

Abstract
Amine-reactive dyes, also known as LIVE/DEAD® fixable dead cell stains, are a class of viability dyes suitable for identifying dead cells in samples that will be fixed. These dyes cross the cell membranes of dead cells, and react with free amines in the cytoplasm. Live cells exclude these dyes because their cell membranes are intact, and free dye is washed away after staining. Notably, the reaction is irreversible; therefore, when cells are fixed and permeabilized (as with intracellular staining procedures), the bound dye remains associated with the dead cells (unlike other viability dyes). Since amine-reactive dyes are fluorescent when excited by lasers, dead cells can be identified by flow cytometry.
This unit describes procedures, troubleshooting, and outcomes for using the two most commonly used amine-reactive dyes, ViViD and Aqua Blue.
Keywords: Amine Reactive Dye, LIVE/DEAD® fixable cell stain, Cell Viability, non-specific binding
Unit Introduction
Amine reactive viability dyes offer an innovative alternative to traditional viability markers. There are eight LIVE/DEAD® fixable dead cells stains available in the market today, covering most of the visible and UV spectrum. Two of the commonly used amine reactive dyes, ViViD and Aqua Blue are described in this protocol. These dyes differ from other viability markers because they react with free amines in the cytoplasm. Live cells exclude these dyes because their cell membranes are intact, and free dye is washed away after staining (Figure 1). Notably, the reaction is irreversible; therefore, when cells are fixed and permeabilized (as with intracellular staining procedures), the bound dye remains associated with the dead cells (unlike other viability dyes). To measure these two dyes, it is critical to determine the correct filter configuration for detecting signal from each dye on the flow cytometer. For example, figure 2 shows the excitation curves (dotted lines) and the emission curves (solid lines) of ViViD and Aqua Blue, which can be excited by a 408nm violet laser (vertical black line). The shaded area over the emission curve shows the range of detection. Hence, ViViD is measured between 425-475nm (450/50 band pass filter), while Aqua Blue is detected between 525-575nm (505nm long pass filter + 515/20nm band pass filter). Additionally, it is important to note that the flow cytometer used to measure amine reactive dyes should be calibrated and standardized as described (Perfetto et al., 2006a).
Figure 1. The Staining theory of amine reactive dyes.
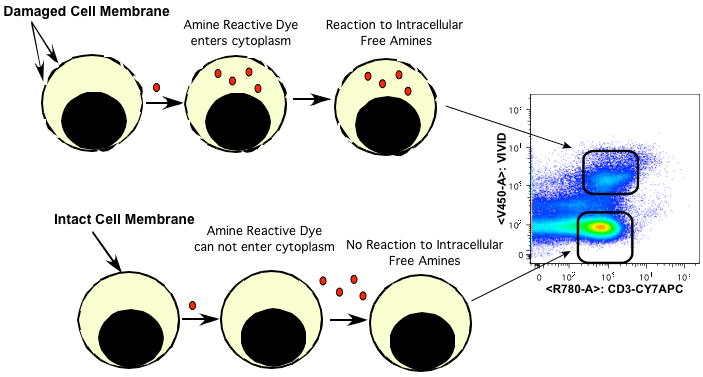
Figure 1 shows the theory behind the staining of amine reactive dyes. The upper pathway shows cells with damaged membranes, in this example the amine reactive dye ViViD enters through these membranes and reacts with the free amines in the cytosol. After binding with free amines this dye complex is positive for fluorescence in the V450 detect (arrow pointing to the upper cell population or the dead cells). In the lower pathway, the dye is excluded by the intact cell membrane the dye remains negative for fluorescence (arrow pointing to the lower cell population or the live cell gate).
Figure 2. The instrument configuration for proper detection of two common amine reactive dyes.
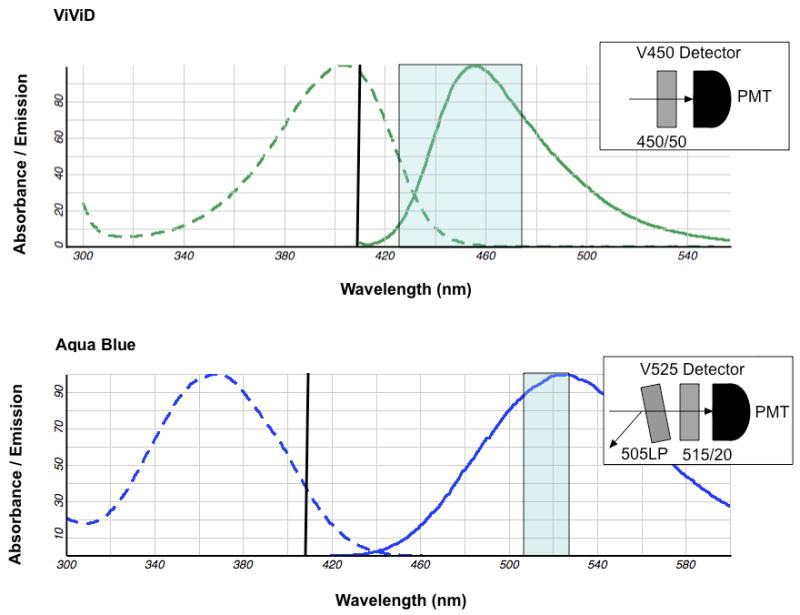
Figure 2 shows the excitation (dotted curve) and emission (solid curve) curves of the ViViD dye (upper panel) and the Aqua Blue dye (lower panel). These dyes are excited by the violet 408nm laser indicated by the black line over the excitation curve. Each rectangle shows band pass filter range of detection for the PMT as shown in each insert. In order to detect fluorescence from each emission curve the V450 detector uses only the 450/50nm band pass filter (upper insert) and the V525 detector uses a 505LP dichrolic filter combined with a 515/20nm band pass filter (lower insert).
