

Abstract
The title compound, C13H11N3O2S, was prepared by reaction of 2-nitrobenzenamine, KOH and 1-isothiocyanatobenzene in an ethanol solution at room temperature. The dihedral angles formed between the thiourea plane and the phenyl rings are 61.9 and 31.0°. The dihedral angle between the two phenyl rings is 78.1°. In the crystal structure, there are weak intermolecular N—H⋯S and C—H⋯S hydrogen-bonding interactions.
Related literature
For related literature, see: Reinbold & Morar (1984 ▶); Xue et al. (2004 ▶); Madan & Taneja (1991 ▶).
Experimental
Crystal data
C13H11N3O2S
M r = 273.31
Monoclinic,
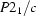
a = 7.3110 (15) Å
b = 24.113 (5) Å
c = 7.4320 (15) Å
β = 90.22 (3)°
V = 1310.2 (5) Å3
Z = 4
Mo Kα radiation
μ = 0.25 mm−1
T = 293 (2) K
0.25 × 0.20 × 0.18 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
Absorption correction: none
2964 measured reflections
2764 independent reflections
2022 reflections with I > 2σ(I)
R int = 0.017
3 standard reflections every 100 reflections intensity decay: none
Refinement
R[F 2 > 2σ(F 2)] = 0.045
wR(F 2) = 0.132
S = 1.09
2764 reflections
173 parameters
H-atom parameters constrained
Δρmax = 0.31 e Å−3
Δρmin = −0.41 e Å−3
Data collection: CAD-4 Software (Enraf–Nonius, 1989 ▶); cell refinement: CAD-4 Software; data reduction: NRCVAX (Gabe et al., 1989 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 1990 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997 ▶); molecular graphics: SHELXTL/PC (Sheldrick, 1990 ▶); software used to prepare material for publication: WinGX (Farrugia, 1999 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536807063374/at2515sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536807063374/at2515Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1A⋯S1i | 0.86 | 2.56 | 3.3744 (19) | 158 |
| C3—H3A⋯S1ii | 0.93 | 2.84 | 3.725 (3) | 159 |
Symmetry codes: (i) 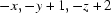 ; (ii)
; (ii) 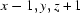 .
.
Acknowledgments
The authors thank the Natural Science Foundation of Shandong Province (grant No. Y2005B04).
supplementary crystallographic information
Comment
Thioureas have been studied for many years because of their broad antibiosis and sterilization properties Recent years study shows that thioureas not only can be used to kill insects and adjust plant growth but also have anti-viral activities (Madan et al., 1991; Reinbold et al.,1984). From our early quantum study on these compounds, we find that they have several active centers and can form polyligand complexes with metals easily (Xue et al., 2004).These complexes are widely used as anticancer medicines Therefore study on thioureas has important impact on the future. In order to search for new compounds with higher bioactivity, the title compound was synthesized and we herein report its crystal structure.
In the title compound, bond lengths and angles are generally normal. The C7—S1 bond length of 1.686 (2)Å is indicative of considerable double-bond character. The dihedral angle between the plane (C6—C8/N1/N2/S1) and the plane (C8—C13/N2) is 31.01°. The torsion angles of S1—C7—N2—C8 and N1—C7—N2—C8 are -1.31 and -179.19°, respectively.
In the crystal structure, there are weak intermolecular C—H···S and N—H···S hydrogen bonding interactions. These interactions stabilize the title structure.
Experimental
The title compound was prepared by reaction of 2-nitrobenzenamine (0.05 mol), KOH (0.15 mol) and 1-isothiocyanatobenzene (0.05 mol) in the ethanol solution (40 ml) at room temperature. Single crystals of the title compound suitable for X-ray measurements was obtained by recrystallization from ethanol/acetone (v/v=1:1) at room temperature.
Refinement
The H atoms were fixed geometrically and were treated as riding on the parent C atoms, with C—H = 0.93 Å and N—H = 0.86 Å, and Uiso=1.2 times Ueq of the parent atoms.
Figures
Fig. 1.

The structure of the title compound showing 30% probability displacement ellipsoids and the atom-numbering scheme.
