

Abstract
This study was undertaken to investigate whether the mechanism of increased Na+–K+–2Cl− (NKCC1) cotransporter activity by osmotic shrinkage involved AMP-activated protein kinase (AMPK) activation. AMPK was found to phosphorylate a recombinant GST-dogfish (1–260) NKCC1 fragment at Ser38 and Ser214, corresponding to Ser77 and Ser242 in human NKCC1, respectively. Incubation of human erythrocytes with 20 μm A769662 AMPK activator increased Ser242 NKCC1 phosphorylation but did not stimulate 86Rb+ uptake. Under hypertonic conditions in human red blood cells (RBCs) incubated with 0.3 m sucrose, NKCC1 activity increased as measured by bumetanide-sensitive 86Rb+ uptake and AMPK was activated. However, there was no effect of AMPKα1 deletion in mouse RBCs on the increased rate of 86Rb+ uptake induced by hyperosmolarity. AMPK activation by osmotic shrinkage of mouse RBCs was abrogated by 10 μm STO-609 CaMKKβ inhibitor, but incubation with STO-609 did not affect the increase in 86Rb+ uptake induced by hyperosmolarity. Osmotic shrinkage of human and mouse RBCs led to activation loop phosphorylation of the STE20/SPS1-related proline/alanine-rich kinase (SPAK) at Thr233, which was accompanied by phosphorylation of NKCC1 at Thr203/207/212, one of which (Thr207) is responsible for cotransporter activation. Therefore, phosphorylation-induced activation of NKCC1 by osmotic shrinkage does not involve AMPK and is likely to be due to SPAK activation.
Introduction
The Na+–K+–2Cl− cotransporter 1 (NKCC1) is a ubiquitously expressed electroneutral Na+-dependent transporter involved in cell volume homeostasis and in the regulation of intracellular K+ and Cl− concentrations. NKCC1 is stimulated by cell shrinkage, metabolic/exercise stress, mechanical stress and ischaemia/hypoxia, and its physiology and pathophysiology in blood, brain and heart have been extensively reviewed (Pedersen et al. 2006). Stimulation of NKCC1 activity by osmotic shrinkage has been proposed to participate in the process of ‘regulatory volume increase’ (RVI) and can be inhibited by the loop diuretic drug, bumetanide. In red blood cells (RBCs), NKCC1 is activated by hyperosmolarity (Lytle, 1997) and by treatment with sodium arsenite (Flatman & Creanor, 1999a) or calyculin A (Lytle, 1997; Flatman & Creanor, 1999b). The STE20/SPS1-related proline/alanine-rich kinase (SPAK) and the homologous oxidative stress response kinase-1 (OSR1) have been shown to bind to NKCC1 and its related gene product, NKCC2 (Piechotta et al. 2002). SPAK activates NKCC1 by phosphorylation (Dowd & Forbush, 2003) at three conserved threonine residues, Thr203, Thr207 and Thr212, in the human sequence (Vitari et al. 2006). Phosphorylation of Thr189 in dogfish NKCC1, equivalent to Thr207 in human NKCC1, is essential for transport activation (Flemmer et al. 2002). Hypertonic activation of NKCC2, which is predominantly expressed in kidney and involved in renal salt re-absorption, requires phosphorylation of all three analogous residues (Thr99, Thr104 and Thr117, rabbit sequence) for a full response (Gimenéz & Forbush, 2005). The human Na+–Cl− cotransporter (NCC) is also activated by phosphorylation by SPAK/OSR1 at three conserved threonine residues (Richardson et al. 2008). Interestingly, the SPAK and OSR1 kinases are downstream of WNK1 (with-no-lysine (K) protein kinase-1), which harbours gain-of-function mutations in Gordon's hypertension syndrome (Vitari et al. 2005). Phosphorylation-induced activation of SPAK and OSR1 by WNK1 occurs in the activation (T-) loop at Thr233 and Thr185, respectively (Vitari et al. 2005). Therefore, activation of WNK isozymes under hyperosmotic conditions leads to the activation of SPAK and OSR1, which in turn phosphorylate and activate ion cotransporters (Richardson & Alessi, 2008). Recently, AMP-activated protein kinase (AMPK) was shown to phosphorylate NKCC2 on Ser126, whose mutation to alanine reduced cotransporter activity (Fraser et al. 2007). Since AMPK activity was increased in kidney in response to salt intake (Fraser et al. 2005), phosphorylation-induced NKCC2 activation was proposed to contribute to renal salt re-absorption (Fraser et al. 2007).
AMPK is a highly conserved eukaryotic serine/threonine protein kinase that acts not only as a sensor of cellular energy status, but also plays a critical role in systemic energy balance (Hardie et al. 1998; Kahn et al. 2005; Witters et al. 2006; Hardie 2007; Steinberg & Kemp, 2009). AMPK is a heterotrimer consisting of a catalytic α subunit and two regulatory subunits, β and γ. Multiple isoforms exist giving 12 possible combinations of holoenzyme with different tissue distribution and subcellular localization. AMPK can be activated by changes in the intracellular AMP:ATP ratio, as occurs for example as a result of anoxia or other stresses, or via an increase in intracellular Ca2+. LKB1 (the Peutz–Jeghers protein) and Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKKβ) are upstream kinases that activate AMPK by phosphorylating Thr172 in the activation loop of the catalytic α-subunits. A rise in AMP allosterically stimulates AMPK activity by binding to the γ-subunits and also prevents dephosphorylation of Thr172. Once activated, AMPK phosphorylates several metabolic targets resulting in a decrease in ATP consumption and stimulation of ATP production.
It is now becoming clear that AMPK function extends beyond metabolic control and energy homeostasis, for example to the control of cell division, cell proliferation, cell architecture, cell polarity and ion transport (Hue & Rider, 2007). Since AMPK is activated by hypertonic shock (Woods et al. 2003) and by arsenite treatment (Corton et al. 1994), both of which increase NKCC1 activity in RBCs (Lytle, 1997; Flatman & Creanor, 1999a), we investigated whether NKCC1 was an AMPK substrate and whether AMPK activation by erythrocyte cell shrinkage could contribute to increased ion transport.
Methods
Isolation of RBCs
Blood was drawn from healthy, consenting, human volunteers or from 8- to 14-week-old male AMPKα1-deficient mice, generated in a mixed C57BL/6 and 129Sv genetic background as described previously (Jorgensen et al. 2004), and their wild-type littermates. Experimental procedures were approved by the local animal welfare committee and were in accordance with the policies of the Journal of Physiology (Drummond, 2009). Blood was collected from animals anaesthetized with Nembutal (40–50 mg kg−1 administered intraperitoneally) by cardiac puncture in heparinized tubes and processed immediately. A total of about 40 mice were used and the animals were killed by interruption of circulation. Mouse or human red cells were sedimented immediately by centrifugation at 1000 g for 10 min at room temperature. After removal of the upper buffy coat, erythrocytes were pelleted and wash twice in phosphate-buffered saline (PBS) supplemented with 5 mm glucose. RBCs were resuspended at 50% haematocrit in Krebs–Ringer solution without bicarbonate, but buffered with 20 mm Hepes pH 7.4, and containing 11 mm glucose. The cells were stored at 4°C for use within hours of collection or stored overnight at 4°C. All incubations were carried out at 37°C and at 5% haematocrit as described in the figure legends.