The utility of amine reactive dyes is best demonstrated in the context of staining panels, which employ various combinations of fluorochrome mAb-conjugates, using samples that have been cryopreserved and/or stimulated with antigen. Such samples often include large numbers of dead cells, which can bind m-Ab conjugates non-specifically. In ICS (Intracytoplasmic Cytokine Staining) and peptide-MHC Class I (“tetramer”) assays, (Chattopadhyay et al., 2008; Perfetto et al., 2006b), where detection of rare events is critical, non-specific binding of mAB-conjugated by dead cells may result in significant overestimation of the proportion of antigen-specific cells. In summary, amine reactive viability dyes can be a powerful substitute for more traditional viability dyes and can be used in a variety of panel designs.
Protocol 1: Titration of Amine Reactive Dyes
The purpose of this protocol is to ensure that an optimal amount of amine reactive dye is used in staining experiments. The optimal concentration is defined as the concentration, which produces the highest signal (MFI) and the lowest background.
To this end, amine reactive dyes should be tested at concentrations above and below the manufacturer's recommended dilution, using samples that contain substantial numbers of dead cells. For example, PBMC samples contain substantial numbers of dead cells when incubated at 37° C for three to five days in RPMI cell culture media (containing 10% serum). Similarly, the frequency of dead cells is high when frozen cells are thawed under sub-optimal conditions (e.g. extended exposure of frozen cells to ambient temperatures). Once titration is completed, new vials from the same lot of amine reactive dye can be used at the same concentration; titration should be performed with each new lot.
Materials
ViViD amine reactive dye (LIVE/DEAD® fixable violet dead cell stain) or Aqua Blue amine reactive dye (LIVE/DEAD® fixable aqua dead cell stain) - Invitrogen
Bead Storage Media - see Reagent and Solution section
Standard Staining Media - see Reagent and Solution section
Phosphate Buffer Saline (PBS) - Becton Dickenson
Fetal Calf Serum (FCS)– Invitrogen or other suppliers
Neonatal calf sera - Invitrogen or other suppliers
RPMI-1640 - GIBCO
Sodium Azide - Sigma Chemical or other suppliers
Flow Cytometer – An analyzer or sorter equipped with a violet laser and optics such as shown in Figure 2. Alternatively, note that the ViViD Aqua Blue dye is also well excited by a UV laser (Figure 2, lower panel).
Titration of amine reactive dyes
Stock of ViViD dye: Each amine reactive dye kit comes with 25ug of lyophilized dye and DMSO. Note: lyophilized dye is stored desiccated and expires at 60 months under these conditions. DMSO must be stored under desiccated conditions.
Thaw DMSO supplied with kit completely using a 37C waterbath until completely thawed (approx. 30 sec). Add 50ul amount of DMSO into a vial of lyophilized dye as indicated in the table below for first dilution.
Make six serial dilutions using 25ul from the first dilution (500ug/ml) into 25ul volumes of DMSO resulting in six stock concentrations as listed in the table below.
Remove 1ul of each stock concentration and add 39ul of dH2O, resulting in six working concentrations as listed in the table below. Note: This DILUTION in dH2O is critical. Loss of fluorescence intensity will occur if this dye is prepared in other media sources containing amino acids.
- Add 5ul of each working dilution into 95ul of cells suspended in PBS. The cell source should contain substantial numbers of dead cells (as described in the introduction to this section). Note: PBS is critical. Loss of fluorescence intensity will occur if this dye is prepared in other media sources containing amino acids. However, standard staining media may be used in subsequent staining steps.
Dilution Dye wt.
(ug)DMSO
(ul)Stock Conc.
(ug/ml)Working Conc.
(ug/ml)Final Conc.
(ug/ml)1 25 50 500 12.50 0.625 2 25 100 250 6.25 0.313 3 25 200 125 3.12 0.156 4 25 400 62.5 1.56 0.078 5 25 800 31.25 0.78 0.039 6 25 1600 15.62 0.39 0.020 Mix and incubate at RT for 20min shielded from light. Note: This reaction time is optimal. Increased time or temperature does not improve staining intensity of the amine reactive dyes.
Wash cells twice in Standard Staining Media and stain with fluorochrome-conjugated mAb mix (e.g., anti-CD3 Cy7APC) according to a standard staining procedure as described In Unit 6.2 and Lamoreaux et al., (2006).
Acquire fluorescence measurements for each dilution using a standardized flow cytometer and a detector configuration, e.g. as described in the unit introduction.
Figure 3 shows data from a titration experiment for ViViD. Figure 3A shows all ViViD dilutions within CD3+ T-cells, note the different MFIs for negative and positive cell populations in dilution 2.5 and dilution 5ug/ml. Figure 3B is a representative histogram of single dilution (2.5ug/ml) showing the ViViD positive (2.6%) and the ViViD negative cell populations. Plotting the medians of positive cell populations verses each dye dilution shows the brightest signal or saturation point at 10ug/ml (Figure 3C). However, when considering the amine reactive dyes, the lowest background signal is more important than the maximum separation. Hence, the best separation with the lowest background is dilution 2.5ug/ml (arrow A) even though dilution 5ug/ml (arrow B) yields the more positive signal, the background is higher and is judged unacceptable (Figure 3D).
Figure 3. Titration of amine reactive dyes.
