

Abstract
The shear stress of flowing blood on the surfaces of endothelial cells that provide the barrier to transport of solutes and water between blood and the underlying tissue modulates the permeability to solutes and the hydraulic conductivity. This review begins with a discussion of transport pathways across the endothelium and then considers the experimental evidence from both in vivo and in vitro studies that shows an influence of shear stress on endothelial transport properties after both acute (minutes to hours) and chronic (hours to days) changes in shear stress. Next, the effects of shear stress on individual transport pathways (tight junctions, adherens junctions, vesicles and leaky junctions) are described, and this information is integrated with the transport experiments to suggest mechanisms controlling both acute and chronic responses of transport properties to shear stress. The review ends with a summary of future research challenges.
Keywords: Shear stress, Permeability, Hydraulic conductivity, Glycocalyx, Nitric oxide
1. Introduction
The endothelium lines all blood vessels from the largest arteries and veins down to the smallest capillaries. This review focuses on the mechanical environment, more specifically the shear stress of flowing blood on the surface of endothelial cells in arteries and the microcirculation. We begin by defining the transport properties that are affected by shear stress and then review the pathways for transport across the endothelium. This is followed by a description of the experiments that have uncovered an influence of shear stress on fluid and solute transport, both in vitro and in vivo and also those that have not detected a shear effect on transport. Subsequent sections describe the influences of shear stress on specific transport pathways and their roles in modulating overall transport in response to shear. Mechanotransduction of fluid shear stress and the role of the surface glycocalyx occupy a central role in the story that unfolds. The review concludes with a section suggesting important areas for future research.
To characterize transport across the endothelium, the following membrane transport equations are often employed.1
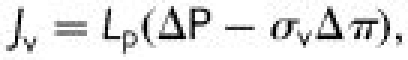 |
1 |
| 2 |
where Jv is the volume flux (volumetric flow rate per unit area) across the endothelium, Js is the solute flux (solute flow rate per unit area), ΔP is the pressure differential across the endothelium, Δπ is the osmotic pressure differential, Cp is the solute concentration in the plasma (assumed much greater than the concentration just beneath the endothelium), σv is the osmotic reflection coefficient, σs is the solute drag reflection coefficient, Lp is the hydraulic conductivity, Po is the diffusive permeability and Pe is the apparent permeability. ΔP and Δπ are driving forces for volume transport (water flow), which convects solute; Cp (ΔC) is the driving force for diffusive solute transport; and Po, Lp, σv and σs are transport properties. The importance of convection relative to diffusion is characterized by the parameter.
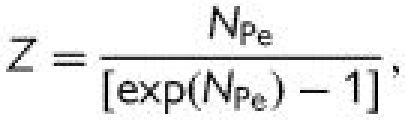 |
3 |
where the Peclet number is defined as:
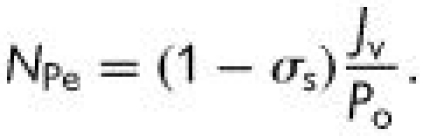 |
4 |
When NPe ≫ 1, Z → 0, and Pe → (1 − σs)Jv—the transport is dominated by convection; when NPe ≪ 1, Z → 1, and Pe→ Po—the transport is dominated by diffusion. It is conventional to think of membrane transport properties as constant, but a remarkable feature of the endothelium is that its transport properties are sensitive to both the chemical and mechanical environment within which it resides.
2. Transport pathways across the endothelium
Figure 1 shows the major transport pathways across the endothelium in a schematic representation including the tight junctions, breaks in the tight junctions, vesicles, leaky junctions, and the glycocalyx. While there are many variants of these pathways expressed in different microcirculatory regions (arterioles, capillaries, venules)2 and in specialized beds such as the fenestrated capillaries of the kidney glomerulus and the tight junctions of the blood–brain barrier, it will be useful to have a simplified model in mind for interpreting shear effects on transport.
Figure 1.

Transport pathways across the endothelium. The major transport pathways are: the tight junctions, breaks in the tight junctions, vesicles, and leaky junctions. The surface glycocalyx covers the entrance to all but the leaky junctions (figure courtesy of Limary Cancel).
We should also note at the outset that much of the literature on shear effects and endothelial transport is based on studies in cultured cells from arteries, veins, and microvascular beds. While a detailed comparison of the properties of in vitro and in vivo microvascular endothelium is beyond the scope of this review, we do note that Lp values both in vitro and in vivo are very close indeed (order 10−7 cm/s/cm H2O). There is, however, greater divergence in solute permeability values. For example, in a review of studies based on tracer uptake experiments in microvessels from the 70s and 80s, Michel (1996) summarized albumin Pe values of order 10−8 cm/s.3 More recent studies from the 90s (reviewed in Ogunrinade et al., 2002)4 and later,5 have reported values of order 10−7 cm/s. Values of Pe for in vitro systems range from 10−6 cm/s for albumin6 to 10−7 cm/s for 70 kDa dextran.7 The modest differences in transport properties between in vivo and in vitro systems suggest the relevance of in vitro studies of shear-dependent transport to the microcirculation.
2.1. Tight junctions
Intercellular junctions consist of an apical tight junction that typically does not allow transport of water soluble solutes above 2 nm in diameter and excludes pressure-driven water flux because of the high hydraulic resistance of the small pathway. Several protein components of the tight junction have been identified, including the occludin and the claudin families, which are believed to play a key role in providing the tight junction seal. Zonnula occludens proteins (ZO-1 and ZO-2) link the tight junction to cytoskeletal filaments including actin.8
2.2. Breaks in the tight junction
Larger water soluble solutes and water flux are accommodated by the ‘breaks’ in the tight junction that open up to the size of the adherens junction (order 20 nm). The adherens junction is a cell–cell adhesive junction that binds cells together and connects the cytoskeleton (actin) to the plasma membrane.9 The adhesive components of adherens junctions are formed by type II cadherins (calcium-dependent adhesive proteins) that belong to the larger cadherin superfamily, of which VE cadherin is prominent.
2.3. Vesicles
Vesicles that are coated with clathrin provide a pathway for endocytotic uptake of specific macromolecules from blood through localized receptors.10 Dissolved solutes in the blood can be taken up by this process of fluid-phase endocytosis as well. These vesicles may form a large pore system for transcytosis if they transport their cargo to the abluminal side of the endothelium.
Capillary endothelium contains 60–80 nm sized plasma-lemmal vesicles or caveolae that invaginate from either the luminal or abluminal plasma membrane.11 Caveolae are coated by a 21-kDa integral membrane protein, caveolin-1, on their cytoplasmic face. Caveolae may also be responsible for transcytosis. There are other vesicular transport pathways not associated with clathrin or caveolin (pinocytosis) that may also contribute to transendothelial transport.
2.4. Leaky junctions
Intercellular junctions are not expected to allow significant passage of LDL (diameter 23 nm) since LDL dimensions are greater than the estimated width of breaks in the tight junction. Weinbaum et al.,12 after work by Gerrity et al.,13 hypothesized that the large pore was a leaky interendothelial cleft associated with a small number of cells in the process of cell division or cell death. Leaky junction dimensions were estimated by Chien et al.14 in rat aortas to have widths ranging between 80 and 1330 nm for mitotic cells, and between 15 and 1000 nm for dead or dying cells. The ‘leaky junction’ is accessible to all solutes and water flux.
2.5. Glycocalyx
The surface of endothelial cells is decorated with a wide variety of membrane-bound macromolecules, which constitute the glycocalyx. This structure is believed to influence the transport of water and larger solutes up to the size of albumin through the breaks in the tight junction by adding a transport resistance to the channel entrance.15 The leaky junction openings are generally too big to be covered by the glycocalyx and the added resistance to this pathway by the glycocalyx is expected to be negligible. The solutes that are small enough to pass through tight junctions are generally much smaller than the fibre spacing in the glycocalyx and therefore there is little additional resistance to transport offered by the glycocalyx.
A cartoon that integrates all of the components of the glycocalyx described in this section is shown in Figure 2 (adapted from Tarbell and Pahakis).16 Glycosaminoglycans (GAGs) are linear polydisperse heteropolysaccharides, characterized by distinct disaccharide unit repeats. Specific combinations of these give rise to different GAG families, such as the heparan sulfate (HS), chondroitin/dermatan sulfate (CS), and hyaluronic acid or hyaluronan (HA) found on ECs. Proteoglycans are proteins that contain specific sites where sulfated GAGs are covalently attached. HS and CS chains vary between 50 and 150 disaccharide units, and have an average molecular weight of about 30 kDa.
