

Abstract
Calcium-activated chloride channels (CaCCs) play important roles in several physiological processes. In vascular smooth muscle, activation of these ion channels by agonist-induced Ca2+ release results in membrane depolarization and vasoconstriction. The molecular identity of vascular CaCCs is not fully defined. Here we present evidence that TMEM16A (or anoctamin 1), a member of the transmembrane 16 (TMEM16) protein family, forms CaCCs in pulmonary artery smooth muscle cells (PASMCs). Patch-clamp analysis in freshly isolated PASMCs revealed strongly outward-rectifying, slowly activating Ca2+-activated Cl− currents sharing a high degree of similarity with heterologous TMEM16A currents. TMEM16A mRNA was identified in rat and human pulmonary arteries and various other vascular smooth muscle cell types. Further analyses revealed that several TMEM16A splice variants were detected in rat PASMCs and that TMEM16F and TMEM16K were also expressed in these cells, while TMEM16B, TMEM16D and TMEM16E were all at least 50 times less abundantly expressed and the remaining TMEM16 family members were absent. Downregulation of TMEM16A gene expression in primary cultures of rat PASMCs, with small interfering RNAs, was accompanied by almost total loss of whole-cell CaCC currents. Based on these results, we propose that TMEM16A is the major constituent of the vascular calcium-activated chloride channel in rat pulmonary artery smooth muscle.
Introduction
Calcium-activated chloride channels (CaCCs) play important roles in several cellular functions. They are of key importance in vascular smooth muscle (VSM), where they are activated by a rise in the intracellular Ca2+ concentration following agonist-induced Ca2+ release from intracellular stores, leading to membrane depolarization and muscle contraction. In addition, CaCCs are activated by Ca2+ released from ryanodine receptors located in the sarcoplasmic reticulum and are responsible for spontaneous transient inward Cl− currents (STICs) observed in several VSM cell types (Large & Wang, 1996; Leblanc et al. 2005).
Two types of CaCC currents (ICaCC) have been identified in VSM, a ‘classic’ Ca2+-dependent current (Byrne & Large, 1987) and a cGMP-dependent ICaCC (Matchkov et al. 2004; Piper & Large, 2004a,b;). Classic ICaCC in VSM exhibits distinctive outward rectification, a small (∼1–3 pS) single-channel conductance, high thiocyanate permeability and cGMP is not mandatory for channel activation (Large & Wang, 1996).
Although CaCCs have been studied for almost three decades, their molecular identity has been elusive (Nilius & Droogmans, 2003; Hartzell et al. 2009). Elucidating the molecular identity of the classic CaCC is an important goal given its ubiquitous presence in vascular (and non-vascular) smooth muscle and its essential role in regulating smooth muscle tone (Large & Wang, 1996; Leblanc et al. 2005). Several molecular candidates have been proposed for vascular CaCCs, including members of the CLCA (Ca2+-activated chloride channel) and bestrophin gene families. Based on RT-PCR analysis, CLCA1 was found to be expressed in mouse portal vein smooth muscle (Britton et al. 2002), and CLCA4 transcripts were detected in many VSMs, including aorta and coronary vessels (Elble et al. 2002). However, several of the biophysical properties of native ICaCC, including single-channel conductance, degree of outward rectification and Ca2+ sensitivity, differ from those of heterologously expressed CLCA channels (Britton et al. 2002). Bestrophin_3 (Best-3) mRNA and protein were found in several VSMs, but appeared to be regulated by both Ca2+ and cGMP (Matchkov et al. 2005, 2008). Gene silencing experiments with small interfering RNA (siRNA) indicated that Best-3 represents the cGMP-activated CaCC, but its involvement in the classic ICaCC is unlikely (Matchkov et al. 2008).
Novel candidates for CaCC have recently been proposed: the TMEM16/anoctamin family (Caputo et al. 2008; Schroeder et al. 2008; Yang et al. 2008). TMEM16A/anoctamin 1 was the first member of this family shown to function as a CaCC. Heterologous expression of TMEM16A, or of the closely related TMEM16B/anoctamin 2 protein, resulted in Cl− currents sensitive to intracellular Ca2+ and with the degree of outward rectification, ion selectivity and pharmacological profile typical of native ICaCC observed in many tissues (Caputo et al. 2008; Schroeder et al. 2008; Yang et al. 2008; Galietta, 2009; Hartzell et al. 2009). It is currently unknown whether other members of the TMEM16 family form CaCCs. Several TMEM16A splice variants have been described, which result in channels with different biophysical properties (Caputo et al. 2008; Ferrera et al. 2009). The alternatively spliced exons code for segments of 116 (segment a) and 22 residues (segment b) at the N-terminus and segments of four (segment c) and 26 residues (segment d) in the first putative intracellular loop.
Motivated by these discoveries, we set out to test the hypothesis that TMEM16A, or other members of the TMEM16 family, are responsible for the classic ICaCC in VSM. The notion that heterologous coexpression of TMEM16A with receptors ubiquitously found in the vascular system, such as endothelin receptor subtype A or angiotensin II receptor subtype 1, giving rise to endothelin- or angiotensin-induced Cl− currents, reinforced the hypothesis that TMEM16A may form CaCCs in VSM (Yang et al. 2008). We focused our studies on pulmonary artery smooth muscle cells (PASMCs) because previous findings suggested that this cell type exhibits a pure classic ICaCC, i.e. neither the cGMP-activated current nor the expression of Best-3 have been detected (Matchkov et al. 2005, 2008); thus PASMCs represent a simple system to assess the properties and molecular identity of ICaCC, in the absence of the closely related cGMP-dependent CaCC current.