Confocal microscopy
To observe erythrocyte morphology, cells were resuspended in Hepes-buffered Krebs solution with or without 0.3 m sucrose at 107 cells ml−1. Drops (20 μl) were smeared onto glass slides and air dried for 30 min for fixation in cold methanol for 2 min. Slides were rinsed 3 times for 5 min with PBS. Images were taken in a Zeiss Axiovert 135M microscope (Carl Zeiss MicroImaging, Oberkochen, Germany) with a water immersion ×63 magnification.
Incubation of RBCs and measurement of NKCC1 cotransporter activity
86Rb+ uptake was measured as described previously (Flatman & Creanor, 1999b). Briefly, well-agitated RBC suspensions at 5% haematocrit in a final volume of 1.5 ml were pre-incubated for 30 min prior to the addition of 5 μCi ml−1 of 86Rb+ with or without 100 μm bumetanide or as described in the figure legends. At various times, 0.1 ml samples of cell suspension were added to tubes containing 0.9 ml of ice-cold PBS and 0.4 ml of di-n-butylphtalate. After vortexing and centrifugation (22 000 g× 20 s), the supernatants and oil were aspirated off the red cell pellets. Sedimented cells were lysed in 1 ml of distilled water and proteins were precipitated with a final concentration of 2.5% (w/v) trichloroacetic acid on ice. Following centrifugation at 22 000 g× 10 min, 86Rb+ uptake was measured on aliquots of supernatant (0.9 ml) by liquid scintillation counting. Rates of 86Rb+ uptake were expressed per mg of haemoglobin, estimated from measurements of protein (by the Lowry method, see below) versus haematocrit.
AMPK assay
Lysates were prepared by adding 0.5 ml of 10 mm Hepes pH 7.4, 2 mm EDTA, 2 mm sodium pyrophosphate, 20 mm NaF, 15 mm 2-mercaptoethanol, 1% (w/v) Triton X-100 to pelleted cells from 2 ml of 5% haematocrit RBC suspensions. AMPKα1-containing heterotrimeric complexes were immunoprecipitated from 750 μg of erythrocyte lysates using anti-α1-AMPK antibody (1 μg) coupled to 40 μl of Protein G-Sepharose suspension (1:1) in lysis buffer. Immunoprecipitates were washed twice in lysis buffer and twice in assay buffer (50 mm Hepes, pH 7.2, 80 mm KCl, 1 mm EDTA, 1 mm DTT, 5 mm MgCl2 and 1% (v/v) glycerol). AMPK was assayed in a final volume of 50 μl of assay buffer with 0.2 mm SAMS peptide, 0.2 mm AMP, and 0.1 mm [γ-32P]MgATP (specific radioactivity: 1000 cpm pmol−1), at 30°C (Davies et al. 1989). After 10 min, 20 μl aliquots were spotted onto Whatman P81 papers for the measurement of 32P incorporation (Roskoski, 1983). One unit of protein kinase activity corresponds to the incorporation of 1 nmol of phosphate into the peptide substrate per minute under the assay conditions.
Immunoblotting
Lysates were prepared as described above for AMPK assay. Extracts (15 μl) were subjected to SDS-PAGE in 7% (w/v) acrylamide gels, to avoid interference from haemglobin subunits, and electroblotted onto nitrocellulose membranes (Trans-Blot Bio-Rad, Hercules, CA, USA). The membranes were probed with primary antibodies and blocked with 5% non-fat milk for detection of immunoreactive bands by enhanced chemiluminescence with protein A-horsedish peroxidase and the SuperSignal chemiluminescence system (Pierce, Rockford, IL, USA). Band intensities were quantified by scanning films and processing image intensities with the program Image J (133 for Mac OS X).
In vitro phosphorylation
Recombinant GST (Glutathione-S-Transferase)-dogfish NKCC1(1–260) or GST-human NCC (1–100) fusion proteins (15 μg) were incubated in phosphorylation buffer (10 mm Mops, pH 7, 10 mm magnesium acetate, 0.5 mm EDTA, 0.1% (v/v) 2-mercaptoethanol) with recombinant activated bacterially expressed α1β1γ1-AMPK (Horman et al. 2008; 3 mU ml−1), 0.2 mm AMP and 0.1 mm[γ-32P]MgATP (specific radioactivity, 300 cpm pmol−1) in a final volume of 50 μl at 30°C for up to 20 min. At the indicated times, aliquots (5 μl) were removed for SDS-PAGE, Coomassie Blue staining and gel drying for quantification of 32P incorporation by phosphorimaging. Protein bands in Coomassie-stained gels were quantified (Bultot et al. 2009) for measurements of stoichiometries of phosphorylation, taking molecular masses of 53 800 Da and 37 810 Da for the GST-NKCC1 and GST-NCC fusion proteins.
Phosphorylation site identification by mass spectrometry
The recombinant GST-dogfish NKCC1 fusion protein (10 μg) was phosphorylated in vitro by AMPK and [γ-32P]MgATP (specific radioactivity, 1000 cpm pmol−1) as described above for 1 h and precipitated with 10% (w/v) TCA for digestion with trypsin overnight at 30°C (Horman et al. 2006). Peptides were separated by reverse-phase narrow-bore HPLC in a linear acetonitrile gradient at a flow rate of 200 μl min−1 and radioactive peaks were analysed by nanoelectrospray ionization tandem mass spectrometry in a LCQ Deca XP Plus ion-trap mass spectrometer (Thermo Finnigan, San Jose, CA, USA).
Other methods
Protein concentration was estimated (Lowry et al. 1951) with bovine serum albumin as standard. Data are expressed as mean ±s.e.m. A Student's two-sided t test was used to assess the statistical significance (P < 0.05) of the data. All statistical analyses were performed using GraphPad Prism version 4.00 (GraphPad Software, San Diego, CA, USA).
