

Abstract
GABAergic synapses on Cajal–Retzius neurons in layer I of the murine neocortex experience GABAB receptor (GABABR)-mediated tonic inhibition. Extracellular GABA concentration ([GABA]o) that determines the strength of GABABR-mediated inhibition is controlled by GABA transporters (GATs). In this study, we hypothesized that the strength ofpresynaptic GABABR activation reflects [GABA]o in the vicinity of synaptic contacts. Slices obtained from two age groups were used, namely postnatal days (P)2–3 and P5–7. GABAergic postsynaptic currents (IPSCs) were recorded using the whole-cell patch-clamp technique. Minimal electrical stimulation in layer I was applied to elicit evoked IPSCs (eIPSCs) using a paired-pulse protocol. Three parameters were selected for comparison: the mean eIPSC amplitude, paired-pulse ratio, and failure rate. When GAT-1 and GAT-2/3 were blocked by NO-711 (10 μm) and SNAP-5114 (40 μm), respectively, no tonic GABABR-mediated inhibition was observed. In order to restore the control levels of GABABR-mediated inhibition, 250 and 125 nm exogenous GABA was required at P2–3 and P5–7, respectively. Addition of 3-mercaptopropionic acid, a glutamate decarboxylase inhibitor, did not significantly change the obtained values arguing against the suggestion that a mechanism different from GATs contributes to [GABA]o control. We conclude that juxtasynaptic [GABA]o is higher (about 250 nm) at P2–3 than at P5–7 (about 125 nm). As both radial cell migration and corticogenesis in general are strongly dependent on [GABA]o and the formation of the last layer 2/3 is finished by P4 in rodents, the observed [GABA]o reduction in layer I might reflect this crucial event in the cortical development.
Introduction
γ-Aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the adult brain. Synaptically released GABA activates postsynaptic GABAA receptors giving rise to inhibitory postsynaptic potentials (IPSPs). GABA diffusion out of the synaptic cleft and/or its removal by GABA transporters is followed by IPSP termination. In addition to ‘phasic’, synaptic transmission, ambient GABA, which is normallypresent in the extracellular space, can activate GABAA receptors (GABAARs) and/or GABAB receptors (GABABRs). Persistent activation of postsynaptic GABAARs leads to a ‘tonic’ inhibitory current that controls the level of excitability of neurons (Brickley et al. 1996; Stell & Mody, 2002; Nusser & Mody, 2002; Semyanov et al. 2004), while tonic activation ofpresynaptic GABABRs can regulate the strength of glutamatergic inputs (Kombian et al. 1996; Dittman & Regehr, 1997) and GABAergic inputs (Le Feuvre et al. 1997; Jensen et al. 2003; Kirmse & Kirischuk, 2006a). Given GABAARs or GABABRs are not saturated by ambient GABA levels, physiological activity and/or manipulations affecting extracellular GABA levels can shape the excitability and plasticity of neuronal networks.
Brain microdialysis has become a frequently used method to study extracellular concentrations of neurotransmitters in specific areas of the living brain (Ungerstedt, 1991; van der Zeyden et al. 2008). However, extracellular GABA levels ([GABA]o) measured using this technique demonstrate drastic variability even if the extracellular liquid was sampled from the same region (for example, in the hippocampus from several nanomolar to several micromolar; Lerma et al. 1986; Biggs et al. 1992; Rowley et al. 1995; Rakovska et al. 1998). Because the size of sampling probe is relatively large (usually ∼250 μm in diameter), one cannot exclude that the observed variability of [GABA]o results from tissue damage. For example, microdialysis studies report ambient glutamate levels of 1–4 μm (Lerma et al. 1986; Baker et al. 2002), while measurements performed in acute brain slices demonstrated much lower extracellular glutamate concentration (Cavelier & Attwell, 2005; Herman & Jahr, 2007).
In our recent study, we have demonstrated that GABAergic inputs to Cajal–Retzius (CR) cells in the marginal zone of mouse neonatal cortex experience tonicpresynaptic GABABR-mediated inhibition. It has been shown that [GABA]o and in turn the strength of GABABR-mediated inhibition is determined by the activity of GABA transporters (GATs): GAT-2/3 operating in the reverse mode releases GABA, while GAT-1 functioning in the uptake mode removes GABA from the extracellular space. In addition, [GABA]o is dependent on the activity of glutamate decarboxylase (GAD), a GABA synthesizing enzyme (Kirmse & Kirischuk, 2006a). In the current work, [GABA]o had firstly been reduced by GAT and/or GAD blockers, and the amount of exogenous GABA required to restore the control level of GABABR-mediated inhibition has been taken as an estimate for [GABA]o. We have found that [GABA]o in the vicinity of GABAergic synapses on CR cells is about 250 nm at postnatal days 2–3 (P2–3) and decreases to 125 nm at P5–7.
Methods
Brain slicepreparation
All experimental procedures were carried out according to the permit given by the State Office of Health and Social Affairs Berlin (Landesamt für Gesundheit und Soziales Berlin, T0121/03), which complies with international and European Union norms (Drummond, 2009). Experiments were designed to minimize the number of animals used. All experiments were conducted with pigmented C57BL/6J mice pups of postnatal days (P)2–3 and 5–7 (the day of birth was designated as P0). Animals were deeply anaesthetized with ether (by inhalation), then decapitated and the brain rapidly removed. The brain was transferred into ice-cold saline that contained (in mm): 125 NaCl, 4 KCl, 10 glucose, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, and 2.5 MgCl2, constantly aerated with a 5% CO2–95% O2 mixture (pH 7.3). Sagittal slices of both hemispheres were cut on a vibrating blade microtome (Campden Instruments Ltd, Loughborough, UK). Afterpreparation, slices (200 μm thick) were stored for at least 1 h at room temperature in artificial cerebrospinal fluid (ACSF) that contained (in mm): 125 NaCl, 4 KCl, 10 glucose, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, and 1 MgCl2; pH was buffered to 7.3 by continuous bubbling with 5% CO2–95% O2 mixture.