Methods
A detailed Methods section is provided in the online Supplemental material.
Human samples
Intralobar human pulmonary arteries were carefully dissected from healthy areas of distal lung sections obtained from two patients undergoing lung resection for lung cancer. Both subjects gave written informed consent. The study was approved by the local research ethics committee (South Manchester Research Ethics Committee).
Cell isolation and culture
Male Sprague–Dawley rats (weight 225–300 g) were killed by cervical dislocation in accordance with the UK Home Office guidelines as outlined by Drummond (2009). The intrapulmonary artery was dissected out, cleaned of connective tissue and cut into rings, which were then used for enzymatic cell isolation. Cells were stored at +4°C and used on the same day or cultured for up to 15 days under standard conditions.
Composition of solutions
For whole-cell recordings, the extracellular solution contained (mm): 150 NaCl, 1 CaCl2, 1 MgCl2, 10 glucose, 10 mannitol and 10 Hepes; pH was adjusted to 7.4 with NaOH. The pipette solution contained (mm): 130 CsCl, 10 EGTA, 1 MgCl2, 10 Hepes, 1 MgATP and 1.0, 6.0, 8.0 or 9.5 mm CaCl2 to obtain ∼17, 225 or 600 nm or 1.5 μm free [Ca2+], respectively (Caputo et al. 2008); pH was adjusted to 7.3 with NaOH. In some experiments, Cl− was substituted by gluconate by replacing NaCl with equimolar sodium gluconate, and liquid junction potential was calculated (Neher, 1992) and corrected off-line.
Small interfering RNA
Small interfering RNAs (Sigma Aldrich, UK) directed against exon 15 or exon 18 of the rat TMEM16A gene were used in this study (Supplemental Table 1). As a negative control, a scrambled siRNA was used. Primary cultured PASMCs were transfected with 10 or 40 nm siRNA using N-TER (Sigma-Aldrich, Gillingham, Dorset, UK) according to the manufacturer's instructions. Cells were used 72 h later for quantitative PCR (qPCR) or patch-clamp studies.
Reverse transcriptase-PCR and RNA quantification
Reverse transcriptase-PCR was used to asses the expression of the following RNAs: (1) rat and human TMEM16A; (2) all of the rat TMEM16 family members; and (3) rat TMEM16A splice variants. Primers were designed to amplify intron-spanning sequences (Supplemental Tables 2 and 3). The RT-PCR products were sequenced (GATC biotech, Konstanz, Germany) to confirm the specificity of the reaction.
Real-time quantitative polymerase chain reaction (qPCR) was performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and carried out with an ABI Prism® 7500 Sequence Detection System (Applied Biosystems). Relative TMEM16x mRNA levels were calculated using the threshold cycle (Ct) value and the 2−ΔΔCt method (Livak & Schmittgen, 2001).
Electrophysiology
All current recordings were performed with the whole-cell configuration of the patch-clamp technique. Currents were filtered at 2 kHz and sampled at 10 kHz, unless stated otherwise.
Current versus voltage relationships were constructed by measuring the current at the end of 1 s voltage steps from −80 to +100 mV in 10 mV increments, elicited every 5 s. Holding potential was 0 mV. Instantaneous tail current versus voltage relationships were constructed by measuring the tail current amplitude at each potential (from −80 to +130 mV in 10 mV increments; pulse duration 0.7 s) after a 1 s depolarizing step to + 100 mV, elicited every 5 s from a holding potential of 0 mV. Tail currents at each potential were fitted with a single exponential function, and the instantaneous tail current amplitude was estimated from extrapolation of the fit to the beginning of the test pulse. The instantaneous tail currents (It) were normalized against the current at the end of the prepulse (Ipp) and plotted as a function of the test potential.
For non-stationary noise analysis (Heinemann & Conti, 1992) 20–100 identical pulses to a test potential of +100 mV (filtered at 6 kHz and sampled at 20 kHz) were applied, and the mean response, I, was calculated. The variance, σ2, was computed from the averaged squared difference of consecutive traces. Background variance at 0 mV was subtracted and the variance-mean plot was fitted by:
| (1) |
with the single channel current, i, and the number of channels, N, as free parameters.
The maximal channel open probability (Po,max) was determined as:
| (2) |
where Imax is the maximal current.
Data analysis
Data were analysed with self-written routines developed in the IGOR Pro (Wavemetrics, Lake Oswego, OR, USA) environment or using custom-written software (Dr M. Pusch, CNR, Genoa, Italy). Data are given as means ±s.e.m. Statistical significance was evaluated using a Student's two-tailed t test and P < 0.05 taken to indicate a significant difference.
Results
Calcium-activated Cl−-currents in freshly isolated rat PASMCs
Pulmonary artery smooth muscle cells exhibit prominent K+ currents, including Ca2+-activated K+ currents. To suppress these endogenous conductances, we replaced K+ with Cs+ in our intracellular solutions. In the first set of experiments, cells were dialysed with intracellular solutions containing 17 or 600 nm free Ca2+ ([Ca2+]i). Figure 1Aa shows that 600 nm[Ca2+]i activated an outward current that increased and stabilized about 200–260 s after rupturing the cell membrane (whole-cell configuration). The time course of this activation was assessed by measuring the current at the end of 2 s voltage ramps from −100 to +100 mV elicited at various time points (Fig. 1Ab). On average, the current increased by about twofold (from 16 ± 3 to 31 ± 4 pA pF−1, n= 6) after 260 s in the whole-cell configuration. In contrast, whole-cell currents measured in the presence of 17 nm[Ca2+]i were much smaller, the current averaging 8 ± 1 pA pF−1 (n= 6), and no significant time-dependent increase was observed on establishing the whole-cell configuration. These findings suggest that Ca2+ is essential for activation of an outwardly rectifying current.