Figure 2.

A cartoon representation of proteoglycans and glycoproteins on the surface of endothelial cells. Caveolin-1 associates with regions high in cholesterol and sphingolipids in the membrane (darker circles, left), and forms cave-like structures, caveolae (right). Glypicans, along with their heparan sulfate chains (blue dotted lines) localize in these regions. Transmembrane syndecans are shown to cluster in the outer edge of caveolae. Besides heparan sulfate, syndecans also contain chondroitin sulfate, lower down the core protein (green dotted lines). A glycoprotein with its short oligosaccharide branched chains and their associated SA ‘caps’ are displayed in the middle part of the figure (green). Hyaluronic acid or hyaluronan is a very long glycosaminoglycans (orange dotted line), which weaves into the glycocalyx and binds with CD44. Transmembrane CD44 can have chondroitin sulfate, heparan sulfate and oligosaccharides attached to it, and localizes in caveolae. Plasma proteins (grey), along with cations and cationic amino acids (red circles) are known to associate with glycosaminoglycans. (A) The cytoplasmic domains of syndecans link it to cytoskeletal elements (red line). (B) Oligomerization of syndecans helps them make direct associations with intracellular signalling effectors. (C) A series of molecules involved with endothelial nitric oxide synthase signalling localize in caveolae. Adapted from Tarbell and Pahakis.16
The most prominent GAGs on the surface of ECs are HS, accounting for more than 50% of the total GAG pool, the rest being comprising CS and HA. The transmembrane syndecans that associate with the cytoskeleton and the membrane-bound glypicans that are enriched in caveolae are the major protein core families of HS proteoglycans found on ECs.
In contrast to CS and HS, HA is a much longer disaccharide polymer, on the order of 1000 kDa, which is synthesized on the cell surface and is not covalently attached to a core protein. It is not sulfated, but obtains its negative charge from carboxyl groups that endow it with exceptional hydration properties. Completing the picture, glycoproteins with short-branched oligosaccharides attached to their core are also found on the surface of ECs. Many important receptors on the cell surface, such as selectins, integrins, and members of the immunoglobulin superfamily, have oligosaccharides attached to them and are classified as glycoproteins. The structure and properties of the glycocalyx are reviewed in much greater detail in Tarbell and Pahakis16 and Weinbaum et al.17
2.6. Transport pathways in arteries and microvessels
Using a bovine aortic endothelial cell (BAEC) monolayer model in vitro and measuring the transport of albumin, LDL and pressure-driven water flux with and without blockers of receptor-mediated vesicular transport, Cancel et al.18 determined the distribution of material flux through three pathways (vesicle, break in the tight junction, and leaky junction) under the reasonable assumptions that LDL is too large to pass through breaks in the tight junction and water does not pass through vesicles (Table 1). The predictions, summarized in Table 1, indicate that the break is the dominant pathway for water (77.7%), but that the leaky junction carries most of the LDL (90.9%) and a substantial portion of the albumin (44%). These predictions are consistent with the in vivo observations that LDL receptors (vesicles) do not contribute significantly to LDL uptake in pressurized arteries19 and that regions of enhanced uptake of Evans blue albumin are predictive of regions of enhanced LDL uptake.20
Table 1.
Summary of three-pore model predictions18
| Component | Vesicle | Break | Leaky junction |
|---|---|---|---|
| Albumin (%) | 20 | 36 | 44 |
| LDL (%) | 9.1 | 0 | 90.9 |
| Water (%) | 0 | 77.7 | 22.3 |
For microvessels, there has not been a quantitative study such as the one summarized in Table 1 for the in vitro system. The consensus, as reviewed by Michel and Curry21 where leaky junctions are not mentioned, seems to be that leaky junctions are not important in normal (non-fenestrated) microvessels and that vesicles and breaks assume a larger fraction of macromolecule transport while water flux is confined largely to breaks in the tight junction. This is consistent with the fact that apoptotic/mitotic cells that are responsible for leaky junctions are very infrequent in stable microvascular beds. Assuming, for example, that one in 3000 capillary endothelial cells is leaky14 and that one endothelial cell covers the circumference of the microvessel, there would be an axial distance of 3000 endothelial cells (order of 3 cm) between leakage sites—much longer than any capillary. However, this is clearly not the case in areas where remodeling (angiogenesis) is occurring such as in the retinal microvasculature of diabetic animals, around coronary infarcts, and in the vicinity of cancer tumors.
3. Shear stress affects endothelial transport properties
The early studies of shear stress effects on endothelial transport were thoroughly reviewed by Tarbell.1 The major results are summarized here briefly and more recent results are described in greater detail. Shear stress can increase Po of albumin and Lp on BAEC monolayers.22,23 The BAEC Lp response is mediated by nitric oxide (NO) production (Figure 3),24 whereas the albumin Po response is independent of NO.25 However, Po is measured in the absence of water flux, and it is not known if albumin transport coupled to water flux is enhanced by shear-induced increases in Lp. Static experiments (no shear stress) in BAECs have shown that albumin Pe increases as Jv increases, but with a negative reflection coefficient (σs), indicating that water and albumin do completely share a common pathway.26 This is all consistent with the more recent findings of Cancel et al.18 indicating that albumin diffusive transport may have a significant contribution from the vesicular pathway whereas water flux is dominated by the break in the tight junction (Table 1). The static transport of 70 kDa dextran across BAECs is characterized by σs of 0.67, suggesting that most if not all of its transport is through a water conducting pathway.7
Figure 3.

Effect of the nitric oxide synthase inhibitor NG-monomethyl-l-arginine on bovine aortic endothelial cell monolayer Lp. *P < 0.05 for NG-monomethyl-l-arginine compared with control (no inhibitor). Data are presented as mean ± SEM (adapted from Chang et al.24).
DePaola et al.27 using BAECs and Seebach et al.28 using HUVECs examined the electrical impedance of endothelial monolayers exposed to shear stress and observed an initial increase in impedance (order 20%) within minutes of shear application followed by a gradual decline in impedance by as much as 40% after 24 h. These changes that occurred during the process of endothelial cell remodeling in response to shear were reversible after removal of shear. McIntire et al.29 exposed bovine brain microvascular endothelial cell monolayers to steady shear stress of 1 or 10 dyn/cm2 for up to 72 h and measured Po of dextrans up to a molecular weight of 2 million. Large increases in Po, up to 76-fold for the largest dextran, were observed between 10 and 30 h with a return nearly to baseline after 48 h. Most recently, Warboys et al.30 exposed porcine aortic endothelial cells in vitro to both acute (1 h) and chronic (7–9 days) steady shear stress in an orbital shaker with a spatially averaged shear stress value of 1.82 dyn/cm2. They measured Po of albumin, and consistent with Jo et al.,22 observed a significant increase in Po after 1 h of shear stress, but a significant decrease, by about one-third, after 7–9 days of exposure. They also recorded a decrease in mitosis rate of a similar magnitude after 7–9 days of exposure.
A study of sinusoidal shear stress in BAECs showed that as long as the amplitude was smaller than the mean, Lp increased in a similar manner to its response to steady shear stress. However, when the amplitude exceeded the mean so that a reversing oscillatory shear was imposed, there was no increase in Lp.31 This unexpected behaviour resulted from a large increase in NO production under the reversing conditions that suppressed the Lp increase. This suggests a biphasic response of Lp to NO in BAECs: increases in NO at low levels increase Lp, but at high levels, increases in NO suppress Lp. This is consistent with Kurose et al.32 and Baldwin et al.33 who showed that when NO production was inhibited by blocking nitric oxide synthase (NOS), venules of the rat mesentery became more leaky to albumin.
The mechanisms by which endothelial NO production controls Lp have been investigated in several studies. He et al.34 described a mechanism wherein shear-induced NO drives cGMP production (Lp increase) that interacts with cAMP production (Lp decrease) through cGMP-dependent phophodiesterases. The balance can be tilted toward Lp increase by cGMP-dependent protein kinases that alter junctional proteins or Lp decrease through cAMP-dependent protein kinases that affect cytoskeletal tension. DeMaio et al.35 observed that the tight junction protein occludin was phosphorylated in response to shear stress in BAECs in tandem with an increase in Lp. The intermediate pathways between the upstream cGMP/cAMP and the downstream phosphorylation of occludin have not been established.