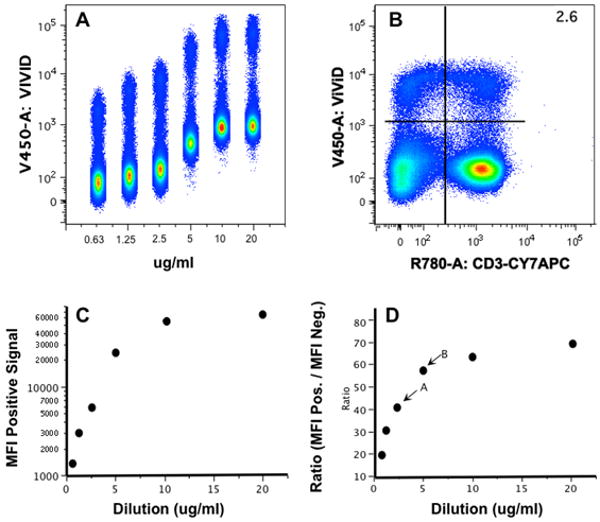
Figure 3 is a representative titration of the ViViD dye used in staining panels. Cells were incubated for 5 days under standard culture conditions and then stained with dilutions of ViViD dye (20, 10, 5, 2.5, 1.25 and 0.625ug/ml). After staining was complete, cells were stained with anti-CD3 using standard staining procedures. Gating on only CD3+ cells (live and dead) all dilutions can be concatenated on one histogram (figure 3A). This display can be used to measure the MFI of the negative and positive cell populations using the appropriate software. Figure 3B is a representative histogram of the working dilution of 2.5ug/ml of ViVID and anti-CD3. When considering the amine reactive dyes, the lowest background signal is more important than the absolute positive signal.
To illustrate the differences in the positive signal relative to the background staining, figure 3C (positive MFI) can be compared to the figure 3D (Ratio = pos. MFI / neg. MFI). The separation is acceptable at either of the lower dilutions (2.5 or 5ug/ml), however since the background is lowest at 2.5 ug/ml (arrow A) as compared to 5ug/ml (arrow B) the lower dilution is best.
Protocol 2: Creating Stable Amine Reactive Dye Labeled Compensation beads
The purpose of this protocol is to create a LIVE/DEAD® fixable dye compensation control for correction of spectral overlap in multicolor flow cytometry. The compensation reagents created are reproducible and stable under long-term storage conditions as described in this protocol.
There is one important consideration for using these controls. When dead cells are excluded in the same channel as other cell types (e.g., CD14+ monocytes or CD20+ B-cells, also known as a “DUMP Channel”), only one compensation control is necessary. Typically, the LIVE/DEAD® fixable dye is the brightest reagent in this detector because only the CD3+ T cells are included in the final gating (negative gating), hence only the ViViD stained beads need to be considered as a compensation control. Another advantage of negative gating can be demonstrated in figure 4, where ViViD is combined with anti-CD20 Pacific Blue (PB) and anti-CD14 Pacific Blue to exclude dead cells, B-cells, and monocytes (respectively) using a single detector. After compensation, five populations can be discriminated based on the ViViD and MAB-conjugates (Figure 4A), as follows: live CD3+ T-cells (1), dead CD3+ T-cells (2), monocytes (3), B-cells (4), and live CD3- CD14- CD20- cells (5). Figures 4B-F shows the overlay of the different populations (1-5) over the lymphocyte gate containing the CD3+ T-cells (1). Each population shows the degree of contamination within the traditional lymphocyte gate, dead cells (C), monocytes (D), B-cells (E) and live CD3-14-20- cells (F). Hence, negative cell gating removes many unwanted cells, which might otherwise fall within the gates of interest.
Figure 4. Dump Channel Analysis.
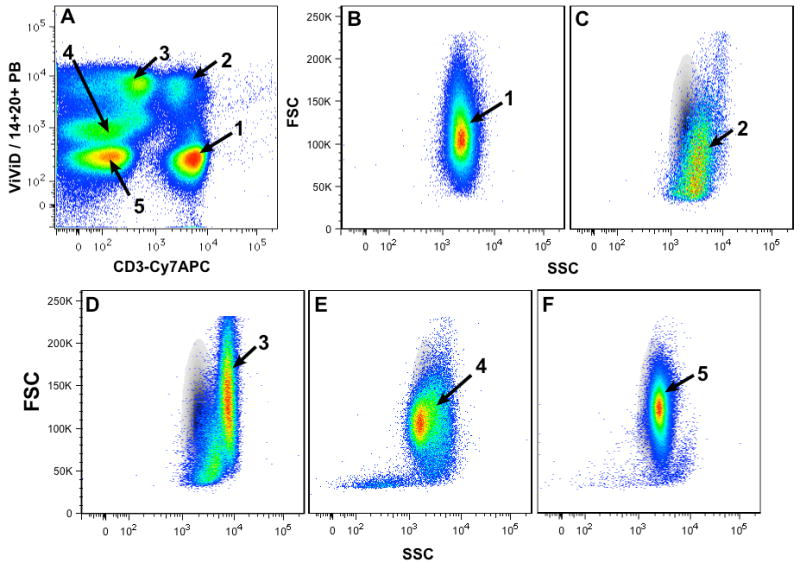
This figure shows the effective use of a “DUMP” channel, which uses negative gating strategy. In this strategy, ViViD is combined with anti-CD20 Pacific Blue (PB) and anti-CD14 PB to exclude dead cells, B-cells, and monocytes (respectively) using a single detector. After compensation, five populations can be discriminated based on the ViViD and MAB-conjugates (Figure 4A), as follows: live CD3+ T-cells (1), dead CD3+ T-cells (2), monocytes (3), B-cells (4), and live CD3- CD14- CD20- cells (5). Figures 4B-F shows the overlay of the different populations (1-5) over the lymphocyte gate containing the CD3+ T-cells (1). Each population shows the degree of contamination within the traditional lymphocyte gate, dead cells (C), monocytes (D), B-cells (E) and live CD3-14-20- cells (F).