Figure 1. Whole-cell Ca2+-activated Cl− currents in rat PASMCs.
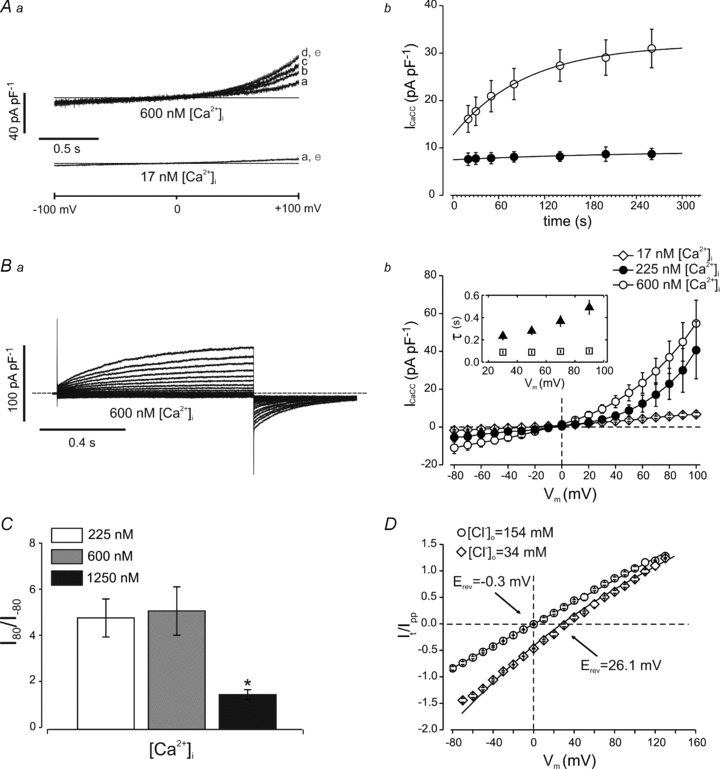
Aa, whole-cell membrane currents recorded from single PASMCs in response to 2 s voltage ramps from −100 to +100 mV. Traces marked from a to e indicate currents recorded at 20, 50, 80, 200 and 260 s from rupturing the membrane (whole-cell configuration), respectively. Calcium ions were added to the intracellular solution as indicated. The dashed line represents the zero-current level. Each recording is representative of five others. Ab, time course of whole-cell current activation at +100 mV in the presence of 17 nm (filled circles, n= 6) or 600 nm[Ca2+]i (open circles, n= 6). Current amplitude was obtained from ramp protocols as described in the main text. The continuous line through the open circles is the best fit to a single exponential function with a time constant of 96 s. The continuous line through the filled circles was drawn by eye. Ba, whole-cell currents recorded in response to 1 s voltage pulses (prepulse) from −80 to +100 mV in 10 mV increments followed by 700 ms pulses to −60 mV, in a single PASMC. The cell was dialysed with a solution containing 600 nm[Ca2+]i. The dashed line represents the zero-current level. Bb, mean current–voltage relationship measured at the end of the prepulse for cells dialysed with 17 (open diamonds, n= 9), 225 (filled circles, n= 5) or 600 nm[Ca2+]i (open circles, n= 11). Inset shows mean time constants of current activation (filled triangles) and deactivation (open squares) in the presence of 600 nm[Ca2+]i (n= 11). C, mean rectification index determined as the ratio of the current measured at +80 and at −80 mV in the presence of various [Ca2+]i, as indicated. The number of cells was 5–11 in each case. *P < 0.05. D, mean tail current amplitude plotted versus the membrane potential for 154 mm (circles) and 34 mm[Cl−]o (diamonds, n= 5). Continuous lines represent fit to the Goldman–Hodgkin–Katz equation. The reversal potential shifted from −0.3 to +26.1 mV with the reduction in Cl− concentration [Cl−]o.
In the presence of 17 nm[Ca2+]i, very small currents were observed in response to depolarizing voltage pulses, while in the presence of 600 nm[Ca2+]i distinct outward currents and deactivating inward tail currents were observed in response to depolarizing steps from −80 to +100 mV followed by repolarization to −60 mV (Fig. 1B); similar results were obtained in the presence of an intermediate [Ca2+]i of 225 nm. The outward currents observed at 225 or 600 nm[Ca2+]i had two components: a small, instantaneous time-independent component, indicating channels that were open at the holding potential (0 mV), followed by a time-dependent current activation. The time-dependent components at various potentials were well fitted with a single exponential function. The time constant of activation (τa) increased mildly (∼twofold) with the voltage, being equal to 234 ± 33 ms (n= 11) at +30 mV versus 491 ± 62 ms (n= 11) at +90 mV in 600 nm[Ca2+]i (P < 0.05; Fig. 1Bb, inset). The tail currents measured upon repolarization to −60 mV were also well fitted with a single exponential that did not change with voltage over the range of +30 to +90 mV (Fig. 1Bb, inset). The time constant (τd) of the deactivating tail current was 108 ± 34 s (n= 11) when preceded by a prepulse to +30 mV and 101 ± 11 s (n= 11) when preceded by a prepulse to +90 mV.