Crystal data
| C13H11N3O2S | F000 = 568 |
| Mr = 273.31 | Dx = 1.386 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 25 reflections |
| a = 7.3110 (15) Å | θ = 1.7–27.0º |
| b = 24.113 (5) Å | µ = 0.25 mm−1 |
| c = 7.4320 (15) Å | T = 293 (2) K |
| β = 90.22 (3)º | Block, yellow |
| V = 1310.2 (5) Å3 | 0.25 × 0.20 × 0.18 mm |
| Z = 4 |
Data collection
| Enraf–Nonius CAD-4 diffractometer | Rint = 0.017 |
| Radiation source: fine-focus sealed tube | θmax = 27.0º |
| Monochromator: graphite | θmin = 1.7º |
| T = 293(2) K | h = 0→8 |
| ω scans | k = 0→28 |
| Absorption correction: none | l = −8→8 |
| 2964 measured reflections | 3 standard reflections |
| 2764 independent reflections | every 100 reflections |
| 2022 reflections with I > 2σ(I) | intensity decay: none |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.045 | w = 1/[σ2(Fo2) + (0.0588P)2 + 0.4395P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.132 | (Δ/σ)max < 0.001 |
| S = 1.09 | Δρmax = 0.31 e Å−3 |
| 2764 reflections | Δρmin = −0.41 e Å−3 |
| 173 parameters | Extinction correction: SHELXL, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.115 (6) |
| Secondary atom site location: difference Fourier map |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.20879 (9) | 0.56603 (3) | 0.96789 (9) | 0.0669 (3) | |
| O1 | −0.0828 (3) | 0.79863 (8) | 1.3139 (4) | 0.1026 (8) | |
| O2 | −0.1250 (3) | 0.71304 (8) | 1.3693 (3) | 0.0785 (6) | |
| N1 | −0.0460 (3) | 0.55266 (7) | 1.2122 (3) | 0.0556 (5) | |
| H1A | −0.0549 | 0.5206 | 1.1619 | 0.067* | |
| N2 | 0.0901 (2) | 0.63776 (7) | 1.2248 (2) | 0.0482 (4) | |
| H2A | 0.0145 | 0.6425 | 1.3119 | 0.058* | |
| N3 | −0.0333 (3) | 0.75057 (8) | 1.3045 (3) | 0.0577 (5) | |
| C1 | −0.1032 (4) | 0.57222 (10) | 1.5305 (3) | 0.0622 (6) | |
| H1B | 0.0220 | 0.5731 | 1.5528 | 0.075* | |
| C2 | −0.2268 (5) | 0.58020 (11) | 1.6701 (4) | 0.0767 (8) | |
| H2B | −0.1840 | 0.5866 | 1.7863 | 0.092* | |
| C3 | −0.4120 (5) | 0.57870 (12) | 1.6372 (5) | 0.0844 (10) | |
| H3A | −0.4939 | 0.5848 | 1.7305 | 0.101* | |
| C4 | −0.4759 (4) | 0.56819 (12) | 1.4666 (5) | 0.0853 (10) | |
| H4A | −0.6013 | 0.5663 | 1.4458 | 0.102* | |
| C5 | −0.3554 (3) | 0.56029 (10) | 1.3246 (4) | 0.0635 (6) | |
| H5A | −0.3990 | 0.5533 | 1.2091 | 0.076* | |
| C6 | −0.1685 (3) | 0.56302 (8) | 1.3585 (3) | 0.0477 (5) | |
| C7 | 0.0815 (3) | 0.58703 (9) | 1.1440 (3) | 0.0462 (5) | |
| C8 | 0.2032 (3) | 0.68340 (8) | 1.1878 (2) | 0.0423 (5) | |
| C9 | 0.3819 (3) | 0.67718 (9) | 1.1219 (3) | 0.0509 (5) | |
| H9A | 0.4277 | 0.6417 | 1.1030 | 0.061* | |
| C10 | 0.4912 (3) | 0.72225 (11) | 1.0846 (3) | 0.0601 (6) | |
| H10A | 0.6086 | 0.7167 | 1.0403 | 0.072* | |
| C11 | 0.4284 (4) | 0.77548 (10) | 1.1125 (3) | 0.0649 (7) | |
| H11A | 0.5015 | 0.8057 | 1.0836 | 0.078* | |
| C12 | 0.2561 (4) | 0.78358 (9) | 1.1834 (3) | 0.0599 (6) | |