Antibodies and other reagents
Anti-phospho-Thr172-AMPKα antibody was from Cell Signaling Technology (Beverly, MA, USA) and T4 mouse monoclonal anti-NKCC antibody was from American Research Products, Inc. (EMELCA Bioscience, Antwerp, Belgium). Anti-α1 AMPK, anti-CaMKKβ and pyruvate dehydrogenase (PDH) antibodies were donated by Prof. D. G. Hardie (Dundee University, Scotland). Recombinant GST-dogfish NKCC1 (1–260), recombinant GST-human NCC (1–100) and anti-phospho-Thr233, anti-phospho-Ser373-SPAK, anti-total SPAK, anti-phospho-Thr203/Thr207/Thr212-NKCC1, anti-WNK and anti-OSR1 antibodies were kindly provided by Prof. D. R. Alessi (University of Dundee, Scotland). Polyclonal antibodies were raised against phosphopeptides corresponding to the sequences surrounding Ser77 and Ser242 of human NKCC1, CPLGPTPpSQSRFQV and CGEKLLRPpSLGEFHD, respectively with an N-terminal cysteine for coupling to keyhole limpet haemocyanin and immunization in sheep (Sugden et al. 1999). Sera were first bound to columns containing the same immobilized peptides that had been used for immunization. The phospho-specific antibodies were eluted (Sugden et al. 1999) then passed through columns containing immobilized dephosphopeptides. Phospho-specific antibodies were tested for immunoreactivity by ELISA with the immunizing antigen and for specificity by immunoblotting. Anti-sheep, anti-mouse and anti-rabbit IgG conjugated to peroxidase were purchased from Sigma (St Louis, MO), Santa Cruz Biotechnology (Santa Cruz, CA, USA) and GE Healthcare (Little Chalfont, UK), respectively. A769662 was kindly given by Dr A. Balandran (AstraZeneca, Mölndal, Sweden). STO-609 was from Calbiochem (La Jolla, CA, USA) and bumetanide was from Biomol Research Laboratories Inc. (Plymouth, PA, USA). Radioactive 86Rb+ (37.0 GBq g−1) and [γ-32P]ATP (111 TBq mmol−1) were from Perkin-Elmer (Billerica, MA, USA). Recombinant bacterially expressed α1β1γ1-AMPK heterotrimers were kindly provided by Dr D. Neumann (ETH Zurich, Switzerland) and activated as described (Horman et al. 2008).
Results
In vitro phosphorylation of NKCC1 by AMPK and phosphorylation site identification
A recombinant N-terminal fragment (residues 1–260 corresponding to the intracellular cytosolic domain) of dogfish NKCC1 fused to GST was phosphorylated in vitro from [γ-32P]MgATP and recombinant activated α1β1γ1-AMPK heterotrimers (Fig. 1A). Phosphorylation was time dependent reaching a maximal stoichiometry of about 0.8 mole of phosphate incorporated/mole of GST-NKCC1 (1–260). However, with a second batch of GST-NKCC1 (1–260), maximal phosphorylation by AMPK reached a stoichiometry of 1.79 ± 0.11 mol mol−1 (n= 4, results not shown), suggesting that two sites could be phosphorylated. The differences in stoichiometry of phosphorylation by AMPK might have been due to differences in the proportion of recombinant GST-NKCC1 protein present in the proper conformation for phosphorylation by AMPK in the two preparations. A recombinant N-terminal fragment (residues 1–100) of the related human NCC transporter was a poor AMPK substrate, whose maximal stoichiometry of phosphorylation was only 0.07 mol mol−1 (Fig. 1A). After maximal phosphorylation of the first GST-dogfish NKCC1 preparation by AMPK and [γ-32P]MgATP, the protein was precipitated, digested with trypsin and the peptides separated by HPLC. The major radioactive peaks (Fig. 1B) were analysed by mass spectrometry. In peaks I and II, Ser214 was identified as the phosphorylation site in the phosphorylated peptides LIRPSLAELHDELDK and LIRPSLAELHDELDKEPFEDGYVNGEESSPAEEA (corresponding to a non-specific tryptic peptide with one missed cleavage), respectively, of the dogfish NKCC1 sequence. Ser214 of dogfish NKCC1 corresponds to Ser242 of the human protein and to the AMPK site previously identified in rabbit renal NKCC2 (Ser126) based on a sequence alignment (Fig. 1C). The phosphorylation site in peak III (Fig. 1B) was identified as Ser38 in the phosphorylated tryptic peptide (SQSGPEPGAGQQEPPPPATPLRPVASQSR) of the dogfish NKCC1 sequence after re-digestion of fractions 43 + 44 by chymotrypsin. This site corresponds to Ser77 of human NKCC1 (Fig. 1C). It is not conserved in rabbit NKCC2 and is not present in human NCC. None of the AMPK sites identified in NKCC1 correspond to the SPAK/OSR1 sites (Thr203/207/212) implicated in human NKCC1 activation.
Figure 1. Phosphorylation of NKCC1 by AMPK and identification of the sites.
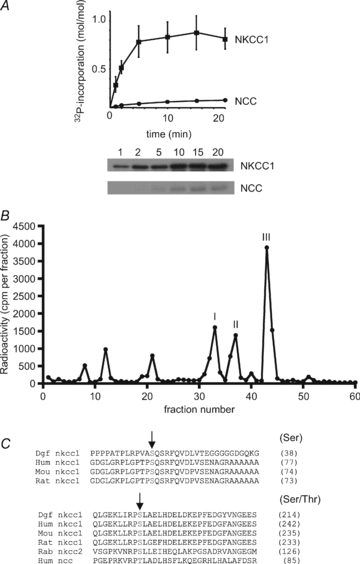
A, recombinant GST-dogfish NKCC1 and GST-human NCC were phosphorylated in vitro with AMPK and [γ-32P]MgATP. At the indicated times, aliquots were removed for SDS-PAGE and phosphorimaging for the measurement of 32P incorporation. The results are the means ±s.e.m. of three separate experiments and representative autoradiograms are shown below. B, after maximal in vitro phosphorylation by AMPK, GST-dogfish NKCC1 was precipitated and digested with trypsin. Peptides were separated by reverse-phase narrow-bore HPLC in an acetonitrile gradient and fractions were counted by Cerenkov radiation. Phosphorylation sites in the three radiolabelled peaks were identified by mass spectrometry. C, an alignment of N-terminal domain sequences surrounding the AMPK sites of dogfish NKCC1 with human, mouse and rat, as well as with sequences of rabbit NKCC2 and human NCC is shown. AMPK phosphorylation sites in dogfish NKCC1 are indicated by arrows and numbers between brackets refer to the residues corresponding to dogfish Ser38 and Ser214 in the different species.
Polyclonal sheep antibodies were raised against phosphorylated peptides corresponding to the sequences surrounding Ser77 and Ser242 of human NKCC1. The specificity of the antibodies was verified by immunoblotting purified GST-dogfish NKCC1 (1–260), phosphorylated with or without activated AMPK and non-radioactive MgATP (Supplemental Fig. S1A).
