

Abstract
The physiological role of the cystic fibrosis transmembrane conductance regulator (CFTR) in cardiomyocytes remains unclear. Using spontaneously beating neonatal ventricular cardiomyocytes from wild-type (WT) or CFTR knockout (KO) mice, we examined the role of CFTR in the modulation of cardiomyocyte contraction rate. Contraction rates of spontaneously beating myocytes were captured by video imaging. Real-time changes in intracellular ([Ca2+]i) and protein kinase A (PKA) activity were measured by fura-2 and fluorescence resonance energy transfer, respectively. Acute inhibition of CFTR in WT cardiomyocytes using the CFTR inhibitor CFTRinh-172 transiently inhibited the contraction rate. By contrast, cardiomyocytes from CFTR KO mice displayed normal contraction rates. Further investigation revealed that acute inhibition of CFTR activity in WT cardiomyoctyes activated L-type Ca2+ channels, leading to a transient increase of [Ca2+]i and inhibition of PKA activity. Additionally, we found that contraction rate normalization following acute CFTR inhibition in WT cardiomyocytes or chronic deletion in cardiomyocytes from CFTR KO mice requires the activation of Ca2+/calmodulin-dependent kinase II (CaMKII) and Ca2+-activated Cl− channels (CaCC) because simultaneous addition of myristoylated-autocamtide-2-related inhibitory peptide or niflumic acid and CFTRinh-172 to WT cardiomyocytes or treatment of cardiomyoctes from CFTR KO mice with these agents caused sustained attenuation of contraction rates. Our results demonstrate that regulation of cardiomyocyte contraction involves CFTR. They also reveal that activation of CaMKII and CaCC compensates for loss of CFTR function. Increased dependence on CaMKII upon loss of CFTR function might leave cystic fibrosis patients at increased risk of heart dysfunction and disease.
Introduction
To sustain proper physiological function, the heart maintains tight control of ion channels and transporters. Four major Cl− currents have been described in the heart, including the cystic fibrosis transmembrane conductance regulator (CFTR), Ca2+-activated Cl− channels (CaCC), swelling-activated Cl− channels and inwardly rectifying Cl− channels. Current thinking is that CFTR, swelling-activated Cl− channels and CaCC share a similar role in maintaining the resting membrane potential and minimizing action potential prolongation associated with stimulation of inward Ca2+ current. As a result, these Cl− channels may be arrythmogenic and block hypercontraction through prevention of intracellular Ca2+ concentration ([Ca2+]i) overload (Yamawake et al. 1992; Collier et al. 1996; Hiraoka et al. 1998).
Shortly after the identification of the CFTR gene from epithelia, a cAMP- or PKA-dependent Cl− channel resembling CFTR was identified in cardiomyocytes (Nagel et al. 1992). Subsequently, expression of a cardiac-specific isoform of CFTR (cCFTR) was reported in atrial and ventricular myocytes of numerous species, including humans (Tilly et al. 1996; Warth et al. 1996; Duan et al. 1999; Gao et al. 2007). This isoform arises from alternative splicing, resulting in loss of a portion of the first intracellular loop, but otherwise retains >95% sequence homology to epithelial CFTR. The cardiac-specificisoform of CFTR behaves in a similar manner to epithelial CFTR and is activated and inhibited by the same agents that affect epithelial CFTR. Additionally, clinical mutations of CFTR, which cause the disease cystic fibrosis (CF), lie outside of this region. While no clear physiological role for CFTR in the heart has been firmly established, it has been suggested that, as a Cl− channel, CFTR may be involved in the autonomic regulation of action potential duration by minimizing action potential prolongation associated with β-adrenergic stimulation (Duan et al. 2005). Studies showing that CFTR is upregulated during ischaemia (Uramoto et al. 2003) and that CFTR KO mice lose the protection of ischaemic preconditioning (Chen et al. 2004) suggest that CFTR plays an important role in cardiomyocytes. While the aforementioned studies have provided important insight into the potential roles of CFTR in the heart, we lack full understanding of the physiological role(s) of CFTR and the mechanism(s) employed to fulfil its functions.
Thus, the goal of the present study was to determine whether loss of CFTR affects ventricular cardiomyocyte function by examining the effects of CFTR loss on spontaneously beating neonatal cardiomyocytes. In addition to contraction rate measurements, we monitored real-time changes in [Ca2+]i and protein kinase A (PKA) activity, and CFTR-dependent changes in Ca2+/calmodulin-dependent kinase II (CaMKII) activity. These experiments showed that CFTR is involved in the regulation of cardiomyocyte contraction rate. However, acute or chronic loss of CFTR was compensated for by activation of CaMKII and CaCC.
Methods
Ethical approval of animal use
FV/B mice were obtained from Charles River Laboratory (Wilmington, MA, USA) and used for generation of wild-type (WT) mice. The CFTR KO mice (strain 2515 B6.129S6-Cftrtm1Kth/J; Zhou et al. 1994) were initially obtained from Dr Deborah Nelson at the University of Chicago (Chicago, IL, USA) and bred in-house. All animals were used within 24 h of birth. In the present study a total of 201 WT and 54 CFTR KO mice were used. Use of animals was approved by the University of Illinois at Urbana-Champaign IACUC and is in accordance with The Journal of Physiology guidelines (Drummond, 2009).
Isolation and culture of neonatal mouse cardiomyocytes
Ventricular myocytes were cultured from neonatal mice using a similar protocol to that previously reported (Xiang et al. 2005). Newborn mice were killed by decapitation with surgical scissors. Decapitation was used so that isolated myocytes would not be affected by anaesthetics or altered environment (redox and pH (CO2)). This procedure is justified to yield viable myocytes after isolation, because both sedation and anaesthetics significantly reduce the survival rate ofisolated heart cells from neonates. Decapitation of neonates avoiding prior anaesthesia was done in compliance with the Report of the AVMA Panel on Euthanasia JACMA, Col 218, No. 5, 1 March 2001, pp. 682–683 (http://www.avma.org/resources/euthanasia.pdf) and the NIH ARAC Guidelines for the Euthanasia of Mouse and Rat Fetuses and Neonates (http://oacu.od.nih.gov/ARAC/euthmous.htm). Neonatal mouse cardiac tissue devoid of atria were exposed to enzymatic digestion. Subsequently, isolated cells were pipetted onto 35 mm Petri dishes precoated with 1.5% gelatin type A or laminin. Cells were plated at high and low density for measurement of contraction rate or real-time imaging experiments, respectively. High-density plating allowed cells to form a syncytium, whereas low-density plating favoured individual quiescent myocytes. Cells were incubated at 37°C for 2–3 days until they displayed appropriate myocyte morphology or formed a syncytium for contraction rate measurements. Culture media (containing serum and 20 mm Hepes for buffering) was replaced daily and at least 1 h prior to experiments.
Measurement of contraction rate
Measurement of myocyte contraction rate was performed as previously reported (Xiang et al. 2002). The 35 mm Petri dishes were placed in a temperature- and CO2-regulated apparatus. Using a CCD camera (Photometrics, Tucson, AZ, USA) and MetaMorph imaging software (Molecular Devices, Downington, PA, USA), 5 s video images were obtained every 2 min. Following acquisition, a specific point within the field was selected and analysed for movement in each x, y and z plane. Movements in the x and y plane were calculated and number of deflections noted as number of contractions. Contraction velocity measurements were also recorded. Each data point represents a single observation from different experiments so that only one data set was obtained from each experiment. Each drug was tested using multiple isolations to ensure that the observed responses were not particular to a single isolation.