In vitro studies of transport properties in response to shear stress have displayed diverse responses in different cell types. Lakshminarayanan et al.36 showed that bovine retinal microvascular endothelial cells (BRECs) responded very much like BAECs, with a sustained increase in Lp in response to steady shear stress that was blocked by a NOS inhibitor. Similar experiments with a HUVEC model, however, showed a transient increase and return to baseline within 2 h,37 although HUVECs produce NO steadily in response to steady shear.38 These diverse shear responses were paralleled by the responses of these cell types to vascular endothelial growth factor (VEGF).6 Clearly the transport properties of endothelial monolayers and their response to shear stress depend on the cell source.
Studies on intact vessels and in vivo preparations have also revealed shear (flow)-dependent endothelial transport properties. The Lp of excised rabbit carotid arteries increased by 30% within 20 min of exposure to 1 dyn/cm2 shear stress.39 This is a relatively small increase in Lp, but the sub-endothelial intimal and medial layers of the artery contribute about 75% of the overall resistance to volume flux in an artery.40
An effect of flow on capillary permeability that had been hypothesized by Crone and Levitt41 has been observed in many recent studies. Shibata and Kamiya42 measured local capillary permeability of Cr-EDTA (MW 341) in the rabbit tenuissimus muscle and observed a tripling of Pe as red cell velocity increased 5-fold. Caldwell et al.43 determined a significant positive relationship between iodophenylpentadecanoic acid uptake in the coronary microcirculation of dogs and local flow rate. Kajimura and Michel44 observed a positive correlation between flow velocity and potassium ion permeability in single perfused venules of rats that was mediated by nitric oxide. Montermini et al.45 showed a similar response of sodium fluorescein permeability to increases in flow that could be abolished with an NOS inhibitor. They, however, interpreted their results in terms of a population of flow (shear) sensitive small pores (0.8 nm) that would not conduct water or larger solutes such as albumin. This seemed to be consistent with the report that Neal and Bates (2002)46 could not find a positive correlation between imposed shear stress and Lp in microvessels of frog mesentery using a double cannulation method. But Yuan et al.47 had measured Pe of albumin in isolated cannulated coronary venules at various intraluminal perfusion velocities and observed a 47% increase in Pe when velocity increased from 7 to 13 mm/s. This variance with the predictions of Montermini et al. and the observations of Neal and Bates may have arisen from differences in the vessel type, species and experimental preparation.
Building upon earlier studies,48,49 Williams50 measured Lp after step changes in shear stress in arterioles, capillaries, and venules of the frog mesentery using the modified Landis technique. The response of the vessels varied across the capillary bed: arteriolar capillaries demonstrated no response, true capillaries a moderate response, and venular capillaries a strong response (5-fold increase) to a step change in shear stress. More recently, Kim et al.51 observed that changes in Lp were positively correlated with the magnitude of acute changes in shear stress in autoperfused microvessels in rat mesenteric tissue. The effect was greater in capillaries compared with terminal arterioles. Very significantly, the shear dependence of Lp could be eliminated by superfusion with a NOS inhibitor, indicating that the measured effects were not an artifact of methodology. One possible explanation for these results is the differential expression of eNOS in the rat mesenteric microcirculation. Kashiwagi et al.52 observed abundant expression of eNOS in capillaries and venules in rat mesenteric tissue in vivo in contrast to little expression of eNOS in small arterioles. However, controversy remains as Adamson et al.53 measured Lp in rat mesentery venular microvessels and did not observe a correlation between shear stress and Lp in vessels perfused in the orthograde direction, but did find a correlation for retrograde perfusion.
4. Intercellular junction shear stress affects transport
The intercellular junction between endothelial cells provides the principal pathway for transendothelial flow driven by a pressure differential (eqn 1). Tarbell et al.54 estimated the fluid shear stresses on the walls of this intercellular junction channel (the break in the tight junction—Figure 1) by assuming either a simple parallel plate geometry with the spacing of the adherens junction (about 20 nm) or a fibre matrix model with 0.6 nm fibre radius and 7 nm spacing. Using reasonable values for the fluid velocity in the slit, they estimated shear stresses of 40 dyn/cm2 (slit) and 50 dyn/cm2 (fibre matrix). These surprisingly high shear stress values, comparable to the shear stress of flowing blood on the endothelial surface, were hypothesized to stimulate the cells. To test this idea Tarbell et al.54 introduced a standard pressure differential of 10 cm H2O across BAEC monolayers and after establishing a baseline Lp, they introduced a step increase in the pressure differential to 20–30 cm H2O to introduce a step increase in the intercellular junction shear stress.
Typical results for this experiment are shown in Figure 4 below. The normalized Jv during the first 55 min shows data for a 10 cm H2O pressure differential. At time 60 min the pressure differential was stepped up to 20 cm H2O. The first 55 min shows the characteristic ‘sealing effect’ in which Jv declines to a stable baseline value. The sealing effect was originally described by Suttorp et al.55 in cultured endothelial cells. Note that the sealing effect does not appear to be substantially affected by pre-treatment with the endothelial NOS (eNOS) inhibitor NG-monomethyl-l-arginine (L-NMMA) that blocks the production of NO. The step to 20 cm H2O initiates another sealing period that is followed by a rise to a new baseline after 300 min. For the untreated monolayers, this new baseline is 250% higher than the original baseline after only a 100% increase in pressure differential. This indicates that the new baseline Lp value is 2.5 times higher than the baseline at 10 cm H2O. However, when the monolayer is pre-treated with L-NMMA, this increase is completely inhibited and the new Lp baseline is essentially the same as the baseline at 10 cm H2O. This data suggests that intercellular junction shear stress induces NO release leading to Lp increase much like shear stress on the luminal endothelial surface increases NO and Lp (Figure 3). Very similar phenomena were observed in the microcirculation of intact animals by Kim et al.51
Figure 4.

Jv (normalized) as function of time for experiments with step change from 10 to 20 cm H2O at 60 min without drug (open circle; n = 12) and with 100 µM NG-monomethyl-l-arginine (closed squares; n = 7). P < 0.05 for t > 210 min (adapted from Tarbell et al.54).
Recognizing that this mechanism of Lp increase in response to increased pressure may play a role in pulmonary edema driven by hypertension, Dull et al.56 performed similar pressurization—Lp experiments using bovine lung microvascular endothelial cells (BLMEC) and also observed an increase in Lp at higher transendothelial pressures that could be attenuated by 70% using an NOS inhibitor (L-NAME). The apparent mechanotransduction mechanism for the intercellular junction shear stress stimulation of endothelial cells uncovered by Dull et al.56 was quite surprising and will be discussed in the next section.
5. Role of the glycocalyx
As described above, several studies, both in vitro and in vivo, have shown that the luminal surface shear-induced increase in endothelial Lp is stimulated by shear-induced NO. It appears that shear-induced NO from endothelial cells is a mechanotransduction event that is mediated by the cell surface glycocalyx. A study in canine femoral arteries showed that the selective depletion of HA with the enzyme hyaluronidase blocked flow-induced NO production,57 whereas an in vitro study in BAECs58 reported that the depletion of HS with heparinase had the same effect (Figure 5 below). A more extensive recent investigation in BAECs confirmed that the depletion of HA and HS blocked shear-induced NO production, but a similar depletion of CS (with chondroitinase) had no effect.59
Figure 5.

Effect of steady shear stress on bovine aortic endothelial cell nitric oxide production after heparinase treatment. Nitric oxide production induced by 20 dyn/cm2 steady shear stress (squares) was significantly inhibited for monolayers that were pre-treated with heparinase III. Data are represented as mean ± SE (adapted from Florian et al.58).
To close the loop on the relationships among shear stress, the glycocalyx, NO production and endothelial Lp, Lopez et al.60 measured the response of BAEC Lp to a step increase in luminal shear stress from 0 to 13.3 dyn/cm2 after pre-treatment of the monolayers with the same GAG-digesting enzymes used by Pahakis et al.59 They observed that heparinase (Figure 6 below) and hyaluronidase, both of which blocked shear-induced NO production, completely blocked shear-induced increases in Lp. Chondroitinase (cleaving CS), which did not block shear-induced NO production, did partially inhibit the shear-Lp response. The heparinase and hyaluronidase results are entirely consistent with a glycocalyx-mediated mechanotransduction of NO production that increases Lp.
Figure 6.

Effect of heparinase treatment on bovine aortic endothelial cell Lp driven by a 10 cm H2O pressure differential imposed at time zero. A shear stress of 20 dyn/cm2 was imposed at 60 min. The shear stress response of Lp was completely inhibited when the cell monolayer was pre-treated with heparinase III (adapted from Lopez et al.60).