Effect of A769662 on AMPK activation, NKCC1 phosphorylation and 86Rb+ uptake in human RBCs
Since treatment of erythrocytes with 5-aminoimidazole-4-carboxamide riboside (AICAR, which is converted in cells to the AMP analogue, ZMP) does not lead to AMPK activation in RBCs (results not shown and B. Viollet, personal communication), human erythrocytes were incubated with the recently discovered small-molecule, non-nucleotide, thienopyridone A769662 AMPK activator (Cool et al. 2006). Incubation with 20 μm A769662 led to a time-dependent increase in AMPK activity reaching a plateau of about 2-fold after 20 min (data not shown) and after 30 min, a significant increase in AMPK Thr172 phosphorylation of about 50% (Fig. 2A) accompanied AMPK activation. Therefore, the phosphorylation state of the two new AMPK sites in NKCC1 was investigated using the phospho-specific antibodies that had been generated in sheep. After 30 min of incubation with A769662, no change in Ser77 NKCC1 phosphorylation in response to A769662 treatment occurred, whereas for the Ser242 NKCC1 site there was a significant 50% increase in phosphorylation, with no change in phosphorylation state of the conserved Thr203/207/212 NKCC1 sites (Fig. 2B), of which Thr207 phosphorylation is required for NKCC1 cotransporter activation. Total 86Rb+ uptake was likewise unaffected (Fig. 2C), suggesting that AMPK activation by A769662 is not linked to NKCC1 activation.
Figure 2. Effect of A769662 treatment on AMPK activation, SPAK and NKCC1 phosphorylation and 86Rb+ uptake in human RBCs.
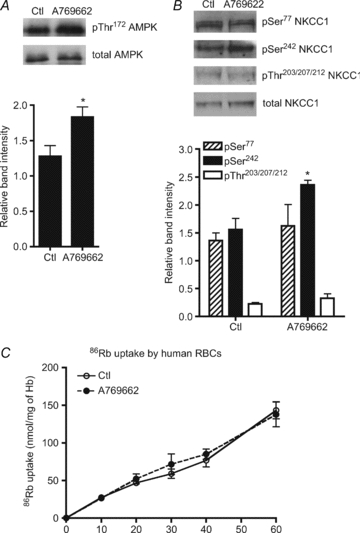
Washed human erythrocytes were incubated with 20 μm A769662 for 30 min and extracts were prepared for immunoblotting as described in Methods. A, upper panel, a representative immunoblot of α-AMPK Thr172 phosphorylation versus total α1-AMPK. B, upper panel, representative immunoblots of NKCC1 Ser77, Ser242 and Thr203/207/212 phosphorylation versus total NKCC1. Densitometric scanning ratios of band intensities obtained with the anti-phospho antibodies relative to those obtained for the loading controls are shown in the lower panels and the results are the means ±s.e.m. of three independent experiments. *Significant difference (P < 0.05, paired t test) with respect to the controls. C, washed RBCs were pre-incubated at 5% haematocrit and 37°C in Hepes-buffered Krebs medium containing 11 mm glucose, without (○) or with (•) 20 μm A-769662. After 20 min, tracer 86Rb+ (5 μCi ml−1) was added to the suspension at t= 0 and uptake was followed at the indicated times over 60 min. The results are the means ±s.e.m. of four independent experiments.
Hyperosmolarity induces shrinkage, AMPK activation and stimulation of bumetanide-sensitive 86Rb+ uptake in human RBCs
Incubation of human erythrocytes to induce hyperosmotic shock with 0.3 m sucrose (added to the Krebs–Ringer solution) led to cell shrinkage as observed by fixation and confocal microscopy (not shown). Under these conditions, AMPK Thr172 phosphorylation increased rapidly becoming significant after 1 min and reaching a maximum at 30 min (Fig. 3A). Accordingly, AMPK activity increased up to 2-fold after 30 min of incubation under hyperosmotic conditions compared to basal activity (Fig. 3B). We then tested whether cell shrinkage would increase NKCC1 activity as measured by bumetanide-sensitive 86Rb+ uptake in human erythrocytes. RBCs were preincubated for 30 min with sucrose to fully activate AMPK with or without the NKCC1 inhibitor bumetanide. 86Rb+ tracer was then added and the rate of ion uptake was measured over 60 min (Fig. 3C). Cell shrinkage with 0.3 m sucrose increased the initial rate of total 86Rb+ uptake about 2-fold compared with the controls (Fig. 3D) and the hyperosmolarity-induced increase in 86Rb+ uptake was inhibited by about 60% at 60 min by incubation with bumetanide (Fig. 3C). Moreover, the stimulation of ion uptake by hyperosmolarity was completely abrogated by bumetanide addition (Fig. 3C and D). Therefore in human erythrocytes, cell shrinkage induced by sucrose addition led to both AMPK activation and an increase in NKCC1 activity.
Figure 3. Effects of hyperosmolarity on AMPK activity and bumetanide-sensitive 86Rb+ uptake in human RBCs.
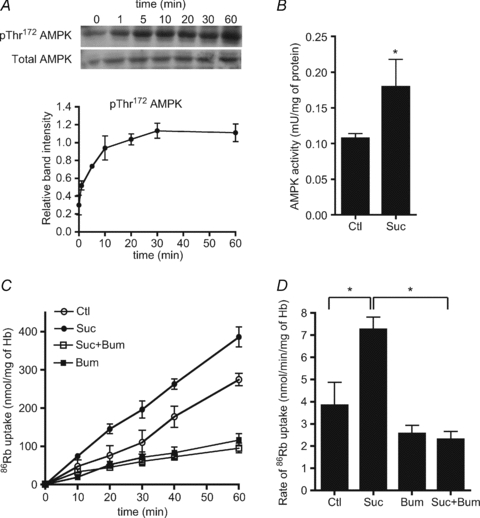
Washed human erythrocytes were incubated with 0.3 m sucrose for the indicated times and extracts were analysed by immunoblotting. A, upper panel, a representative immunoblot of α-AMPK Thr172 phosphorylation versus total α1-AMPK. Densitometric scanning ratios of phospho/total AMPK are shown in the lower panel and the results are the means ±s.e.m. of three independent experiments. B, extracts from erythrocytes incubated with 0.3 m sucrose for 30 min were immunoprecipitated with anti-AMPK α1-subunit antibody for AMPK assay. The results are the means ±s.e.m. of three independent experiments. *Significant difference (P < 0.05, paired t test) with respect to the controls. C, washed human RBCs were incubated at 5% haematocrit and 37°C in Hepes-buffered Krebs medium containing 11 mm glucose, without (○) or with (•) 0.3 m sucrose or with 100 μm bumetanide (□) or sucrose + bumetanide (▪). After 30 min, tracer 86Rb+ (5 μCi ml−1) was added to the suspension at t= 0 and uptake was followed at the indicated times. D, initial influx rates (calculated over 60 min) are the means ±s.e.m. of five independent experiments. *Significant difference (P < 0.05, paired t test).