Measurement of [Ca2+]i
Quiescent ventricular myocytes were cultured at low density as described in ‘Isolation and culture of neonatal mouse cardiomyocytes’. Upon formation of myocyte morphology (24–48 h after culture), culture media was removed and cells were washed with 1× Dulbecco's phosphate-buffered solution (DPBS), after which cells were loaded with 5 μm fura-2 AM dissolved in 0.01% pluronic F-127 plus 0.1% DMSO in DPBS at room temperature for 30 min. Following loading, cells were washed with 1× DPBS or Ca2+-free DPBS [with EGTA (500 μm) to remove residual free Ca2+] and left untreated for 30 min to remove any unloaded dye and allow for complete de-esterification of fura-2 AM into fura-2, its active form. For Ca2+-free experiments, experiments were performed in Ca2+-free DPBS with EGTA. Changes in [Ca2+]i were monitored by fura-2 excitation at 340 and 380 nm and emission at 500 nm. Since the ratio of the light emitted at 340 nm excitation to that emitted at 380 nm excitation (340/380 ratio) is a direct index of the level of [Ca2+]i, data are expressed as the 340/380 ratio following subtraction of background fluorescence.
For monitoring of Ca2+ transients, we used fura-2 in a similar manner to that described above. In order to capture full transients, myocytes were plated at a density that allowed spontaneous contraction, but facilitated visualization of a single layer of myocytes. Myocytes cultured in this manner retained their spontaneous beating at regular intervals, but at a substantially reduced rate.
Measurement of real-time PKA or cAMP activity via fluorescence resonance energy transfer (FRET)
Following 24 h in culture, neonatal cardiomyocytes were infected with an adenovirus containing AKAR2.2 (PKA) or ICUE3 (cAMP; 100 multiplicity of infection (MOI), 24 h) in a similar manner to that previously published (Soto et al. 2009). Between 24 and 48 h postinfection cells were washed and stored in DPBS for FRET recordings. Dual emission ratio imaging was acquired for 200 s at 20 s intervals. Images in both channels were subjected to background subtraction, and ratios of yellow fluorescent protein (YFP) to cyan fluorescent protein (CFP) were calculated at different time points and then normalized to baseline signals. For AKAR2.2, phosphorylation of the PKA consensus site leads to an increase in the YFP/CFP ratio (Soto et al. 2009), whereas the binding of cAMP to the Epac site of ICUE3 causes a decrease in FRET (DiPilato et al. 2004).
Measurement of phospholamban phosphorylation
Neonatal cardiomyocytes, pretreated with 20 μm CFTRinh-172 for various times, were chilled, washed, and harvested in lysis buffer [20 mm Hepes pH 7.5, 2 mm EDTA pH 8.0, 150 mm NaCl, 10% glycerol, 0.6% NP4O, 20 mm Na4P2O7, 50 mm NaF, 2.5 mm Na3VO4 and Protease Inhibitor Single-Use Cocktail (Thermo Scientific, Pierce, Rockford, IL, USA)]. The lysates were clarified at 13,200g for 20 min at 4°C. The samples were resolved on an SDS-PAGE gel. Phospholamban phosphorylation was detected with a phospho-threonine 17 antibody (PLB-Thr17, Badrilla Ltd, Leeds, UK). Additionally, total phospholamban was probed with an antiphospholamban antibody (PLB, Badrilla Ltd) and γ-tubulin with an anti-γ-tubulin antibody (Sigma, St Louis, MO, USA). Primary antibodies were visualized with IRDye 680CW goat antimouse or with IRDye 800CW goat antirabbit secondary antibodies using an Odyssey scanner (Li-cor biosciences, Lincoln, NE, USA). The signals obtained from PLB-Thr17 were corrected and plotted as the increase over internal control (γ-tubulin).
Chemicals
Niflumic acid, thapsigargin, nifedipine and myristoylated-PKA inhibitor fragment 14–22 (PKI) were obtained from Sigma-Aldrich (St Louis, MO, USA). KB-R7943 came from Tocris Bioscience (Ellisville, MO, USA). BayK-8644 was purchased from Calbiochem (San Diego, CA, USA), while CFTRinh-172 was obtained from Calbiochem or the Cystic Fibrosis Foundation Therapeutics compound collection (Rosalind Franklin University of Medicine and Science, Chicago, IL, USA). Myristoylated-autocamtide-2-related inhibitory peptide (AIP) was obtained from Biomol (Plymouth Meeting, PA, USA). Fura-2 AM was purchased from Invitrogen (Carlsbad, CA, USA). Ryanodine came from Alomone Labs (Jerusalem, Israel). Niflumic acid, CFTRinh-172, ryanodine, thapsigargin, and fura-2 AM were dissolved in DMSO, while PKI and AIP were dissolved in water. All drugs were made as 1:1000 stock solutions and diluted on the day of the experiment.
Statistical analysis
For presentation of changes in contraction rate, [Ca2+]i or PKA activity, peak responses were subtracted from their respective baseline values. For percentage baseline measurements, the last measured contraction rates were divided by baseline values. Normalization was performed by GraphPad Prism software (GraphPad Software, La Jolla, CA, USA) by setting the first baseline measurement to 100% and the value of 0 to 0%. For comparison between two groups of data, Student's unpaired t test was used to determine significance. For multiple comparisons, one-way ANOVA was performed. Time course curves were compared by two-way ANOVA. Results were considered significant at P < 0.05 and are denoted accordingly.
Results
Effect of pharmacological inhibition and genetic deletion of CFTR on cardiomyocyte contraction rate
To examine the potential role of CFTR in the regulation of ventricular cardiomyocyte contraction rate, we used syncytial neonatal mouse ventricular myocytes, which form a single spontaneously beating unit. In control experiments (DMSO or H2O), we found that cardiomyocytes held a steady rhythm of beating over time (Figs 1, 4 and 5). To determine the acute effect of CFTR inhibition on cardiomyocyte contraction rate, we used the selective CFTR inhibitor, CFTRinh-172 (Ma et al. 2002). Application of CFTRinh-172 (10 and 20 μm) resulted in a transient, but significant (P < 0.05), dose-dependent inhibition of contraction rate thatsubsequently returned to baseline levels (Fig. 1). Addition of CFTRinh-172 (20 μm) to cardiomyocytes from CFTR KO myocytes failed to significantly inhibit the contraction rate when compared with baseline or DMSO (P > 0.05), demonstrating specificity of action of CFTRinh-172 and the ability of CFTR KO cardiomyocytes to function normally without CFTR. Examination of deflection magnitude and contraction velocity, indirect indicators of contractility, showed no significant changes upon addition of CFTRinh-172 (P > 0.05; data not shown). As seen from the representative trace in Fig. 1C, CFTRinh-172predominantly increased the interval between contractions. Thus, CFTR is involved in the physiological regulation of cardiomyocyte contraction rate; however, both acute and chronic loss of CFTR activity can be compensated for.
Figure 1. Pharmacological inhibition, but not genetic deletion, of CFTR attenuates cardiomyocyte contraction rate.
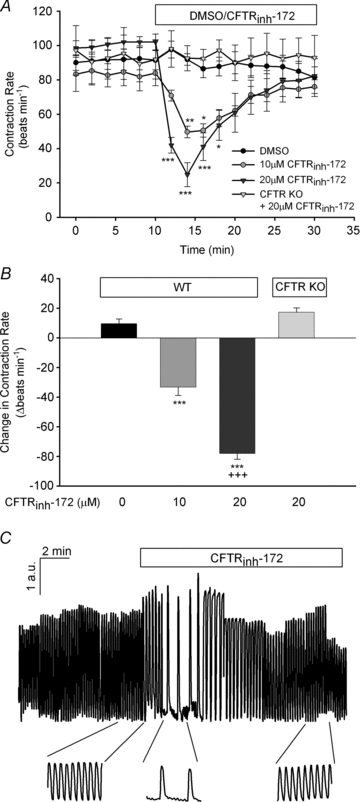
A, syncytial WT ventricular myocytes were treated with DMSO (n≥ 6) or CFTRinh-172 (10 μm or 20 μm; n= 7 each) for 20 min following a 10 min baseline period. Separately, the effect of CFTRinh-172 (20 μm; n= 5) on ventricular myocytes obtained from CFTR KO mice was also examined. *P < 0.05; **P < 0.01; ***P < 0.001 vs. DMSO or CFTR KO mice by two-way ANOVA. B, comparison of the maximal inhibitory effect of CFTRinh-172 on WT and CFTR KO cardiomyocyte contraction rate. ***P < 0.001 vs. DMSO by one-way ANOVA; +++P < 0.001 vs. 10 μm CFTRinh-172 by Student's unpaired t test. C, normalized representative trace of spontaneously beating WT ventricular myocyte with original traces prior to and after CFTRinh-172 (20 μm) addition. Deflection in the y-axis is expressed as arbitrary units (a.u.).