Electrophysiological recordings in acute slices
For recordings, slices were placed into a recording chamber (∼0.4 ml volume) on the microscope stage (Axioscope FS, Zeiss) equipped with phase contrast optics. Slices were submerged with a constant flow of oxygenated ACSF. Flow rate was set to 1 ml min−1. A 40× water immersion objective was used in all experiments. Cajal–Retzius (CR) cells were visually selected according to morphological criteria: (1) location in layer I; (2) horizontal orientation; (3) large ovoid soma; and (4) one thick tapered dendrite typically extended in parallel to the pial surface.
Ten micromolar 6,7-dinitroquinoxaline-2,3-dione (DNQX; an AMPA/kainate receptor antagonist) and 50 μm dl-2-amino-5-phosphonopentanoic acid (APV; an NMDA receptor blocker) were added to the ACSF to block glutamatergic currents. GABAergic postsynaptic currents (IPSCs) were recorded using the whole-cell configuration of the patch-clamp technique. Intra-pipette solution contained (in mm): 100 potassium gluconate, 50 KCl, 5 NaCl, 0.5 CaCl2, 5 EGTA, 25 Hepes, 2 MgATP, 0.3 GTP; pH was set to 7.2 with KOH. Pipette resistance was 3–5 MΩ, when filled with the above saline. Electrophysiological signals were acquired using an EPC-7 amplifier (HEKA Elektronik, Lambrecht, Germany), a 16-bit AD/DA board (ITC-16), and TIDA 4.11 software (HEKA Elektronik). The signals were filtered at 3 kHz and sampled at a rate of 10 kHz. Hyperpolarizing pulses (10 mV) were applied to control the access resistance during experiments. Only recordings with a series resistance below 40 MΩ were accepted. Series resistance compensation was not applied. Cells exhibiting more than 20% changes in the access resistance during an experiment were discarded. The chloride reversal potential was about −20 mV. The holding potential was set to −70 mV.
All experiments were performed at room temperature (22–25°C). 1-[2-[Tris(4-methoxyphenyl)methoxy]ethyl]-(S)-3-piperid inecarboxylic acid (SNAP-5114) and 1,2,5,6-tetrahydro-1-[2-[[(diphenylmethylene)amino]oxy] ethyl]-3-pyridinecarboxylic acid hydrochloride (NO-711) were from Tocris (Bristol, UK). All other chemicals were obtained from Sigma-Aldrich.
Electrical stimulation
Evoked GABAergic postsynaptic currents (eIPSCs) were elicited by focal electrical stimulation through a glass pipette filled with the ACSF (about 10 MΩ). In this case, N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide (QX 314, 2 mm) was added to the intracellular solution toprevent generation of action potentials in the tested neurons. An isolated stimulation unit was used to generate rectangular electrical pulses. Pulse duration was set to 0.5 ms. Pulse intensity was adjusted to activate a unitary synaptic input (minimal stimulation). Stimulation was accepted as minimal if the following criteria were satisfied: (1) eIPSC latency remained stable (<20% fluctuations); (2) lowering stimulus intensity by 20% resulted in a complete failure of eIPSCs; (3) an increase in stimulus intensity by 20% changed neither mean eIPSC amplitude nor eIPSC shape (Kirmse et al. 2007). Typical pulse intensity required for minimal stimulation was between 1 and 2 μA.
CR cells receive two types of GABAergic inputs characterized as fast and slowly rising eIPSCs. Because inputs generating the slowly rising eIPSCs experience weak tonic GABABR-mediated inhibition (Kirmse et al. 2007), only fast rising eIPSCs (10–90% rise time less than 1 ms, the mean value 0.7 ms) have been selected in this study.
Paired-pulse stimulation with an inter-stimulus interval of 50 ms was applied at 0.2 Hz. At least 40 responses were recorded under each experimental condition. Three parameters were taken to assess the strength of GABABR-mediated inhibition: (1) the mean amplitude of the first eIPSC, (2) the paired-pulse ratio (PPR), i.e. the mean amplitude of the second eIPSC divided by the mean amplitude of the first eIPSC, and (3) the failure rate, i.e. the percentage of trials in which the first stimulus failed to elicit an eIPSC. As the mean eIPSC amplitudes dramatically fluctuates from cell to cell (from tens to hundreds of picoamperes), for each cell the mean eIPSC amplitudes obtained under experimental conditions (GABA, SNAP-5114, etc.) were normalized to the corresponding mean eIPSC amplitudes recorded under control conditions.
Because postsynaptic IPSCs are suggested to report the strength ofpresynaptic inhibition, it is of importance that the postsynaptic site is not modified by experimental treatments. In this study, even the highest [GABA] used (2.5 μm) failed to influence the following basic parameters of postsynaptic cells: the resting potential (−46 ± 3 versus−43 ± 3 mV, P > 0.15, n= 21), the membrane resistance (320 ± 36 versus 342 ± 41 MΩ, P > 0.2, n= 21), and the holding current (−17 ± 8 versus−19 ± 9 pA, P > 0.3, n= 21, data not shown). Definitely, we cannot exclude the possibility that exogenous GABA results in a partial desensitization of GABAA receptors (Overstreet et al. 2000). To inspect this, miniature IPSCs (mIPSCs) were recorded in thepresence of 1 μm tetrodotoxin, a blocker of voltage-dependent Na+ channels. To increase mIPSC frequency, N-ethylmaleimide (NEM, 50 μm) waspre-applied for 5 min. In CR cells, NEM was shown to affect neither mIPSC amplitudes nor their kinetics (Kirmse & Kirischuk, 2006b). Because intact, synaptically located GATs can decrease [GABA] in the synaptic cleft as compared to exogenous [GABA] applied, SNAP-5114 (40 μm) and NO-711 (10 μm) were added to block GAT-2/3 and GAT-1, respectively (Borden, 1996). No change of the median mIPSC amplitude was detected in thepresence of 1 μm GABA. The corresponding values were 26.9 ± 2.5 and 27.5 ± 2.8 pA in control and in thepresence of 1 μm GABA, respectively (Fig. 1A–C, n= 8, P > 0.4, Student's paired t test). Although a slight increase of membrane noise was observed in thepresence of 2.5 μm exogenous GABA (Fig. 1D), the median amplitude of mIPSCs was also not affected (32.1 ± 1.9 versus 30.5 ± 2.1 pA in control and in thepresence of 2.5 μm GABA, respectively, Fig. 1D–F, n= 8, P > 0.5, paired Student's t test). In addition, neither rise times nor decay kinetics of mIPSCs were influenced by exogenous GABA (data not shown). In these experiments mIPSCs were recorded from four P2–3 and four P5–7 CR cells. As we did not observe any difference between these two age groups, the data were pooled together. We conclude that low concentrations of exogenous GABA used in this study do not influence postsynaptic GABAARs in thispreparation.