The current versus voltage relationship measured at the end of 1 s voltage steps from −80 to +100 mV in the presence of 600 nm[Ca2+]i (or 225 nm[Ca2+]i) exhibited a pronounced outward rectification (Fig. 1Bb), while in the presence of higher (1.25 μm) [Ca2+]i this relationship was almost linear (not shown). We quantified the degree of current rectification as the ratio between the current measured in response to 1 s voltage pulses to +80 and to −80 mV (rectification index, I+80/I−80). In the presence of 225 or 600 nm[Ca2+]i the I+80/I−80 index was 4.9 ± 0.8 (n= 5) and 5.2 ± 1.0 (n= 11), respectively, but when [Ca2+]i was raised to 1.25 μm this changed to 1.5 ± 0.2 (n= 5) (P < 0.05), indicating a loss of outward rectification (Fig. 1C).
The reversal potentials of these Ca2+-activated currents (∼0 mV) were near to the equilibrium potential for Cl− in our ionic conditions. To assess the value of the reversal potential more precisely, we determined the instantaneous current versus voltage relationship in the presence of 600 nm[Ca2+]i by measuring the amplitude of tail currents (It) at various potentials, as described in the Methods. The instantaneous tail currents in five separate experiments were averaged and plotted as a function of the test potential (Fig. 1D). The fit of the data to the Goldman–Hodgkin–Katz equation yielded a value of −0.3 ± 0.1 mV (n= 5) for the reversal potential. Lowering the extracellular Cl− concentration ([Cl−]o) (from 154 to 34 mm) by replacing Cl− with gluconate resulted in a shift of the reversal potential to 26 ± 2 mV (n= 5). The reversal potential expected for a perfectly selective Cl− channel is 33.7 mV. This small discrepancy may be attributed to a small permeability to gluconate of the CaCC channels in PASMCs. We conclude that these currents are ICaCC because they are carried by Cl−, activated by Ca2+, show time-dependent activation and deactivation at depolarizing and hyperpolarizing potentials, respectively, and exhibit a loss of inward rectification in the presence of high [Ca2+]i, as observed for ICaCC in other cell types (Kuruma & Hartzell, 2000).
Finally, we used non-stationary noise analysis to determine the single-channel conductance of the CaCC in PASMCs (Supplemental Fig. 1). The average i (measured at +100 mV) in six separate experiments was 0.35 ± 0.06 pA, and Po,max was 0.65 ± 0.06.
TMEM16A is expressed in VSM
The biophysical characteristics described above are broadly similar to those reported for heterologous TMEM16A currents (Caputo et al. 2008; Schroeder et al. 2008; Yang et al. 2008). We therefore set out to test whether TMEM16A is expressed in PASMCs and various other VSM cell types. Using RT-PCR with gene-specific primers we identified mRNA expression of TMEM16A in rat PASMCs, rat aorta and two VSM cell lines (A7R5 and A10 cells; Fig. 2A). We also assessed the presence of TMEM16A mRNA in human pulmonary arteries from two donors. Figure 2A illustrates that TMEM16A is expressed in human pulmonary arteries.
Figure 2. TMEM16A is expressed in vascular smooth muscle.
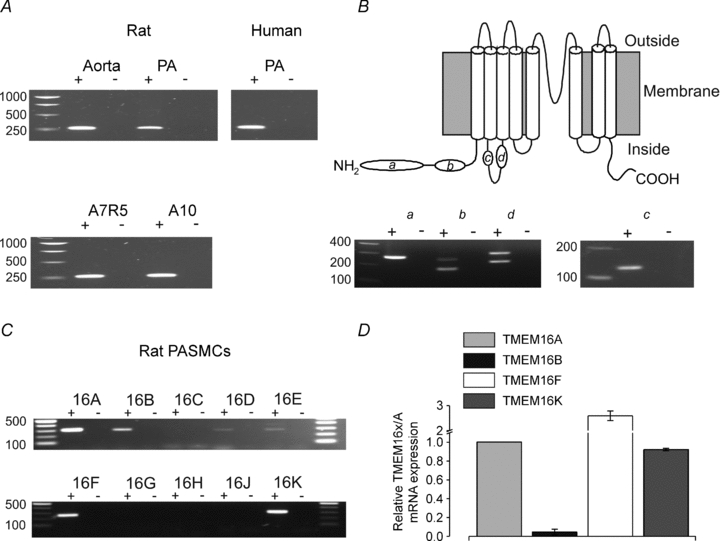
A, RT-PCR analysis of TMEM16A in rat and human pulmonary artery (PA), rat aorta, A7R5 and A10 cells using cDNA samples (+) or the corresponding negative controls (−) as starting material. The expected product size was 305 bp. For rat aorta and PA, similar results were obtained from samples collected from a total of three animals; for human PA, similar results were obtained from samples obtained from a total of two donors; for A7R5 and A10 cells, similar results were obtained from samples obtained from a total of three separate batches. B, topology diagram of the TMEM16A protein illustrating the position of the alternative spliced exons (a, b, c and d; upper panel) and RT-PCR analysis of TMEM16A splice variants in rat PASMCs (lower panel). The expected product sizes are reported in Supplemental Table 2. Similar results were obtained from samples collected from a total of three rats. C, RT-PCR analysis of TMEM16 family members in rat PASMCs using cDNA samples (+) or negative controls as starting materials (−). The expected product size for each family member is reported in Supplemental Table 3. Similar results were obtained from samples obtained from a total of three rats. D, mean TMEM16x mRNA expression, measured via qPCR, relative to the expression of TMEM16A in rat PASMCs (n= 3).
