

Abstract
Th cells that produce IL-17 (Th17 cells) are a distinct subset of Th cells implicated in several autoimmune diseases. Although CD28–B7 interaction has been shown to be involved in Th17 differentiation in vitro, the role of CTLA-4 in controlling Th17 development is completely unknown. We report in this paper that blocking the CTLA-4–B7 interaction potentiates Th17 cell differentiation in vitro and in vivo. Furthermore, blocking CTLA-4–B7 interaction in vivo confers the susceptibility of experimental autoimmune myocarditis to CD28−/− mice or increases the severity of experimental autoimmune myocarditis in wild-type mice. The enhanced disease susceptibility is mediated by heightened Th17 responses. With these results, we are the first to demonstrate that CTLA-4–B7 interaction inhibits Th17 differentiation in vitro and in vivo and suppresses Th17-mediated autoimmunity.
The range of identified effector CD4+ T cell lineages recently has expanded with the description of a distinct IL-17–producing subset, called Th17, which develops via cytokine signals antagonized by products of the Th1 and Th2 lineages (1, 2). By secreting IL-17, IL-17F, and IL-22, Th17 cells induce a massive tissue reaction as a result of the broad distribution of the IL-17R and IL-22R. Recent data in humans and mice suggest that Th17 cells play a critical role in the pathogenesis of a diverse group of immune-mediated diseases (2).
The CD28 costimulatory signal has been found to be important for the differentiation of CD4+ Th cells into Th2 cells (3). It has also been shown that both CD28 and ICOS are involved in Th17 cell differentiation (4). However, the direct role of CTLA-4 in Th17 cell differentiation has not been elucidated. In this study, we are the first to demonstrate that CTLA-4–B7 interaction inhibits Th17 cell differentiation in vitro and in vivo and suppresses the development of Th17- mediated autoimmunity.
Materials and Methods
Mice
Wild-type (WT) BALB/c, C57BL/6 (B6), Rag-1−/−, and CD28−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME). CTLA-4−/−/B7-1−/−/B7-2−/− mice on the B6 background were obtained from Dr. J. Bluestone (University of California, San Francisco, CA). All of the mice were used for experiments at ages of 6–10 wk.
In vitro cytokine production
CD4+ T cells were isolated using mouse CD4+ T cell isolation kits (R&D Systems, Minneapolis, MN) and stimulated with anti-CD3 (2 μg/ml) and irradiated T-depleted splenocytes as feeder cells in the presence of human (h) CTLA-4-Ig (Abatacept; Bristol-Myers Squibb, Princeton, NJ), recombinant human (rh)IgG1-Fc (R&D Systems), hamster IgG (Sigma-Aldrich, St. Louis, MO), or anti–CTLA-4 (UC10-4F10; eBioscience, San Diego, CA) for 48 h. The supernatants collected were subjected to cytokine detection by ELISA (eBioscience).
In vitro and in vivo Th17 differentiation
Naive CD4+ T cells isolated by mouse naive CD4+ T cell isolation kits (R&D Systems) were stimulated with soluble anti-CD3 (2 μg/ml) in the presence of irradiated T-depleted syngeneic splenocytes with hCTLA-4-Ig (12.5 μg/ml), rhIgG1-Fc (12.5 μg/ml), anti–CTLA-4 (5 μg/ml), or hamster IgG (5 μg/ml) underTh17 conditions: hTGF-β (5 ng/ml) (R&D Systems) plus IL-6 (20 ng/ml) (PeproTech, Rocky Hill, NJ), anti–IL-4 (11B.11;10 μg/ml), and anti–IFN-γ (XMG1.2; 15 μg/ml). For Th17 differentiation in vivo, Rag-1−/− mice (n = 10) were i.v. injected with purified naive CD4+ T cells fromCD28−/− or WT mice and administered i.p. with hCTLA-4-Ig or rhIgG1-Fc (150 μg) every other day for 10 d orwith anti–CTLA-4 or hamster IgG(100μg) for 5 consecutive days and analyzed on day 10.
