Madura et al., 1995; Gabdoulline and Wade, 1998; Elcock et al., 1999) or in the manner of the Langevin dynamics (Tan et al., 2006; Prabhu et al., 2008; Gordon et al., 2009), which includes an additional inertia term. These approaches have become standard techniques in generalized molecular dynamics methods and have been applied to the simulation of ion channels and other biomolecular systems.
Yet another interesting issue concerns the possible use of high-order curvature definitions in solvation and surface modeling. The Willmore flow (Willmore, 1997) captures the deviation of a surface from the local sphericity and minimizes the difference of the square of the mean curvature and the Gauss curvature. As a generalization of the Willmore energy functional, the Canham–Helfrich curvature energy functional (Canham, 1970; Helfrich, 1973) allows the aforementioned difference to be minimized with respect to an arbitrary ratio. This theory has been widely used in the membrane bending analysis (Ou-Yang and Helfrich, 1989; Iwamoto et al., 2006). There are still many other high-order geometric flow models, such as those proposed in our earlier work (Wei, 1999; Bates et al., 2009). Unlike the mean curvature operator that clearly minimizes the surface area as well as the surface free energy of macromolecules, the exact physical role of various high-order curvature models in solvation and surface modeling is yet to be carefully analyzed and explored.
Moreover, the inclusion of a quantum mechanical description is important for the understanding of the formation and break-up of chemical bonds, reaction rates, and selectivity (Nottale and Auffray, 2008). This extension would also add an extra scale to the present differential geometry based multiscale theory. In fact, we have incorporated the quantum mechanical description into the electrostatic system in our effect of modeling nano-electronic devices (Chen and Wei, 2009). The continuous miniaturization of nano-scale electronic devices, such as metal oxide semiconductor field effect transistors (MOSFETs), has given rise to a pressing demand in the new theoretical understanding and practical tactics for dealing with quantum mechanical effects, such as channel tunneling and gate leaking, in integrated circuits. We have developed a variation framework to put the nanoscale description of electrostatic interactions and the microscopic description of electronic dynamics on an equal footing. By energy optimization, we have derived self-consistently coupled Poisson–Schrödinger equations for the electron structure of nano devices. In a similar manner, the incorporation of the quantum mechanical description into the present multiscale formalism can be pursued. This development is undertaken and will be published elsewhere.
Further, there are more dramatic extensions of the present multiscale formalism. First, we can incorporate integral equation approaches of solvation analysis (Tully-Smith and Reiss, 1970; Fries and Patey, 1985; Beglov and Roux, 1997) into the differential geometry based multiscale description. There are many existing theories in the integral equation approaches, including (molecular) density functional theory, hyper-netted chain (HNC), and Percus–Yevick (PY) equations. These approaches emphasize the distortion of the solvent distribution near the solvent-solute boundary (Akiyama and Hirata, 1998). Additionally, a differential geometry based solvent-solute boundary can be introduced to the polarizable continuum model (PCM) (Tomasi et al., 2005) to improve its surface description. This approach has already included a quantum description of the solute and is multiscale in nature. The PCM often calculates energies and gradients by using the Hartree–Fock and/or the density functional theory (Parr and Yang, 1989; Yang and Lee, 1995). Moreover, the combination of the present theory with the generalized Born (GB) model (Feig and Brooks III, 2004) is feasible too. As the solvent-solute boundary can dramatically impact the result of theoretical predictions, the aforementioned generalizations should be very significant to the current practice in solvation analysis.
Furthermore, there has been a long and interesting history of developing mechanical models for morphogenesis of biological systems at cellular and/or organismic levels (Oster et al., 1985; Collier et al., 1996). This development is based on the fundamental laws of physics, and has considerably enriched our understanding of biological systems. Recently, Navier–Stokes and Stokes models for visco-elastic systems have yielded impressive successes in the description of tetrapod embryogenesis (Fleury, 2009; le Noble et al., 2005). The construction of multiscale models in the present work follows the same principle, i.e., utilizing the fundamental laws of physics to describe biological phenomena and experimental measurements. However, the subject of interest described in the present work is much more elementary, i.e., biomolecular systems, rather than cellular systems and organisms. It is hoped that multiscale models at macromolecular levels might complement the earlier mechanical models.
Finally, we note that computational feasibility, numerical efficiency, simulation strategy, and model validation are very important aspects of multiscale modeling and simulation. More detailed investigations of the solvation process, including alternative models, extensive numerical examples of both small molecules and macromolecules, specific studies of geometric flow surfaces, as well as comparisons with experimental data and other existing models, have been carried out and will be published elsewhere (Chen et al., 2010). A Poisson–Boltzmann model based implicit molecular dynamics method has also been developed in our recent work (Geng and Wei, 2008). The further incorporation of fluid dynamics into the implicit molecular dynamics is under our consideration and will be published elsewhere. The extension of the present multiscale models for the investigation of ion channels is in progress.