We used RT-PCR amplification to assess which TMEM16A splicing variants are expressed in rat PASMCs. The single band amplified by primers targeting the a and c segments indicated that these exons are constitutively expressed in rat PASMCs (n= 3). Primers targeting either b or d exons gave rise to two bands, indicating the presence of TMEM16A transcripts that either contain or lack these alternatively spliced exons in rat PASMCs (Fig. 2B). Thus, a combination of TMEM16A channel types are expected to be expressed in these cells.
We next examined whether other members of the TMEM16 family were expressed in rat PASMCs. Reverse transcriptase-PCR analysis demonstrated the presence of transcripts for TMEM16B, TMEM16F and TMEM16K, while TMEM16D and TMEM16E appeared to be expressed at very low levels and expression of the remaining members was not detected (n= 3; Fig. 2C). We quantified the relative amounts of mRNA for TMEM16A, TMEM16B, TMEM16F and TMEM16K via qPCR but did not attempt a precise quantification of TMEM16D and TMEM16E because qPCR of transcripts expressed at very low level is unreliable due to low signal to background noise ratio. TMEM16B was found to be expressed at very low levels, 49 ± 21 (n= 3) times less than TMEM16A, while TMEM16A, TMEM16F and TMEM16K were expressed at approximately the same levels (Fig. 2D).
TMEM16A protein produces ICaCC in primary cultured PASMCs
At present, TMEM16A and TMEM16B are the only TMEM family members that have been shown to form CaCCs; but, as described above, TMEM16B is expressed at very low levels in rat PASMCs. We therefore explored the possibility that TMEM16A mediates ICaCC in these cells by using a siRNA approach. Primary cultured PASMCs were transfected with non-related (scrambled) siRNA or with one of two different siRNAs (TMEM16A-siRNAs) directed against exon 15 or exon 18 of the TMEM16A gene. Transfection of primary cultured rat PASMCs with 10 or 40 nm of TMEM16A-siRNA (exon 15) significantly (P < 0.05) reduced TMEM16A expression in a dose-dependent manner to 57 ± 5 (n= 4) and 30 ± 1% (n= 7) of non-transfected control cells, respectively (Fig. 3A). Similarly, TMEM16A-siRNA against exon 18 (40 nm) reduced TMEM16A expression to 16 ± 9% (n= 3) of non-transfected control cells. In contrast, in cells that were exposed to 10 or 40 nM scrambled siRNA the level of TMEM16A mRNA was unchanged (n= 5, P > 0.05; Fig. 3A).
Figure 3. Small interfering RNA treatment induces knockdown of TMEM16A and suppresses CaCC currents in cultured rat PASMCs.
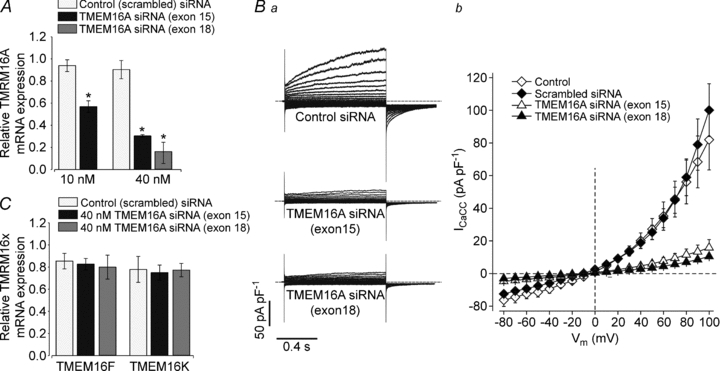
A, mean TMEM16A mRNA expression, measured via qPCR, in cells transfected with 10 or 40 nm scrambled siRNAs or siRNAs directed against exon 15 of TMEM16A or with 40 nm siRNAs directed against exon 18 of TMEM16A, relative to the expression in non-treated cells (n= 3–7). *P < 0.05. Ba, representative whole-cell ICaCC measured in single cultured PASMCs treated with scrambled siRNAs or TMEM16-siRNAs, as indicated. The [Ca2+]i was 600 nm. Each dashed line represents the zero-current level. Bb, mean whole-cell current versus voltage relationships from untreated cultured PASMCs (open diamonds, n= 6), and cells treated with 40 nm of scrambled siRNAs (filled diamonds, n= 5), TMEM16A-siRNAs (exon 15, open triangles, n= 10) or TMEM16A-siRNAs (exon 18, filled triangles, n= 5). C, mean TMEM16x mRNA expression, measured via qPCR, in cells transfected with scrambled siRNAs or with siRNAs directed against exon 15 or exon 18 of TMEM16A (40 nm), relative to the expression in non-treated cells (n= 3).
The downregulation of TMEM16A expression was mirrored by a reduction in ICaCC (Fig. 3Ba). In cells treated with 40 nm TMEM16A-siRNA against exon 15 or exon 18 and studied with 600 nm[Ca2+]i, ICaCC at +100 mV was 16 ± 5 (n= 10) and 10.5 ± 0.8 pA pF−1 (n= 5), respectively i.e. ∼84 and ∼90% less than the current measured in cells treated with the scrambled siRNA control (102 ± 22 pA pF−1, n= 5, P < 0.05; Fig. 3Bb). In contrast, treatment with scrambled siRNA had no significant effect on ICaCC amplitude. The current density at +100 mV averaged 82 ± 18 (n= 5) and 102 ± 22 pA pF−1 (n= 6) for non-treated cells and cells exposed to 40 nm scrambled siRNA, respectively (Fig. 3Bb).