In vitro Th1/Th2 differentiation
Naive CD4+ T cells were cultured with anti-CD3 (2 μg/ml) and irradiated T-depleted splenocytes as feeder cells under Th1 (anti–IL-4 [11B.11; 10 μg/ml] plus mouse IL-12 [5 ng/ml] [R&D Systems]) and Th2 (anti–IFN-γ [XMG1.2; 10 μg/ml] and anti–IL-12 [10 μg/ml] [eBioscience] plus mouse IL-4 [5 ng/ml] [R&D Systems]) conditions for 4 d, and live CD4+ T cells were isolated, washed thoroughly, and cultured in the presence of hIL-2 (50 U/ml; Sigma-Aldrich) in Th1 or Th2 conditions for an additional 2 d. Then the cells were restimulated in the presence of immobilized anti-CD3 (1 μg/ml) and Golgi-Stop for 5 h and subjected to intracellular staining.
Induction of experimental autoimmune myocarditis
Mice were immunized twice at 7-d intervals with 100 μg murine α-myosin peptide MYHC-α peptide (614–629[Ac-SLKLMATLFSTYASAD-OH]) emulsified 1:1 in PBS/CFA (1 mg/ml; Difco Laboratories, Detroit, MI) as described previously (5). On days 0 and 5, mice received an i.p. injection of 500 ng pertussis toxin (List Biological Laboratories, Campbell, CA). For hCTLA-4-Ig or anti–CTLA-4 treatment, mice were injected i.p. with hCTLA-4-Ig or rhIgG1-Fc (150 μg) every other day starting from day 2 or with anti–CTLA-4 or hamster IgG (100 μg) on days 0, 3, and 6. For neutralizing IL-17 in vivo, MYHC-α–immunized CD28−/− mice were administered i.p. with a neutralizing anti–IL-17 Ab (100 μg) on days 9, 12, and 15. All mice were sacrificed on day 22. Myocarditis was scored on H&E-stained sections using grades from 0 to 4: 0, no inflammatory infiltrates; 1, small foci of in- flammatory cells between myocytes; 2, larger foci of >100 inflammatory cells; 3, >10% of a cross-section involved; and 4, >30% of a cross-section involved (5). Heart-infiltrating cells were isolated as previously described (6) and stained with fluorescence-conjugated Abs to CD4, CD3, and CD45 (BD Biosciences).
Intracellular staining of cytokines and flow cytometric analysis
For intracellular cytokine staining, draining lymph node cells, infiltrating lymphocytes isolated from the heart, or differentiated cells (in vitro Th17) were stimulated with PMA (50 ng/ml; Sigma-Aldrich) and ionomycin (1 μg/ml; Sigma-Aldrich) for 5 h in culture medium in the presence of GolgiStop (BD Biosciences). After staining of surface markers, cells were fixed and permeabilized by fixation/permeabilization buffer (eBioscience), followed by staining with Abs against mouse IL-17, IL-4, and IFN-γ (BD Biosciences) and analyzed on an LSRII flow analyzer (BD Biosciences) using FlowJo (Tree Star, Ashland, OR) software.
Data analysis
Data are shown as mean ± SEM, and statistical significance was based on p < 0.05 (Student t test).
Results and Discussion
CTLA-4–B7 interaction inhibits Th17 cell differentiation in vitro and in vivo