Acknowledgements
The author thanks Nathan Baker, Zhan Chen, and Weitao Yang for useful discussions of solvation modeling. This work was supported in part by NSF grants DMS-0616704 and CCF-0936830, and NIH grants CA-127189 and GM-090208.
Footnotes
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Abaid N, Eisenberg RS, Liu W. Asymptotic expansions of i-v relations via a Poisson–Nernst–Planck system. SIAM J. Appl. Dyn. Syst. 2008;7:1507–1526. [Google Scholar]
- Abraham FF, Broughton JQ, Bernstein N, Kaxiras E. Spanning the continuum to quantum length scales in a dynamic simulation of brittle fracture. Europhys. Lett. 1998;44:783–787. [Google Scholar]
- Akiyama R, Hirata F. Theoretical study for water structure at highly ordered surface: effect of surface structure. J. Chem. Phys. 1998;108:4904–4911. [Google Scholar]
- Alavi S, Wei GW, Snider RF. Chain relations of reduced distribution functions and their associated correlation functions. J. Chem. Phys. 1997;108:706–714. [Google Scholar]
- Ashbaugh HS. Convergence of molecular and macroscopic continuum descriptions of ion hydration. J. Phys. Chem. B. 2000;104(31):7235–7238. [Google Scholar]
- Auffray C, Nottale L. Scale relativity theory and integrative systems biology, 1: founding principles and scale laws. Prog. Biophys. Mol. Biol. 2008;97:79–114. doi: 10.1016/j.pbiomolbio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Baker NA. Poisson-Boltzmann methods for biomolecular electrostatics. Methods Enzymol. 2004;383:94–118. doi: 10.1016/S0076-6879(04)83005-2. [DOI] [PubMed] [Google Scholar]
- Baker NA. Improving implicit solvent simulations: a Poisson-centric view. Curr. Opin. Struct. Biol. 2005;15(2):137–43. doi: 10.1016/j.sbi.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bates PW, Wei GW, Zhao S. The minimal molecular surface. 2006 doi: 10.1002/jcc.20796. arXiv:q-bio/0610038v1. [DOI] [PubMed]
- Bates PW, Wei GW, Zhao S. Minimal molecular surfaces and their applications. J. Comput. Chem. 2008;29(3):380–91. doi: 10.1002/jcc.20796. [DOI] [PubMed] [Google Scholar]
- Bates PW, Chen D, Sun YH, Wei GW, Zhao S. Geometric and potential driving formation and evolution of biomolecular surfaces. J. Math. Biol. 2009;59:193–231. doi: 10.1007/s00285-008-0226-7. [DOI] [PubMed] [Google Scholar]
- Beglov D, Roux B. An integral equation to describe the solvation of polar molecules in liquid water. J. Phys. Chem. B. 1997;101(39):7821–6. [Google Scholar]
- Bertozzi AL, Greer JB. Low-curvature image simplifiers: global regularity of smooth solutions and Laplacian limiting schemes. Commun. Pure Appl. Math. 2004;57(6):764–790. [Google Scholar]
- Blomgren P, Chan TF. Color TV: total variation methods for restoration of vector-valued images. IEEE Trans. Image Process. 1998;7(3):304–309. doi: 10.1109/83.661180. [DOI] [PubMed] [Google Scholar]
- Bobenko AI, Schröder P. Discrete Willmore flow. Symp. on Geometry Processing. 2005 2005 July;:101–110. [Google Scholar]
- Boschitsch AH, Fenley MO. Hybrid boundary element and finite difference method for solving the nonlinear Poisson–Boltzmann equation. J. Comput. Chem. 2004;25(7):935–955. doi: 10.1002/jcc.20000. [DOI] [PubMed] [Google Scholar]
- Boschitsch AH, Fenley MO. A new outer boundary formulation and energy corrections for the nonlinear Poisson–Boltzmann equation. J. Comput. Chem. 2007;28(5):909–21. doi: 10.1002/jcc.20565. [DOI] [PubMed] [Google Scholar]
- Bostrom M, Tavares FW, Bratko D, Ninham BW. Specific ion effects in solutions of globular proteins: comparison between analytical models and simulation. J. Phys. Chem. B. 2005;109(51):24489–94. doi: 10.1021/jp0551869. [DOI] [PubMed] [Google Scholar]
- Budiono, Byun D, Nyugen VD, Kim J, Ko HS. Free surface transition and momentum augmentation of liquid flow in micro/nano-scale channels with hydrophobic and hydrophilic surfaces. J. Mech. Sci. Technol. 2008;22:2554–2562. [Google Scholar]
- Canham PB. The minimum energy of bending as a possible explanation of the biconcave shape of the human red blood cell. J. Theor. Biol. 1970;26:61–81. doi: 10.1016/s0022-5193(70)80032-7. [DOI] [PubMed] [Google Scholar]