Figure 1. Low concentrations of exogenous GABA do not influence the median mIPSC amplitudes.
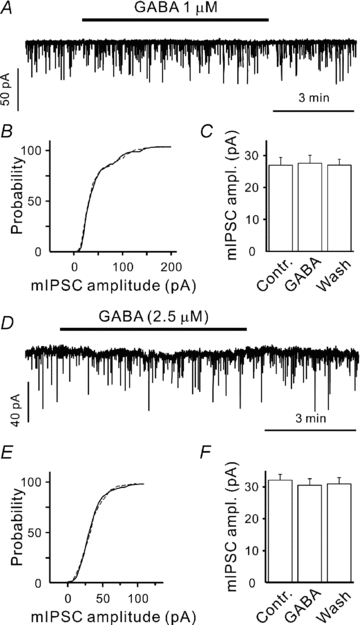
A and D, sample traces showing mIPSC recordings in control and in thepresence of 1 μm (A) or 2.5 μm (D) exogenous GABA. B and E, cumulative histograms demonstrating that 1 μm (B) and 2.5 μm (E) exogenous GABA do not affect mIPSC distribution (continuous line – control, dashed line – in thepresence of GABA). C and F, statistical data showing that there are no effects of 1 and 2.5 μm exogenous GABA on the median mIPSC amplitudes (n= 8 in both cases). In these experiments, GAT-1 and GAT-2/3 were blocked by NO-711 (10 μm) and SNAP-5114 (40 μm), respectively.
Data evaluation and statistics
Data were evaluated off-line using Tida 4.11 (HEKA Elektronik) or PeakCount V3.2 software (C. Henneberger, Institute of Neurophysiology, Berlin). All results arepresented as means ±s.e.m. The error bars in all figures indicate s.e.m. Differences between means were tested for significance using Student's paired t test, unless otherwise stated.
Results
Blockade of GAT-1 and GAT-2/3
In ourprevious work (Kirmse & Kirischuk, 2006a), we have shown that GABA released through GATs suppresses GABAergic synaptic transmission in neocortical layer I via activation ofpresynaptic GABABRs. Figure 2 shows that co-application of NO-711 (10 μm), a GAT-1 antagonist, and SNAP-5114 (40 μm), a GAT-2/3 blocker, completely eliminated GABABR-mediated inhibition of GABA release. In this set of experiments we recorded from four P2–3 and four P5–7 CR cells. As we did not observe any difference between these two age groups, the data were pooled. CGP55845 (1 μm) failed to influence the mean eIPSC amplitude (0.87 ± 0.03 and 0.89 ± 0.05 of control, n= 8, P > 0.4), PPR (0.85 ± 0.05 and 0.86 ± 0.04, n= 8, P > 0.75) and failure rate (0.15 ± 0.02 and 0.17 ± 0.02, n= 8, P > 0.2, in thepresence of NO-711 plus SNAP-5114 and NO-711 plus SNAP-5114 plus CGP55845, respectively). The observed decrease of the mean eIPSC amplitude in thepresence of NO-711 and SNAP-5114 reflects the fact that GAT-1 provides GABA for the filling ofpresynaptic vesicles. GAT-1 blockade reduces the median mIPSC amplitude (to 0.82 ± 0.05 and to 0.75 ± 0.06 of control at P2–3 (this study, data not shown) and at P5–7 (Kirmse & Kirischuk, 2006a), respectively). Thus, these results suggest that GABABR activation is mainly determined by GAT activity.
Figure 2. Blockade of GAT-1 and GAT-2/3 eliminatespresynaptic GABABR-mediated inhibition.
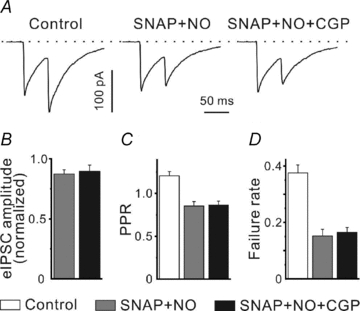
A, sample traces showing eIPSCs elicited by paired-pulse stimulation in control, in thepresence of SNAP-5114 (SNAP, 40 μm) plus NO-711 (NO, 10 μm) and SNAP-5114 plus NO-711 plus CGP55845 (CGP, 1 μm). Inter-stimulus interval was set to 50 ms. B–D, statistical data demonstrating that CGP55845 fails to influence the mean eIPSC amplitude (B), PPR (C) and failure rate (D) in thepresence of GAT blockers.