Materials
ViViD amine reactive dye (LIVE/DEAD® fixable violet dead cell stain) or Aqua Blue amine reactive dye (LIVE/DEAD® fixable aqua dead cell stain) - Invitrogen
Bead Storage Media - see Reagent and Solution section
Standard Staining Media - see Reagent and Solution section
Phosphate Buffer Saline (PBS) - Becton Dickenson or other suppliers
Fetal Calf Serum (FCS)– Invitrogen or other suppliers
Neonatal calf sera - Invitrogen or other suppliers
RPMI-1640 - GIBCO or other suppliers
Sodium Azide - Sigma Chemical or other suppliers
Flow Cytometer – An analyzer or sorter equipped with a violet (or UV) laser and optics such as shown in Figure 2.
R-NH2 Beads (SMPLX Amine active beads) – Bangs Laboratories*
Creating a stable compensation control for the amine reactive dyes
Pre-labeled R-NH2 COMP Beads with Matching Negative Control: These beads are pre-labeled with amine dye and contain the matching negative control. Expiration time is one year. Note: since the concentration of amines on the beads is in large excess relative to the dye concentration the beads will require dilution against the highest dye concentration to determine the correct number of beads per vial of dye. The following steps are used to create this control.
Bead Stock: Dilute the stock beads to a conc. of 8.0 ×107 beads/ml in PBS (e.g. use 10ul of R-NH2 bead stock at 2.8 ×109 beads/ml in a total volume of 350ul in PBS).
Perform a serial dilution of beads with PBS down to approx. 2.5×106 beads using total volumes of 175ul per dilution.
Wash all dilutions twice in PBS. Centrifuge 1× at 400×g for 3min and dry pellet.
Add 5ul of pre-diluted (reconstituted vial) amine dye at the highest concentration to each bead dilution (50ul of Dye into 50ul of PBS to a final conc. = 250ug/ml) to all beads tubes, keeping the dye concentration constant with variable beads concentration).
Incubate at RT for 60 min and shield from light.
Wash twice in PBS. Centrifuge 1× at 400×g for 3min. Re-suspend in 100ul of bead staining media (see section Reagents and Solutions).
Add 100ul of equal conc. of unstained beads in bead staining media (see section Reagents and Solutions).
Run on a standardized Flow Cytometer and determine the bead dilution with maximum MFI (e.g. in our tests, 10×106 beads produced the highest MFI per one vial of dye stock conc.).
Using the bead: dye ratio determined in step 9, a larger batch of beads can be created following the procedure above. As an example, if the ideal bead concentration was determined to be 10×106 beads per one vial of dye stock concentration (250ug/ml) then 20×106 beads would require 2 vial, etc. This means it requires 1.25ug of dye to stain 10×106 beads to produce the maximum MFI.
Figure 5 shows the results of negative and positive beads labeled with the ViViD dye (left panel) and the Aqua Blue dye (right panel) as prepared by this protocol.
Figure 5. Pre-labeled compensation Controls used in cell staining panels.
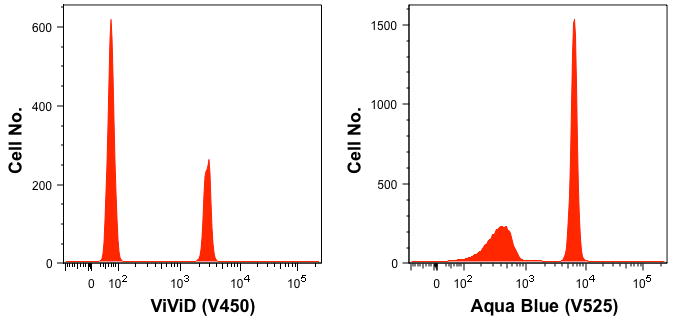
Figure 5 illustrates two pre-labeled compensation controls used in staining panels, one labeled with amine reactive dye, ViViD and the other labeled with amine reactive dye, Aqua Blue. These controls contain both the negative control beads (lower intensity) and the positive control beads (high intensity).
Protocol 3: Staining, Gating and Analysis of Amine Reactive dyes
The purpose of this protocol is to demonstrate how to use amine reactive dyes in multicolor cell panels to identify and remove dead cells from the gating analysis. As previously discussed, dead cells can nonspecifically bind mAb-conjugates, which can result in artifacts and erroneous labeling of cell populations (Perfetto et al., 2006b). Hence, only viable cells should be used in most gating analysis strategies. In this protocol, an example of the standard gating strategy using a ViViD dump channel (negative gating) and anti-CD3 will illustrate the utility of the ViViD amine reactive dye to describe gating and analysis strategies.
Materials
ViViD amine reactive dye (LIVE/DEAD® fixable violet dead cell stain) or Aqua Blue amine reactive dye (LIVE/DEAD® fixable aqua dead cell stain) - Invitrogen
Bead Storage Media - see Reagent and Solution section
Standard Staining Media - see Reagent and Solution section
Phosphate Buffer Saline (PBS) - Becton Dickenson or other suppliers
Fetal Calf Serum (FCS)– Invitrogen or other suppliers
Neonatal calf sera -– Invitrogen or other suppliers
RPMI-1640 -– GIBCO or other suppliers
Sodium Azide - Sigma Chemical or other suppliers
Flow Cytometer – An analyzer or sorter equipped with a violet (or UV) laser and optics such as shown in Figure 2.
Staining
Thaw DMSO using a 37C water bath until completely thawed (approx. 30 sec). Add the amount of DMSO into a vial of lyophilized dye as determined in the titration protocol (see Protocol 1: Titration of amine reactive dyes). Note: lyophilized dye is stored desiccated and expires at 60 months under these conditions. DMSO must be stored under desiccated conditions.
Mix thoroughly with a pipet tip (depending on the dye used DMSO will turn from clear to colored, indicating the dye is dissolved).
Store at −20° C. These vials can be thawed and frozen until aliquot is empty. DO NOT store as aliquots in buffered media or media containing amino acids and do not use longer then 3 months.
Add 1ul of stock concentration into 39 ul of dH2O (see protocol: Titration of amine reactive dyes). Note: Dilution in dH2O is critical. Loss of fluorescent intensity will occur if this dye is prepared in other media sources containing amino acids.