Specificity of TMEM16A-directed siRNAs
A potential problem associated with gene silencing mediated by siRNAs is the risk of non-specific effects due to knockdown of transcripts other than the intended one, owing to partial sequence complementarity between a transcript and the siRNA molecules used (Jackson & Linsley, 2010). We tested for potential unwanted target effects by using both an in silico and an experimental approach. We first searched the available databases for any sequence similarity between the siRNAs and unwanted targets. A search conducted on the Genbank database using the BLAST algorithm revealed that TMEM16A-siRNAs against either exon 15 or exon 18 did not present identity with any other TMEM16 family members. Furthermore, TMEM16A-siRNA against exon 18 did not present sequence identity with the sequence of any other ion channel gene, while the siRNA against exon 15 had 84% identity with the voltage-gated sodium channel α-subunit 10a (Scn10a) gene and 68% with the transient receptor potential subfamily M, member 6 (Trpm6) gene. However, voltage-gated sodium currents are not present in pulmonary artery smooth muscle and Trpm6 is a Mg2+-permeable channel that cannot account for the Cl− current seen in this study.
We then tested experimentally whether exposure of rat PASMCs to 40 nm TMEM16A-siRNAs (exon 15 and exon 18) affected the expression of TMEM16F or TMEM16K, by using qPCR. We detected no change in the level of expression of these TMEM16 family members compared with cells treated with 40 nm scrambled siRNA. However, treatment with 40 nm scrambled siRNA caused an overall ∼20% reduction of the expression of these mRNAs (Fig. 3C, n= 3).
Discussion
The key finding of this paper is the observation that siRNA directed against TMEM16A resulted in a dramatic reduction in mRNA level and concomitant reduction in ICaCC in rat PASMCs, while control (scrambled) siRNA had no effect. Since TMEM16A was found to be expressed in both rat and human PASMCs, as well as in several VSM cell types, it can be argued that this channel may be responsible for ICaCC in the smooth muscle of several vascular beds and across species.
TMEM16A as the CaCC of rat PASMCs
Our whole-cell experiments revealed an ICaCC in rat PASMCs with electrophysiological features similar to those reported for ICaCC in coronary artery, portal vein and other VSM cells (Leblanc et al. 2005). The time constant of current activation measured at +70 mV was 400 ms, compared with a range of 200–300 ms in rabbit pulmonary artery, coronary artery and portal vein (Greenwood et al. 2001). The ICaCC deactivation time constant was 108 ms, very close to the values measured in rabbit pulmonary artery, coronary artery and portal vein (90–100 ms range; Greenwood et al. 2001). These analogies suggest that common, or similar, ion channel proteins mediate ICaCC in VSM of different vascular beds.
The overall characteristics of rat PASMC ICaCC are similar to those of heterologous TMEM16A currents (Caputo et al. 2008; Schroeder et al. 2008; Yang et al. 2008). Both currents were potently activated by 225 or 600 nm[Ca2+]i and exhibited comparable degrees of outward rectification (Caputo et al. 2008). Furthermore, the current kinetic properties are similar, the ICaCC deactivation time constant being in the 100 ms range in both cases (Caputo et al. 2008). In contrast, heterologous TMEM16B channels display much faster kinetics of current activation (4.4 ms at +100 mV) and deactivation (7.1 ms at −100 mV; Pifferi et al. 2009). Therefore, the kinetic characteristics of ICaCC in PASMCs closely resemble those of TMEM16A but not those of TMEM16B. Although rat PASMCs also express other TMEM16 family members, none of them has yet been shown to function as a CaCC.
The unitary conductance of CaCC in rat PASMC measured via noise analysis was ∼3 pS. A similar value was obtained from single-channel recordings in rabbit PASMCs and other VSM cell types (Large & Wang, 1996; Leblanc et al. 2005). This value is lower than the single-channel conductance of TMEM16A heterologously expressed in HEK cells (∼8 pS; Yang et al. 2008). It is tempting to speculate that this discrepancy may reflect association of TMEM16A with one or more of the other TMEM16 family members found to be expressed in rat PASMCs. It could also reflect association with accessory subunits or post-translational modifications in native VSM cells. A precise comparison between native and heterologously expressed TMEM16A currents is greatly complicated by the fact that several splicing variants are present in rat PASMCs. Native currents in rat PASMCs could therefore arise from several TMEM16A channel types, possibly expressed at different levels. A recent report showed that the various TMEM16A splicing variants give rise to ICaCC with substantially different biophysical properties, including the degree of outward rectification, kinetics of activation and Ca2+ sensitivity (Ferrera et al. 2009).
Rat PASMCs express multiple TMEM16 family members
We found that three members of the TMEM16 family are predominately expressed in rat PASMCs, namely TMEM16A, TMEM16F and TMEM16K. Furthermore, Gritli-Linde et al. (2009) reported expression of the same TMEM16 proteins along the walls of the dorsal aorta and of other blood vessels in mice. Thus, expression of TMEM16A, TMEM16F and TMEM16K may be ubiquitous in the vasculature. At present, TMEM16A is the only one of them that has been shown to function as a CaCC. The protein sequence of rat TMEM16F presents an overall identity of less than ∼20% with the sequence of rat TMEM16A. However, the degree of identity between TMEM16A and TMEM16F is much higher (∼55%) in the region comprising the fifth and sixth putative transmembrane domains and the loop in between them, which were suggested to form part of the pore of the channel (Yang et al. 2008). Furthermore, some of the residues of TMEM16A that are important for regulating channel selectivity (R701 and K725) are conserved in TMEM16F. These considerations may suggest that TMEM16F is capable of forming CaCCs. In contrast, the primary structure of rat TMEM16K presents much less similarity with TMEM16A (less than 20%). Furthermore, TMEM16K lacks a part (25 amino acids) of the putative pore-forming loop that includes R701, while lysine 725 is replaced with a serine. These considerations may indicate that TMEM16K has functions other than channel activity.