Next, exogenous GABA was applied in thepresence of NO-711 and SNAP-5114. Because NO-711 induces a GABABR-independent decrease of the mean eIPSC amplitude, the latter obtained under control conditions cannot be directly used as a measure of GABABR activation in thepresence of NO-711. However, Fig. 3A shows that 125 nm GABA significantly decreased the mean eIPSC amplitude at P5–7 (to 0.69 ± 0.08 of control, n= 5, P < 0.05), while 250 nm GABA was required to induce a similar eIPSC reduction at P2–3 (0.67 ± 0.04 of control, n= 6, P < 0.01, one population Student's t test). PPRs and failure rates, which can be assumed to be less sensitive to the NO-711-induced reduction of the quantal amplitude as compared to the mean eIPSC amplitude, confirmed this observation. At P2–3, neither PPR (1.18 ± 0.06 and 1.13 ± 0.05) nor the failure rates (0.27 ± 0.03 and 0.27 ± 0.02, n= 6, minimal P > 0.5) significantly differed in control and in thepresence of 250 nm GABA (Fig. 3B and C). At P5–7, both PPR (1.18 ± 0.08 and 1.20 ± 8) and the failure rates (0.32 ± 0.04 and 0.33 ± 0.05, n= 5, minimal P > 0.6) were similar in control and in thepresence 125 nm GABA (Fig. 3B and C).
Figure 3. eIPSCs at P2–3 and P5–7 differ in their sensitivity to exogenous GABA when both GAT-1 and GAT-2/3 are blocked.
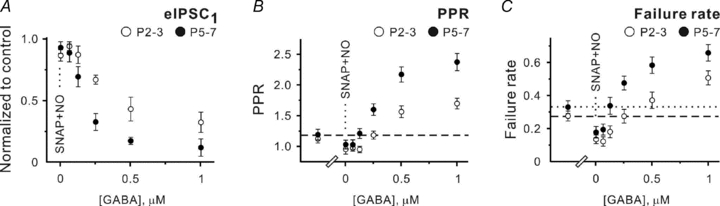
A–C, statistical data showing the effects of SNAP-5114 (40 μm) plus NO-711 (10 μm) and different GABA concentrations applied in thepresence of SNAP-5114 plus NO-711 on the mean eIPSC amplitude (A), PPR (B) and failure rates (C) at P2–3 (open symbols) and P5–7 (filled symbols). Each point represents a mean value obtained in at least five experiments.
However, one cannot exclude the possibility that extracellular GABA diffusion and/or degradation undergoes developmental changes. Consequently, juxtasynaptic [GABA] might be similar at P2–3 and P5–7, but the applied GABA concentrations would differ. To address this possibility, baclofen, a specific GABAB receptor agonist, was applied in thepresence of SNAP-5114 (40 μm) and NO-711 (10 μm). Figure 4 shows that like for GABA, dose–response curves for baclofen differ at P2–3 and P5–7. Baclofen at 250 nm significantly inhibited GABA release at P5–7, while 500 nm baclofen was required at P2–3 to produce similar effects. Thus, these results support the hypothesis that [GABA]o decreases during the first postnatal week. We conclude that 250 and 125 nm exogenous GABA mimics the control [GABA]o at P2–3 and P5–7, respectively, when GAT-1 and GAT-2/3 are blocked.
Figure 4. Dose–response curves for baclofen differ at P2–3 and P5–7.
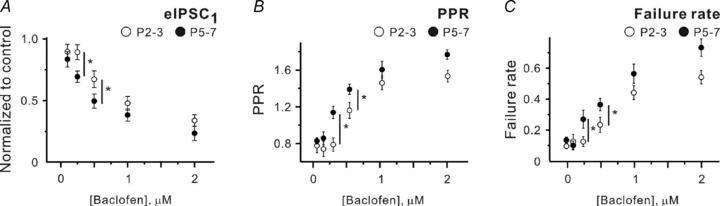
A–C, statistical data showing the effects of different baclofen concentrations applied in thepresence of SNAP-5114 plus NO-711 on the mean eIPSC amplitude (A), PPR (B) and failure rates (C) at P2–3 (open symbols) and P5–7 (filled symbols). Each point represents a mean value obtained in at least five experiments. *P < 0.05 (unpaired Student's t test).
Blockade of glutamate decarboxylase (GAD) and GATs
Definitely, one cannot exclude the possibility that an unidentified mechanism(s) contributes to [GABA]o setting in addition to GATs. To inspect this suggestion, we performed an additional set of experiments applying SNAP-5114 plus NO-711 in thepresence of 3-mercaptopropionic acid (MPA), a GAD inhibitor. The following protocol was used in this set of experiments: (1) brain slices werepre-incubated in the ACSF supplemented with 1 mm MPA for >1 h, and (2) the ACSF contained 100 μm MPA throughout the experiments. Because of the reasonably long time for MPApre-incubation required to induce [GABA]o reduction (about 1 h; Golan & Grossman, 1996), it was technically not possible to obtain the control values. However, PPRs (0.81 ± 0.06 at P2–3 and 0.86 ± 0.03 at P5–7) and failure rates (0.17 ± 0.02 at P2–3 (n= 9) and 0.15 ± 0.02 at P5–7 (n= 10)) in thepresence of MPA were significantly lower than the control values obtained inprevious experiments (Fig. 3). Moreover, application of SNAP-5114 and NO-711 in thepresence of MPA failed to affect PPRs and failure rates (Fig. 5B and C). These results confirm that MPA treatment reduced [GABA]i and suppressed GAT-mediated GABA release. If a hypothesized mechanism of GABA release depends on [GABA]i, the amount of exogenous GABA required to mimic the control conditions in thepresence of MPA plus NO-711 plus SNAP-5114 should deviate from the values obtained in thepresence of NO-711 and SNAP-5114 only. However, it was not the case. Figure 5A shows that 125 nm exogenous GABA significantly decreased the mean eIPSC amplitude at P5–7 (to 0.68 ± 0.05 of control, n= 8, P < 0.05), while 250 nm GABA was required to induce a similar effect at P2–3 (0.68 ± 0.03 of control, n= 6, P < 0.01). Similarly, PPRs and failure rates were close to the control values taken as the mean values from experiments with SNAP-5114 plus NO-711 and SNAP-5114 (Figs 3 and 5, see below). At P5–7, the corresponding numbers were 1.21 ± 0.06 (n= 18) versus 1.22 ± 0.06 (n= 8) and 0.29 ± 0.03 (n= 18) versus 0.27 ± 0.05 (n= 8) for PPRs and failure rates in control and in thepresence of 125 nm GABA, respectively (minimal P > 0.7, unpaired Student's t test, Fig. 5B and C). At P2–3, the appropriate values were 1.23 ± 0.04 (n= 22) versus 1.25 ± 0.07 (n= 7) and 0.28 ± 0.04 (n= 22) versus 0.27 ± 0.04 (n= 7) for PPRs and failure rates in control and in thepresence of 250 nm GABA, respectively (minimal P > 0.7, unpaired Student's t test, Fig. 5B and C). Thus, we conclude that an existence of another GABA releasing mechanism is rather unlikely in thispreparation and the ambient [GABA]o is about 250 and 125 nm at P2–3 and P5–7, respectively.