AMPKα1 deletion in mice has no effect on hyperosmolarity-induced NKCC1 activation
Since the major AMPK catalytic subunit isoform in RBCs is α1-AMPK (Föller et al. 2009 and our data not shown), we took advantage of a mouse model in which the AMPKα1 subunit isoform had been deleted to explore the potential link between AMPK activation and the stimulation of NKCC1 activity during hyperosmotic stress. The cell shrinkage-induced increase in 86Rb+ uptake was compared in erythrocytes from wild-type (Fig. 4A) and AMPKα1-deficient mice (Fig. 4B). No effect of AMPKα1 deletion on the rate of basal 86Rb+ uptake, the hyperosmolarity-induced increase in total 86Rb+ uptake or the bumetanide-sensitive increase in uptake by hyperosmolarity was seen (Fig. 4C).
Figure 4. Effect of hyperosmolarity induced by sucrose treatment on 86Rb+ uptake in erythrocytes from wild-type and AMPKα1-deficient mice.
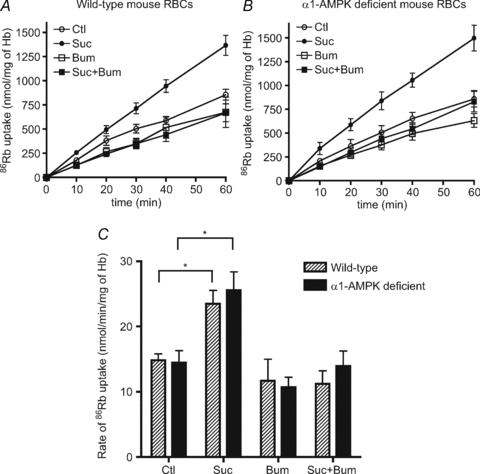
Washed erythrocytes from wild-type and AMPKα1-deficient mice were incubated with and without 0.3 m sucrose, with and without 100 μm bumetanide for 86Rb+ uptake measurements at the indicated times. The results are the means ±s.e.m. of five independent experiments. C, initial influx rates of 86Rb+ uptake were measured in the presence and absence of 100 μm bumetanide (see legend to Fig. 3). *Significant difference (P < 0.05, paired t test).
Effect of STO-609 on hyperosmolarity-induced AMPK activation and 86Rb+ uptake in mouse RBCs
When mouse erythrocytes were incubated with the STO-609 selective CaMKKβ inhibitor, AMPK activation by 0.3 m sucrose was abrogated (Fig. 5A), suggesting that hyperosmolarity-induced AMPK activation involves a rise in intracellular Ca2+ and phosphorylation of the α-subunit Thr172 residue by CaMKKβ. Both mouse and human RBCs were found to contain both CaMKKβ and LKB1 as detected by immunoblotting of full lysates (Fig. S1B). Although incubation with STO-609 abrogated AMPK activation by cell shrinkage, it did not significantly reduce the hyperosmolarity-induced increase in total 86Rb+ uptake (Fig. 5B) and an increase in bumetanide-sensitive uptake was still seen in the presence of sucrose and STO-609. Therefore, together with results obtained in α1-AMPK deficient mice, the findings suggest that AMPK activation and stimulation of NKCC1 activity by hyperosmolarity are parallel events that are not causatively linked.
Figure 5. Effect of the STO-609 CaMKKβ inhibitor on hyperosmolarity-induced AMPK activation and 86Rb+ uptake in mouse RBCs.
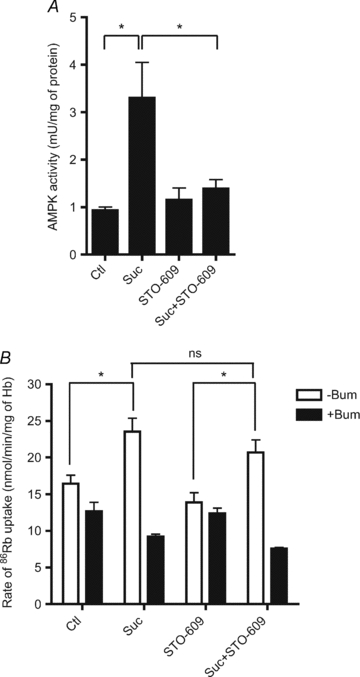
Washed mouse erythrocytes were incubated at 5% haematocrit and 37°C in Hepes-buffered Krebs medium containing 11 mm glucose, with or without 10 μm STO-609 and with or without 0.3 m sucrose for 30 min. A, extracts were immunoprecipated with anti-AMPK α1-subunit antibody for AMPK assay. The results are the means ±s.e.m. of three independent experiments. B, after 30 min, initial rates of 86Rb+ uptake were calculated from measurements made at 10 and 30 min in the presence and absence of 100 μm bumetanide. The results are the means ±s.e.m. of six independent experiments. *Significant difference (P < 0.05, paired t test).
Effects of A769662 and hyperosmolarity on SPAK/NKCC1phosphorylation and 86Rb+ uptake in mouse RBCs
Mouse and human RBCs were found to contain WNK1 and OSR1, as detected by immunoblotting, as well as CaMKKβ and LKB1 (Fig. S1B). To assess possible contamination from non-RBCs, we immunoblotted extracts from washed erythrocytes using an antibody raised against human pyruvate dehyrogenase (PDH), a mitochondrial enzyme marker, to indicate contamination by white cells. No signal could be detected in red cell lysates, whereas a strong band was detected in hepatocyte extracts as positive control (Fig. S1C). Therefore, contamination of our preparations by non-red cells was minimal.
Incubation of human RBCs with 0.3 m sucrose led to a time-dependent increase in SPAK activation loop Thr233 phosphorylation, along with the second site on SPAK phosphorylated by WNK1, namely Ser373, which is not required for activation (Fig. S2A). Moreover, SPAK activation by osmotic shrinkage correlated closely with NKCC1 phosphorylation on the three conserved Thr residues (Fig. S2B). Therefore, mouse RBCs were incubated for different times with A769662 and sucrose to further investigate the possible correlation between NKCC1 phosphorylation and stimulation of bumetanide-sensitive 86Rb+ uptake. Incubation with A769662 or sucrose led to AMPK Thr172 phosphorylation, as observed in human RBCs (Figs 2A and 3A), and AMPK activation by A769662 treatment was greater at 60 min of incubation than at 5 min (Fig. 6A). Incubation with A769662 had no effect on SPAK Thr233, SPAK Ser373 (Fig. 6B) and NKCC1 Thr203/207/212 phosphorylation (Fig. 7A) and was without effect on bumetanide-sensitive 86Rb+ uptake (Fig. 7B) either at 5 or at 60 min of incubation. In response to hyperosmolarity, SPAK Thr233 activation loop phosphorylation was maximal at 5 and 60 min of incubation (Fig. 6B) whereas the level of NKCC1 Thr203/207/212 phosphorylation was greater at 60 min than at 5 min (Fig. 7A). Bumetanide-sensitive 86Rb+ uptake was slightly greater at 60 min than at 5 min of incubation with sucrose (Fig. 7B) and so the extent of NKCC1 phosphorylation did not directly correlate with NKCC1 activation by hyperosmolarity in mouse RBCs and other factors are likely involved.