Figure 4. Calcium/calmodulin-dependent kinase II activity is required to maintain normal contraction rates following pharmacological inhibition or genetic deletion of CFTR.
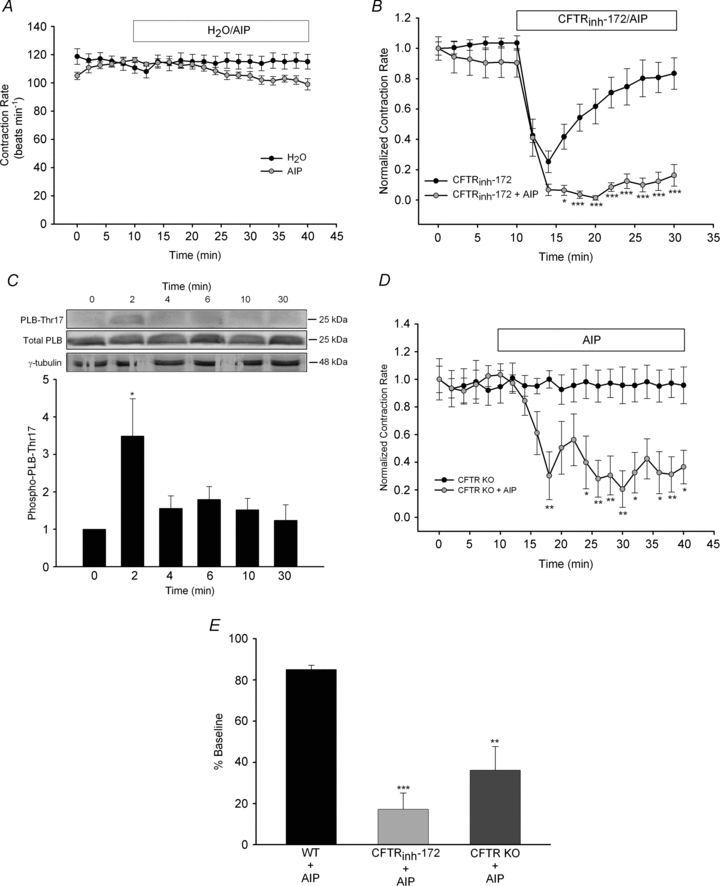
A, syncytial WT ventricular myocytes were treated with H2O (n= 5) or AIP (1 μm, n= 6) for 30 min following a 10 min baseline period. B, separately, the effect of simultaneous addition of AIP plus CFTRinh-172 (20 μm; n= 8) for 20 min was examined and compared with responses by CFTRinh-172 alone (from Fig. 1A). *P < 0.05; ***P < 0.001 vs. CFTRinh-172 alone by two-way ANOVA. C, WT cardiomyocytes were exposed to CFTRinh-172 (20 μm) for various lengths of time (n= 3–5 each) and then harvested to examine PLB-Thr17 phosphorylation. All results were quantified and normalized. A representative blot is shown. *P < 0.05 vs. no treatment by Student's unpaired t test. D, cardiomyocytes from CFTR KO mice (n= 8) were treated with AIP (1 μm) for 30 min following baseline measurements. Contraction rates were compared with those of cardiomyocytes from CFTR KO mice shown in Fig. 1A. *P < 0.05; **P < 0.01 vs. cardiomyocytes from CFTR KO alone by two-way ANOVA. E, a comparison of changes in contraction rate due to AIP in WT cardiomyocytes (n= 6), WT cardiomyocytes plus CFTRinh-172 (n= 8) and cardiomyocytes from CFTR KO mice (n= 8). **P < 0.01; ***P < 0.001 vs. WT by Student's unpaired t test.
Figure 5. Calcium-activated Cl− channel plays an important role in cardiomyocyte contraction rate following CFTR inhibition or KO.
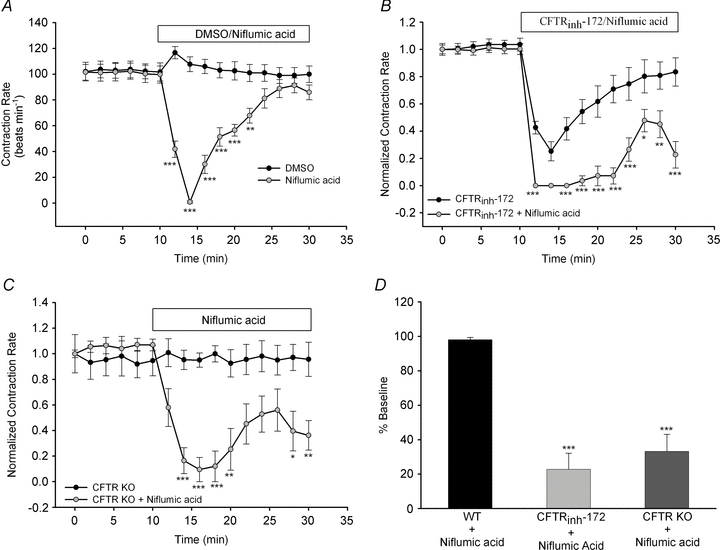
A, syncytial WT ventricular myocytes were treated with DMSO (n= 6) or niflumic acid (100 μm; n= 7) for 30 min following a 10 min baseline period. B, separately, the effect of simultaneous addition of niflumic acid (100 μm) plus CFTRinh-172 (20 μm; n= 8) for 20 min was examined and compared with the effect of CFTRinh-172 alone (from Fig. 1A). C, ventricular myocytes from CFTR KO mice (n= 8) were treated with niflumic acid (100 μm) for 20 min after baseline measurements. Contraction rates were compared with those of the cardiomyocytes from the CFTR KO mice shown in Fig. 1A. *P < 0.05; **P < 0.01; ***P < 0.001 vs. cardiomyocytes from CFTR KO alone by two-way ANOVA. D, a comparison of changes in contraction rate due to niflumic acid in WT cardiomyocytes (n= 7), WT cardiomyocytes plus CFTRinh-172 (n= 8) and cardiomyocytes from CFTR KO mice (n= 8). ***P < 0.001 vs. WT by Student's unpaired t test.
Effect of CFTRinh-172 on [Ca2+]i and real-time PKA activity
The contraction rate of spontaneously beating cardiomyocytes is dependent on changes in [Ca2+]i and PKA activity (Vinogradova et al. 2006). Therefore, to understand the mechanism whereby CFTR blockade transiently inhibits cardiomyocyte contraction rate, we investigated whether CFTRinh-172 alters [Ca2+]i and/or PKA activity. We first examined [Ca2+]i by monitoring changes in fura-2 fluorescence, which is a direct index of [Ca2+]i, in cardiomyocytes from WT and CFTR KO mice. In both quiescent and spontaneously beating WT myocytes, CFTRinh-172 (20 μm) significantly increased [Ca2+]i and [Ca2+]i transients when compared with DMSO-treated myocytes (P < 0.001; Fig. 2). In contrast, in cardiomyocytes from CFTR KO mice CFTRinh-172 failed to elicit a significant increase in [Ca2+]i compared with DMSO (P > 0.05). In spontaneously beating myocytes, CFTRinh-172 increased both systolic and diastolic Ca2+ (Fig. 2B). To understand the source of this [Ca2+]i increase, we performed additional experiments targeting extracellular and/or intracellular Ca2+ stores and the responsible channels/transporters. Since spontaneously beating cardiomyocytes are sensitive to pharmacological modulation of Ca2+ channels/transporters (data not shown), we used quiescent cardiomyocytes for these experiments. Removal of extracellular Ca2+ completely prevented the CFTRinh-172-induced increase in [Ca2+]i (Fig. 2C). In contrast, inhibition of sarcoplasmic reticulum Ca2+ release by pretreatment with thapsigargin (1 μm) and ryanodine (10 μm) prior to CFTRinh-172 did not have any significant influence on [Ca2+]i changes (Fig. 2C). While many routes of Ca2+ entry in cardiomyocytes exist, L-type Ca2+ channels and the Na+–Ca2+ exchanger are the most prominent. Therefore, we examined whether one or both of these proteins are involved in mediating the CFTRinh-172-induced increases in [Ca2+]i. Pretreatment with the L-type Ca2+ channel inhibitor nifedipine (10 μm), but not the Na+–Ca2+ exchange inhibitor KB-R7943 (10 μm), completely prevented CFTRinh-172-induced increases in [Ca2+]i (Fig. 2C). Thus, CFTR inhibition causes L-type Ca2+ channel activation, resulting in a transient increase in [Ca2+]i.