Figure 5. GAD blockade by mercaptopropionic acid (MPA) does not change the sensitivity of eIPSCs to exogenous GABA when both GAT-1 and GAT-2/3 are blocked.
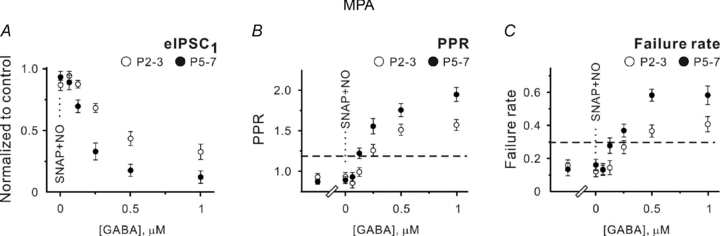
A–C, statistical data showing the effects of SNAP-5114 (40 μm) plus NO-711 (10 μm) and different GABA concentrations applied in thepresence of SNAP-5114 plus NO-711 on the mean eIPSC amplitude (A), PPR (B) and failure rates (C) at P2–3 (open symbols) and P5–7 (filled symbols). Each point represents a mean value obtained in at least five experiments. MPA (100 μm) waspresent in the extracellular saline throughout the experiments. Dashed lines show the control values taken from the experiments with SNAP-5114 plus NO-711 and SNAP-5114.
Blockade of GAT-2/3 with SNAP-5114
During perinatal development GAT-2/3 expression is much higher than that of GAT-1, although expression levels of all GATs rise during the first postnatal weeks (Conti et al. 2004). To examine if there is any change in relative contributions of GATs to [GABA]o control, only GAT-2/3 was blocked by 40 μm SNAP-5114 in the next set of experiments. In both age groups, SNAP-5114 application increased the mean amplitude of eIPSCs. Corresponding normalized values were 1.78 ± 0.16 at P2–3 (n= 10, P < 0.001) and 1.69 ± 0.13 at P5–7 (n= 9, P < 0.01, one population Student's t test). PPR decreased from 1.19 ± 0.08 to 0.71 ± 0.05 at P2–3 (n= 10, P < 0.001) and from 1.23 ± 0.09 to 0.81 ± 0.10 at P5–7 (n= 9, P < 0.001). Failure rates decreased from 0.3 ± 0.08 to 0.09 ± 0.02 at P2–3 (n= 10, P < 0.01) and from 0.27 ± 0.03 to 0.08 ± 0.03 at P5–7 (n= 9, P < 0.01, Fig. 6A–C). Addition of 0.5 μm GABA to the extracellular solution returned all three parameters close to their control values. The mean eIPSC amplitudes were 0.99 ± 0.17 of control at P2–3 (n= 6, P= 0.87) and 0.92 ± 0.07 at P5–7 (n= 6, P= 0.17, one population Student's t test). PPRs were 1.23 ± 0.09 (1.17 ± 0.11 in control, n= 6, P= 0.57) at P2–3 and 1.16 ± 0.09 (1.15 ± 0.09 in control, n= 6, P= 0.81) at P5–7. The failure rates were 0.34 ± 0.04 versus 0.31 ± 0.12 in control at P2–3 (n= 6, P= 0.74) and 0.25 ± 0.04 versus 0.23 ± 0.03 in control at P5–7 (n= 6, P= 0.59). Moreover, in thepresence of 250 nm exogenous GABA, all three parameters were significantly (P < 0.05) different from their control values (Fig. 6A–C). These results demonstrate that GAT-1 can significantly decrease [GABA]o. Moreover, comparing the data obtained with SNAP-5114 (Fig. 6) and SNAP-5114 plus NO-711 (Fig. 3), one may notice that GAT-1 blockade shifts the dose–response curves to the left stronger at P5–7 than at P2–3 suggesting that the balance between GAT-mediated GABA release and uptake is developmentally regulated and might underlie the observed [GABA]o decrease at P5–7.
Figure 6. GAT-2/3-mediated GABA release can be mimicked by 0.5 μm exogenous GABA.
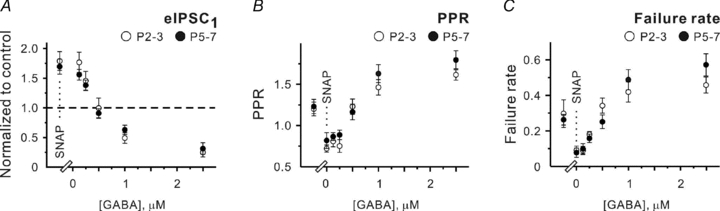
A–C, statistical data showing the effects of SNAP-5114 (SNAP, 40 μm) and different GABA concentrations applied in thepresence of SNAP-5114 on the mean eIPSC amplitude (C), PPR (D) and failure rates (E) at P2–3 (open symbols) and P5–7 (filled symbols). Each point represents a mean value obtained in at least six experiments.