Surprisingly, the intercellular junction shear stress (driven by the pressure differential across the monolayer) that induced an increase in Lp in BLMEC was completely blocked by pre-treatment of the cell monolayers with heparinase.56 This suggests that the HS proteoglycans of the glycocalyx that fill the entrance of the normal inter-endothelial junction (not the leaky junction) are acting as the mechanotransducers for intercellular junction shear stress. This may explain why the responses of endothelial cell monolayers to increases in luminal shear stress and intercellular junction shear stress are very similar. It must be remembered in this context that changes in the normal force of fluid pressure on the glycocalyx that is distributed over the entire cell surface do not lead to acute mechnaotransduction,61 but rather it is the pressure driven shear force of fluid flow that is confined to the intercellular junction region. In a subsequent paper, Giantsos et al.62 demonstrated that certain cationic copolymers containing methacrylamidopropyl trimethylammonium chloride were able to substantially block the pressure-induced increase in BLMEC Lp and therefore may have potential as infusible therapeutics for pulmonary edema. The mechanism of this effect appears to be through inhibition of the mechanotransduction of intercellular junction shear stress by restriction of the motion of proteoglycans (GAGs) in the glycocalyx.
It should be noted that shear stress-induced Lp responses that have been observed using in vivo and ex vivo models39,49–51 are inherently based on endothelium that have been acclimated to shear stress, whereas the in vitro endothelial layers of many other studies showing a shear stress-induced Lp response23,60 were not exposed to shear stress before experiencing a step increase at the beginning of the experiment. That both the in vivo and in vitro systems exhibit an increase in Lp in response to an increase in shear stress suggests either that a change in shear stress, whether from zero (in vitro) or a homeostatic value (in vivo), initiates an endothelial remodeling process that is characterized by an increase in Lp as endothelial junctions remodel, or that endothelial mechanotransduction leading to an NO increase that drives an Lp increase proceeds independent of remodeling. It has been suggested elsewhere,16 but not proven, that glypican core proteins of the glycocalyx mediate the NO response to shear stress and the subsequent Lp response (time scale of minutes to hours), whereas syndecan core proteins mediate the remodeling response over longer time scales (order of hours to days). This would imply that the initial NO (Lp) response to shear stress is uncoupled from the remodeling response. Additional studies will be required to test this hypothesis directly. The mechanism of the long time transport response is also complicated by the fact that leaky junctions, which can accommodate water flux (Table 1), are affected by shear stress over time scales of hours to days (see below).
6. Shear stress effects on transport pathways
The shear stress effects on Lp and Pe that have been described are mediated by transport pathways that are affected by shear. This subject was reviewed by Tarbell1 in 2003 and is updated in this section.
6.1. Tight junctions
DeMaio et al.35 determined in BAECs that steady shear stress increased occludin phosphorylation after only 5 min of exposure, and continued to induce phosphorylation out to the 3 h of total exposure. The phosphorylation event occurs earliest and is associated with alterations in Lp before there is a change in occludin content, suggesting that the phosphorylation event is central to the transport response. Subsequent studies demonstrated that VEGF increases occludin phosphorylation in BRECs within 15 min63 and increases Lp and 70 kDa dextran permeability while maintaining constant reflection coefficient.64,65 These studies suggest that the tight junction disassembles by increasing either the number of breaks or the length of breaks in the tight junction as occludin is phophorylated, but the glycocalyx, which serves as the solute filter, remains unaffected. It is plausible that shear stress alters the tight junction in a similar manner.
More recently Colgan et al.66 studied the regulation of bovine brain microvascular endothelial cell tight junctions in vitro after longer shear stress exposures of 24 h. They observed the classical actin reorganization and cell alignment in the direction of shear along with a substantial increase in occludin mRNA and protein expression and in occludin-ZO-1 association. Both occludin and ZO-1 displayed strong localization to the cell border after 24 h. They also found that the diffusive permeability (Po) of 40 kDa dextran was reduced compared with a static control, whereas Po for the much smaller solute, sucrose was not affected by shear exposure. These observations are not inconsistent with the results of McIntire et al.29 and Warboys et al.30 discussed earlier. They suggest that the breaks in the tight junction are being reduced by the incorporation of new protein into the junction, but that the tight junction seal itself is not being substantially enhanced (recall Figure 1). Siddharthan et al.67 exposed human brain microvascular endothelial cells to low levels of shear stress (1–2 dyn/cm2) for 5 days and observed an upregulation of ZO-1 and a reduction in 70 kDa dextran permeability. Collins et al.68 subjected BAECs to cyclic strain at 1 Hz for 24 h and observed an increase in both occludin and ZO-1 and occludin-ZO-1 association. All of these studies suggest that exposure of endothelial cells to mechanical forces for an extended period of time (days) results in a strengthening of the tight junction.
6.2. Adherens junctions
HUVEC monolayers were exposed to steady shear stress and the junctional intensity of VE cadherin, plakoglobin and platelet endothelial cell adhesion molecule 1 (PECAM-1) were examined by Schnittler et al.69 This study showed that in normal Ca2+ media, maintenance of intercellular adhesion under shear stress requires cadherins associated with plakoglobin in the junction. When endothelial monolayers that display a cuboidal, cobblestone morphology in static culture are exposed to steady shear stress for 24–48 h, they elongate and align in the shear direction.70,71 Noria et al.72 investigated intercellular junction remodeling during this process and observed a disruption of junctional VE cadherin, alpha and beta catenin, and plakoglobin at 8.5 h with a recovery of functional continuity of all proteins at 48 h. Because intercellular adhesion at adherens junctions is required for the assembly of tight junctions,73 their break down and reassembly likely follow adherens junction remodeling. McIntire et al.29 showed maximal increases in endothelial transport after 10–30 h of exposure to shear stress and a return to baseline values by 48 h, consistent with a transient disruption and reassembly of intercellular junctions. But recall that DeMaio et al.35 showed occludin phosphorylation as early as 5 min after exposure to shear stress, suggesting that tight junction remodeling may be initiated before the adherens junction is altered.
Thi et al.74 determined the reorganization of junction proteins of rat fat pad microvascular endothelial cells after exposure to 10.5 dyn/cm2 for 5 h. They observed a disruption of the intense border staining for F-actin, vinculin, connexin 43 and ZO-1 at this time point as the cells showed signs of junctional remodeling. These experiments were carried out in media containing serum or BSA. The fascinating result in this paper was the observation that this early stage junction remodeling to shear stress was completely blocked when heparinase was added to the protein-containing media or when protein-free media was employed for the shear experiments. Heparinase degrades the HS component of the glycocalyx (Figure 2) and protein-free media leads to collapse of the glycocalyx.75 Yao et al.76 exposed BAECs to shear stress of 15 dyn/cm2 for 24 h and observed that the classic endothelial remodeling response of cell elongation and alignment in the direction of shear was completely blocked by treatment of the cells with heparinase. This reinforced the role of the glycocalyx in endothelial junction remodeling to shear stress. It has been suggested elsewhere16 that this remodeling response is likely mediated by syndecan components of the glycocalyx since they have transmembrane core proteins that are linked to the actin cytoskeleton (Figure 2), whereas the NO response and the acute increases in permeability are mediated by glypicans that are linked to the plasma membrane in caveolae where eNOS and other signaling molecules are concentrated (Figure 2).
6.3. Leaky junctions
Leaky junctions are associated with cell proliferation or turnover (mitosis) and cell death (apoptosis) as described earlier. Mitosis and apoptosis are affected by shear stress as many studies have demonstrated.38,77–85 The basic observations are that higher shear stress suppresses mitosis and apoptosis while low shear stress supports both processes. Taken together, these studies suggest that endothelial cell turnover rates and apoptosis rates, and by association the prevalence of leaky junctions, will be greater in low shear stress regions of arteries where atherosclerosis is localized. Cancel and Tarbell86 recently demonstrated a strong positive correlation between apoptosis rate and LDL permeability in BAEC monolayers in vitro. However, at least one study has indicated that extremely high shear stresses (50–100 dyn/cm2) may actually stimulate, rather than suppress, endothelial cell proliferation.87
It has been shown recently that the suppression of endothelial cell (BAEC) turnover by shear stress over a 24 h period is completely blocked by treatment of the cells with heparinase to disrupt the glycocalyx.76 This suggests that the presence of an intact glycocayx is critical for the control of the leaky junction and LDL permeability in the circulation.