Figure 2. Inhibition of CFTR increases [Ca2+]i via L-type Ca2+ channels.
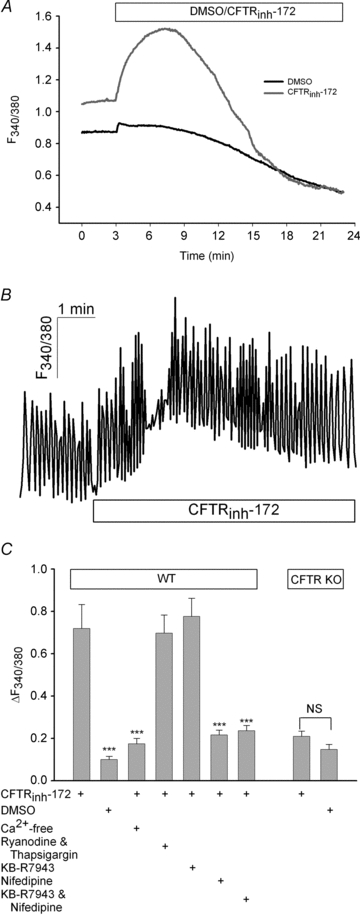
A, quiescent WT ventricular myocytes were loaded with fura-2 to monitor changes in [Ca2+]i. Following a 3 min baseline period, either CFTRinh-172 (20 μm, n= 32) or DMSO (n= 19) was added, and subsequent effects were measured for 20 min. B, representative trace (n= 12) of spontaneously beating cardiomyocyte that was loaded with fura-2 to measure Ca2+ transients prior to and after CFTRinh-172 (20 μm) addition. Separate experiments with DMSO showed no change in diastolic or systolic Ca2+ (data not shown; n= 4). C, quiescent WT cardiomyocytes were bathed in Ca2+-free solution (n= 25), pretreated with ryanodine (10 μm) and thapsigargin (1 μm; n= 26), KB-R7943 (10 μm; n= 23), nifedipine (10 μm; n= 26) or KB-R7943 and nifedipine (n= 23) for 30 min prior to [Ca2+]i measurements. Additionally, in myocytes from CFTR KO mice, fura-2 fluorescence was monitored upon CFTRinh-172 (20 μm; n= 23) or DMSO (n= 24) addition. ***P < 0.001 vs. CFTRinh-172 by Student's unpaired t test; NS, not significant.
Subsequently, we sought to determine the effect of CFTRinh-172 on PKA activity by using FRET to monitor PKA activity in real time. Compared with DMSO, CFTRinh-172 (20 μm) significantly inhibited PKA activity, in both magnitude and duration (P < 0.001; Fig. 3A). This inhibition was transient and recovered to baseline levels within 20 min, resembling the pattern of inhibition observed on contraction rates. The CFTRinh-172-induced inhibition of PKA was not the result of upstream alterations in adenylyl cyclase activity, since separate experiments measuring cAMP showed that CFTRinh-172 caused no change in cAMP levels (DMSO 0.17 ± 0.02 vs. CFTRinh-172 0.20 ± 0.01 –ΔYFP/CFP, P > 0.05 vs. DMSO). To understand how CFTR inhibition leads to a decrease in PKA activity we examined whether there was a link between the [Ca2+]i change and PKA activity. In these experiments, inhibition of the L-type Ca2+ channel with nifedipine pretreatment (10 μm) completelyprevented CFTRinh-172-induced attenuation of PKA activity (Fig. 3B), indicating that activation of L-type Ca2+ channels was responsible for the CFTRinh-172-induced inhibition of PKA activity. Similarly, direct activation of L-type Ca2+ channels by BayK-8644 (1 μm) caused a significant decrease in PKA activity (Fig. 3B), providing further evidence that L-type Ca2+ channel activation can negatively affect PKA activity. To verify that inhibition of PKA activity leads directly to functional attenuation of contraction rate, we examined the effect of myristoylated PKI (20 μm), a selective membrane-permeable peptididic inhibitor of PKA, on cardiomyocyte contraction rate. In agreement with previous reports (Vinogradova et al. 2006; Soto et al. 2009), we found that PKA inhibition caused a significant decrease in contraction rate (P < 0.001; Fig. 3C). As seen in both FRET and contraction rate experiments, CFTRinh-172 did not cause a further decrease in PKA activity or spontaneous contractions when combined with PKI, which elicited a more severe depression in PKA activity and spontaneous contractions compared with CFTRinh-172 (Fig. 3C). In separate experiments, we found that direct activation of L-type Ca2+ channels by BayK-8644 (1 μm) also led to a significant inhibition of contraction rate (H2O 11 ± 3 vs. BayK-8644 −133 ± 5 Δbeats min−1, P < 0.001 vs. H2O, n= 6). Taken together, our data indicate that CFTR blockage activates L-type Ca2+ channels, causing a transient increase in [Ca2+]i and hence, a transient inhibition of PKA activity, thereby leading to a decrease in contraction rate.
Figure 3. CFTRinh-172-induced activation of L-type Ca2+ channels causes inhibition of PKA and contraction rate.
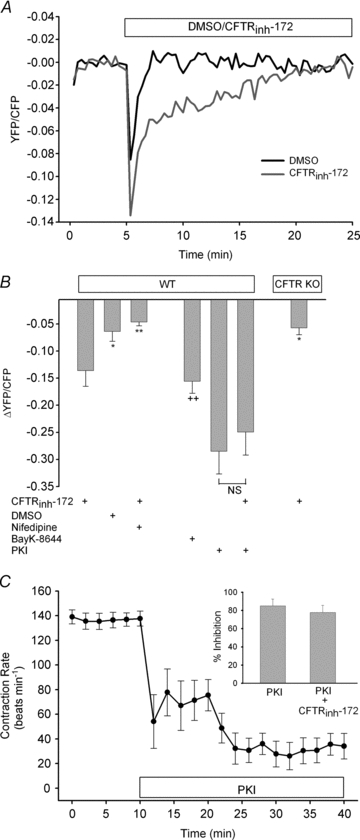
A, quiescent WT ventricular myocytes were infected with AKAR2.2 to monitor real-time changes in PKA activity. Following a 5 min baseline period, CFTRinh-172 (20 μm; n= 24) or DMSO (n= 18) was added, and subsequent effects were measured for 20 min. B, WT cardiomyocytes were pretreated with nifedipine (10 μm; n= 24) for 30 min prior to CFTRinh-172 addition. Additionally, PKA activity following CFTRinh-172 (20 μm; n= 19) was monitored in myocytes from CFTR KO mice. In separate experiments, the effects of BayK-8644 (1 μm; n= 15), PKI (20 μm; n= 14) or myristoylated PKI plus CFTRinh-172 (n= 14) on PKA activity in WT cardiomyocytes were measured. *P < 0.05; **P < 0.01 vs. CFTRinh-172; ++P < 0.01 vs. DMSO by Student's unpaired t test. C, syncytial WT ventricular myocytes were treated with PKI (20 μm; n= 8) for 30 min following a 10 min baseline period. Inset shows a comparison of syncytial WT cardiomyocytes treated with PKI alone or with PKI plus CFTRinh-172 (20 μm; n= 7).