Discussion
Local [GABA]o has been proposed to play an important role in corticogenesis. The majority of quantitative [GABA]o measurements were performed using a brain microdialysis technique. The latter is a powerful method, but because of a relatively large size of the sampling probe, it can provide only a spatially averaged [GABA]o. Nevertheless, data obtained have demonstrated that [GABA]o could vary from several nanomolar to several micromolar (Lerma et al. 1986; Biggs et al. 1992; Rowley et al. 1995; Rakovska et al. 1998). Alternatively, one can use so-called ‘sniffer cells’, i.e. a non-neuronal cell line expressing high affinity GABAARs, to measure [GABA]o. Using this approach, [GABA]o was reported to be about 0.5 μm in the cortical organotypic cultures and acutelyprepared slices from E14.5 mouse embryos (Cuzon et al. 2006). The spatial resolution of this method is determined by the ‘sniffer cell’ size, but, on the other hand, [GABA]o can be measured only near the surface of the slice and, definitely, the obtained values can significantly differ from [GABA]o within the slice. In this study, we suggested usingpresynaptic GABABRs as a detector of [GABA]o in the marginal zone/layer I of the neocortex. The results obtained show that [GABA]o in the vicinity of GABAergic synapses on CR cells amounts to 250 nm at P2–3 and 125 nm at P5–7.
Validity of the applied approach
The first question is whether GABABRs are sensitive enough to correctly report [GABA]o. This seems to be the case, while the EC50 of GABA for GABABRs was reported to be about 0.3 μm (Chu et al. 1990; Asay & Boyd, 2006). This value is quite close to the [GABA]o levels observed in this study. Another concern is the amount of endogenous GABA remaining in the extracellular space in thepresence of GAT blockers. Because CGP55845 failed to affect GABAergic transmission in thepresence SNAP-5114 plus NO-711 (Fig. 2), GABABRs appear to be not activated under these conditions. However, in all sets of experiments small (about 100 nm) changes in exogenous GABA concentration strongly influenced the strength of GABAergic transmission. In addition, GAD blockade with MPA strongly decreased [GABA]i and in turn [GABA]o, but did not modify the dose–response curves obtained in thepresence of SNAP-5114 and NO-711. A shift to the left would be expected if the residual [GABA]o levels were decreased in thepresence of MPA. Definitely, the residual [GABA]o is not nil even in thepresence of both GAT and GAD blockers, but it looks reasonable to suggest that in the worst case the residual [GABA]o is comparable with the lowest GABA concentration applied (about 100 nm).
Secondly, low concentrations of exogenous GABA may influence postsynaptic GABAA receptors resulting, for instance, in their partial desensitization (Overstreet et al. 2000). However, Fig. 1 shows that even the highest GABA concentration used (2.5 μm) failed to decrease the median mIPSC amplitude suggesting that this effect is rather small. Thirdly, as all experiments were performed on GABAergic synapses, not only exogenous but also synaptically released GABA can activatepresynaptically located GABABRs (Kirmse & Kirischuk, 2006a) and in turn influence PPR. This effect is expected to be stronger when GATs are blocked. However, two other selected parameters (the mean eIPSC amplitude and failure rate) should not be dependent on this autocrine GABA action. As all three parameters demonstrated similar dependence on exogenous GABA concentration, we suggest that the autocrine action of GABA is rather minor. Fourthly, NO-711-induced decrease in the median mIPSC amplitude may have a consequence that small synaptic events will fall below the detection threshold. This can potentially affect all three selected parameters. However, the mean amplitudes of eIPSCs were not particularly small at P2–3 and P5–7 both in controls (147 ± 17, n= 22, and 131 ± 21 pA, n= 19, respectively) and in thepresence of SNAP-5114 plus NO-711 (123 ± 23, n= 12, and 106 ± 18 pA, n= 10, respectively). Moreover, despite the fact that the NO-711-induced reduction of the quantal size was comparable at P2–3 and P5–7, the dependence of selected parameters on exogenous GABA and baclofen concentrations differed in two age groups (Figs 3–5) suggesting that the sensitivity of our approach was only slightly influenced by NO-711-induced reduction of the quantal amplitude. Finally, all experiments were performed at room temperature. Because the activity of GATs is strongly dependent on temperature, one can ask whether the obtained [GABA]o values are physiologically relevant. However, in ourprevious study we demonstrated that the strength of GABABR-mediated inhibition of GABA release was comparable at room and near physiological temperatures (Kirmse & Kirischuk, 2006a). These results allow the suggestion that the [GABA]o levels at room and near physiological temperatures are also comparable.
[GABA]o in the neocortical layer I
In theprevious study (Kirmse & Kirischuk, 2006a), we have shown that GAT-1 and GAT-2/3 play different roles in controlling GABAergic synaptic transmission in neocortical layer I. GAT-2/3 blockade reduces PPR and does not influence the median mIPSC amplitude suggesting that GAT-2/3 releases GABA and is located perisynaptically. GAT-1 blockade also decreases PPR, but, in contrast to GAT-2/3, this effect is accompanied by a decrease of the median mIPSC amplitude. As low [GABA]o does not desensitize postsynaptic GABAARs in thispreparation, the observed reduction of mIPSC amplitude indicates that at leastpresynaptic GAT-1 operates in the uptake mode and provides GABA for vesicle filling. On the other hand, the GAT-1 blockade-induced reduction in PPR allows the suggestion that GAT-1 releases GABA, i.e. operates in the reverse mode. This discrepancy could be explained by suggesting that there are two different locations of GAT-1. One is neuronal andpresynaptic; the other may be glial (Minelli et al. 1995). Unfortunately, theprecise spatial distribution of GAT-2/3 and GAT-1 is still unknown.