Endothelial solid strain induced by the pressure pulse has also been shown to influence apoptosis rates.88 Li et al.89 showed that BAECs cultured on compliant substrates had twice the protection against oxidant stress-induced apoptosis as cells cultured on non-compliant surfaces. Haga et al.90 demonstrated that BAECs exposed to 10% cyclic strain suppressed apoptosis due to serum withdrawal compared with rigid substrate controls. This would suggest that compliant vessels will have fewer leaky junctions than rigid vessels and be less likely to accumulate LDL.
6.4. Vesicles
An early study by Davies et al.91 in BAECs found that continuous exposure to steady shear stresses stimulated time- and shear stress level-dependent increases in pinocytotic rate that returned to control levels after several hours of application. Elevated steady shear stress has also been shown to increase LDL endocytosis92 and increased intracellular uptake of albumin.93 None of these studies of vesicle rates measured the overall transport rates across the endothelium, and therefore, we cannot be sure that an increase in pinocytotic rate can be equated with an increase in transendothelial transport. The study by Davies et al.91 does suggest that changes in shear stress might elevate vesicular transport transiently.
Rizzo et al.94 reported that shear stress alters caveolin expression and distribution, increases cavolae density, and leads to enhanced mechanosensitivity to subsequent changes in shear stress in cultured endothelial cells. Their data suggest that cultured endothelial cells respond to a sustained flow environment by directing caveolae to the cell surface where they serve to mediate, at least in part, mechanotransduction responses. Because important glycocalyx components reside in caveolae (Figure 2), including glypican-1 that has been hypothesized to be the mechanosensor for eNOS phosphorylation and NO formation,60 this mechanism of caveolae recruitment to the luminal surface provides a vehicle for sustaining or even amplifying shear-induced NO formation95 that subsequently affects transport pathways.
6.5. Physiological relevance of shear-dependent permeability
This review has highlighted the many systems, both in vitro and in vivo, that have displayed a shear-dependent endothelial permeability response. As suggested by several investigators,41,42,45 the physiological significance of acute responses to changes in shear stress (flow) is most important in the microcirculation as they dictate how the cardiovascular system responds to changes in activity level or the demands of specific organs that occur on a minute-to-minute basis. This can be seen most clearly, for example, in the capillaries of skeletal muscle during exercise. The muscle demands increased blood flow to accommodate metabolite exchange, but unless the transport barrier allows increased transvascular flux of solutes the increased delivery by blood flow would not be fully utilized. Elevated water flux (Lp) may also be useful for flushing out interstitial space and preventing the accumulation of higher molecular weight products (e.g. growth factors, MMPs, etc.) that can be convected out of the interstitium. This would not necessarily upset osmotic balance across the vessel wall as long as albumin transport is simultaneously enhanced.
Chronic responses are most relevant to the variation in endothelial permeability characteristics that arise in regions of the circulation with diverse shear stress environments. For example the inner walls of bifurcations and the outer walls of curvatures in arteries are exposed to relatively high shear stress whereas the outer walls of bifurcations and the inner walls of curvatures are exposed to low shear stress. The low shear stress regions are more permeable to macromolecules (LDL) and more susceptible to atherosclerosis.1
7. Future research challenges
In this concluding section, the unresolved research problems that were identified at various points within the review are brought together in a brief summary of future research challenges.
The intracellular signaling pathways that link activation of second messengers such as NO-cGMP and cAMP with intercellular junction protein activation and mobilization are undoubtedly complex and remain to be determined.
Controversy continues over the shear-dependent response of microvascular Lp in vivo with several groups reporting a pronounced response50,51 and others46,53 not observing a response. There are differences in methods including single or double cannulation, autoperfusion, nature of the perfusate as well as differences in species and microvascular region that remain to be sorted out.
Cells in culture display diverse responses of transport properties to shear stress as well. For example, HUVECs are not responsive whereas BAECs are.96 The differences likely derive from divergent intracellular signaling pathways (1 above) and their understanding may be useful in unraveling the diversity of responses in the microcirculation in vivo (2 above).
Acute responses of the transport barrier to shear stress are mediated by mechanotransduction events and the autocrine production of biomolecules that affect transport pathways across the endothelium, most clearly the paracellular pathway through the breaks in the tight junction. Longer term responses over many hours likely involve contributions from endothelial junction remodeling, alterations in apoptosis and mitosis rates that affect leaky junctions and the continued production of autocrine factors driven by mechanotransduction events. These interactions remain to be deconstructed.
While in roads have been made in our understanding of shear effects on the intercellular junction and leaky junction pathways, very little is known about shear effects on vesicular transport and in particular the separation of intracellular vs. transcellular transport. Studies of these processes would be very valuable.
Funding
This review was supported by National Heart Lung and Blood Institute grant RO1 HL-57093.
Acknowledgements
Thanks to Limary Cancel for composing Figure 1 and for helpful comments. Thanks to Zhong-Dong Shi for help with the final manuscript preparation.
Conflict of interest: none declared.
References
- 1.Tarbell JM. Mass transport in arteries and the localization of atherosclerosis. Annu Rev Biomed Eng. 2003;5:79–118. doi: 10.1146/annurev.bioeng.5.040202.121529. doi:10.1146/annurev.bioeng.5.040202.121529. [DOI] [PubMed] [Google Scholar]
- 2.Simionescu M, Simionescu N. Handbook of Physiology The Cardiovascular System Microcirculation. IV, Chapter 3. Bethesda, MD: American Physiological Society; 1984. Ultrastructure of the microvessel wall: functional correlations; pp. 41–101. [Google Scholar]
- 3.Michel CC. Transport of macromolecules through microvascular walls. Cardiovasc Res. 1996;32:644–653. [PubMed] [Google Scholar]
- 4.Ogunrinade O, Kameya GT, Truskey GA. Effect of fluid shear stress on the permeability of the arterial endothelium. Ann Biomed Eng. 2002;30:430–446. doi: 10.1114/1.1467924. doi:10.1114/1.1467924. [DOI] [PubMed] [Google Scholar]
- 5.Fu BM, Shen S. Acute VEGF effect on solute permeability of mammalian microvessels in vivo. Microvasc Res. 2004;68:51–62. doi: 10.1016/j.mvr.2004.03.004. doi:10.1016/j.mvr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Chang YS, Munn LL, Hillsley MV, Dull RO, Yuan J, Lakshminarayanan S, et al. Effect of vascular endothelial growth factor on cultured endothelial cell monolayer transport properties. Microvasc Res. 2000;59:265–277. doi: 10.1006/mvre.1999.2225. doi:10.1006/mvre.1999.2225. [DOI] [PubMed] [Google Scholar]