| H12A | 0.2143 | 0.8193 | 1.2058 | 0.072* | |
| C13 | 0.1449 (3) | 0.73817 (8) | 1.2216 (3) | 0.0465 (5) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0690 (4) | 0.0677 (4) | 0.0644 (4) | −0.0224 (3) | 0.0339 (3) | −0.0228 (3) |
| O1 | 0.0986 (16) | 0.0629 (12) | 0.147 (2) | 0.0267 (11) | 0.0178 (14) | −0.0059 (12) |
| O2 | 0.0682 (11) | 0.0700 (12) | 0.0977 (14) | 0.0081 (9) | 0.0318 (10) | −0.0063 (10) |
| N1 | 0.0613 (12) | 0.0468 (10) | 0.0589 (11) | −0.0139 (8) | 0.0251 (9) | −0.0121 (8) |
| N2 | 0.0478 (10) | 0.0446 (9) | 0.0523 (10) | −0.0057 (7) | 0.0190 (8) | −0.0050 (7) |
| N3 | 0.0621 (12) | 0.0547 (11) | 0.0562 (11) | 0.0099 (10) | −0.0038 (9) | −0.0074 (9) |
| C1 | 0.0589 (14) | 0.0683 (15) | 0.0595 (15) | −0.0087 (11) | 0.0147 (11) | −0.0049 (11) |
| C2 | 0.104 (2) | 0.0671 (16) | 0.0595 (15) | −0.0087 (15) | 0.0338 (15) | −0.0044 (12) |
| C3 | 0.092 (2) | 0.0666 (17) | 0.095 (2) | 0.0051 (15) | 0.0597 (19) | 0.0042 (15) |
| C4 | 0.0514 (15) | 0.0791 (19) | 0.126 (3) | 0.0074 (13) | 0.0338 (16) | 0.0107 (18) |
| C5 | 0.0527 (14) | 0.0594 (14) | 0.0787 (17) | 0.0021 (11) | 0.0120 (12) | 0.0061 (12) |
| C6 | 0.0504 (12) | 0.0376 (10) | 0.0552 (12) | −0.0051 (8) | 0.0195 (9) | 0.0013 (8) |
| C7 | 0.0441 (11) | 0.0480 (11) | 0.0466 (11) | −0.0048 (9) | 0.0117 (8) | −0.0016 (9) |
| C8 | 0.0460 (11) | 0.0449 (10) | 0.0362 (10) | −0.0060 (8) | 0.0024 (8) | 0.0019 (8) |
| C9 | 0.0464 (11) | 0.0558 (12) | 0.0505 (12) | −0.0058 (9) | 0.0070 (9) | −0.0018 (9) |
| C10 | 0.0529 (13) | 0.0753 (17) | 0.0522 (13) | −0.0214 (11) | 0.0042 (10) | −0.0002 (11) |
| C11 | 0.0758 (17) | 0.0615 (15) | 0.0575 (14) | −0.0303 (13) | −0.0017 (12) | 0.0054 (11) |
| C12 | 0.0816 (17) | 0.0441 (11) | 0.0540 (13) | −0.0103 (11) | −0.0096 (12) | 0.0012 (9) |
| C13 | 0.0517 (12) | 0.0484 (11) | 0.0394 (10) | −0.0013 (9) | −0.0041 (9) | −0.0005 (8) |
Geometric parameters (Å, °)
| S1—C7 | 1.686 (2) | C3—H3A | 0.9300 |
| O1—N3 | 1.216 (3) | C4—C5 | 1.391 (4) |
| O2—N3 | 1.226 (3) | C4—H4A | 0.9300 |
| N1—C7 | 1.348 (3) | C5—C6 | 1.390 (3) |
| N1—C6 | 1.433 (3) | C5—H5A | 0.9300 |
| N1—H1A | 0.8600 | C8—C9 | 1.405 (3) |
| N2—C7 | 1.364 (3) | C8—C13 | 1.411 (3) |
| N2—C8 | 1.404 (2) | C9—C10 | 1.378 (3) |
| N2—H2A | 0.8600 | C9—H9A | 0.9300 |
| N3—C13 | 1.474 (3) | C10—C11 | 1.379 (4) |
| C1—C6 | 1.381 (4) | C10—H10A | 0.9300 |
| C1—C2 | 1.392 (3) | C11—C12 | 1.381 (4) |
| C1—H1B | 0.9300 | C11—H11A | 0.9300 |
| C2—C3 | 1.375 (5) | C12—C13 | 1.394 (3) |
| C2—H2B | 0.9300 | C12—H12A | 0.9300 |
| C3—C4 | 1.373 (5) | ||
| C7—N1—C6 | 127.83 (17) | C1—C6—C5 | 120.8 (2) |
| C7—N1—H1A | 116.1 | C1—C6—N1 | 121.0 (2) |
| C6—N1—H1A | 116.1 | C5—C6—N1 | 118.1 (2) |
| C7—N2—C8 | 129.97 (16) | N1—C7—N2 | 114.60 (17) |
| C7—N2—H2A | 115.0 | N1—C7—S1 | 119.45 (16) |
| C8—N2—H2A | 115.0 | N2—C7—S1 | 125.92 (15) |