In thepresence of SNAP-5114, 0.5 μm exogenous GABA was required to mimic the control strength of GABABR-mediated inhibition in both investigated age groups (Fig. 6). Similar (0.5 μm) [GABA]o levels have been reported in embryonic cortical slices using the ‘sniffer cell’ technique (Cuzon et al. 2006). Because ‘sniffer cells’ are larger than synapses, this similarity might mean that extrasynaptic GAT-2/3 controls the global [GABA]o in the neocortex, while synaptically located GAT-1 tunes [GABA]o more locally. Indeed, when both GATs were blocked, the amount of exogenous GABA required to mimic the control levels of GABABR-mediated inhibition was significantly smaller. Moreover, it was age dependent (250 and 125 nm at P2–3 and P5–7, respectively). Definitely, one cannot exclude that there is another, as yet unknown, mechanism that releases or takes up GABA. However, it is reasonable to suggest that if a mechanism exists, its strength should be dependent on [GABA]i. However, MPA failed to change exogenous GABA concentrations necessary to mimic the controls in thepresence of SNAP-5114 and NO-711 (Figs 3, 4). As tonic GABABR-mediated inhibition persists in thepresence of tetrodotoxin, an antagonist of voltage-sensitive Na+ channels (Kirmse & Kirischuk, 2006a), synaptic activity seems to play a minor role in controlling [GABA]o in thispreparation as well. Thus, we conclude that [GABA]o is mainly determined by the activities of GAT-2/3 and GAT-1. [GABA]o in the vicinity of GABAergic synapses on CR cells amounts to 250 nm at P2–3 and 125 nm at P5–7. As [GABA]o in both age groups appears to be controlled by GATs, we suggest that the equilibrium between GAT-mediated release and uptake is developmentally regulated. Mechanisms underlying the observed developmental decrease of [GABA]o are unclear. GAT-1 and GAT-2/3 expressions may be differently regulated during the first postnatal week leading to a relative potentiation of GABA uptake (Conti et al. 2004). Alternatively, as GATs co-transport GABA with Na+ and Cl−, developmental regulation of chloride transporters may differentially influence GAT-1 and GAT-2/3 activities altering the balance between GABA release and uptake. Further experiments are required to have this question answered.
Physiological implication
GABA is not only the main inhibitory neurotransmitter in the adult mammalian brain, but also functions as a trophic factor during neuronal maturation even before synapses are formed (for review see Owens & Kriegstein, 2002; Represa & Ben-Ari, 2005). In the rodent neocortex, GABA is alreadypresent at embryonic day 14 (Van Eden et al. 1989).Precursor cells in the neocortical proliferative zone express functional GABAA receptors which are activated by endogenous GABA (LoTurco et al. 1995). Pharmacological block of various GABA receptors (GABAA, GABAC and GABAB) influences the motility of migrating cells suggesting that GABA receptors are functional at this developmental stage (Behar et al. 1996; Manent et al. 2005). Local in vivo application of the GABAA antagonist bicuculline methiodide or the agonist muscimol via cortical surface Elvax implants has been shown to induce prominent alterations in the cortical architecture. Both bicuculline- and muscimol-treated animals revealed heterotopic cell clusters in the upper layers and a complete loss of the cortical lamination in the region underlying the Elvax implant (Heck et al. 2007). These data show that at least radial cell migration in the neocortex requires that [GABA]o in the marginal zone/layer I is high enough to activate GABAA receptors but not too high to desensitize them. If such conditions are achieved, [GABA]o seems to serve as a ‘stop’ signal for radial cell migration. As radial cell migration is finished by P4 in rodents, we hypothesize that the observed [GABA]o decrease at P5–7 might reflect an elimination of the ‘stop’ signal which is not necessary any longer.
Acknowledgments
The technical assistance of Mrs Kerstin Rückwardt is highly appreciated. This study was supported by Charité (Personal Grant) and Deutsche Forschungsgemeinschaft (DFG, KI1093/1-2) to S.K.
Glossary
Abbreviations
- CR cell
Cajal–Retzius cell
- GAD
glutamate decarboxylase
- GAT
GABA transporter
- PPR
paired-pulse ratio
Author contributions
A.D., O.M. and P.U. contributed to data collection and analysis. K.K. and S.K. contributed to the conception and design of experiments, and the drafting of the article as well as revising it critically for important intellectual content. All authors have approved the final version of the manuscript.
References
- Asay MJ, Boyd SK. Characterization of the binding of [3H]CGP54626 to GABAB receptors in the male bullfrog (Rana catesbeiana) Brain Res. 2006;1094:76–85. doi: 10.1016/j.brainres.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar TN, Li YX, Tran HT, Ma W, Dunlap V, Scott C, Barker JL. GABA stimulates chemotaxis and chemokinesis of embryonic cortical neurons via calcium-dependent mechanisms. J Neurosci. 1996;16:1808–1818. doi: 10.1523/JNEUROSCI.16-05-01808.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs CS, Pearce BR, Fowler LJ, Whitton PS. The effect of sodium valproate on extracellular GABA and other amino acids in the rat ventral hippocampus: an in vivo microdialysis study. Brain Res. 1992;594:138–142. doi: 10.1016/0006-8993(92)91038-g. [DOI] [PubMed] [Google Scholar]
- Borden LA. GABA transporter heterogeneity: pharmacology and cellular localization. Neurochem Int. 1996;29:335–356. doi: 10.1016/0197-0186(95)00158-1. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Cull-Candy SG, Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J Physiol. 1996;497:753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelier P, Attwell D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J Physiol. 2005;564:397–410. doi: 10.1113/jphysiol.2004.082131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu DC, Albin RL, Young AB, Penney JB. Distribution and kinetics of GABAB binding sites in rat central nervous system: a quantitative autoradiographic study. Neurosci. 1990;34:341–357. doi: 10.1016/0306-4522(90)90144-s. [DOI] [PubMed] [Google Scholar]