Calcium/calmodulin-dependent kinase II in the recovery of CFTRinh-172-induced contraction rate depression
Protein kinase A and CaMKII regulate overlapping signalling cascades, leading to complementary regulation of proteins (i.e. phospholamban) involved in cardiomyocyte contraction (Wang et al. 2004). We therefore sought to determine whether CFTR inhibition alters CaMKII activity. We first examined the effect of CaMKII inhibition on cardiomyocyte contraction rate. In cardiomyocytes from WT mice, addition of AIP (1 μm), a selective CaMKII inhibitor, alone reproducibly caused a minor, statistically insignificant (P > 0.05) decrease in contraction rate compared with control cardiomyocytes (Fig. 4A). However, in separate experiments, when we added CFTRinh-172 (20 μm) together with AIP, cardiomyocyte contraction rate was not able to recover to baseline in a manner comparable to CFTRinh-172 alone (Fig. 4B). To explore further the role of CaMKII, we measured the effect of CFTRinh-172 on PLB-Thr17 phosphorylation, a specific CaMKII phosphorylation site on phospholamban. In agreement with our functional data, CFTRinh-172 (20 μm) caused a significant increase in PLB-Thr17 phosphorylation (Fig. 4C). Although this phosphorylation occurred very rapidly (within 2 min), it was transient, as phosphorylation returned to near-baseline levels within 4 min (Fig. 4C). Thus, while CFTRinh-172 induces a transient increase in [Ca2+]i that causes a decrease in contraction rate, it also activates CaMKII, which facilitates recovery from CFTR inhibition. To determine whether CaMKII is also important in conditions of chronic CFTR loss, we examined the effects of CaMKII inhibition on cardiomyocytes from CFTR KO mice. Figure 4D and E demonstrates that inhibition of CaMKII by AIP profoundly attenuated the contraction rate of cardiomyocytes from CFTR KO mice and that this effect was significantly greater than the action of AIP on the contraction rate of WT cardiomyocytes. Therefore, although cardiomyocytes from CFTR KO mice produce normal contraction rates, they have increased dependence on CaMKII activity to maintain these rates.
Role of CaCC in the regulation of cardiomyocyte contraction rate
Subsequently, we sought to determine how CaMKII activation facilitates an increase in contraction rate for recovery from CFTR inhibition. Since PLB-Thr17 phosphorylation is transient (Fig. 4C), we hypothesized that contraction rate recovery is not purely a result of altered PLB phosphorylation. Loss of CFTR activity is known to lead to increased CaCC activity as a compensatory Cl−-conductive pathway (Grubb et al. 1994; Hartzell et al. 2005). With our observations that CFTR inhibition increases [Ca2+]i and CaMKII activity, we sought to determine whether activation of CaCC might compensate for loss of CFTR activity. To investigate the role of CaCC in the regulation of cardiomyocyte contraction rate, we used niflumic acid (100 μm) to inhibit CaCC directly. Addition of niflumic acid alone resulted in a rapid, but transient, decrease in contraction rate, which recovered to baseline (Fig. 5A). Thus, inhibition of either CFTR or CaCC resulted in similar functional effects on cardiomyocyte contraction rate, indicating that, in ventricular cardiomyocytes, CFTR and CaCC may have complementary roles. To determine whether CaCC activity is necessary for recovery from CFTR inhibition, we performed experiments in which we simultaneously applied niflumic acid (100 μm) and CFTRinh-172 (20 μm) to cardiomyocytes from WT and CFTR KO mice. We found that inhibition of both CFTR and CaCC in WT cardiomyocytes strongly abrogated the contraction rate andprevented full recovery to baseline (Fig. 5B). Examination of CaCC inhibition in cardiomyocytes from CFTR KO mice also showed a significant inhibitory effect of niflumic acid on cardiomyocyte contraction rate (Fig. 5C). Like AIP (Fig. 4E), niflumic acid-induced inhibition of contraction rate in cardiomyocytes from CFTR KO mice was significantly enhanced when compared with that in cardiomyocytes from WT mice (Fig. 5D). This result indicates an increased reliance on CaCC. Thus, CaCC is important for recovery from CFTR inhibition and constitutes an important downstream effector of CaMKII activation.
Discussion
Since the first electrophysiological recording of CFTR in the heart (Nagel et al. 1992), there has been little dispute regarding it being responsible for the cardiac PKA-stimulated Cl− channel. While there have been conflicting reports about CFTR expression and/or activity in humans (Warth et al. 1996; Berul et al. 1997; Du et al. 2000), studies have shown that CFTR is expressed, and electrophysiological recordings have been made from rats (Uramoto et al. 2003), guinea-pigs (Hume et al. 1994), rabbits (Hart et al. 1996), swine (Gao et al. 2007), dogs (Hume et al. 1994), primates (Warth et al. 1996) and, most pertinent to our study, mice (Lader et al. 2000). Despite these studies, questions remain regarding the role of CFTR in the heart. Thus, the primary goal of our study was to determine whether CFTR plays a physiological role in the regulation of heart function.
For our studies, we used neonatal ventricular cardiomyocytes. Unlike adult ventricular myocytes, neonatal ventricular myocytes spontaneously beat as a single syncytial unit when cultured appropriately. This property allowed us to examine myocyte function without exogenous perturbation. While it may be tempting to compare these cells to adult pacemaker or atrial cells, which also spontaneously contract, it is more likely that neonatal ventricular myocytes are analogous to adult ventricular myocytes. In a comparison of their electrophysiological properties, Nuss & Marban (1994) showed that mouse neonatal and adult ventricular myocytes express similar ion channels and have comparable action potential durations. In previous exploratory studies, we found that CFTR blockade in paced adult mouse ventricular myocytes transiently inhibited contractility in a manner similar to the effects observed in thepresent study using neonatal mouse ventricular cardiomyocytes (Dr. Dagoberto Soto, University of Illinois, Urbana-Champaign, unpublished results). While the exact role of CFTR in adult ventricular cardiomyocytes remains to be determined, we have shown that CFTR plays an active role in the regulation of heart function, as measured by CFTRinh-172-induced depression of neonatal cardiomyocyte contraction rate. These results are in agreement with cardiomyocyte simulation studies, which showed that decreased CFTR current density lengthens action potential duration (Kuzumoto et al. 2008). In our study, we found that inhibition of CFTR causes a transient increase in [Ca2+]i due to activation of L-type Ca2+ channels, whose importance in action potential duration and contraction regulation are well established. We did not dissect out the mechanism whereby inhibition of CFTR activates L-type Ca2+ channels, but a potential mechanism is through alterations in membrane potential (see Fig. 6). While the effects of CFTR inhibition on neonatal cardiomyocyte membrane potential have not been examined, the simulation studies of Kuzumoto et al. (2008) predict that decreased CFTR activity causes increased depolarization during action potential repolarization. Additionally, Xu et al. found that inhibition of CaCCs prolongs early action potential repolarization (Xu et al. 2002). Our findings that CFTRinh-172 and niflumic acid have similar effects on contraction rate suggest that CFTR may also affect membrane potential and action potential duration. However, further studies are required to explore this possibility.
Figure 6. Proposed model for the effect of CFTR disruption on cardiomyocyte signalling and ion transport.
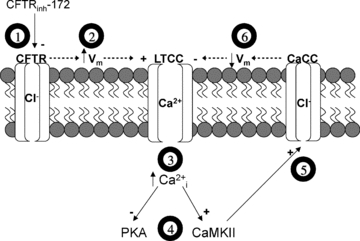
Acute inhibition of CFTR by CFTRinh-172 (1) probably causes an increase in membrane potential (Vm), which leads to increased activation of L-type Ca2+ channels (LTCC) (2). Increased LTCC activation raises intracellular Ca2+ (3), resulting in inhibition of PKA and activation of CaMKII (4). Inhibition of PKA causes a decrease in contraction rate. Activation of CaMKII stimulates Ca2+-activated Cl− channels (CaCC) (5), which probably serves to restore changes in membrane potential and decrease LTCC hyperactivity (6). This decrease in [Ca2+]i probably removes the inhibition on PKA activity, thus restoring contraction rate.