Add 5 ul of the working conc. into cells suspended in 95 ul of PBS. Note: Because some mAb-fluorochromes and free amino acids in media can compete with amine reactive dyes all other surface stains should be added in a separate step to avoid the loss of dye effectiveness. In addition, PBS is used in this staining step but all other wash steps can use media containing HI-FCS or other enriched media containing amino acids.
Mix and incubate at RT for 20 min shielded from light. Note: This reaction time is critical. Increased time or temperature does not improve staining intensity of the amine reactive dyes.
Wash 1× in standard staining medium or medium containing HI-FCS (Heat Inactivated Fetal Calf Sera). After wash, fluorochrome mAb conjugates (typically prepared in a master mix brought to a volume of 100ul/test). Mix thoroughly and incubate according to each panel standard operating procedure (See Unit 6.2 or Chattopadhyay et al.,.[2008] or Lamoreaux et al., [2006]). Note: PERM/FIX reagents such as Cytofix/Cytoperm or 0.5% PFA will not change the stability of this dye nor will the dye leak out of the cells.
Run compensation controls and all stained cells using a standardized flow cytometer. Figure 6 shows a typical gating strategy when collecting cell samples to verify correct gate position and efficiency of the viability stain. The first histogram uses the measurements of forward scatter area and forward scatter height to determine the single cells (inside the diagonal gate -A) from the doublets (above the diagonal gate -A). From the single cell gate the viability gate (DUMP channel using ViViD, anti-CD14 and anti-CD19) is setup with anti-CD3 to identify the live CD3+ T cells (B), which are gated for further analysis depending on the staining panel characteristics.
Figure 6. Common gating strategy to remove doublets and dead cells in the cell analysis.
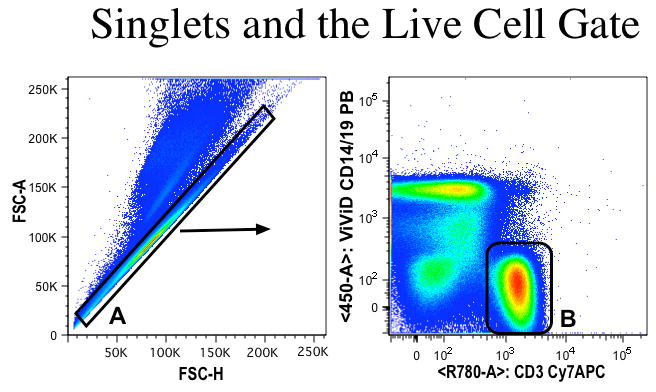
Figure 6 shows the gating strategy used to gate on the live CD3+ T cells. The first histogram uses the measurements of forward scatter area and forward scatter height to determine the single cells (inside the diagonal gate) from the doublets (above the diagonal gate). From the single cell gate the viability gate (DUMP channel using ViViD, CD14 and CD19) is setup with CD3. This negative gate contains only the live CD3+ T cells while excluding dead cells, monocytes, B-cells and live cells not stained with CD14,19 or CD3.
Reagents and Solutions
This section contains all recipes for the protocols, media, beads, etc.
Bead Storage Media:
| Reagent | Stock Conc. | Volume | Final Conc. |
|---|---|---|---|
| Fetal calf sera (HI-FCS*) | na | 1ml | 1% |
| Sodium Azide (NaN3) | 5% | 400ul | 0.02% |
| PBS | na | 98.6ml | na |
| Total | 100 ml | ||
Heat inactivation = thawed FCS is placed at 56°C for one hour.
Standard Staining Media:
| Reagent | Volume | Stock Conc. | Final |
|---|---|---|---|
| Neonatal calf sera (HI NCS*) | 20 ml | 100% | 4% |
| Sodium Azide (NaN3) | 0.5ml | 20% | 0.02% |
| RPMI 1640** | 174 ml | n/a | n/a |
| Total | 500 ml | ||
Heat inactivation (HI) = thawed sera is placed at 56°C for one hour.
RPMI 1640 formula #00-0327DK
Commentary
Background Information
Immune monitoring and vaccine immunogenicity studies often require the measurement of low frequency cell populations in samples that have been cryopreserved. This inevitably leads to questions of sensitivity and reproducibility, since dead cells in the sample may non-specifically bind monoclonal antibody-conjugates (mAb-conjugates) and cause significant artifacts (Maecker et al., 2005; O'Brien and Bolton, 1995; Perfetto et al., 2004a; Schmid et al., 1999). Fortunately, viability dyes may be used to exclude dead cells from analysis. Historically, viability dyes were employed that enter damaged cells via compromised cell membranes, and then intercalate into DNA; propidium iodide (PI) is an example of such a dye. However, PI may leak out of cells within a short period of time, leading to significant signal loss (Clarke and Pinder, 1998; Costantino et al., 1995; Desrues et al., 1989). This is particularly problematic when permeablization reagents are used to stain intracellular molecules (such as cytokines), as is often the case in immune monitoring and immunogenecity studies. To avoid this problem, ethidium monoazide (EMA) may be used. This dye covalently binds to DNA after exposure to ultraviolet (UV) light. Although this dye can resolve dead cell populations and is unaffected by intracellular treatments, the need for a UV light source is inconvenient (Riedy et al., 1991). Also, the degree of membrane damage in apoptotic cells can be variable, leaving some Annexin V+ cells with intermediate levels of PI or EMA staining (Matteucci et al., 1999; Waters et al., 2002). The amine reactive dyes as discussed in this protocol avoid many of disadvantages of these traditional viability markers. Hence, these are a good alternative for measuring and removing dead cells from the cell analysis.