| O1—N3—O2 | 121.2 (2) | N2—C8—C9 | 122.26 (18) |
| O1—N3—C13 | 118.8 (2) | N2—C8—C13 | 121.35 (18) |
| O2—N3—C13 | 120.00 (18) | C9—C8—C13 | 116.35 (18) |
| C6—C1—C2 | 119.3 (3) | C10—C9—C8 | 121.8 (2) |
| C6—C1—H1B | 120.4 | C10—C9—H9A | 119.1 |
| C2—C1—H1B | 120.4 | C8—C9—H9A | 119.1 |
| C3—C2—C1 | 120.3 (3) | C9—C10—C11 | 120.7 (2) |
| C3—C2—H2B | 119.8 | C9—C10—H10A | 119.7 |
| C1—C2—H2B | 119.8 | C11—C10—H10A | 119.7 |
| C4—C3—C2 | 120.0 (2) | C10—C11—C12 | 119.6 (2) |
| C4—C3—H3A | 120.0 | C10—C11—H11A | 120.2 |
| C2—C3—H3A | 120.0 | C12—C11—H11A | 120.2 |
| C3—C4—C5 | 120.8 (3) | C11—C12—C13 | 120.0 (2) |
| C3—C4—H4A | 119.6 | C11—C12—H12A | 120.0 |
| C5—C4—H4A | 119.6 | C13—C12—H12A | 120.0 |
| C6—C5—C4 | 118.8 (3) | C12—C13—C8 | 121.5 (2) |
| C6—C5—H5A | 120.6 | C12—C13—N3 | 116.3 (2) |
| C4—C5—H5A | 120.6 | C8—C13—N3 | 122.17 (18) |
| C6—C1—C2—C3 | 0.3 (4) | N2—C8—C9—C10 | 179.4 (2) |
| C1—C2—C3—C4 | 1.3 (4) | C13—C8—C9—C10 | −2.8 (3) |
| C2—C3—C4—C5 | −1.6 (4) | C8—C9—C10—C11 | 0.5 (3) |
| C3—C4—C5—C6 | 0.2 (4) | C9—C10—C11—C12 | 1.9 (4) |
| C2—C1—C6—C5 | −1.6 (3) | C10—C11—C12—C13 | −1.9 (4) |
| C2—C1—C6—N1 | −177.8 (2) | C11—C12—C13—C8 | −0.6 (3) |
| C4—C5—C6—C1 | 1.4 (3) | C11—C12—C13—N3 | 177.4 (2) |
| C4—C5—C6—N1 | 177.6 (2) | N2—C8—C13—C12 | −179.37 (19) |
| C7—N1—C6—C1 | −63.3 (3) | C9—C8—C13—C12 | 2.9 (3) |
| C7—N1—C6—C5 | 120.4 (3) | N2—C8—C13—N3 | 2.8 (3) |
| C6—N1—C7—N2 | −1.0 (3) | C9—C8—C13—N3 | −174.95 (18) |
| C6—N1—C7—S1 | −179.05 (19) | O1—N3—C13—C12 | 9.1 (3) |
| C8—N2—C7—N1 | −179.2 (2) | O2—N3—C13—C12 | −167.8 (2) |
| C8—N2—C7—S1 | −1.3 (3) | O1—N3—C13—C8 | −172.9 (2) |
| C7—N2—C8—C9 | −31.8 (3) | O2—N3—C13—C8 | 10.2 (3) |
| C7—N2—C8—C13 | 150.6 (2) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···S1i | 0.86 | 2.56 | 3.3744 (19) | 158 |
| C3—H3A···S1ii | 0.93 | 2.84 | 3.725 (3) | 159 |
Symmetry codes: (i) −x, −y+1, −z+2; (ii) x−1, y, z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: AT2515).
References
- Enraf–Nonius (1989). CAD-4 Software Version 5.0. Enraf–Nonius, Delft, The Netherlands.
- Farrugia, L. J. (1999). J. Appl. Cryst.32, 837–838.
- Gabe, E. J., Le Page, Y., Charland, J.-P., Lee, F. L. & White, P. S. (1989). J. Appl. Cryst.22, 384–387.
- Madan, V. K. & Taneja, A. D. (1991). J. Indian Chem. Soc.68, 162–163.
- Reinbold, A. M. & Morar, G. V. (1984). Ser. Biol. Khim. Nauk.4, 75–77.
- Sheldrick, G. M. (1990). SHELXTL/PC Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
- Sheldrick, G. M. (1997). SHELXL97 and SHELXS97 University of Göttingen, Germany.
- Xue, S. J., Duan, L. P. & Xe, S. Y. (2004). Chin. J. Struct. Chem.23, 441–444.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536807063374/at2515sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536807063374/at2515Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report