Figure 6. Effect of hyperosmolarity and A769662 treatment on AMPK and SPAK phosphorylation in mouse RBCs.
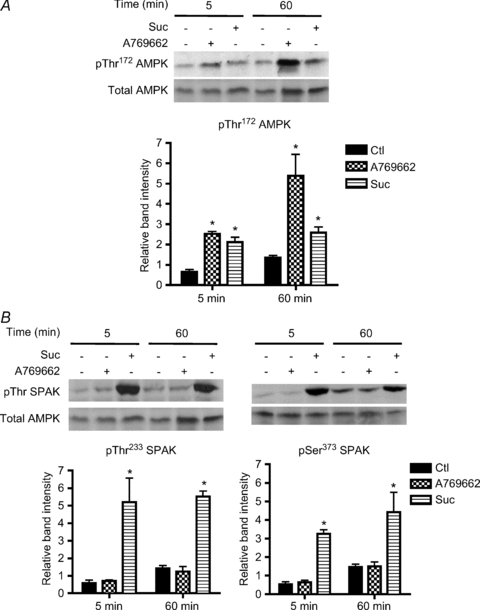
Washed mouse erythrocytes were incubated at 5% haematocrit and 37°C in Hepes-buffered Krebs medium containing 11 mm glucose, with or without 20 μm A769662 and with or without 0.3 m sucrose for 5 or 60 min. A, upper panel, a representative immunoblot of α-AMPK Thr172 phosphorylation versus total α1-AMPK. B, upper panels, representative immunoblots of SPAK Thr233 and Ser373 phosphorylation versus total AMPK as a loading control. Lower panels, densitometric scanning ratios of band intensities obtained with the anti-phospho antibodies relative to those obtained for the loading controls; the results are the means ±s.e.m. of three independent experiments. *Significant difference (P < 0.05, paired t test) with respect to the controls.
Figure 7. Effect of hyperosmolarity and A769662 treatment on NKCC1 phosphorylation and bumetanide-sensitive 86Rb+ uptake in mouse RBCs.
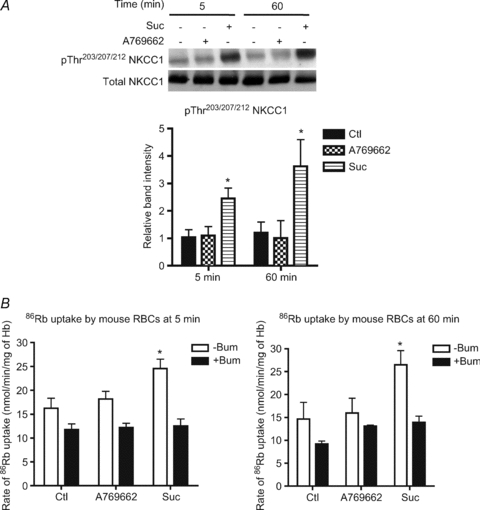
Washed mouse erythrocytes were incubated at 5% haematocrit and 37°C in Hepes-buffered Krebs medium containing 11 mm glucose, with or without 20 μm A769662 and with or without 0.3 m sucrose for 5 or 60 min. A, upper panel, a representative immunoblot of NKCC1 Thr203/207/212 phosphorylation versus total NKCC1 Densitometric scanning ratios of band intensities obtained with the anti-phospho antibodies relative to those obtained for the loading controls are shown in the lower panel. B, initial rates of 86Rb+ uptake were measured in the presence and absence of 100 μm bumetanide (see legend to Fig. 3). The results are the means ±s.e.m. of three independent experiments. *Significant difference (P < 0.05, paired t test) with respect to the controls.
Discussion
At the outset of this work, little was known about the AMPK and WNK/SPAK systems in RBCs and the molecular mechanism by which cell shrinkage activates NKCC1 in erythrocytes had not been clarified. Our aim was to investigate the mechanisms by which hyperosmolarity increases NKCC1 cotransporter activity and particularly whether AMPK activation could be implicated. We show that similar to NKCC2, NKCC1 can be phosphorylated by AMPK. Phosphorylation of NKCC1 occurred at Ser38 and Ser214 (dogfish sequence) corresponding to Ser77 and Ser242 in human NKCC1. The AMPK phosphorylation recognition motif is φ(Xβ)XXS/TXXXφ where φ is a hydrophobic residue (M, V, L, I or F), β is a basic residue (R, K or H) and the parentheses indicate that the order of residues at the P-4 and P-3 positions is not critical (Dale et al. 1995). The sequence surrounding Ser242 of human NKCC1 loosely fits the AMPK consensus, with an Arg residue at the P-2 rather than P-3 position. The corresponding residue in human NCC is Thr85 and flanked by Arg residues at P-2 and P-5 plus a Lys at P-4, explaining its poor rate of phosphorylation by AMPK. Ser38 of dogfish NKCC1 lies in a perfect context for phosphorylation by AMPK, which is not as favourable in the mammalian sequences due to the absence of a basic residue. However, the corresponding Ser77 residue of human NKCC1 was found to be phosphorylated in a phosphoproteomics study of HeLa cells subjected to cell cycle arrest (Dephoure et al. 2008).
Although the increase in 86Rb+ uptake by cell shrinkage in RBCs was associated with AMPK activation, phosphorylation of NKCC1 by AMPK is unlikely to be involved for the following reasons: (i) incubation of RBCs with A769662 to activate AMPK did not increase 86Rb+ uptake from either human or mouse cells, although phosphorylation at Ser242 of NKCC1 was increased in human erythrocytes; (ii) AMPKα1 deletion in mouse RBCs had no effect on the increase in rate of bumetanide-sensitive 86Rb+ uptake by hyperosmolarity; (iii) STO-609 treatment of mouse RBCs abrogated AMPK activation by hyperosmolarity but did not significantly reduce bumetanide-sensitive 86Rb+ uptake. Although human RBCs clearly express CaMKKβ (Fig. S1B), it remains to be seen whether STO-609 would block hyperosmolarity-induced AMPK activation, as observed in mouse RBCs. AMPK activation by A769662 involves allosteric stimulation of activity and inhibition of dephosphorylation by protein phosphatases, which increases the extent of its activation loop Thr172 phosphorylation (Sanders et al. 2007). Extracts were routinely prepared with direct lysis without washing the cells and AMPK activation was assessed after immunoprecipitation (Fig. S2B), which washes away the AMPK activator and so activation could not have occurred after lysis. AMPK activation was also assessed by immunoblotting (Fig. 2A), clearly showing that Thr172 phosphorylation was increased and that the AMPK activator was effective in the intact cells. Therefore, lack of permeation of A769662 could not explain the lack of effect on ion transport. Moreover, in freshly isolated rat cardiomyoctes treated with A769662 or oligomycin and in rat hepatocytes incubated with A769662 or AICA riboside to activate AMPK, there was no stimulation of 86Rb+ uptake (Fig. S3).