- Carstensen V, Kimmel R, Sapiro G. Geodesic active contours. Int. J. Comput. Vis. 1997;22:61–79. [Google Scholar]
- Cecil Thomas. A numerical method for computing minimal surfaces in arbitrary dimension. J. Comput. Phys. 2005;206(2):650–660. [Google Scholar]
- Cerutti DS, Baker NA, McCammon JA. Solvent reaction field potential inside an uncharged globular protein: a bridge between implicit and explicit solvent models? J. Chem. Phys. 2007;127(15):155101. doi: 10.1063/1.2771171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Conlisk AT. Electroosmotic flow and particle transport in micro/nano nozzles and diffusers. Biomed. Microdevices. 2008;10:289–289. doi: 10.1007/s10544-007-9135-6. [DOI] [PubMed] [Google Scholar]
- Chen D, Wei GW. Modeling and simulation of nano-electronic devices. J. Comput. Phys. 2009:1–44. doi: 10.1016/j.jcp.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DP, Eisenberg RS, Jerome JW, Shu CW. Hydrodynamic model of temperature change in open ionic channels. Biophys. J. 1995;69:2304–2322. doi: 10.1016/S0006-3495(95)80101-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Baker NA, Wei GW. Differential geometry based solvation models, I: Eulerian formulation. J. Comput. Phys. 2010 doi: 10.1016/j.jcp.2010.06.036. to be submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LT, Dzubiella J, McCammon AJ, Li B. Application of the level-set method to the implicit solvation of nonpolar molecules. J. Chem. Phys. 2007;127(8) doi: 10.1063/1.2757169. [DOI] [PubMed] [Google Scholar]
- Cheng XL, Ivanov I, Wang HL, Sine SM, McCammon JA. Molecular dynamics simulations of a prokaryotic homologue of the nicotinic acetylcholine receptor. Biophys. J. 2009;96:4502–4513. doi: 10.1016/j.bpj.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopp DL. Computing minimal surfaces via level set curvature flow. J. Comput. Phys. 1993;106(1):77–91. [Google Scholar]
- Chorny I, Dill KA, Jacobson MP. Surfaces affect ion pairing. J. Phys. Chem. B. 2005;109(50):24056–60. doi: 10.1021/jp055043m. [DOI] [PubMed] [Google Scholar]
- Chu KT, Bazant MZ. Nonlinear electrochemical relaxation around conductors. Phys. Rev. E. 2006;74:011501. doi: 10.1103/PhysRevE.74.011501. [DOI] [PubMed] [Google Scholar]
- Coifman RR, Lafon S, Lee AB, Maggioni M, Warner F, Zucker S. Geometric diffusions as a tool for harmonic analysis and structure definition of data: diffusion maps. Proceedings of the National Academy of Sciences. 2005:7426–7431. doi: 10.1073/pnas.0500334102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier JR, Monk NAM, Maini PK, Lewis JH. Pattern formation by lateral inhibition with feedback: a mathematical model of delta-notch intercellular signalling. J. Theor. Biol. 1996;183:429–446. doi: 10.1006/jtbi.1996.0233. [DOI] [PubMed] [Google Scholar]
- Connolly ML. Analytical molecular surface calculation. J. Appl. Crystallogr. 1983;16(5):548–558. [Google Scholar]
- Davis ME, McCammon JA. Electrostatics in biomolecular structure and dynamics. Chem. Rev. 1990;94:509–21. [Google Scholar]
- Desbrun M, Meyer M, Schröder P, Barr AH. Implicit fairing of irregular meshes using diffusion and curvature flow. ACM SIGGRAPH. 1999:317–324. [Google Scholar]
- Dinh HQ, Yezzi A, Turk G. Texture transfer during shape transformation. ACM Trans. Graph. 2005;24(2):289–310. [Google Scholar]
- Dong F, Olsen B, Baker NA. Computational methods for biomolecular electrostatics. Methods Cell Biol. 2008;84:843–70. doi: 10.1016/S0091-679X(07)84026-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Q, Liu C, Wang XQ. A phase field approach in the numerical study of the elastic bending energy for vesicle membranes. J. Comput. Phys. 2004;198:450–468. [Google Scholar]
- Dzubiella J, Swanson JMJ, McCammon JA. Coupling hydrophobicity, dispersion, and electrostatics in continuum solvent models. Phys. Rev. Lett. 2006a;96:087802. doi: 10.1103/PhysRevLett.96.087802. [DOI] [PubMed] [Google Scholar]
- Dzubiella J, Swanson JMJ, McCammon JA. Coupling nonpolar and polar solvation free energies in implicit solvent models. J. Chem. Phys. 2006b;124:084905. doi: 10.1063/1.2171192. [DOI] [PubMed] [Google Scholar]