In addition to alterations in [Ca2+]i, we have also shown that CFTR inhibition leads to a transient decrease in PKA activity, as measured by A-kinase activity receptor (AKAR) phosphorylation. This study, along with others (Vinogradova et al. 2006; Soto et al. 2009), has shown that PKA is an important regulator of contraction. We did not attempt to determine the exact mechanism whereby CFTR blockade leads to PKA inhibition. However, we did show that this inhibition was caused by L-type Ca2+ channel-mediated Ca2+ influx. Cardiac myocytes contain Ca2+-sensitive adenylyl cyclases (AC V and AC VI) that can be inhibited by increases in [Ca2+]i (Willoughby & Cooper, 2007). However, our finding that cAMP remains unchanged following CFTR inhibition makes this unlikely. An additional possibility is that Ca2+-sensitive phosphatases, such as calcineurin, are involved in the inhibition we observed. It has been shown that calcineurin antagonizes PKA activity through dephosphorylation of PKA effectors (Santana et al. 2002) and even directly inhibits PKA itself (Orie et al. 2009). The lack of effect on cAMP also suggests that CFTR inhibition probably does not alter hyperpolarization-activated cyclic nucleotide-gated channels (HCN), which are known to be involved in contraction rate regulation (Z.M.S., unpublished results; Er et al. 2003), although we cannot rule out an indirect effect of membrane potential alteration. Altogether, we have documented how inhibition of CFTR activity can alter ion channel and intracellular signalling activities to affect cardiomyocyte function.
Importantly, we found that CFTR-dependent depression of contraction rate was transient and was restored to normal rates. Additionally, we found that cardiomyocytes from CFTR KO mice displayed normal contraction rates, further indicating that CFTR activity or expression is not critical to maintaining cardiomyocyte function. This fact is not surprising given that mutations in CFTR do not lead to early embryonic death in CF patients or mice. The ability of cardiomyocytes to compensate for transient or long-term loss of CFTR does not discount its significance, but rather underscores the adaptability of the heart to maintain its vital function. It is well accepted that gene knockout causes adaptation, but we have shown that short-term and chronic inhibition of CFTR leads to compensatory changes in cardiac myocyte function. Our data demonstrate that CaMKII and CaCC activity are necessary for recovery from acute CFTR inhibition in WT cardiomyocytes and maintenance of normal contraction rates in cardiomyocytes from CFTR KO mice. Interplay between CFTR and CaCC has been noted in several tissues that express both channels (Hartzell et al. 2005). In airway epithelia, loss of CFTR, either in CF patients or in CFTR KO mice, leads to compensatory increases in CaCC activity (Grubb et al. 1994). Additionally, in bovine pulmonary artery endothelial cells (Wei et al. 2001), Xenopus oocytes (Kunzelmann et al. 1997) and mouse parotid acinar cells (Perez-Cornejo & Arreola, 2004), expression of CFTR leads to a negative regulation of CaCC. While niflumic acid is a well-documented inhibitor of CaCC (Hartzell et al. 2005), it is not specific and can also affect CFTR (Scott-Ward et al. 2004) and K+ channels (Wang et al. 1997; Zhou et al. 2007). Although we cannot rule out effects on non-CFTR targets, the effects of niflumic acid on the contraction rate of cardiomyocytes from CFTR KO mice make us confident that our results are not due to additional effects on CFTR. This, coupled with the involvement of [Ca2+]i and CaMKII, both of which activate CaCC, strengthens our view about the importance of CaCC during loss of CFTR activity. However, since Ca2+ and CaMKII also affect many other ion channels and kinases important in contraction, further studies are required.
Calcium/calmodulin-dependent kinase II has been identified as an important intracellular signalling molecule in cardiac myocytes involved in regulating numerous cellular processes, including activation of ion channels, modulation of action potential, hypertrophy and apoptosis (Anderson, 2007). In our study, we found that CaMKII is important in the recovery of cardiomyocyte contraction rate caused by inhibition of CFTR. Its role in this process appears to be, in part, due to its ability to activate CaCC, since inhibition of CaCC or CaMKII prevented recovery from CFTR blockade. Calcium/calmodulin-dependent kinase II has also been shown to decrease the inactivation rate of L-type Ca2+ channels (Guo & Duff, 2006), which may aid in contraction rate recovery. Another important finding is that, while cardiomyocytes from CFTR KO mice display normal baseline contraction rates, in the presence of CaMKII inhibition these rates were severely diminished. In contrast, CaMKII inhibition in WT mice showed only minimal attenuation of baseline contraction rate. These data indicate that cardiomyocytes from CFTR KO mice have an increased dependence on CaMKII. These findings are of particular interest, since cardiac myocytes with increased CaMKII expression or activity are prone to pathological cardiac conditions. Calcium/calmodulin-dependent kinase II has been linked to heart failure; hearts from mice with heart failure have increased expression of CaMKIIδ, and overexpression of CaMKII causes heart failure in mice (Hoch et al. 1999; Maier, 2005). In addition, increased CaMKII activity may lead to prolonged action potential duration and cause long QT syndrome by increasing late-inactivating or slowly inactivating Na+ currents (Bers, 2008). At this time, we cannot say whether the increased dependence on CaMKII is due to long-term loss of CFTR expression or function. However, it would be intriguing to determine whether CF mutations, which disrupt CFTR processing and trafficking (e.g. ΔF508; Kopito, 1999) or perturb CFTR channel gating (e.g. G551D; Li et al. 1996) also display increased dependence on CaMKII activity. Thus, our findings and the observations that CFTR is downregulated in ventricular tissue from heart failure patients (Solbach et al. 2008) and that heart damage occurs in some CF patients (Wiebicke et al. 1993; Zebrak et al. 2000) warrant future investigation of the role of CaMKII in CF hearts.
There have been scattered reports of cardiac abnormalities in CF patients, including alterations in left ventricular function, tachycardia, cardiomyopathy, myocardial fibrosis and necrosis (Ambrosi et al. 1993; Wiebicke et al. 1993; Florea et al. 2000; Zebrak et al. 2000). More recently, it was found that 51% of CF patients with severe lung infection had significantly decreased ejection fraction during exercise (Nash et al. 2007). It is unclear whether these findings are due to primary cardiac defects or are secondary effects from altered pulmonary function. Additionally, with the involvement of CFTR in smooth muscle contraction and vascular reactivity (Robert et al. 2005), vascular effects on the heart must also be considered. The physiological relevance of CFTR in the heart requires investigation, since recent advances in patient care have extended the average lifespan of CF patients to the mid-30s, with some individuals with milder mutations achieving normal lifespans (70–80 years; http://www.cff.org). Therefore, it is likely that as therapeutic advances continue to be made and individuals live longer, more active lives, CF patients will enter the age range where cardiovascular abnormalities become apparent and may exhibit significant clinical cardiac symptoms. A more complete understanding of the role of CFTR in the heart may lead to the identification of potential cardiac defects in CF patients and thus, increase the quality of care for these patients.
Our present study presents findings that further our understanding regarding the physiological role of CFTR in cardiomyocytes. Importantly, we found that CFTR is involved in the regulation of cardiomyocyte contraction. Acute loss of CFTR activity leads to a reduction in the spontaneous beating of neonatal ventricular myocytes through alterations in [Ca2+]i and PKA. However, CaMKII and CaCC activity compensate for this loss and restore myocyte function. While cardiomyocytes from CFTR KO mice can maintain normal spontaneous contractions, they exhibit increased dependence on both CaMKII and CaCC.
Acknowledgments
The authors would like to thank Dr Deborah Nelson (University of Chicago) for providing CFTR KO mice and extensive discussions regarding experimental results. We would also like to thank Dr Robert Bridges (Rosalind Franklin University of Medicine and Science) and Cystic Fibrosis Foundation Therapeutics (CFFT) for the gift of CFTRinh-172 from CFFT's compound collection. This study was supported by the American Heart Association (0815670G to Z.M.S.; 0635331N to Y.X.), the National Institutes of Health (HL082846 to Y.X.) and the University of Illinois (P.M.B.).