Time Consideration
The time needed for cell staining with viability dyes and multicolor antibody conjugates is typically no more than 30 minutes. It would be tempting to shorten this procedure by mixing the mAb-conjugates with the viability dye: however, this is not recommended because the staining medium used for antibody staining often contains proteins. Moreover, some antibody preparations typically contain high levels of proteins as stabilizers. These proteins introduce free amines, which compete with free amines from the cytosol of dead cells to bind to the amine reactive dyes. This competition significantly decreases the fluorescence intensity of the dye, and increases background staining. Therefore, we recommend pre-staining cells (resuspended in PBS) with amine reactive dyes, washing the cell sample, and then incubating with multicolor antibodies in the staining medium of choice.
Anticipated Results
The problem of non-specific binding is particularly notable when analyzing rare events within cryopreserved samples, since these samples are likely to contain significant numbers of dead cells. When viability markers are not included in these analyses, the frequency of the cells of interest can be dramatically misrepresented because dead cells can non-specifically bind reagents. For example, in figure 7, a rare population (CMV-specific CD8+ T-cells) was examined in bone marrow samples. Data was analyzed with and without excluding cells in the dump channel, which included antibodies against CD14 and CD19 (conjugated to Pacific Blue) and ViViD. The top row shows a “traditional” gating strategy (without dump channels) that might be used on a six-parameter flow cytometer tetramer+ cells are identified in a bivariate plot with CD8 after gating to identify T cells on the basis of CD3 expression. The second row shows a polychromatic gating strategy to eliminate aberrant binding events. In this analysis, live CD3+ T cells were distinguished from dead cells, monocytes, and B cells, which could bind tetramer and mAbs nonspecifically. Notably, CD8+ T cells specific for the CMV epitope could be cleanly identified with the polychromatic gating strategy (second row, right panel, 0.62%), whereas tetramer-binding cells are overrepresented in the top panel (1.57%). Backgating analysis shows that many of these cells are binding reagents in the dump channel, suggesting that much of the binding is non-specific (bottom panel; tetramer+ events are shown in blue overlaid on the total population in a bivariate CD3 versus CD14/CD19/ViViD plot). Thus, it is possible that sample-to-sample variation in viability, or in the frequency of B-cells and monocytes, could have a profound impact on the proportion of antigen-specific cells identified, thereby skewing study results. This demonstrates very clearly the need for amine reactive dyes in cell-staining panels.
Figure 7. An example of gating strategy used to eliminate aberrant binding events due to dead cells that may change tetramer analysis.
This figure shows an example of the utility of using amine reactive dyes to eliminate erroneous frequencies due to non-specific binding of dead cells. In this example, CMV antigen specific CD8+ T cells are measured in a staining panel containing a specific CMV tetramer reagent. The first row shows a traditional gating strategy. The final gate indicates that 1.57% of the CD8+ T cells are tetramer positive. However, when including the viability gate (second row) the CD8+ tetramer + T cells were determined to be at a lower frequency (0.62%) than reported using traditional gating. Overlaying the total CD8+ tetramer + T cells from the first row onto the viability gate (bottom row) shows that the majority of the cells are in fact ViViD + and hence should not be included in the final analysis.
Troubleshooting and Critical Parameters
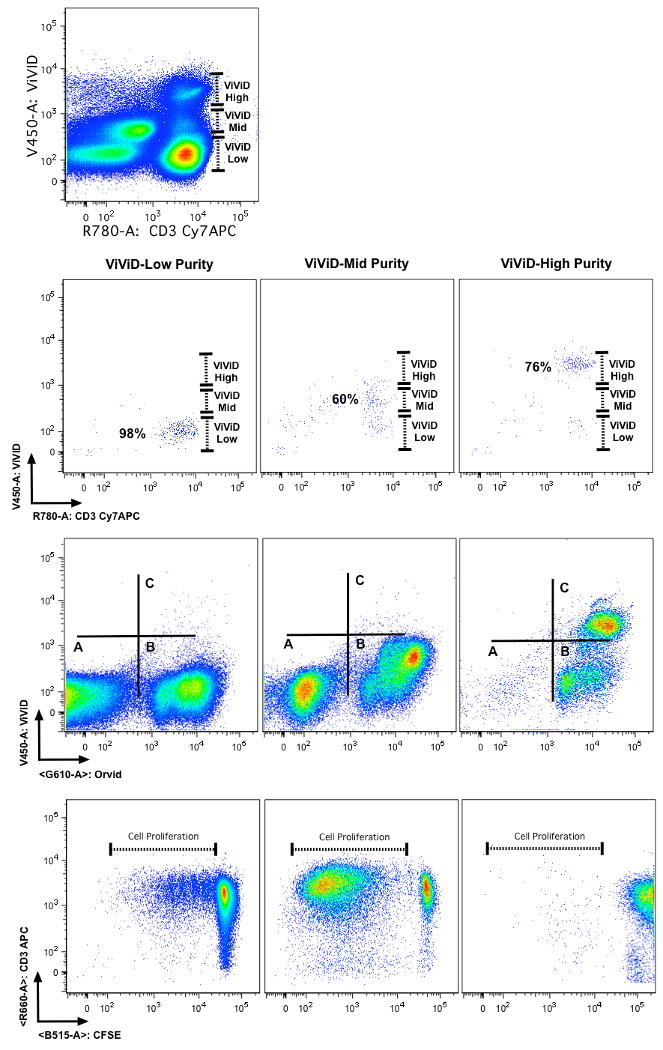
In some cases, greater intensity of the amine reactive dye can be seen above background but below expected positive intensity. In these situations it is important to determine if these cells should be excluded because they are dead or to include them because they are viable. To this end, the immunophenotype, cytokine production, or proliferation of the ViViD dim and negative cells may be compared. If the vivid dim population shows a similar expression profile as the negative cells, then the former should be included in the analysis. For example, we compared three cell populations sorted from the viability histogram in the first row of figure 8; these include: live CD3+ T cells (ViViD-Low), dim ViViD+ CD3+ cells (ViViD-Mid) and ViViD+ CD3+ dead cells (ViViD-High). In addition to cell surface stains, the cells were loaded with the proliferation marker CFSE. The sorted cells (figure 8, 2nd row) were then cultured in the presence of SEB for five days using standard culture procedures. After this incubation, cells were re-stained for CD3 (anti-CD3-APC) and OrViD (a second amine reactive dye) and measured on the flow cytometer. OrViD positive cells died as a result of the 5-day incubation process and were excluded from the analysis (figure 8, 3rd row). The sorted population of cells that was ViViD-Mid progressed through as many (or more) rounds of division as the ViViD negative population (ViViD-Low), suggesting that these cells should be included in the final analysis (figure 8, 4th row). Notably, ViViD staining is reduced in proliferating cells, presumably as a result of dilution of bound dye into the daughter cells. These results suggest that protein expression is increased for cell populations that are activated in vivo or in vitro resulting in a slightly increased ViViD binding (ViVid Mid) and fluorescence.