- Engquist WNEB, Li X, Ren W, Vanden-Eijnden E. Heterogeneous multiscale methods: a review. Commun. Comput. Phys. 2007;2:367–450. [Google Scholar]
- Elcock AH, Gabdoulline RR, Wade RC, McCammon JA. Computer simulation of protein-protein association kinetics: acetylcholinesterase-fasciculin. J. Mol. Biol. 1999;291(1):149–162. doi: 10.1006/jmbi.1999.2919. [DOI] [PubMed] [Google Scholar]
- Evans LC, Spruck J. Motion of level sets by mean curvature. J. Differ. Geom. 1991;33:635–681. [Google Scholar]
- Federer H. Curvature measures. Trans. Am. Math. Soc. 1959;93:418–491. [Google Scholar]
- Feig M, Brooks CL., III Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr. Opin. Struct. Biol. 2004;14:217–224. doi: 10.1016/j.sbi.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Fixman M. The Poisson–Boltzmann equation and its application to polyelectrolytes. J. Chem. Phys. 1979;70(11):4995–5005. [Google Scholar]
- Fleury V. Clarifying tetrapod embryogenesis a physicist's point of view. Eur. Phys. J. Appl. Phys. 2009;45:30101. [Google Scholar]
- Fogolari F, Brigo A, Molinari H. The Poisson–Boltzmann equation for biomolecular electrostatics: a tool for structural biology. J. Mol. Recognit. 2002;15(6):377–92. doi: 10.1002/jmr.577. [DOI] [PubMed] [Google Scholar]
- Forsman J. A simple correlation-corrected Poisson–Boltzmann theory. J. Phys. Chem. B. 2004;108(26):9236–45. [Google Scholar]
- Franco AA, Schott P, Jallut C, Maschke B. A dynamic mechanistic model of an electrochemical interface. J. Electrochem. Soc. 2006;153:A1053–A1061. [Google Scholar]
- Fries PH, Patey GN. The solution of the hypernetted-chain approximation for fluids of nonspherical particles. a general method with application to dipolar hard spheres. J. Chem. Phys. 1985;82:429–440. [Google Scholar]
- Gabdoulline RR, Wade RC. Brownian dynamics simulation of protein–protein diffusional encounter. Methods Enzymol. 1998;14(3):329–341. doi: 10.1006/meth.1998.0588. [DOI] [PubMed] [Google Scholar]
- Gallicchio E, Levy RM. AGBNP: an analytic implicit solvent model suitable for molecular dynamics simulations and high-resolution modeling. J. Comput. Chem. 2004;25(4):479–499. doi: 10.1002/jcc.10400. [DOI] [PubMed] [Google Scholar]
- Geng W, Yu S, Wei GW. Treatment of charge singularities in implicit solvent models. J. Phys. Chem. 2007;127:114106. doi: 10.1063/1.2768064. [DOI] [PubMed] [Google Scholar]
- Geng WH, Wei GW. Interface technique based implicit solvent molecular dynamics. J. Comput. Phys. 2008 submitted. [Google Scholar]
- Geng WH, Wei GW. Matched interface and boundary implementation of Poisson–Boltzmann based molecular dynamics. J. Comput. Phys. 2009 doi: 10.1016/j.jcp.2010.09.031. solicited submission. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa G, Sochen N, Zeevi YY. Image sharpening by flows based on triple well potentials. J. Math. Imaging Vis. 2004;20:121–131. [Google Scholar]
- Gilson MK, Davis ME, Luty BA, McCammon JA. Computation of electrostatic forces on solvated molecules using the Poisson–Boltzmann equation. J. Phys. Chem. 1993;97(14):3591–3600. [Google Scholar]
- Gomes J, Faugeras OD. Using the vector distance functions to evolve manifolds of arbitrary codimension. Lect. Notes Comput. Sci. 2001;2106:1–13. [Google Scholar]
- Gordon D, Krishnamurthy V, Chung SH. Generalized Langevin models of molecular dynamics simulations with applications to ion channels. J. Chem. Phys. 2009;131:134102. doi: 10.1063/1.3233945. [DOI] [PubMed] [Google Scholar]
- Greer JB, Bertozzi AL. H-1 solutions of a class of fourth order nonlinear equations for image processing. Discrete Contin. Dyn. Syst. 2004a;10:349–366. [Google Scholar]
- Greer JB, Bertozzi AL. Traveling wave solutions of fourth order pdes for image processing. SIAM J. Math. Anal. 2004b;36:38–68. [Google Scholar]
- Grigoryev SA, Arya G, Correll S, Woodcock CL, Schlick T. Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proc. Natl. Acad. Sci. USA. 2009;106:13317–13322. doi: 10.1073/pnas.0903280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinspun E, Hirani AN, Desbrun M, Schröder P. Discrete shells. ACM SIGGRAPH Symp. on Computer Animation. 2003:62–67. [Google Scholar]