- Conti F, Minelli A, Melone M. GABA transporters in the mammalian cerebral cortex: localization, development and pathological implications. Brain Res Brain Res Rev. 2004;45:196–212. doi: 10.1016/j.brainresrev.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Cuzon VC, Yeh PW, Cheng Q, Yeh HH. Ambient GABA promotes cortical entry of tangentially migrating cells derived from the medial ganglionic eminence. Cereb Cortex. 2006;16:1377–1388. doi: 10.1093/cercor/bhj084. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Mechanism and kinetics of heterosynaptic depression at a cerebellar synapse. J Neurosci. 1997;17:9048–9059. doi: 10.1523/JNEUROSCI.17-23-09048.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan H, Grossman Y. Block of glutamate decarboxylase decreases GABAergic inhibition at the crayfish synapses: possible role ofpresynaptic metabotropic mechanisms. J Neurophysiol. 1996;75:2089–2098. doi: 10.1152/jn.1996.75.5.2089. [DOI] [PubMed] [Google Scholar]
- Heck N, Kilb W, Reiprich P, Kubota H, Furukawa T, Fukuda A, Luhmann HJ. GABA-A receptors regulate neocortical neuronal migration in vitro and in vivo. Cereb Cortex. 2007;17:138–148. doi: 10.1093/cercor/bhj135. [DOI] [PubMed] [Google Scholar]
- Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. J Neurosci. 2007;27:9736–9741. doi: 10.1523/JNEUROSCI.3009-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Chiu CS, Sokolova I, Lester HA, Mody I. GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J Neurophysiol. 2003;90:2690–2701. doi: 10.1152/jn.00240.2003. [DOI] [PubMed] [Google Scholar]
- Kirmse K, Dvorzhak A, Henneberger C, Grantyn R, Kirischuk S. Cajal–Retzius cells in the mouse neocortex receive two types ofpre- and postsynaptically distinct GABAergic inputs. J Physiol. 2007;585:881–895. doi: 10.1113/jphysiol.2007.145003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmse K, Kirischuk S. Ambient GABA constrains the strength of GABAergic synapses at Cajal-Retzius cells in the developing visual cortex. J Neurosci. 2006a;26:4216–4227. doi: 10.1523/JNEUROSCI.0589-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmse K, Kirischuk S. N-Ethylmaleimide increases release probability at GABAergic synapses in layer I of the mouse visual cortex. Eur J Neurosci. 2006b;24:2741–2748. doi: 10.1111/j.1460-9568.2006.05179.x. [DOI] [PubMed] [Google Scholar]
- Kombian SB, Zidichouski JA, Pittman QJ. GABAB receptorspresynaptically modulate excitatory synaptic transmission in the rat supraoptic nucleus in vitro. J Neurophysiol. 1996;76:1166–1179. doi: 10.1152/jn.1996.76.2.1166. [DOI] [PubMed] [Google Scholar]
- Le Feuvre Y, Fricker D, Leresche N. GABAA receptor-mediated IPSCs in rat thalamic sensory nuclei: patterns of discharge and tonic modulation by GABAB autoreceptors. J Physiol. 1997;502:91–104. doi: 10.1111/j.1469-7793.1997.091bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J, Herranz AS, Herreras O, Abraira V, Martin del Rio R. In vivo determination of extracellular concentration of amino acids in the rat hippocampus. A method based on brain dialysis and computerized analysis. Brain Res. 1986;384:145–155. doi: 10.1016/0006-8993(86)91230-8. [DOI] [PubMed] [Google Scholar]
- LoTurco JJ, Owens DF, Heath MJ, Davis MB, Kriegstein AR. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15:1287–1298. doi: 10.1016/0896-6273(95)90008-x. [DOI] [PubMed] [Google Scholar]
- Manent JB, Demarque M, Jorquera I, Pellegrino C, Ben-Ari Y, Aniksztejn L, Represa A. A noncanonical release of GABA and glutamate modulates neuronal migration. J Neurosci. 2005;25:4755–4765. doi: 10.1523/JNEUROSCI.0553-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minelli A, Brecha NC, Karschin C, DeBiasi S, Conti F. GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J Neurosci. 1995;15:7734–7746. doi: 10.1523/JNEUROSCI.15-11-07734.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Mody I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J Neurophysiol. 2002;87:2624–2628. doi: 10.1152/jn.2002.87.5.2624. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Jones MV, Westbrook GL. Slow desensitization regulates the availability of synaptic GABAA receptors. J Neurosci. 2000;20:7914–7921. doi: 10.1523/JNEUROSCI.20-21-07914.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- Rakovska A, Giovannini MG, Della CL, Kalfin R, Bianchi L, Pepeu G. Neurotensin modulation of acetylcholine and GABA release from the rat hippocampus: an in vivo microdialysis study. Neurochem Int. 1998;33:335–340. doi: 10.1016/s0197-0186(98)00036-9. [DOI] [PubMed] [Google Scholar]
- Represa A, Ben-Ari Y. Trophic actions of GABA on neuronal development. Trends Neurosci. 2005;28:278–283. doi: 10.1016/j.tins.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Rowley HL, Martin KF, Marsden CA. Determination of in vivo amino acid neurotransmitters by high-performance liquid chromatography with o-phthalaldehyde-sulphite derivatisation. J Neurosci Methods. 1995;57:93–99. doi: 10.1016/0165-0270(94)00132-z. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABAA receptors: modulating gain and maintaining the tone. Trends Neurosci. 2004;27:262–269. doi: 10.1016/j.tins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Stell BM, Mody I. Receptors with different affinities mediate phasic and tonic GABAA conductances in hippocampal neurons. J Neurosci. 2002;22:RC223. doi: 10.1523/JNEUROSCI.22-10-j0003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerstedt U. Microdialysis: principles and applications for studies in animals and man. J Intern Med. 1991;230:365–373. doi: 10.1111/j.1365-2796.1991.tb00459.x. [DOI] [PubMed] [Google Scholar]
- Van Der Zeyden M, Oldenziel WH, Rea K, Cremers TI, Westerink BH. Microdialysis of GABA and glutamate: Analysis, interpretation and comparison with microsensors. Pharmacol Biochem Behav. 2008;90:135–147. doi: 10.1016/j.pbb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Van Eden CG, Mrzljak L, Voorn P, Uylings HB. Prenatal development of GABA-ergic neurons in the neocortex of the rat. J Comp Neurol. 1989;289:213–227. doi: 10.1002/cne.902890204. [DOI] [PubMed] [Google Scholar]