A recent study showed that tracheal epithelia from mice in which the TMEM16A gene was deleted (knockouts) had greatly reduced (of about 60%) CaCC activity (Rock et al. 2009). In this tissue, TMEM16J, TMEM16F and TMEM16K mRNAs were detected, and it was speculated that these TMEM family members may account for the small residual response to UTP observed in TMEM16A knockout mice. In rat PASMCs, however, we noted that treatment with 40 nm scrambled siRNA caused a ∼20% reduction of the expression of TMEM16F and TMEM16K but had no effect on ICaCC amplitude. This suggests that these two TMEM family members do not form CaCCs in rat PASMCs. Moreover, TMEM16F and TMEM16K are ubiquitously found (Galietta, 2009), even in tissues that do not display CaCC activity. Thus, it is possible to speculate that they play some cellular functions other than CaCC.
Physiological significance
In VSM, [Cl−]i ranges between 30 and 60 mm (Large & Wang, 1996; Chipperfield & Harper, 2000; Leblanc et al. 2005). Thus, the reversal potential for Cl− in VSM varies between −20 and −30 mV. As a consequence, activation of Cl− conductances leads to membrane depolarization, increased Ca2+ entry through L-type Ca2+ channels and ultimately enhanced contraction. It is unclear whether CaCCs contribute to the resting membrane potentials in VSM. The resting levels of [Ca2+]i in the cytoplasm vary between 40 and 140 nm, and only a small fraction of TMEM16A channels are expected to be active in those conditions. However, the observation that replacement of extracellular Cl− with SCN−, an anion which is highly permeable through CaCCs, resulted in a pronounced outward current in rabbit PASMCs is consistent with the idea that CaCCs contribute to the resting membrane potential in this vascular bed (Hogg et al. 1993). The notion that conditions associated with an increase in [Cl−]i, such as rat deoxycorticosterone acetate (DOCA)–salt hypertension, result in a more depolarized resting membrane potential also supports the idea that Cl− conductances may contribute to the resting membrane potential in VSM (Davis et al. 1993).
TMEM16A channels are expected to play a role in a variety of pathological conditions that are associated with an increase in [Ca2+]i, such as pulmonary hypertension or sepsis. Furthermore, upregulation of 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS)-sensitive chloride currents was associated with PASMC proliferation, which occurs in pulmonary hypertension (Liang et al. 2009). This is in agreement with the observation that TMEM16A is upregulated in cancer and may participate in cell proliferation (Hartzell et al. 2009).
The functional importance of TMEM16A in vivo will be best understood by studying cardiovascular changes in knockout mice. Such mice were generated prior to the discovery that TMEM16A functions as a CaCC (Rock et al. 2008). However, homozygous knockout mice die shortly after birth, probably due to malformation of tracheal cartilage rings (Rock et al. 2008). This short lifespan precludes the use of this model for studying the long-term effects of TMEM16A deletion on VSM function and blood pressure regulation. Thus, generation of tissue-specific knockouts for TMEM16A will be crucial for elucidating the role of TMEM16A channels in the vasculature.
In conclusion, our data suggest that TMEM16A appears to be at least an essential component of CaCCs in rat PASMCs.
Acknowledgments
We thank Drs S. D. Singh and J. Plumb (University of Manchester, Northwest Lung Centre, Wythenshawe Hospital, Manchester) for providing human samples. We are grateful to Professor Alison Gurney, Dr Peter Brown and Professor David Eisner for critical reading of the manuscript. P.T. is a Research Council UK fellow. This work was supported by BBSRC, Wellcome Trust and Royal Society grants to P.T.
Glossary
Abbreviations
- Best-3
bestrophin-3
- CaCC
calcium-activated chloride channel
- ICaCC
calcium-activated chloride current
- PASMC
pulmonary artery smooth muscle cell
- qPCR
real-time quantitative polymerase chain reaction
- siRNA
small interfering RNA
- STIC
spontaneous transient inward chloride currents
- VSM
vascular smooth muscle
Author contributions
P.T. designed the experiments; all authors performed the experiments and analysed the data; P.T. drafted the manuscript; and all authors critically reviewed and approved the final version. All experiments were carried out at the University of Manchester.