- Grochowski P, Trylska J. Continuum molecular electrostatics, salt effects and counterion binding. A review of the Poisson–Boltzmann theory and its modifications. Biopolymers. 2007;89(2):93–113. doi: 10.1002/bip.20877. [DOI] [PubMed] [Google Scholar]
- Gurau V, Mann JA. A critical overview of computational fluid dynamics multiphase models for proton exchange membrane fuel cells. SIAM J. Appl. Math. 2009;70:410–454. [Google Scholar]
- Helfrich W. Elastic properties of lipid bilayers: theory and possible experiments. Z. Naturforsch. C. 1973;28:693–703. doi: 10.1515/znc-1973-11-1209. [DOI] [PubMed] [Google Scholar]
- Holst MJ. Multilevel Methods for the Poisson–Boltzmann Equation. University of Illinois at Urbana-Champaign, Numerical Computing Group; Urbana-Champaign: 1993. [Google Scholar]
- Huang DM, Geissler PL, Chandler D. Scaling of hydrophobic solvation free energies. J. Phys. Chem. B. 2001;105(28):6704–6709. [Google Scholar]
- Hwang H, Schatz GC, Ratner MA. Ion current calculations based on three dimensional Poisson–Nernst–Planck theory for a cyclic peptide nanotube. J. Phys. Chem. B. 2006;110:6999–7008. doi: 10.1021/jp055740e. [DOI] [PubMed] [Google Scholar]
- Im W, Beglov D, Roux B. Continuum solvation model: electrostatic forces from numerical solutions to the Poisson–Boltzmann equation. Comput. Phys. Commun. 1998;111(1-3):59–75. [Google Scholar]
- Iwamoto M, Liu F, Ou-Yang ZC. Shape and stability of two-dimensional lipid domains with dipole-dipole interactions. J. Chem. Phys. 2006;125:224701. doi: 10.1063/1.2402160. [DOI] [PubMed] [Google Scholar]
- Knap J, Ortiz M. Spanning the continuum to quantum length scales in a dynamic simulation of brittle fracture. J. Mech. Phys. Solids. 2001;49:1899–1923. [Google Scholar]
- Kobbelt L. Discrete fairing and variational subdivision for freeform surface design. Vis. Comput. 2000;16(3–4):142–158. [Google Scholar]
- Lapidus LJ, Yao S, McGarrity K, Hertzog D, Tubman E, Bakajin O. Protein hydrophobic collapse and early folding steps observed in a microfluidic mixer. Biophys. J. 2007;93:218–224. doi: 10.1529/biophysj.106.103077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Noble F, Fleury V, Pries A, Corvol P, Eichmann A, Reneman RS. Control of arterial branching morphogenesis in embryogenesis: go with the flow. Eur. Phys. J., Appl. Phys. 2005;65:619–628. doi: 10.1016/j.cardiores.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Levy RM, Zhang LY, Gallicchio E, Felts AK. On the nonpolar hydration free energy of proteins: surface area and continuum solvent models for the solute-solvent interaction energy. J. Am. Chem. Soc. 2003;125(31):9523–9530. doi: 10.1021/ja029833a. [DOI] [PubMed] [Google Scholar]
- Li Y, Santosa F. A computational algorithm for minimizing total variation in image restoration. IEEE Trans. Image Process. 1996;5(6):987–95. doi: 10.1109/83.503914. [DOI] [PubMed] [Google Scholar]
- Liu JG, Shu CW. A high-order discontinuous Galerkin method for 2d incompressible flows. J. Comput. Phys. 2000;160:577–596. [Google Scholar]
- Lu Q, Luo R. A Poisson–Boltzmann dynamics method with nonperiodic boundary condition. J. Chem. Phys. 2003;119(21):11035–11047. [Google Scholar]
- Lu BZ, Chen WZ, Wang CX, Xu XJ. Protein molecular dynamics with electrostatic force entirely determined by a single Poisson–Boltzmann calculation. Proteins. 2002;48(3):497–504. doi: 10.1002/prot.10172. [DOI] [PubMed] [Google Scholar]
- Luo R, David L, Gilson MK. Accelerated Poisson–Boltzmann calculations for static and dynamic systems. J. Comput. Chem. 2002;23(13):1244–53. doi: 10.1002/jcc.10120. [DOI] [PubMed] [Google Scholar]
- Lysaker M, Lundervold A, Tai XC. Noise removal using fourth-order partial differential equation with application to medical magnetic resonance images in space and time. IEEE Trans. Image Process. 2003;12:1579–1590. doi: 10.1109/TIP.2003.819229. [DOI] [PubMed] [Google Scholar]
- Madura JD, Briggs JM, Wade RC, Davis ME, Luty BA, Ilin A, Antosiewicz J, Gilson MK, Bagheri B, Scott LR, McCammon JA. Electrostatics and diffusion of molecules in solution—simulations with the University of Houston Brownian Dynamics program. Comput. Phys. Commun. 1995;91(1–3):57–95. [Google Scholar]