- 7.DeMaio L, Tarbell JM, Scaduto RC, Jr, Gardner TW, Antonetti DA. A transmural pressure gradient induces mechanical and biological adaptive responses in endothelial cells. Am J Physiol Heart Circ Physiol. 2004;286:H731–H741. doi: 10.1152/ajpheart.00427.2003. doi:10.1152/ajpheart.00427.2003. [DOI] [PubMed] [Google Scholar]
- 8.Cereijido M, Anderson J. Tight Junctions. 2nd ed. Boca Raton: CRC; 2001. [Google Scholar]
- 9.Eelkema R, Cowin P. General themes in cell–cell junctions and cell adhesion. In: Cereijido M, Anderson J, editors. Tight Junctions. 2nd ed. Boca Raton: CRC; 2001. [Google Scholar]
- 10.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. 5th ed. New York: Garland Science; 2008. [Google Scholar]
- 11.Palade GE, Bruns RR. Structural modulations of plasmalemmal vesicles. J Cell Biol. 1968;37:633–649. doi: 10.1083/jcb.37.3.633. doi:10.1083/jcb.37.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinbaum S, Tzeghai G, Ganatos P, Pfeffer R, Chien S. Effect of cell turnover and leaky junctions on arterial macromolecular transport. Am J Physiol Heart Circ Physiol. 1985;248:H945–H960. doi: 10.1152/ajpheart.1985.248.6.H945. [DOI] [PubMed] [Google Scholar]
- 13.Gerrity RG, Richardson M, Somer JB, Bell FP, Schwartz CJ. Endothelial cell morphology in areas of in vivo Evans blue uptake in the aorta of young pigs. II. Ultrastructure of the intima in areas of differing permeability to proteins. Am J Pathol. 1977;89:313–334. [PMC free article] [PubMed] [Google Scholar]
- 14.Chen YL, Jan KM, Lin HS, Chien S. Ultrastructural studies on macromolecular permeability in relation to endothelial cell turnover. Atherosclerosis. 1995;118:89–104. doi: 10.1016/0021-9150(95)05596-o. doi:10.1016/0021-9150(95)05596-O. [DOI] [PubMed] [Google Scholar]
- 15.Fu B, Curry FR, Adamson RH, Weinbaum S. A model for interpreting the tracer labeling of interendothelial clefts. Ann Biomed Eng. 1997;25:375–397. doi: 10.1007/BF02648050. doi:10.1007/BF02648050. [DOI] [PubMed] [Google Scholar]
- 16.Tarbell JM, Pahakis MY. Mechanotransduction and the glycocalyx. J Intern Med. 2006;259:339–350. doi: 10.1111/j.1365-2796.2006.01620.x. doi:10.1111/j.1365-2796.2006.01620.x. [DOI] [PubMed] [Google Scholar]
- 17.Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121–167. doi: 10.1146/annurev.bioeng.9.060906.151959. doi:10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- 18.Cancel LM, Fitting A, Tarbell JM. In vitro study of LDL transport under pressurized (convective) conditions. Am J Physiol Heart Circ Physiol. 2007;293:H126–H132. doi: 10.1152/ajpheart.01188.2006. doi:10.1152/ajpheart.01188.2006. [DOI] [PubMed] [Google Scholar]
- 19.Wiklund O, Carew TE, Steinberg D. Role of the low density lipoprotein receptor in penetration of low density lipoprotein into rabbit aortic wall. Arteriosclerosis. 1985;5:135–141. doi: 10.1161/01.atv.5.2.135. [DOI] [PubMed] [Google Scholar]
- 20.Schwartz CJ, Valente AJ, Sprague EA, Kelley JL, Nerem RM. The pathogenesis of atherosclerosis: an overview. Clin Cardiol. 1991;14:I1–I16. doi: 10.1002/clc.4960141302. [DOI] [PubMed] [Google Scholar]
- 21.Michel CC, Curry FE. Microvascular permeability. Physiol Rev. 1999;79:703–761. doi: 10.1152/physrev.1999.79.3.703. [DOI] [PubMed] [Google Scholar]
- 22.Jo H, Dull RO, Hollis TM, Tarbell JM. Endothelial albumin permeability is shear dependent, time dependent, and reversible. Am J Physiol. 1991;260:H1992–H1996. doi: 10.1152/ajpheart.1991.260.6.H1992. [DOI] [PubMed] [Google Scholar]
- 23.Sill HW, Chang YS, Artman JR, Frangos JA, Hollis TM, Tarbell JM. Shear stress increases hydraulic conductivity of cultured endothelial monolayers. Am J Physiol Heart Circ Physiol. 1995;268:H535–H543. doi: 10.1152/ajpheart.1995.268.2.H535. [DOI] [PubMed] [Google Scholar]
- 24.Chang YS, Yaccino JA, Lakshminarayanan S, Frangos JA, Tarbell JM. Shear-induced increase in hydraulic conductivity in endothelial cells is mediated by a nitric oxide-dependent mechanism. Arterioscler Thromb Vasc Biol. 2000;20:35–42. [PubMed] [Google Scholar]
- 25.Chang YS. The Mechanism of Shear-induced Increases in Endothelial Transport Properties (thesis) University Park, PA: The Penn. State Univ; 1998. [Google Scholar]
- 26.Dull RO, Jo H, Sill H, Hollis TM, Tarbell JM. The effect of varying albumin concentration and hydrostatic pressure on hydraulic conductivity and albumin permeability of cultured endothelial monolayers. Microvasc Res. 1991;41:390–407. doi: 10.1016/0026-2862(91)90037-c. doi:10.1016/0026-2862(91)90037-C. [DOI] [PubMed] [Google Scholar]
- 27.DePaola N, Phelps JE, Florez L, Keese CR, Minnear FL, Giaever I, et al. Electrical impedance of cultured endothelium under fluid flow. Ann Biomed Eng. 2001;29:648–656. doi: 10.1114/1.1385811. doi:10.1114/1.1385811. [DOI] [PubMed] [Google Scholar]
- 28.Seebach J, Donnert G, Kronstein R, Werth S, Wojciak-Stothard B, Falzarano D, et al. Regulation of endothelial barrier function during flow-induced conversion to an arterial phenotype. Cardiovasc Res. 2007;75:596–607. doi: 10.1016/j.cardiores.2007.04.017. doi:10.1016/j.cardiores.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 29.McIntire L, Wagner J, Whitson P. Effect of flow on macromolecular transport across bovine brain endothelial cell monolayers. ASME/BED Bioeng Conf. 1995;29:79–80. [Google Scholar]
- 30.Warboys CM, Berson RE, Mann GE, Pearson JD, Weinberg PD. Acute and chronic exposure to shear stress have opposite effects on endothelial permeability to macromolecules. Am J Physiol Heart Circ Physiol. 2010;298:H1850–H1856. doi: 10.1152/ajpheart.00114.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hillsley MV, Tarbell JM. Oscillatory shear alters endothelial hydraulic conductivity and nitric oxide levels. Biochem Biophys Res Commun. 2002;293:1466–1471. doi: 10.1016/S0006-291X(02)00410-2. doi:10.1016/S0006-291X(02)00410-2. [DOI] [PubMed] [Google Scholar]
- 32.Kurose I, Kubes P, Wolf R, Anderson DC, Paulson J, Miyasaka M, et al. Inhibition of nitric oxide production. Mechanisms of vascular albumin leakage. Circ Res. 1993;73:164–171. doi: 10.1161/01.res.73.1.164. [DOI] [PubMed] [Google Scholar]
- 33.Baldwin AL, Thurston G, al Naemi H. Inhibition of nitric oxide synthesis increases venular permeability and alters endothelial actin cytoskeleton. Am J Physiol Heart Circ Physiol. 1998;274:H1776–H1784. doi: 10.1152/ajpheart.1998.274.5.H1776. [DOI] [PubMed] [Google Scholar]
- 34.He P, Zeng M, Curry FE. Dominant role of cAMP in regulation of microvessel permeability. Am J Physiol Heart Circ Physiol. 2000;278:H1124–H1133. doi: 10.1152/ajpheart.2000.278.4.H1124. [DOI] [PubMed] [Google Scholar]
- 35.DeMaio L, Chang YS, Gardner TW, Tarbell JM, Antonetti DA. Shear stress regulates occludin content and phosphorylation. Am J Physiol Heart Circ Physiol. 2001;281:H105–H113. doi: 10.1152/ajpheart.2001.281.1.H105. [DOI] [PubMed] [Google Scholar]
- 36.Lakshminarayanan S, Gardner TW, Tarbell JM. Effect of shear stress on the hydraulic conductivity of cultured bovine retinal microvascular endothelial cell monolayers. Curr Eye Res. 2000;21:944–951. doi: 10.1076/ceyr.21.6.944.6985. doi:10.1076/ceyr.21.6.944.6985. [DOI] [PubMed] [Google Scholar]
- 37.Lakshminarayanan S, Tarbell J. Shear stress and VEGF increase hydraulic conductivity in cultured retinal microvascular endothelial cells (abstract) FASEB J. 1999;13:A3. [Google Scholar]