Glossary
Abbreviations
- AIP
myristoylated-autocamtide-2-related inhibitory peptide
- AKAR2.2
A-kinase associated receptor 2.2
- CaCC
Ca2+-activated Cl− channel
- CaMKII
Ca2+/calmodulin-dependent kinase II
- cCFTR
cardiac isoform of CFTR
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- DPBS
Dulbecco's phosphate-buffered solution
- FRET
fluorescence resonance energy transfer
- ICUE3
indicator of cAMP using Epac 3
- KO
knockout
- PKA
protein kinase A
- PKI
myristoylated-PKA inhibitor fragment 14–22
- PLB-Thr17
threonine 17 of phospholamban
- WT
wild-type
Author contributions
Z.M.S. was responsible for the conception, design and performance of all experiments. He also drafted and finalized the manuscript. V.DeA. performed the CaMKII Western blot experiments and contributed to the manuscript preparation and finalization. Y.X. and P.M.B. contributed to the conception and design of experiments, intellectual content and finalization of the manuscript. All experiments were performed at the University of Illinois at Urbana-Champaign.
References
- Ambrosi P, Chazalettes JP, Viard L, Raynaud M, Faugere G, Noirclerc M, Bernard PJ. Left ventricular involvement in mucoviscidosis after 2 years of age. Arch Fr Pediatr. 1993;50:653–656. [PubMed] [Google Scholar]
- Anderson ME. Multiple downstream proarrhythmic targets for calmodulin kinase II: moving beyond an ion channel-centric focus. Cardiovasc Res. 2007;73:657–666. doi: 10.1016/j.cardiores.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Bers DM. Calcium cycling and signalling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- Berul CI, Sweeten T, Vetter VL, Morad M. Lack of cystic fibrosis transmembrane regulator-type chloride current in pediatric human atrial myocytes. Life Sci. 1997;60:189–197. doi: 10.1016/s0024-3205(96)00615-7. [DOI] [PubMed] [Google Scholar]
- Chen H, Liu LL, Ye LL, McGuckin C, Tamowski S, Scowen P, Tian H, Murray K, Hatton WJ, Duan D. Targeted inactivation of cystic fibrosis transmembrane conductance regulator chloride channel gene prevents ischemic preconditioning in isolated mouse heart. Circulation. 2004;110:700–704. doi: 10.1161/01.CIR.0000138110.84758.BB. [DOI] [PubMed] [Google Scholar]
- Collier ML, Levesque PC, Kenyon JL, Hume JR. Unitary Cl− channels activated by cytoplasmic Ca2+ in canine ventricular myocytes. Circ Res. 1996;78:936–944. doi: 10.1161/01.res.78.5.936. [DOI] [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signalling within discrete subcellular compartments. Proc Natl Acad Sci U S A. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in the The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du XY, Finley J, Sorota S. Paucity of CFTR current but modest CFTR immunoreactivity in non-diseased human ventricle. Pflugers Arch. 2000;440:61–67. doi: 10.1007/s004240000254. [DOI] [PubMed] [Google Scholar]
- Duan D, Ye L, Britton F, Miller LJ, Yamazaki J, Horowitz B, Hume JR. Purinoceptor-coupled Cl− channels in mouse heart: a novel, alternative pathway for CFTR regulation. J Physiol. 1999;521:43–56. doi: 10.1111/j.1469-7793.1999.00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan DY, Liu LL, Bozeat N, Huang ZM, Xiang SY, Wang GL, Ye L, Hume JR. Functional role of anion channels in cardiac diseases. Acta Pharmacol Sin. 2005;26:265–278. doi: 10.1111/j.1745-7254.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- Er F, Larbig R, Ludwig A, Biel M, Hofmann F, Beuckelmann DJ, Hoppe UC. Dominant-negative suppression of HCN channels markedly reduces the native pacemaker current If and undermines spontaneous beating of neonatal cardiomyocytes. Circulation. 2003;107:485–489. doi: 10.1161/01.cir.0000045672.32920.cb. [DOI] [PubMed] [Google Scholar]
- Florea VG, Florea ND, Sharma R, Coats AJ, Gibson DG, Hodson ME, Henein MY. Right ventricular dysfunction in adult severe cystic fibrosis. Chest. 2000;118:1063–1068. doi: 10.1378/chest.118.4.1063. [DOI] [PubMed] [Google Scholar]
- Gao Z, Sun HY, Lau CP, Chin-Wan Fung P, Li GR. Evidence for cystic fibrosis transmembrane conductance regulator chloride current in swine ventricular myocytes. J Mol Cell Cardiol. 2007;42:98–105. doi: 10.1016/j.yjmcc.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Grubb BR, Vick RN, Boucher RC. Hyperabsorption of Na+ and raised Ca2+-mediated Cl− secretion in nasal epithelia of CF mice. Am J Physiol. 1994;266:1478–1483. doi: 10.1152/ajpcell.1994.266.5.C1478. [DOI] [PubMed] [Google Scholar]
- Guo J, Duff HJ. Calmodulin kinase II accelerates L-type Ca2+ current recovery from inactivation and compensates for the direct inhibitory effect of [Ca2+]i in rat ventricular myocytes. J Physiol. 2006;574:509–518. doi: 10.1113/jphysiol.2006.109199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart P, Warth JD, Levesque PC, Collier ML, Geary Y, Horowitz B, Hume JR. Cystic fibrosis gene encodes a cAMP-dependent chloride channel in heart. Proc Natl Acad Sci U S A. 1996;93:6343–6348. doi: 10.1073/pnas.93.13.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol. 2005;67:719–758. doi: 10.1146/annurev.physiol.67.032003.154341. [DOI] [PubMed] [Google Scholar]
- Hiraoka M, Kawano S, Hirano Y, Furukawa T. Role of cardiac chloride currents in changes in action potential characteristics and arrhythmias. Cardiovasc Res. 1998;40:23–33. doi: 10.1016/s0008-6363(98)00173-4. [DOI] [PubMed] [Google Scholar]
- Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of δ-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999;84:713–721. doi: 10.1161/01.res.84.6.713. [DOI] [PubMed] [Google Scholar]
- Hume JR, Hart P, Levesque PC, Collier ML, Geary Y, Warth J, Chapman T, Horowitz B. Molecular physiology of CFTR Cl− channels in heart. Jpn J Physiol. 1994;44(Suppl 2):S177–S182. [PubMed] [Google Scholar]
- Kopito RR. Biosynthesis and degradation of CFTR. Physiol Rev. 1999;79:S167–S173. doi: 10.1152/physrev.1999.79.1.S167. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Mall M, Briel M, Hipper A, Nitschke R, Ricken S, Greger R. The cystic fibrosis transmembrane conductance regulator attenuates the endogenous Ca2+-activated Cl− conductance of Xenopus oocytes. Pflugers Arch. 1997;435:178–181. doi: 10.1007/s004240050498. [DOI] [PubMed] [Google Scholar]
- Kuzumoto M, Takeuchi A, Nakai H, Oka C, Noma A, Matsuoka S. Simulation analysis of intracellular Na+ and Cl− homeostasis during β1-adrenergic stimulation of cardiac myocyte. Prog Biophys Mol Biol. 2008;96:171–186. doi: 10.1016/j.pbiomolbio.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Lader AS, Wang Y, Jackson GR, Jr, Borkan SC, Cantiello HF. cAMP-activated anion conductance is associated with expression of CFTR in neonatal mouse cardiac myocytes. Am J Physiol Cell Physiol. 2000;278:C436–C450. doi: 10.1152/ajpcell.2000.278.2.C436. [DOI] [PubMed] [Google Scholar]
- Li C, Ramjeesingh M, Wang W, Garami E, Hewryk M, Lee D, Rommens JM, Galley K, Bear CE. ATPase activity of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1996;271:28463–28468. doi: 10.1074/jbc.271.45.28463. [DOI] [PubMed] [Google Scholar]