- Mikula K, Sevcovic D. A direct method for solving an anisotropic mean curvature flow of plane curves with an external force. Math. Methods Appl. Sci. 2004;27(13):1545–1565. [Google Scholar]
- Mumford D, Shah J. Optimal approximations by piecewise smooth functions and associated variational problems. Commun. Pure Appl. Math. 1989;42(5):577–685. [Google Scholar]
- Nottale L, Auffray C. Scale relativity theory and integrative systems biology, 2: macroscopic quantum-type mechanics. Prog. Biophys. Mol. Biol. 2008;97:115–157. doi: 10.1016/j.pbiomolbio.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Osher S, Rudin LI. Feature-oriented image enhancement using shock filters. SIAM J. Numer. Anal. 1990;27(4):919–940. [Google Scholar]
- Osher S, Sethian JA. Fronts propagating with curvature-dependent speed: algorithms based on Hamilton–Jacobi formulations. J. Comput. Phys. 1988;79(1):12–49. [Google Scholar]
- Oster GF, Murray JD, Maini PK. A model for chondrogenic condensations in the developing limb: the role of extracellular matrix and cell tractions. J. Embryol. Exp. Morphol. 1985;89:93–112. [PubMed] [Google Scholar]
- Ou-Yang ZC, Helfrich W. Bending energy of vesicle membranes: general expressions for the first, second, and third variation of the shape energy and applications to spheres and cylinders. Phys. Rev. A. 1989;39:5280–5288. doi: 10.1103/physreva.39.5280. [DOI] [PubMed] [Google Scholar]
- Parr R, Yang W. Density Functional Theory of Atoms and Molecules. 1989.
- Peskin CS. Numerical analysis of blood flow in the heart. J. Comput. Phys. 1977;25(3):220–52. [Google Scholar]
- Pinkall U, Polthier K. Computing discrete minimal surfaces and their conjugates. Exp. Math. 1993;2(1):15–36. [Google Scholar]
- Prabhu NV, Zhu P, Sharp KA. Implementation and testing of stable, fast implicit solvation in molecular dynamics using the smooth-permittivity finite difference Poisson–Boltzmann method. J. Comput. Chem. 2004;25(16):2049–2064. doi: 10.1002/jcc.20138. [DOI] [PubMed] [Google Scholar]
- Prabhu NV, Panda M, Yang QY, Sharp KA. Explicit ion, implicit water solvation for molecular dynamics of nucleic acids and highly charged molecules. J. Comput. Chem. 2008;29:1113–1130. doi: 10.1002/jcc.20874. [DOI] [PubMed] [Google Scholar]
- Promislow K, Wetton B. Pem fuel cells: a mathematical overview. SIAM J. Appl. Math. 2009;70:369–409. [Google Scholar]
- Richards FM. Areas, volumes, packing, and protein structure. Annu. Rev. Biophys. Bioeng. 1977;6(1):151–176. doi: 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- Roux B, Simonson T. Implicit solvent models. Biophys. Chem. 1999;78(1–2):1–20. doi: 10.1016/s0301-4622(98)00226-9. [DOI] [PubMed] [Google Scholar]
- Rudin LI, Osher S, Fatemi E. Nonlinear total variation based noise removal algorithms.. Proceedings of the Eleventh Annual International Conference of the Center for Nonlinear Studies on Experimental Mathematics: Computational Issues in Nonlinear Science; Elsevier, North-Holland, Amsterdam. 1992. pp. 259–268. [Google Scholar]
- Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Sapiro G, Ringach DL. Anisotropic diffusion of multivalued images with applications to color filtering. IEEE Trans. Image Process. 1996;5(11):1582–1586. doi: 10.1109/83.541429. [DOI] [PubMed] [Google Scholar]
- Sarti A, Malladi R, Sethian JA. Subjective surfaces: a geometric model for boundary completion. Int. J. Comput. Vis. 2002;46(3):201–221. [Google Scholar]
- Sbert C, Solé AF. 3d curves reconstruction based on deformable models. J. Math. Imaging Vis. 2003;18(3):211–223. [Google Scholar]
- Sciubba E. Flow exergy as a Lagrangian for the Navier–Stokes equations for incompressible flow. Int. J. Thermodyn. 2004;7:115–122. [Google Scholar]
- Sethian JA. Evolution, implementation, and application of level set and fast marching methods for advancing fronts. J. Comput. Phys. 2001;169(2):503–555. [Google Scholar]
- Sharp KA, Honig B. Electrostatic interactions in macromolecules—theory and applications. Annu. Rev. Biophys. Biophys. Chem. 1990;19:301–332. doi: 10.1146/annurev.bb.19.060190.001505. [DOI] [PubMed] [Google Scholar]