- 38.Gooch KJ, Dangler CA, Frangos JA. Exogenous, basal, and flow-induced nitric oxide production and endothelial cell proliferation. J Cell Physiol. 1997;171:252–258. doi: 10.1002/(SICI)1097-4652(199706)171:3<252::AID-JCP3>3.0.CO;2-N. doi:10.1002/(SICI)1097-4652(199706)171:3<252::AID-JCP3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 39.Lever MJ, Tarbell JM, Caro CG. The effect of luminal flow in rabbit carotid artery on transmural fluid transport. Exp Physiol. 1992;77:553–563. doi: 10.1113/expphysiol.1992.sp003619. [DOI] [PubMed] [Google Scholar]
- 40.Dabagh M, Jalali P, Tarbell JM. The transport of LDL across the deformable arterial wall: the effect of endothelial cell turnover and intimal deformation under hypertension. Am J Physiol Heart Circ Physiol. 2009;297:H983–H996. doi: 10.1152/ajpheart.00324.2009. doi:10.1152/ajpheart.00324.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crone C, Levitt D. Handbook of Physiology. Vol IV. Bethesda, MD: American Physiological Society; 1984. Capillary permeability to small solutes. The cardiovascular system. Microcirculation; pp. 411–466. [Google Scholar]
- 42.Shibata M, Kamiya A. Blood flow dependence of local capillary permeability of Cr-EDTA in the rabbit skeletal muscle. Jpn J Physiol. 1992;42:631–639. doi: 10.2170/jjphysiol.42.631. doi:10.2170/jjphysiol.42.631. [DOI] [PubMed] [Google Scholar]
- 43.Caldwell JH, Martin GV, Raymond GM, Bassingthwaighte JB. Regional myocardial flow and capillary permeability-surface area products are nearly proportional. Am J Physiol Heart Circ Physiol. 1994;267:H654–H666. doi: 10.1152/ajpheart.1994.267.2.H654. [DOI] [PubMed] [Google Scholar]
- 44.Kajimura M, Michel CC. Flow modulates the transport of K+ through the walls of single perfused mesenteric venules in anaesthetised rats. J Physiol. 1999;521:665–677. doi: 10.1111/j.1469-7793.1999.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Montermini D, Winlove CP, Michel C. Effects of perfusion rate on permeability of frog and rat mesenteric microvessels to sodium fluorescein. J Physiol. 2002;543:959–975. doi: 10.1113/jphysiol.2002.023010. doi:10.1113/jphysiol.2002.023010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neal CR, Bates DO. Measurement of hydraulic conductivity of single perfused Rana mesenteric microvessels between periods of controlled shear stress. J Physiol. 2002;543:947–957. doi: 10.1113/jphysiol.2002.026369. doi:10.1113/jphysiol.2002.026369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan Y, Granger HJ, Zawieja DC, Chilian WM. Flow modulates coronary venular permeability by a nitric oxide-related mechanism. Am J Physiol Heart Circ Physiol. 1992;263:H641–H646. doi: 10.1152/ajpheart.1992.263.2.H641. [DOI] [PubMed] [Google Scholar]
- 48.Williams DA. A shear stress component to the modulation of capillary hydraulic conductivity (Lp) Microcirculation. 1996;3:229–232. doi: 10.3109/10739689609148293. doi:10.3109/10739689609148293. [DOI] [PubMed] [Google Scholar]
- 49.Williams DA. Network assessment of capillary hydraulic conductivity after abrupt changes in fluid shear stress. Microvasc Res. 1999;57:107–117. doi: 10.1006/mvre.1998.2128. doi:10.1006/mvre.1998.2128. [DOI] [PubMed] [Google Scholar]
- 50.Williams DA. Intact capillaries sensitive to rate, magnitude, and pattern of shear stress stimuli as assessed by hydraulic conductivity (Lp) Microvasc Res. 2003;66:147–158. doi: 10.1016/s0026-2862(03)00038-4. doi:10.1016/S0026-2862(03)00038-4. [DOI] [PubMed] [Google Scholar]
- 51.Kim MH, Harris NR, Tarbell JM. Regulation of capillary hydraulic conductivity in response to an acute change in shear. Am J Physiol Heart Circ Physiol. 2005;289:H2126–H2135. doi: 10.1152/ajpheart.01270.2004. doi:10.1152/ajpheart.01270.2004. [DOI] [PubMed] [Google Scholar]
- 52.Kashiwagi S, Kajimura M, Yoshimura Y, Suematsu M. Nonendothelial source of nitric oxide in arterioles but not in venules: alternative source revealed in vivo by diaminofluorescein microfluorography. Circ Res. 2002;91:e55–e64. doi: 10.1161/01.res.0000047529.26278.4d. doi:10.1161/01.RES.0000047529.26278.4D. [DOI] [PubMed] [Google Scholar]
- 53.Adamson R, Sarai R, Weinbaum S, Curry F. Retrograde shear sress modulates rat mesentery microvessel permeability and endothelial adhesion structures. FASEB J. 2009 Abstract 950. [Google Scholar]
- 54.Tarbell JM, Demaio L, Zaw MM. Effect of pressure on hydraulic conductivity of endothelial monolayers: role of endothelial cleft shear stress. J Appl Physiol. 1999;87:261–268. doi: 10.1152/jappl.1999.87.1.261. [DOI] [PubMed] [Google Scholar]
- 55.Suttorp N, Hessz T, Seeger W, Wilke A, Koob R, Lutz F, et al. Bacterial exotoxins and endothelial permeability for water and albumin in vitro. Am J Physiol. 1988;255:C368–C376. doi: 10.1152/ajpcell.1988.255.3.C368. [DOI] [PubMed] [Google Scholar]
- 56.Dull RO, Mecham I, McJames S. Heparan sulfates mediate pressure-induced increase in lung endothelial hydraulic conductivity via nitric oxide/reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1452–L1458. doi: 10.1152/ajplung.00376.2006. doi:10.1152/ajplung.00376.2006. [DOI] [PubMed] [Google Scholar]
- 57.Mochizuki S, Vink H, Hiramatsu O, Kajita T, Shigeto F, Spaan JA, et al. Role of hyaluronic acid glycosaminoglycans in shear-induced endothelium-derived nitric oxide release. Am J Physiol Heart Circ Physiol. 2003;285:H722–H726. doi: 10.1152/ajpheart.00691.2002. [DOI] [PubMed] [Google Scholar]
- 58.Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res. 2003;93:e136–e142. doi: 10.1161/01.RES.0000101744.47866.D5. doi:10.1161/01.RES.0000101744.47866.D5. [DOI] [PubMed] [Google Scholar]
- 59.Pahakis MY, Kosky JR, Dull RO, Tarbell JM. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem Biophys Res Commun. 2007;355:228–233. doi: 10.1016/j.bbrc.2007.01.137. doi:10.1016/j.bbrc.2007.01.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lopez-Quintero SV, Amaya R, Pahakis M, Tarbell JM. The endothelial glycocalyx mediates shear-induced changes in hydraulic conductivity. Am J Physiol Heart Circ Physiol. 2009;296:H1451–H1456. doi: 10.1152/ajpheart.00894.2008. doi:10.1152/ajpheart.00894.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shin HY, Gerritsen ME, Bizios R. Regulation of endothelial cell proliferation and apoptosis by cyclic pressure. Ann Biomed Eng. 2002;30:297–304. doi: 10.1114/1.1458595. doi:10.1114/1.1458595. [DOI] [PubMed] [Google Scholar]
- 62.Giantsos KM, Kopeckova P, Dull RO. The use of an endothelium-targeted cationic copolymer to enhance the barrier function of lung capillary endothelial monolayers. Biomaterials. 2009;30:5885–5891. doi: 10.1016/j.biomaterials.2009.06.048. doi:10.1016/j.biomaterials.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 63.Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem. 1999;274:23463–23467. doi: 10.1074/jbc.274.33.23463. doi:10.1074/jbc.274.33.23463. [DOI] [PubMed] [Google Scholar]
- 64.Demaio L, Antonetti DA, Tarbell JM. Pathways mediating VEGF-induced increases in transendothelial transport (abstract) Ann Biomed Eng. 2000;28:5.77. [Google Scholar]
- 65.Lakshminarayanan S, Antonetti DA, Gardner TW, Tarbell JM. Effect of VEGF on retinal microvascular endothelial hydraulic conductivity: the role of NO. Invest Ophthalmol Vis Sci. 2000;41:4256–4261. [PubMed] [Google Scholar]
- 66.Colgan OC, Ferguson G, Collins NT, Murphy RP, Meade G, Cahill PA, et al. Regulation of bovine brain microvascular endothelial tight junction assembly and barrier function by laminar shear stress. Am J Physiol Heart Circ Physiol. 2007;292:H3190–H3197. doi: 10.1152/ajpheart.01177.2006. doi:10.1152/ajpheart.01177.2006. [DOI] [PubMed] [Google Scholar]