- Ma T, Thiagarajah JR, Yang H, Sonowane ND, Folli C, Galietta LJ, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier LS. CaMKIIδ overexpression in hypertrophy and heart failure: cellular consequences for excitation-contraction coupling. Braz J Med Biol Res. 2005;38:1293–1302. doi: 10.1590/s0100-879x2005000900002. [DOI] [PubMed] [Google Scholar]
- Nagel G, Hwang TC, Nastiuk KL, Nairn AC, Gadsby DC. The protein kinase A-regulated cardiac Cl− channel resembles the cystic fibrosis transmembrane conductance regulator. Nature. 1992;360:81–84. doi: 10.1038/360081a0. [DOI] [PubMed] [Google Scholar]
- Nash EF, Coonar AS, Stephenson AL, Delgado DH, Singer LG, Tullis E, Chapparro-Mutis C. Cardiac dysfunction in adult CF patients with severe lung disease—a retrospective cohort study. Pediatr Pulmonol. 2007:399. [Google Scholar]
- Nuss HB, Marban E. Electrophysiological properties of neonatal mouse cardiac myocytes in primary culture. J Physiol. 1994;479:265–279. doi: 10.1113/jphysiol.1994.sp020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orie NN, Thomas AM, Perrino BA, Tinker A, Clapp L. Ca2+/calcineurin regulation of cloned vascular KATP channels: crosstalk with the protein kinase A pathway. Br J Pharmacol. 2009;157:554–564. doi: 10.1111/j.1476-5381.2009.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Cornejo P, Arreola J. Regulation of Ca2+-activated chloride channels by cAMP and CFTR in parotid acinar cells. Biochem Biophys Res Commun. 2004;316:612–617. doi: 10.1016/j.bbrc.2004.02.097. [DOI] [PubMed] [Google Scholar]
- Robert R, Norez C, Becq F. Disruption of CFTR chloride channel alters mechanical properties and cAMP-dependent Cl− transport of mouse aortic smooth muscle cells. J Physiol. 2005;568:483–495. doi: 10.1113/jphysiol.2005.085019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana LF, Chase EG, Votaw VS, Nelson MT, Greven R. Functional coupling of calcineurin and protein kinase A in mouse ventricular myocytes. J Physiol. 2002;544:57–69. doi: 10.1113/jphysiol.2002.020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Ward TS, Li H, Schmidt A, Cai Z, Sheppard DN. Direct block of the cystic fibrosis transmembrane conductance regulator Cl− channel by niflumic acid. Mol Membr Biol. 2004;21:27–38. doi: 10.1080/09687680310001597758. [DOI] [PubMed] [Google Scholar]
- Solbach TF, Paulus B, Weyand M, Eschenhagen T, Zolk O, Fromm MF. ATP-binding cassette transporters in human heart failure. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:231–243. doi: 10.1007/s00210-008-0279-6. [DOI] [PubMed] [Google Scholar]
- Soto D, De Arcangelis V, Zhang J, Xiang Y. Dynamic protein kinase A activities induced by β-adrenoceptors dictate signalling propagation for substrate phosphorylation and myocyte contraction. Circ Res. 2009;104:770–779. doi: 10.1161/CIRCRESAHA.108.187880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly BC, Bezstarosti K, Boomaars WE, Marino CR, Lamers JM, de Jonge HR. Expression and regulation of chloride channels in neonatal rat cardiomyocytes. Mol Cell Biochem. 1996;157:129–135. doi: 10.1007/BF00227891. [DOI] [PubMed] [Google Scholar]
- Uramoto H, Takahashi N, Dutta AK, Sabirov RZ, Ando-Akatsuka Y, Morishima S, Okada Y. Ischemia-induced enhancement of CFTR expression on the plasma membrane in neonatal rat ventricular myocytes. Jpn J Physiol. 2003;53:357–365. doi: 10.2170/jjphysiol.53.357. [DOI] [PubMed] [Google Scholar]
- Vinogradova TM, Lyashkov AE, Zhu W, Ruknudin AM, Sirenko S, Yang D, Deo S, Barlow M, Johnson S, Caffrey JL, Zhou YY, Xiao RP, Cheng H, Stern MD, Maltsev VA, Lakatta EG. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ Res. 2006;98:505–514. doi: 10.1161/01.RES.0000204575.94040.d1. [DOI] [PubMed] [Google Scholar]
- Wang HS, Dixon JE, McKinnon D. Unexpected and differential effects of Cl− channel blockers on the Kv4.3 and Kv4.2 K+ channels. Implications for the study of the Ito2 current. Circ Res. 1997;81:711–718. doi: 10.1161/01.res.81.5.711. [DOI] [PubMed] [Google Scholar]
- Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP, Cheng H. Sustained β1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signalling pathway. Circ Res. 2004;95:798–806. doi: 10.1161/01.RES.0000145361.50017.aa. [DOI] [PubMed] [Google Scholar]
- Warth JD, Collier ML, Hart P, Geary Y, Gelband CH, Chapman T, Horowitz B, Hume JR. CFTR chloride channels in human and simian heart. Cardiovasc Res. 1996;31:615–624. [PubMed] [Google Scholar]
- Wei L, Vankeerberghen A, Cuppens H, Cassiman JJ, Droogmans G, Nilius B. The C-terminal part of the R-domain, but not the PDZ binding motif, of CFTR is involved in interaction with Ca2+-activated Cl− channels. Pflugers Arch. 2001;442:280–285. doi: 10.1007/s004240100531. [DOI] [PubMed] [Google Scholar]
- Wiebicke W, Artlich A, Gerling I. Myocardial fibrosis–a rare complication in patients with cystic fibrosis. Eur J Pediatr. 1993;152:694–696. doi: 10.1007/BF01955251. [DOI] [PubMed] [Google Scholar]
- Willoughby D, Cooper DM. Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol Rev. 2007;87:965–1010. doi: 10.1152/physrev.00049.2006. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Devic E, Kobilka B. The PDZ binding motif of the β1-adrenergic receptor modulates receptor trafficking and signalling in cardiac myocytes. J Biol Chem. 2002;277:33783–33790. doi: 10.1074/jbc.M204136200. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Naro F, Zoudilova M, Jin SL, Conti M, Kobilka B. Phosphodiesterase 4D is required for β2-adrenoceptor subtype-specific signalling in cardiac myocytes. Proc Natl Acad Sci U S A. 2005;102:909–914. doi: 10.1073/pnas.0405263102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Dong PH, Zhang Z, Ahmmed GU, Chiamvimonvat N. Presence of a calcium-activated chloride channel current in mouse ventricular myocytes. Am J Physiol Heart Circ Physiol. 2002;283:H302–H314. doi: 10.1152/ajpheart.00044.2002. [DOI] [PubMed] [Google Scholar]
- Yamawake N, Hirano Y, Sawanobori T, Hiraoka M. Arrhythmogenic effects of isoproterenol-activated Cl− current in guinea-pig ventricular myocytes. J Mol Cell Cardiol. 1992;24:1047–1058. doi: 10.1016/0022-2828(92)91871-2. [DOI] [PubMed] [Google Scholar]
- Zebrak J, Skuza B, Pogorzelski A, Ligarska R, Kopytko E, Pawlik J, Rutkiewicz E, Witt M. Partial CFTR genotyping and characterisation of cystic fibrosis patients with myocardial fibrosis and necrosis. Clin Genet. 2000;57:56–60. doi: 10.1034/j.1399-0004.2000.570108.x. [DOI] [PubMed] [Google Scholar]
- Zhou L, Dey CR, Wert SE, DuVall MD, Frizzell RA, Whitsett JA. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science. 1994;266:1705–1708. doi: 10.1126/science.7527588. [DOI] [PubMed] [Google Scholar]
- Zhou SS, Zhang LB, Sun WP, Xiao FC, Zhou YM, Li YJ, Li DL. Effects of monocarboxylic acid-derived Cl− channel blockers on depolarization-activated potassium currents in rat ventricular myocytes. Exp Physiol. 2007;92:549–559. doi: 10.1113/expphysiol.2007.037069. [DOI] [PubMed] [Google Scholar]