Surprisingly, incubation of human RBCs with sodium arsenite did not increase 86Rb+ uptake, contrary to its stimulatory effect in ferret red cells (Flatman & Creanor 1999a), and did not lead to AMPK activation (data not shown) as reported in rat hepatocytes (Corton et al. 1994). The lack of effect of arsenite in human compared with ferret RBCs could imply that arsenite treatment does not lead to activation of the signalling pathway responsible for NKCC1 activation in human cells or that this toxic compound is ‘neutralized’ in human but not ferret erythrocytes. Also, there is variation in the responses of RBCs from different species due to differences in the transporters present, the regulatory apparatus, and expression of membrane proteins and their association with glycolytic enzymes (Pasini et al. 2010). Indeed, AMPK activities and rates of 86Rb+ uptake in human erythrocytes (Fig. 3) were somewhat less than in mouse RBCs (Fig. 5). Also, the responses of RBC subpopulations may be different, for example in ageing erythrocytes enzyme activities can decrease as the cells become smaller.
Osmotic shrinkage led to SPAK activation loop phosphorylation in mouse and human RBCs, which was accompanied by phosphorylation of the three conserved Thr residues of NKCC1. Therefore, activation of the WNK1/SPAK pathway by hyperosmolarity could explain the increase in 86Rb+ uptake. One effect of adding 0.3 m sucrose to red cells is that the intracellular Cl− (as well as Na+ and K+) concentration would be increased by shrinkage. The increase in cellular Cl− concentration should in turn shift the Donnan equilibrium between intracellular Cl− and OH− ions and alkalinize the cells slightly. To the best of our knowledge, there is no information in the literature on the pH–activity profiles of WNK or SPAK. It is possible that cytosolic alkalinization might reinforce or oppose the effects of cell shrinkage on the activities of the WNK1/SPAK kinases involved in NKCC1 activation.
In conclusion, increased 86Rb+ uptake by osmotic shrinkage seems to be due to activation of the WNK1/SPAK, rather than the AMPK pathway. AMPK was activated by hyperosmolarity in RBCs, probably secondary to a rise in Ca2+ via CaMKKβ. Although NKCC1 phosphorylation by AMPK does not appear to directly control cotransporter activity, the AMPK Ser77 site was detected as being phosphorylated in vivo in a phosphoproteomics study (Dephoure et al. 2008) and treatment of human RBCs with A769662 led to an increase in phosphorylation of Ser242, the second AMPK site we identified. Interestingly, two-dimensional phosphopeptide mapping of RBCs indicated that NKCC1 acquires five phosphates going from an inactive state to an active state in shrunken cells (Lytle, 1997). However, we were unable to see changes in Ser77 or Ser242 phosphorylation of NKCC1 during hyperosmolarity. Phosphorylation of Ser77/242 by AMPK could affect NKCC1 function other than its ion transport activity (Fig. 8). NKCC1 has many interacting partners including PP1, p38 MAPK, MLCK, PKCδ and SPAK, and is also regulated by the F-actin cytoskeleton (Hoffmann et al. 2007, 2009).
Figure 8. Activation of the WNK1/SPAK and CaMKKβ/AMPK pathways by osmotic shrinkage in RBCs.
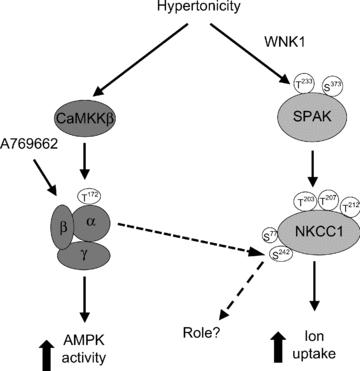
Cell shrinkage by sucrose treatment in RBCs leads to activation of α1-AMPK and SPAK via the CaMKKβ and WNK1 pathways, respectively. SPAK activation correlates with NKCC1 Thr203/207/212 phosphorylation and is likely to be responsible for its activation and increased 86Rb+ uptake induced by hyperosmolarity. AMPK activation by hyperosmolarity does not affect NKCC1 activity. Treatment with A-769662 activates AMPK in RBCs and increases Ser242 NKCC1 phosphorylation, whose function is at present unknown. The proposed scheme is based on the present data.
Acknowledgments
We thank Marie-Agnès Gueuning and Nusrat Hussain for their expert technical assistance, Dr Pierre-Paul Prevot for help with animal procedures and Prof. Louis Hue for critically reading the manuscript. The help of Prof. Dario Alessi, for freely providing reagents to carry out the work, is also gratefully acknowledged. Lastly, we thank Prof. Grahame Hardie for production of the anti-phospho Ser77 and anti-phospho Ser242 antibodies in sheep within the framework of the EXGENESIS Consortium. The work was supported by the Interuniversity Attraction Poles Program – Belgian Science Policy (P6/28), the ‘Action de Recherche Concertée’, Université catholique de Louvain, the Fund for Medical Scientific Research (Belgium) and the EXGENESIS Integrated Project (LSHM-CT-2004-005272) from the European Commission.
Glossary
Abbreviations
- AICAR
5-aminoimidazole-4-carboxamide riboside
- AMPK
AMP-activated protein kinase
- CaMKKβ
Ca2+/calmodulin-dependent protein kinase kinase-β
- GST
Glutathione-S-Transferase
- NCC
Na+–Cl− cotransporter
- NKCC1
Na+–K+–2Cl− cotransporter
- OSR1
oxidative stress resonse protein kinase-1
- PDH
pyruvate dehydrogenase
- SPAK
STE20/SPS1-related proline/alanine-rich kinase
- WNK
with-no-lysine (K) protein kinase
Author contributions
B.S. conducted most of the experimental work and was involved in conception and design, analysis and interpretation of the data and drafting the article. L.M. carried out some immunoblot and statistical analyses. D.V. carried out phosphorylation site identification by mass spectrometry. B.V. provided AMPKα1-deficient mice and was involved in critically revising the manuscript for final approval. M.R. was responsible for conception and design, interpretation of data and drafting the article. All work was performed in the laboratory of M.R., de Duve Institute, Brussels.