- 67.Siddharthan V, Kim YV, Liu S, Kim KS. Human astrocytes/astrocyte-conditioned medium and shear stress enhance the barrier properties of human brain microvascular endothelial cells. Brain Res. 2007;1147:39–50. doi: 10.1016/j.brainres.2007.02.029. doi:10.1016/j.brainres.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Collins NT, Cummins PM, Colgan OC, Ferguson G, Birney YA, Murphy RP, et al. Cyclic strain-mediated regulation of vascular endothelial occludin and ZO-1: influence on intercellular tight junction assembly and function. Arterioscler Thromb Vasc Biol. 2006;26:62–68. doi: 10.1161/01.ATV.0000194097.92824.b3. doi:10.1161/01.ATV.0000194097.92824.b3. [DOI] [PubMed] [Google Scholar]
- 69.Schnittler HJ, Puschel B, Drenckhahn D. Role of cadherins and plakoglobin in interendothelial adhesion under resting conditions and shear stress. Am J Physiol Heart Circ Physiol. 1997;273:H2396–H2405. doi: 10.1152/ajpheart.1997.273.5.H2396. [DOI] [PubMed] [Google Scholar]
- 70.Langille BL, Adamson SL. Relationship between blood flow direction and endothelial cell orientation at arterial branch sites in rabbits and mice. Circ Res. 1981;48:481–488. doi: 10.1161/01.res.48.4.481. [DOI] [PubMed] [Google Scholar]
- 71.Dewey CF, Jr, Bussolari SR, Gimbrone MA, Jr, Davies PF. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomech Eng. 1981;103:177–185. doi: 10.1115/1.3138276. doi:10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- 72.Noria S, Cowan DB, Gotlieb AI, Langille BL. Transient and steady-state effects of shear stress on endothelial cell adherens junctions. Circ Res. 1999;85:504–514. doi: 10.1161/01.res.85.6.504. [DOI] [PubMed] [Google Scholar]
- 73.Dejana E, Corada M, Lampugnani MG. Endothelial cell-to-cell junctions. Faseb J. 1995;9:910–918. [PubMed] [Google Scholar]
- 74.Thi MM, Tarbell JM, Weinbaum S, Spray DC. The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a ‘bumper-car’ model. Proc Natl Acad Sci USA. 2004;101:16483–16488. doi: 10.1073/pnas.0407474101. doi:10.1073/pnas.0407474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Adamson RH, Clough G. Plasma proteins modify the endothelial cell glycocalyx of frog mesenteric microvessels. J Physiol. 1992;445:473–486. doi: 10.1113/jphysiol.1992.sp018934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yao Y, Rabodzey A, Dewey CF., Jr Glycocalyx modulates the motility and proliferative response of vascular endothelium to fluid shear stress. Am J Physiol Heart Circ Physiol. 2007;293:H1023–H1030. doi: 10.1152/ajpheart.00162.2007. doi:10.1152/ajpheart.00162.2007. [DOI] [PubMed] [Google Scholar]
- 77.Chien S. Role of shear stress direction in endothelial mechanotransduction. Mol Cell Biomech. 2008;5:1–8. [PubMed] [Google Scholar]
- 78.Cho A, Mitchell L, Koopmans D, Langille BL. Effects of changes in blood flow rate on cell death and cell proliferation in carotid arteries of immature rabbits. Circ Res. 1997;81:328–337. doi: 10.1161/01.res.81.3.328. [DOI] [PubMed] [Google Scholar]
- 79.Dimmeler S, Haendeler J, Rippmann V, Nehls M, Zeiher AM. Shear stress inhibits apoptosis of human endothelial cells. FEBS Lett. 1996;399:71–74. doi: 10.1016/s0014-5793(96)01289-6. doi:10.1016/S0014-5793(96)01289-6. [DOI] [PubMed] [Google Scholar]
- 80.Freyberg MA, Kaiser D, Graf R, Buttenbender J, Friedl P. Proatherogenic flow conditions initiate endothelial apoptosis via thrombospondin-1 and the integrin-associated protein. Biochem Biophys Res Commun. 2001;286:141–149. doi: 10.1006/bbrc.2001.5314. doi:10.1006/bbrc.2001.5314. [DOI] [PubMed] [Google Scholar]
- 81.Haidekker MA, White CR, Frangos JA. Analysis of temporal shear stress gradients during the onset phase of flow over a backward-facing step. J Biomech Eng. 2001;123:455–463. doi: 10.1115/1.1389460. doi:10.1115/1.1389460. [DOI] [PubMed] [Google Scholar]
- 82.Hoffmann J, Haendeler J, Aicher A, Rossig L, Vasa M, Zeiher AM, et al. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: important role of nitric oxide. Circ Res. 2001;89:709–715. doi: 10.1161/hh2001.097796. doi:10.1161/hh2001.097796. [DOI] [PubMed] [Google Scholar]
- 83.Kadohama T, Nishimura K, Hoshino Y, Sasajima T, Sumpio BE. Effects of different types of fluid shear stress on endothelial cell proliferation and survival. J Cell Physiol. 2007;212:244–251. doi: 10.1002/jcp.21024. doi:10.1002/jcp.21024. [DOI] [PubMed] [Google Scholar]
- 84.Levesque MJ, Nerem RM, Sprague EA. Vascular endothelial cell proliferation in culture and the influence of flow. Biomaterials. 1990;11:702–707. doi: 10.1016/0142-9612(90)90031-k. doi:10.1016/0142-9612(90)90031-K. [DOI] [PubMed] [Google Scholar]
- 85.Tricot O, Mallat Z, Heymes C, Belmin J, Leseche G, Tedgui A. Relation between endothelial cell apoptosis and blood flow direction in human atherosclerotic plaques. Circulation. 2000;101:2450–2453. doi: 10.1161/01.cir.101.21.2450. [DOI] [PubMed] [Google Scholar]
- 86.Cancel LM, Tarbell JM. The role of apoptosis in LDL transport through cultured endothelial cell monolayers. Atherosclerosis. 2010;208:335–341. doi: 10.1016/j.atherosclerosis.2009.07.051. doi:10.1016/j.atherosclerosis.2009.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Metaxa E, Meng H, Kaluvala SR, Szymanski MP, Paluch RA, Kolega J. Nitric oxide-dependent stimulation of endothelial cell proliferation by sustained high flow. Am J Physiol Heart Circ Physiol. 2008;295:H736–H742. doi: 10.1152/ajpheart.01156.2007. doi:10.1152/ajpheart.01156.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chien S. Molecular basis of rheological modulation of endothelial functions: importance of stress direction. Biorheology. 2006;43:95–116. [PubMed] [Google Scholar]
- 89.Li M, Chiou KR, Bugayenko A, Irani K, Kass DA. Reduced wall compliance suppresses Akt-dependent apoptosis protection stimulated by pulse perfusion. Circ Res. 2005;97:587–595. doi: 10.1161/01.RES.0000181432.73920.b1. doi:10.1161/01.RES.0000181432.73920.b1. [DOI] [PubMed] [Google Scholar]
- 90.Haga M, Chen A, Gortler D, Dardik A, Sumpio BE. Shear stress and cyclic strain may suppress apoptosis in endothelial cells by different pathways. Endothelium. 2003;10:149–157. doi: 10.1080/10623320390233463. doi:10.1080/713715223. [DOI] [PubMed] [Google Scholar]
- 91.Davies PF, Dewey CF, Jr, Bussolari SR, Gordon EJ, Gimbrone MA., Jr Influence of hemodynamic forces on vascular endothelial function. In vitro studies of shear stress and pinocytosis in bovine aortic cells. J Clin Invest. 1984;73:1121–1129. doi: 10.1172/JCI111298. doi:10.1172/JCI111298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sprague EA, Steinbach BL, Nerem RM, Schwartz CJ. Influence of a laminar steady-state fluid-imposed wall shear stress on the binding, internalization, and degradation of low-density lipoproteins by cultured arterial endothelium. Circulation. 1987;76:648–656. doi: 10.1161/01.cir.76.3.648. [DOI] [PubMed] [Google Scholar]
- 93.Kudo S, Ikezawa K, Ikeda M, Oka K, Tanishita K. Albumin concentration profile inside cultured endothelial cells exposed to shear stress. ASME/BED Proc Bioeng Conf. 1997;35:547. [Google Scholar]
- 94.Rizzo V, Morton C, DePaola N, Schnitzer JE, Davies PF. Recruitment of endothelial caveolae into mechanotransduction pathways by flow conditioning in vitro. Am J Physiol Heart Circ Physiol. 2003;285:H1720–H1729. doi: 10.1152/ajpheart.00344.2002. [DOI] [PubMed] [Google Scholar]
- 95.Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Physiol Cell Physiol. 2003;285:C499–C508. doi: 10.1152/ajpcell.00122.2003. [DOI] [PubMed] [Google Scholar]
- 96.Pang Z, Antonetti DA, Tarbell JM. Shear stress regulates HUVEC hydraulic conductivity by occludin phosphorylation. Ann Biomed Eng. 2005;33:1536–1545. doi: 10.1007/s10439-005-7786-0. doi:10.1007/s10439-005-7786-0. [DOI] [PubMed] [Google Scholar]