Figure 8. CD3+ T cells that are dimly stained with amine reactive dyes should be included in cell analysis.
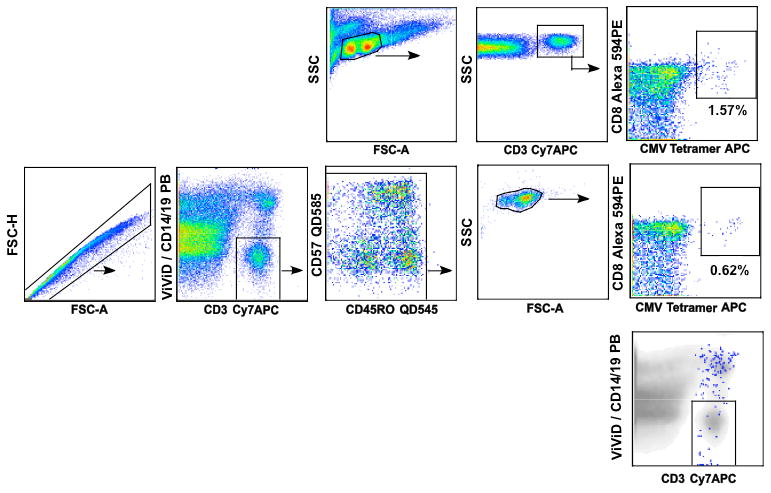
Figure 8 shows the proliferative capacity of three CD3+ ViViD stained populations, ViViV-Low, ViViD-Mid and ViViD-High after staining with CFSE, ViViD and anti-CD3-Cy7APC. The 2nd row shows the sort purity results for each of the sorted cell populations as illustrated in the histogram in the 1st row. The 3rd and 4th rows show results of post incubation for five days in the presence of SEB (Streptococcus Enterotoxin B) followed by staining with OrViD (amine reactive dye measured in G610 detector) and anti-CD3-APC. These additional stains were added to measure dead cells as a result of a 5-day incubation and the total number of CD3+ T cells. In the 3rd row three populations can be described, live cells (A, ViViD-OrViD-), dead cells as a result of incubation (B, ViViD-OrViD+) and dead cell from the original sort (C, ViViD+OrViD+). In both the ViViD-Low and ViViD-Mid cell sorts, live cells were observed and these cells were also proliferating (4th row). The dead cells from the ViViD-High cell sort show no cell proliferation (4th row).
The amine reactive dyes have distinct advantages over traditional viability staining. Firstly, they are simple to use, they are stable and they can be used with other mAb-conjugates after the complete interaction with free amines in the cytosol of the dead cell. Secondly, the amine reactive dyes are sold with a variety of emission and excitation wavelengths and can therefore be included in many cells staining panels. This flexibility allows for many traditional mAb-conjugates combinations to remain while “fitting in” a viability dye. These protocols illustrate the utility of the amine reactive dyes in cell biology.
Critical Parameters
Protocol 1: Titration of amine reactive dyes
Note 1: lyophilized dye is stored desiccated and expires at 60 months under these conditions. DMSO must be stored under desiccated conditions.
Note 2: When making the working dye concentration, the dilution in dH2O is critical. Loss of fluorescence intensity will occur if this dye is prepared in other media sources containing amino acids.
Note 3: It is critical to stain cells with the amine reactive dye in PBS. Loss of fluorescence intensity will occur if this dye is prepared in other media sources containing amino acids. However, standard staining media may be used in subsequent staining steps.
Note 4: This reaction time is optimal. Increased time or temperature does not improve staining intensity of the amine reactive dyes.
Protocol 2: Creating a stable compensation control for the amine reactive dyes
Note 1: Since the beads are in large excess relative to the dye concentration, the beads will require dilution against the highest dye concentration to determine correct number of beads per vial of dye.
Protocol 3: Staining, Gating and Analysis of Amine Reactive dyes
Note 1: Lyophilized dye is stored desiccated and expires at 60 months under these conditions. DMSO must be stored under desiccated conditions.
Note 2: Dilution in dH2O is critical. Loss of fluorescent intensity will occur if this dye is prepared in other media sources containing amino acids.
Note 3: Some mAb-fluorochromes reagents and free amino acids in media can compete with amine reactive dyes all other surface stains should be added in a separate step to avoid the loss of dye effectiveness.
Note 4: Reaction time is critical. Increased time or temperature does not improve staining intensity of the amine reactive dyes.
Note 5: PERM/FIX reagents such as Cytofix/Cytoperm or the use of 0.5% PFA will not change the stability of amine reactive dye nor will the dye leak out of the cells.
Troubleshooting
| Problem | Possible Cause | Solution |
|---|---|---|
| Dead cells are dimly fluorescent and appear separate poorly from the live cell population. |
|
|
| Poor amine reactive dye compensation control. Dim staining as compared to dead cells stained with amine reactive dye. |
|
|
| Poor DUMP compensation control. Amine reactive dye appears not to be subtracted |
|
|
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH; the National Cancer Institute, NIH, under contract No. HHSN261200800001E.