Although it has been well documented that CTLA-4 functions as a negative regulator of T cell activation and tolerance induction (7, 8), the direct role of CTLA-4 in Th17 development has not been defined. Previously, we have demonstrated that blocking CTLA-4–B7 interaction partially restores defective proliferation of CD28−/− T cells (9). To assess the effect of CTLA-4 on Th1, Th2, and Th17 cytokine production, WT and CD28−/− CD4+ T cells were stimulated with anti-CD3 in the presence of WT irradiated T-depleted splenocytes together with hCTLA-4-Ig or rhIgG1-Fc. The advantage of this system is that CTLA-4-Ig would mainly block CD28–B7 interaction in WT T cell culture, whereas it would block CTLA-4–B7 interaction in CD28−/− T cell culture. Addition of hCTLA-4-Ig significantly suppressed the production of IL-4, IFN-γ, and IL-17 by WT CD4+ T cells, whereas it enhanced production of IL-17 but not IL-4 or IFN-γ by CD28−/−CD4+ T cells (Fig. 1A). To verify the role of CD28 and CTLA-4 in the regulation of cytokine production, we used CTLA-4−/−/B7-1−/−/B7-2−/− mice because CTLA-4−/− mice develop fatal lymphoproliferative disease and die at 3– 5 wk of age (10–12), and the T cells from CTLA-4−/− mice are spontaneously activated in vivo (10–12). CD4+ T cells from CTLA-4−/−/B7-1−/−/B7-2−/− mice are naive (13) and produced high levels of IL-4, IFN-γ, and IL-17 in the presence of irradiated T cell-depleted B6 splenocytes (Supplemental Fig. 1). The addition of hCTLA-4-Ig into the CTLA-4−/−/B7-1−/−/B7-2−/− T cell culture abrogated heightened cytokine production (Supplemental Fig. 1), indicating that heightened CD28 activity in the CTLA-4−/− T cells may be the reason for the heightened cytokine production. It has been shown that CTLA-4-Ig can activate APCs through B7 and induce IDO, which inhibits T cell activation (14). However, if CTLA-4-Ig interacts with B7 and induces a reversal signal through B7s on APCs to induce IDO production, one would expect that this treatment should lead to suppression rather than potentiation of CD28−/− T cell activation. In support of this notion, an anti–CTLA-4 blocking Ab selectively enhanced production of IL-17 by CD28−/− T cells, whereas this treatment augmented IL-4, IFN-γ, and IL-17 production by WT T cells (Fig. 1A), because blocking CTLA-4–B7 interaction by anti– CTLA-4 Ab results in heightened CD28 costimulatory function (15). These data suggest that blocking CTLA-4–B7 interaction potentiates Th17 differentiation in vitro.
FIGURE 1.
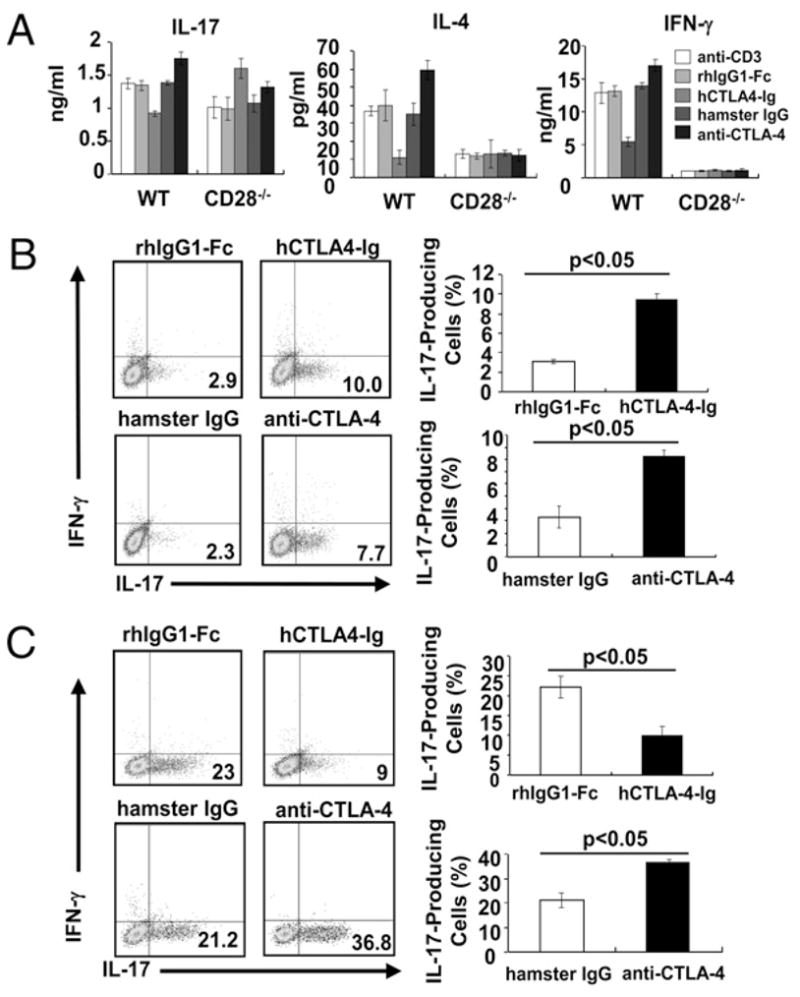
CTLA-4–B7 interaction inhibits Th17 differentiation in vitro. A, Purified naive CD4+ T cells from spleens and lymph nodes of CD28−/− or WT mice were cultured in the presence of irradiated T cell-depleted BALB/c splenocytes and anti-CD3 (2 μg/ml) with rhIgG1-Fc (12.5 μg/ml), hCTLA-4-Ig (12.5 μg/ml), hamster IgG (5 μg/ml), or anti–CTLA-4 (5 μg/ml) for 48 h. The production of IL-17, IL-4, and IFN-γ was determined by ELISA. B and C, Naive CD4+ T cells from CD28−/− mice (B) or WT mice (C) were activated with anti-CD3 and irradiated T-depleted splenocytes as feeders under Th17-polarized condition in the presence of hCTLA-4-Ig, rhIgG1-Fc, anti–CTLA-4, or hamster IgG. The values represent mean ± SEM. The data are representative of three independent experiments.