Supplemental material
Fig. S1
Fig. S2
Fig. S3
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Bultot L, Horman S, Neumann D, Walsh MP, Hue L, Rider MH. Myosin light chains are not a physiological substrate of AMPK in the control of cell structure changes. FEBS Lett. 2009;583:25–28. doi: 10.1016/j.febslet.2008.11.022. [DOI] [PubMed] [Google Scholar]
- Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hardie DG. Role of the AMP-activated protein kinase in the cellular stress response. Curr Biol. 1994;4:315–324. doi: 10.1016/s0960-9822(00)00070-1. [DOI] [PubMed] [Google Scholar]
- Dale S, Wilson WA, Edelman AM, Hardie DG. Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1, and mammalian calmodulin-dependent protein kinase I. FEBS Lett. 1995;361:191–195. doi: 10.1016/0014-5793(95)00172-6. [DOI] [PubMed] [Google Scholar]
- Davies SP, Carling D, Hardie DG. Tissue distribution of the AMP-activated protein kinase, and lack of activation by cyclic-AMP-dependent protein kinase, studied using a specific and sensitive peptide assay. Eur J Biochem. 1989;186:123–128. doi: 10.1111/j.1432-1033.1989.tb15185.x. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd BF, Forbush B. PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1) J Biol Chem. 2003;278:27347–27353. doi: 10.1074/jbc.M301899200. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matter in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatman PW, Creanor J. Regulation of Na+-K+-2Cl− cotransport by protein phosphorylation in ferret erythrocytes. J Physiol. 1999a;517:699–708. doi: 10.1111/j.1469-7793.1999.0699s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatman PW, Creanor J. Stimulation of Na+-K+-2Cl− cotransport by arsenite in ferret erythrocytes. J Physiol. 1999b;519:143–152. doi: 10.1111/j.1469-7793.1999.0143o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemmer AW, Gimenez I, Dowd BF, Darman RB, Forbush B. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem. 2002;277:37551–37558. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- Föller M, Sopjani M, Koka S, Gu S, Mahmud H, Wang K, Floride E, Schleicher E, Schulz E, Münzel T, Lang F. Regulation of erythrocyte survival by AMP-activated protein kinase. FASEB J. 2009;23:1072–1080. doi: 10.1096/fj.08-121772. [DOI] [PubMed] [Google Scholar]
- Fraser SA, Gimenez I, Cook N, Jennings I, Katerelos M, Katsis F, Levidiotis V, Kemp BE, Power DA. Regulation of the renal-specific Na+-K+-2Cl− co-transporter NKCC2 by AMP-activated protein kinase (AMPK) Biochem J. 2007;405:85–93. doi: 10.1042/BJ20061850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser S, Mount P, Hill R, Levidiotis V, Katsis F, Stapleton D, Kemp BE, Power DA. Regulation of the energy sensor AMP-activated protein kinase in the kidney by dietary salt intake and osmolality. Am J Physiol Renal Physiol. 2005;288:578–586. doi: 10.1152/ajprenal.00190.2004. [DOI] [PubMed] [Google Scholar]
- Giménez I, Forbush B. Regulatory phosphorylation sites in the NH2 terminus of the renal Na-K-Cl cotransporter (NKCC2) Am J Physiol Renal Physiol. 2005;289:1341–1345. doi: 10.1152/ajprenal.00214.2005. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Schettino T, Marshall WS. The role of volume-sensitive ion transport systems in regulation of epithelial transport. Comp Biochem Physiol A Mol Integr Physiol. 2007;148:29–43. doi: 10.1016/j.cbpa.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89:193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L, Rider MH. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase α-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem. 2006;281:5335–5340. doi: 10.1074/jbc.M506850200. [DOI] [PubMed] [Google Scholar]
- Horman S, Morel N, Vertommen D, Hussain N, Neumann D, Beauloye C, El Najjar N, Forcet C, Viollet B, Walsh MP, Hue L, Rider MH. AMP-activated protein kinase phosphorylates and desensitizes smooth muscle myosin light chain kinase. J Biol Chem. 2008;283:18505–18512. doi: 10.1074/jbc.M802053200. [DOI] [PubMed] [Google Scholar]
- Hue L, Rider MH. The AMP-activated protein kinase: more than an energy sensor. Essays Biochem. 2007;43:121–137. doi: 10.1042/BSE0430121. [DOI] [PubMed] [Google Scholar]
- Jørgensen SB, Viollet B, Andreelli F, Frøsig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside but not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Lytle C. Activation of the avian erythrocyte Na-K-Cl cotransport protein by cell shrinkage, cAMP, fluoride, and calyculin-A involves phosphorylation at common sites. J Biol Chem. 1997;272:15069–15077. doi: 10.1074/jbc.272.24.15069. [DOI] [PubMed] [Google Scholar]
- Pasini EM, Lutz HU, Mann M, Thomas AW. Red blood cell (RBC) membrane proteomics – Part II: Comparative proteomics and RBC patho-physiology. J Proteomics. 2010;73:421–435. doi: 10.1016/j.jprot.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, O’Donnell ME, Anderson SE, Cala PM. Physiology and pathophysiology of Na+/H+ exchange and Na+-K+-2Cl− cotransport in the heart, brain, and blood. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1–25. doi: 10.1152/ajpregu.00782.2005. [DOI] [PubMed] [Google Scholar]
- Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1) J Biol Chem. 2002;277:50812–50819. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- Richardson C, Alessi DR. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J Cell Sci. 2008;121:3293–3304. doi: 10.1242/jcs.029223. [DOI] [PubMed] [Google Scholar]
- Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR. Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci. 2008;121:675–684. doi: 10.1242/jcs.025312. [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr Assays of protein kinase. Methods Enzymol. 1983;99:3–6. doi: 10.1016/0076-6879(83)99034-1. [DOI] [PubMed] [Google Scholar]
- Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem. 2007;282:32539–32548. doi: 10.1074/jbc.M706543200. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- Sugden C, Crawford RM, Halford NG, Hardie DG. Regulation of spinach SNF1-related (SnRK1) kinases by protein kinases and phosphatases is associated with phosphorylation of the T loop and is regulated by 5′-AMP. Plant J. 1999;19:433–439. doi: 10.1046/j.1365-313x.1999.00532.x. [DOI] [PubMed] [Google Scholar]
- Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J. 2005;391:17–24. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitari AC, Thastrup J, Rafiqi FH, Deak M, Morrice NA, Karlsson HK, Alessi DR. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J. 2006;397:223–231. doi: 10.1042/BJ20060220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witters LA, Kemp BE, Means AR. Chutes and Ladders: the search for protein kinases that act on AMPK. Trends Biochem Sci. 2006;31:13–16. doi: 10.1016/j.tibs.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Woods A, Vertommen D, Neumann D, Türk R, Bayliss J, Schlattner U, Wallimann T, Carling D, Rider MH. Identification of phosphorylation sites in AMP-activated protein kinase (AMPK) for upstream AMPK kinases and study of their roles by site-directed mutagenesis. J Biol Chem. 2003;278:28434–28442. doi: 10.1074/jbc.M303946200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.