- Simonson T. Macromolecular electrostatics: continuum models and their growing pains. Curr. Opin. Struct. Biol. 2001;11(2):243–252. doi: 10.1016/s0959-440x(00)00197-4. [DOI] [PubMed] [Google Scholar]
- Simonson T. Electrostatics and dynamics of proteins. Rep. Prog. Phys. 2003;66(5):737–87. [Google Scholar]
- Sitkoff D, Ben-Tal N, Honig B. Calculation of alkane to water solvation free energies using continuum solvent models. J. Phys. Chem. 1996;100:2744–2752. [Google Scholar]
- Smereka P. Semi-implicit level set methods for curvature and surface diffusion motion. J. Sci. Comput. 2003;19(1):439–456. [Google Scholar]
- Snider RF. Quantum-mechanical modified Boltzmann equation for degenerate internal states. J. Chem. Phys. 1960;32:1051–1060. [Google Scholar]
- Snider RF, Wei GW, Muga JG. Moderately dense gas quantum kinetic theory: aspects of pair correlations. J. Chem. Phys. 1996a;105:3057–3065. [Google Scholar]
- Snider RF, Wei GW, Muga JG. Moderately dense gas quantum kinetic theory: transport coefficient expressions. J. Chem. Phys. 1996b;105:3066–3078. [Google Scholar]
- Sochen N, Kimmel R, Malladi R. A general framework for low level vision. IEEE Trans. Image Process. 1998;7(3):310–318. doi: 10.1109/83.661181. [DOI] [PubMed] [Google Scholar]
- Sun YH, Wu PR, Wei GW, Wang G. Evolution operator based single-step method for image processing. Int. J. Biomed. Imaging. 2006a;83847:1–27. doi: 10.1155/IJBI/2006/83847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YH, Zhou YC, Li SG, Wei GW. A windowed Fourier spectral scheme for hyperbolic conservation laws. J. Comput. Phys. 2006b;214:466–490. [Google Scholar]
- Swanson JMJ, Wagoner JA, Baker NA, McCammon JA. Optimizing the Poisson dielectric boundary with explicit solvent forces and energies: lessons learned with atom-centered dielectric functions. J. Chem. Theory Comput. 2007;3(1):170–83. doi: 10.1021/ct600216k. [DOI] [PubMed] [Google Scholar]
- Tadmor EB, Ortiz M, Phillips R. Quasicontinuum analysis of defects in crystals. Philos. Mag. A. 1996;76:1529–1564. [Google Scholar]
- Tan C, Yang L, Luo R. How well does Poisson–Boltzmann implicit solvent agree with explicit solvent? A quantitative analysis. J. Phys. Chem. B. 2006;110(37):18680–18687. doi: 10.1021/jp063479b. [DOI] [PubMed] [Google Scholar]
- Tang S, Hou TY, Liu WK. A pseudo-spectral multiscale method: interfacial conditions and coarse grid equations. J. Comput. Phys. 2006;213:57–85. [Google Scholar]
- Taubin G. A signal processing approach to fair surface design. Proc. of SIGGRAPH. 1995 1995 August;:351–358. [Google Scholar]
- Tomasi J, Mennucci B, Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Tully-Smith DM, Reiss H. Further development of scaled particle theory of rigid sphere fluids. J. Chem. Phys. 1970;53(10):4015–25. [Google Scholar]
- Valentini P, Schwartzentruber TE. Large-scale molecular dynamics simulations of normal shock waves in dilute argon. Phys. Fluids. 2009;21:066101. [Google Scholar]
- Vlassiouk I, Smirnov S, Siwy Z. Ionic selectivity of single nanochannels. Nano Lett. 2008;8:1978–1985. doi: 10.1021/nl800949k. [DOI] [PubMed] [Google Scholar]
- Wagoner JA, Baker NA. Assessing implicit models for nonpolar mean solvation forces: the importance of dispersion and volume terms. Proc. Natl. Acad. Sci. USA. 2006;103(22):8331–6. doi: 10.1073/pnas.0600118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann L. Die Boltzmann-Gleichung für Gase mit rotierenden Molekulen. Z. Naturforsch. A. 1957;12:660–662. [Google Scholar]
- Wan DC, Patnaik BSV, Wei GW. Discrete singularconvolution-finite subdomain method for the solution of incompressible viscous flows. J. Comput. Phys. 2002;180:229–255. [Google Scholar]
- Wang W, Shu CW. The wkb local discontinuous Galerkin method for the simulation of Schrodinger equation in a resonant tunneling diode. J. Sci. Comput. 2009;40:360–374. [Google Scholar]
- Warshel A, Papazyan A. Electrostatic effects in macromolecules: fundamental concepts and practical modeling. Curr. Opin. Struct. Biol. 1998;8(2):211–7. doi: 10.1016/s0959-440x(98)80041-9. [DOI] [PubMed] [Google Scholar]