To directly address whether CTLA-4 blockade selectively enhances Th17 cell differentiation, we performed in vitro Th1, Th2, and Th17 differentiation assays using naive CD28−/− CD4+ T cells in the presence or absence of hCTLA-4-Ig or anti–CTLA-4. The addition of hCTLA-4-Ig or anti–CTLA-4 led to a significantly heightened differentiation of CD28−/− T cells into Th17 cells (Fig. 1B), whereas this blockade had no effect on Th1 and Th2 differentiation (Supplemental Fig. 2). Consistent with the previous report (4), blocking CD28–B7 interaction impaired the differentiation of WT T cells into Th17 cells (Fig. 1C), whereas anti–CTLA-4 treatment, which facilitates CD28–B7 interaction, augmented Th17 cell differentiation of WT T cells as reported previously (4, 16) (Fig. 1C). In support of this, the expression of the master transcription factors for Th17 cell differentiation, ROR-γt and ROR-α, was augmented as shown by real-time RT-PCR analysis (Supplemental Fig. 3A). IL-17F but not IFN-γ production was significantly enhanced in the presence of CTLA-4 blockade under Th17-polarizing condition (Supplemental Fig. 3B), whereas IL-4 production was undetectable under this condition (data not shown). Furthermore, the potentiation of Th17 cell differentiation by blocking CTLA-4–B7 interaction in CD28−/− T cells may not be due to the heightened proliferation as revealed by CFSE labeling (Supplemental Fig. 4).
It has been shown that BALB/c CD4+ T cells, which scarcely produced IL-17 before transfer, differentiate spontaneously into Th17 cells when they are adoptively transferred into Rag−/− mice (17). We then decided to test whether blocking CTLA-4–B7 interaction potentiates Th17 cell differentiation in vivo in Rag-1−/− mice receivingCD28−/−CD4+Tcells. Treating Rag- 1−/− mice receiving CD28−/−CD4+ T cells with hCTLA-4-Ig or anti–CTLA-4 greatly enhanced Th17 cell differentiation (Fig. 2A, Supplemental Fig. 5). In contrast, treating Rag-1−/− mice receiving CD4+ T cells from WT BALB/c mice with hCTLA-4-Ig significantly suppressed differentiation of Th17 cells, whereas treating these mice with anti–CTLA-4 augmented Th17 cell differentiation (Fig. 2B). These data support the notion that although CD28is required for Th17 differentiation, CTLA-4–B7 interaction inhibits this process.
FIGURE 2.