Footnotes
For proprietary reasons, this reagent can only be ordered by phone, and is not available in the Bangs Laboratories catalog.
Disclaimer: The views expressed here are the opinions of the authors and are not to be considered as official or reflecting the views or policies of the Vaccine Research Center / National Institutes of Health / Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Literature Cited
- Chattopadhyay PK, Melenhorst JJ, Ladell K, Gostick E, Scheinberg P, Barrett AJ, Wooldridge L, Roederer M, Sewell AK, Price DA. Techniques to improve the direct ex vivo detection of low frequency antigen-specific CD8+ T cells with peptide-major histocompatibility complex class I tetramers. Cytometry A. 2008;73:1001–9. doi: 10.1002/cyto.a.20642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RG, Pinder AC. Improved detection of bacteria by flow cytometry using a combination of antibody and viability markers. J Appl Microbiol. 1998;84:577–84. doi: 10.1046/j.1365-2672.1998.00384.x. [DOI] [PubMed] [Google Scholar]
- Costantino PJ, Budd DE, Gare NF. Enumeration of viable Candida albicans blastospores using tetrabromofluorescein (eosin Y) and flow cytometry. Cytometry. 1995;19:370–5. doi: 10.1002/cyto.990190413. [DOI] [PubMed] [Google Scholar]
- Desrues B, Collet B, Rame MP, Bourel D, Bourguet P, Martin A, Delaval P, Toujas L, Dazord L. Distribution of radiolabelled monoclonal antibody Po66 after intravenous injection into nude mice bearing human lung cancer grafts. Cancer Immunol Immunother. 1989;30:295–9. doi: 10.1007/BF01744897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamoreaux L, Roederer M, Koup R. Intracellular cytokine optimization and standard operating procedure. Nature Protocols. 2006;1:1507–1516. doi: 10.1038/nprot.2006.268. [DOI] [PubMed] [Google Scholar]
- Maecker HT, Rinfret A, D'Souza P, Darden J, Roig E, Landry C, Hayes P, Birungi J, Anzala O, Garcia M, Harari A, Frank I, Baydo R, Baker M, Holbrook J, Ottinger J, Lamoreaux L, Epling CL, Sinclair E, Suni MA, Punt K, Calarota S, El-Bahi S, Alter G, Maila H, Kuta E, Cox J, Gray C, Altfeld M, Nougarede N, Boyer J, Tussey L, Tobery T, Bredt B, Roederer M, Koup R, Maino VC, Weinhold K, Pantaleo G, Gilmour J, Horton H, Sekaly RP. Standardization of cytokine flow cytometry assays. BMC Immunol. 2005;6:13. doi: 10.1186/1471-2172-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteucci C, Grelli S, De Smaele E, Fontana C, Mastino A. Identification of nuclei from apoptotic, necrotic, and viable lymphoid cells by using multiparameter flow cytometry. Cytometry. 1999;35:145–53. doi: 10.1002/(sici)1097-0320(19990201)35:2<145::aid-cyto6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- O'Brien MC, Bolton WE. Comparison of cell viability probes compatible with fixation and permeabilization for combined surface and intracellular staining in flow cytometry. Cytometry. 1995;19:243–55. doi: 10.1002/cyto.990190308. [DOI] [PubMed] [Google Scholar]
- Perfetto S, Ambrozak D, Nguyen R, Chattopadhyay P, Roederer M. Quality assurance for polychromatic flow cytometry. 2006a;1:1522–30. doi: 10.1038/nprot.2006.250. Unknown. [DOI] [PubMed] [Google Scholar]
- Perfetto S, Chattopadhyay P, Lamoreaux L, Nguyen R, Ambrozak D, Koup R, Roederer M. Amine reactive dyes: An effective tool to discriminate live and dead cells in polychromatic flow cytometry. Journal of Immunological Methods. 2006b;313:199–208. doi: 10.1016/j.jim.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Perfetto SP, Ambrozak DR, Roederer M, Koup RA. Viable infectious cell sorting in a BSL-3 facility. Methods Mol Biol. 2004a;263:419–24. doi: 10.1385/1-59259-773-4:419. [DOI] [PubMed] [Google Scholar]
- Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004b;4:648–55. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- Riedy MC, Muirhead KA, Jensen CP, Stewart CC. Use of a photolabeling technique to identify nonviable cells in fixed homologous or heterologous cell populations. Cytometry. 1991;12:133–9. doi: 10.1002/cyto.990120206. [DOI] [PubMed] [Google Scholar]
- Schmid I, Ferbas J, Uittenbogaart CH, Giorgi JV. Flow cytometric analysis of live cell proliferation and phenotype in populations with low viability. Cytometry. 1999;35:64–74. doi: 10.1002/(sici)1097-0320(19990101)35:1<64::aid-cyto9>3.3.co;2-p. [DOI] [PubMed] [Google Scholar]
- Waters WR, Harkins KR, Wannemuehler MJ. Five-color flow cytometric analysis of swine lymphocytes for detection of proliferation, apoptosis, viability, and phenotype. Cytometry. 2002;48:146–52. doi: 10.1002/cyto.10122. [DOI] [PubMed] [Google Scholar]
Web Site References
- Invitrogen web site of LIVE/DEAD® fixable dyes: http://www.invitrogen.com/site/us/en/home/Global/invitrogen-search-results.html?searchTerm=live+%2F+dead+fixable&searchRows=15&searchTypes=meta.collection%3Acmgtbuyproduct%3A&zeroCategories=meta.collection%3Amrdb%3A
- Invitrogen Spectra – viewer http://www.invitrogen.com/site/us/en/home/support/Research-Tools/Fluorescence-SpectraViewer.html