- Warshel A, Sharma PK, Kato M, Parson WW. Modeling electrostatic effects in proteins. Biochim. Biophys. Acta, Proteins Proteomics. 2006;1764(11):1647–76. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Weeks JD, Chandler D, Andersen HC. Role of repulsive forces in determining the equilibrium structure of simple liquids. J. Chem. Phys. 1971;54(12):5237–47. [Google Scholar]
- Wei GW. Generalized Perona–Malik equation for image restoration. IEEE Signal Process. Lett. 1999;6(7):165–7. [Google Scholar]
- Wei GW. A new algorithm for solving some mechanical problems. Comput. Methods Appl. Mech. Eng. 2001a;190:2017–2030. [Google Scholar]
- Wei GW. Vibration analysis by discrete singular convolution. J. Sound Vib. 2001b;244:535–553. [Google Scholar]
- Wei GW. Oscillation reduction by anisotropic diffusions. Comput. Phys. Commun. 2002;144:417–342. [Google Scholar]
- Wei GW, Jia YQ. Synchronization-based image edge detection. Europhys. Lett. 2002;59(6):814. [Google Scholar]
- Wei GW, Zhao YB, Xiang Y. Discrete singular convolution and its application to the analysis of plates with internal supports, I: theory and algorithm. Int. J. Numer. Methods Eng. 2002;55:913–946. [Google Scholar]
- Wei GW, Sun YH, Zhou YC, Feig M. Molecular multiresolution surfaces. 2005:1–11. arXiv:math-ph/0511001v1.
- Willmore TJ. Riemannian Geometry. Oxford University Press; Oxford: 1997. [Google Scholar]
- Wolfgang K. Differential Geometry: Curves-Surface-Manifolds. American Mathematical Society; Providence: 2002. [Google Scholar]
- Xu G, Pan Q, Bajaj CL. Discrete surface modeling using partial differential equations. Comput. Aided Geom. Des. 2006;23(2):125–145. doi: 10.1016/j.cagd.2005.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WT, Lee TS. A density matrix divide and conquer approach for electronic structure calculations of large molecules. J. Chem. Phys. 1995;103:5674–5678. [Google Scholar]
- Yu S, Wei GW. Three-dimensional matched interface and boundary (MIB) method for treating geometric singularities. J. Comput. Phys. 2007;227:602–632. [Google Scholar]
- Yu S, Geng W, Wei GW. Treatment of geometric singularities in implicit solvent models. J. Chem. Phys. 2007a;126:244108. doi: 10.1063/1.2743020. [DOI] [PubMed] [Google Scholar]
- Yu S, Zhou Y, Wei GW. Matched interface and boundary (MIB) method for elliptic problems with sharp-edged interfaces. J. Comput. Phys. 2007b;224(2):729–756. [Google Scholar]
- Zhang Y, Xu G, Bajaj C. Quality meshing of implicit solvation models of biomolecular structures. Comput. Aided Geom. Des. 2006;23(6):510–30. doi: 10.1016/j.cagd.2006.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Wei GW. High-order FDTD methods via derivative matching for maxwell's equations with material interfaces. J. Comput. Phys. 2004;200(1):60–103. [Google Scholar]
- Zheng Z, Hansford DJ, Conlisk AT. Effect of multivalent ions on electroosmotic flow in micro-and nanochannels. Electrophoresis. 2003;24:3006–3017. doi: 10.1002/elps.200305561. [DOI] [PubMed] [Google Scholar]
- Zhou YC, Wei GW. High-resolution conjugate filters for the simulation of flows. J. Comput. Phys. 2003;189:150–179. [Google Scholar]
- Zhou YC, Wei GW. On the fictitious-domain and interpolation formulations of the matched interface and boundary (MIB) method. J. Comput. Phys. 2006;219(1):228–246. [Google Scholar]
- Zhou YC, Zhao S, Feig M, Wei GW. High order matched interface and boundary method for elliptic equations with discontinuous coefficients and singular sources. J. Comput. Phys. 2006;213(1):1–30. [Google Scholar]
- Zhou YC, Feig M, Wei GW. Highly accurate biomolecular electrostatics in continuum dielectric environments. J. Comput. Chem. 2008a;29:87–97. doi: 10.1002/jcc.20769. [DOI] [PubMed] [Google Scholar]
- Zhou YC, Holst MJ, McCammon JA. A nonlinear elasticity model of macromolecular conformational change induced by electrostatic forces. J. Math. Anal. Appl. 2008b;340:135–164. doi: 10.1016/j.jmaa.2007.07.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YC, Lu BZ, Huber GA, Holst MJ, McCammon JA. Continuum simulations of acetylcholine consumption by acetylcholinesterase: a Poisson–Nernst–Planck approach. J. Phys. Chem. B. 2008c;112:270–275. doi: 10.1021/jp074900e. [DOI] [PubMed] [Google Scholar]