Supplemental material
Table 1
Table 2
Table 3
Suppl. Figure 1
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Britton FC, Ohya S, Horowitz B, Greenwood IA. Comparison of the properties of CLCA1 generated currents and ICl(Ca) in murine portal vein smooth muscle cells. J Physiol. 2002;539:107–117. doi: 10.1113/jphysiol.2001.013170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne NG, Large WA. Membrane mechanism associated with muscarinic receptor activation in single cells freshly dispersed from the rat anococcygeus muscle. Brit J Pharmacol. 1987;92:371–379. doi: 10.1111/j.1476-5381.1987.tb11333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- Chipperfield AR, Harper AA. Chloride in smooth muscle. Progr Biophys Mol Biol. 2000;74:175–221. doi: 10.1016/s0079-6107(00)00024-9. [DOI] [PubMed] [Google Scholar]
- Davis JP, Chipperfield AR, Harper AA. Accumulation of intracellular chloride by (Na-K-Cl) co-transport in rat arterial smooth muscle is enhanced in deoxycorticosterone acetate (DOCA)/salt hypertension. J Mol Cell Cardiol. 1993;25:233–237. doi: 10.1006/jmcc.1993.1029. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elble RC, Ji G, Nehrke K, DeBiasio J, Kingsley PD, Kotlikoff MI, Pauli BU. Molecular and functional characterization of a murine calcium-activated chloride channel expressed in smooth muscle. J Biol Chem. 2002;277:18586–18591. doi: 10.1074/jbc.M200829200. [DOI] [PubMed] [Google Scholar]
- Ferrera L, Caputo A, Ubby I, Bussani E, Zegarra-Moran O, Ravazzolo R, Pagani F, Galietta LJ. Regulation of TMEM16A chloride channel properties by alternative splicing. J Biol Chem. 2009;284:33360–33368. doi: 10.1074/jbc.M109.046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galietta LJ. The TMEM16 protein family: a new class of chloride channels? Biophys J. 2009;97:3047–3053. doi: 10.1016/j.bpj.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood IA, Ledoux J, Leblanc N. Differential regulation of Ca2+-activated Cl− currents in rabbit arterial and portal vein smooth muscle cells by Ca2+–calmodulin-dependent kinase. J Physiol. 2001;534:395–408. doi: 10.1111/j.1469-7793.2001.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritli-Linde A, Vaziri Sani F, Rock JR, Hallberg K, Iribarne D, Harfe BD, Linde A. Expression patterns of the Tmem16 gene family during cephalic development in the mouse. Gene Expr Patterns. 2009;9:178–191. doi: 10.1016/j.gep.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Hartzell HC, Yu K, Xiao Q, Chien LT, Qu Z. Anoctamin/TMEM16 family members are Ca2+-activated Cl− channels. J Physiol. 2009;587:2127–2139. doi: 10.1113/jphysiol.2008.163709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann SH, Conti F. Nonstationary noise analysis and application to patch clamp recordings. Methods Enzymol. 1992;207:131–148. doi: 10.1016/0076-6879(92)07009-d. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Wang Q, Helliwell RM, Large WA. Properties of spontaneous inward currents in rabbit pulmonary artery smooth muscle cells. Pflugers Arch. 1993;425:233–240. doi: 10.1007/BF00374172. [DOI] [PubMed] [Google Scholar]
- Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- Kuruma A, Hartzell HC. Bimodal control of a Ca2+-activated Cl− channel by different Ca2+ signals. J Gen Physiol. 2000;115:59–80. doi: 10.1085/jgp.115.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. Am J Physiol Cell Physiol. 1996;271:C435–C454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Leblanc N, Ledoux J, Saleh S, Sanguinetti A, Angermann J, O’Driscoll K, Britton F, Perrino BA, Greenwood IA. Regulation of calcium-activated chloride channels in smooth muscle cells: a complex picture is emerging. Can J Physiol Pharmacol. 2005;83:541–556. doi: 10.1139/y05-040. [DOI] [PubMed] [Google Scholar]
- Liang W, Ray JB, He JZ, Backx PH, Ward ME. Regulation of proliferation and membrane potential by chloride currents in rat pulmonary artery smooth muscle cells. Hypertension. 2009;54:286–293. doi: 10.1161/HYPERTENSIONAHA.109.130138. [DOI] [PubMed] [Google Scholar]
-
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the
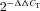 Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar] - Matchkov VV, Aalkjaer C, Nilsson H. A cyclic GMP-dependent calcium-activated chloride current in smooth-muscle cells from rat mesenteric resistance arteries. J Gen Physiol. 2004;123:121–134. doi: 10.1085/jgp.200308972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matchkov VV, Aalkjaer C, Nilsson H. Distribution of cGMP-dependent and cGMP-independent Ca2+-activated Cl− conductances in smooth muscle cells from different vascular beds and colon. Pflugers Arch. 2005;451:371–379. doi: 10.1007/s00424-005-1472-9. [DOI] [PubMed] [Google Scholar]
- Matchkov VV, Larsen P, Bouzinova EV, Rojek A, Boedtkjer DM, Golubinskaya V, Pedersen FS, Aalkjaer C, Nilsson H. Bestrophin-3 (vitelliform macular dystrophy 2-like 3 protein) is essential for the cGMP-dependent calcium-activated chloride conductance in vascular smooth muscle cells. Circ Res. 2008;103:864–872. doi: 10.1161/CIRCRESAHA.108.178517. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiologica Scandinavica. 2003;177:119–147. doi: 10.1046/j.1365-201X.2003.01060.x. [DOI] [PubMed] [Google Scholar]
- Pifferi S, Dibattista M, Menini A. TMEM16B induces chloride currents activated by calcium in mammalian cells. Pflugers Arch. 2009;458:1023–1038. doi: 10.1007/s00424-009-0684-9. [DOI] [PubMed] [Google Scholar]
- Piper AS, Large WA. Single cGMP-activated Ca2+-dependent Cl− channels in rat mesenteric artery smooth muscle cells. J Physiol. 2004a;555:397–408. doi: 10.1113/jphysiol.2003.057646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper AS, Large WA. Direct effect of Ca2+–calmodulin on cGMP-activated Ca2+-dependent Cl− channels in rat mesenteric artery myocytes. J Physiol. 2004b;559:449–457. doi: 10.1113/jphysiol.2004.070045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JR, Futtner CR, Harfe BD. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev Biol. 2008;321:141–149. doi: 10.1016/j.ydbio.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Rock JR, O’Neal WK, Gabriel SE, Randell SH, Harfe BD, Boucher RC, Grubb BR. Transmembrane protein 16A (TMEM16A) is a Ca2+-regulated Cl− secretory channel in mouse airways. J Biol Chem. 2009;284:14875–14880. doi: 10.1074/jbc.C109.000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.