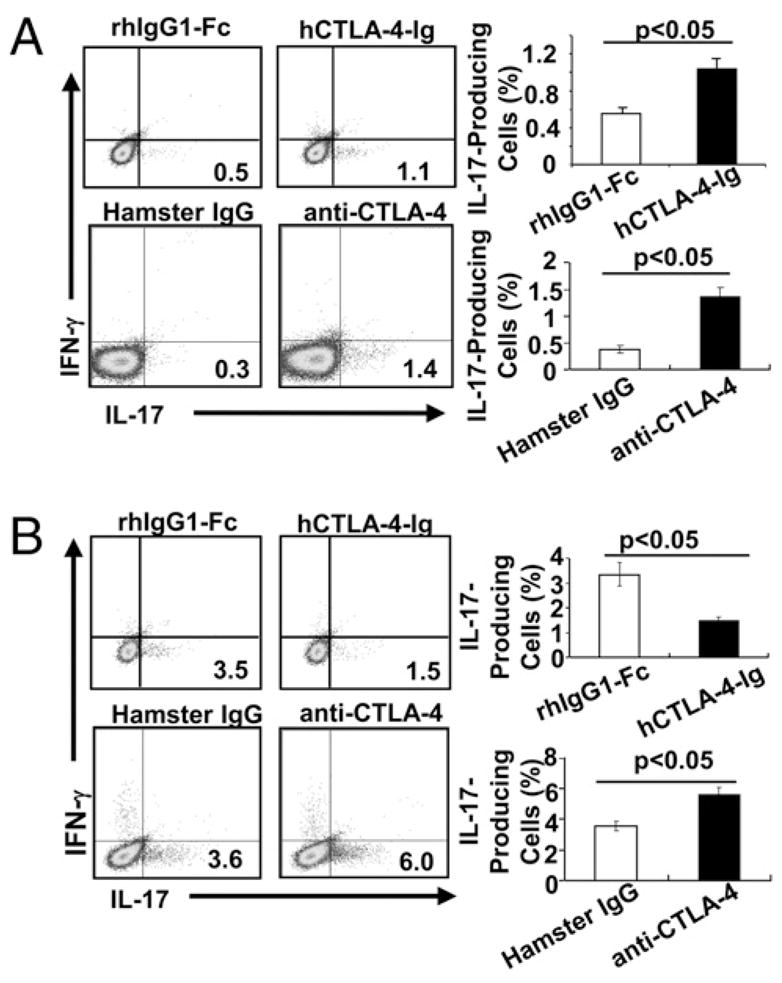
BlockingCTLA-4–B7 interaction facilitates Th17 differentiation in vivo. Rag-1−/− mice receiving naiveCD4+ T cells from CD28−/− mice (A) or WT mice (B) were i.p. injected with rhIgG1-Fc or hCTLA-4-Ig (150 μg/mouse) every other day for 10 d or hamster IgG or anti–CTLA-4 (100 μg/mouse) for 5 consecutive days. On day 10, IFN-γ– and IL-17–producing cells were determined by surface-staining splenocytes with anti-CD4 and intracellular staining with anti–IFN-γ and anti–IL-17 after PMA plus ionomycin stimulation. The data shown are representative of a group of 10 mice in two independent experiments, and the values are shown as mean ± SEM.
Blocking CTLA-4–B7 interaction potentiates Th17-mediated autoimmunity
To define whether blocking CTLA-4–B7 interaction regulates Th17-mediated autoimmune responses, we chose experimental autoimmune myocarditis (EAM) as a model, because it has been shown to be mediated by Th17 but not Th1 or Th2 (18). CD28−/− mice were immunized with MYHC-α peptide in CFA in the presence of hCTLA-4-Ig. As expected, although few CD28−/− mice developed EAM, the histological scores in these mice were very mild (Fig. 3A, 3C). In contrast, all WT mice developed EAM with higher histological scores (Fig. 3). Intriguingly, hCTLA-4-Ig treatment significantly increased the incidence and severity of myocarditis in CD28−/− mice (Fig. 3A, 3C). In support of a critical role for CTLA-4 in the development of EAM, anti–CTLA-4 treatment significantly exacerbated the disease in WT mice (Fig. 3B, 3C).
FIGURE 3.

Blocking CTLA-4–B7 interaction confers the susceptibility of autoimmune myocarditis to CD28−/− mice or exacerbates the disease in WT mice. A, CD28−/− mice (n = 10) were immunized with 100 μg MYHC-α peptide in CFA with rhIgG1-Fc or hCTLA-4-Ig in the presence or absence of a neutralizing anti–IL-17. WT BALB/c mice immunized with MYHC-α peptide (n = 10) were used as a positive control. All mice were sacrificed on day 22. H&E-stained sections of heart from immunized mice (original magnification ×200). a, WT mice; b, CD28−/− mice treated with rhIgG1-fc treatment; and c, CD28−/− mice receiving hCTLA-4-Ig treatment. d, CD28−/− mice receiving hCTLA-4-Ig treatment and anti–IL-17 neutralization. B, H&E-stained sections of heart (original magnification ×200) of WT BALB/c mice (n = 10) were immunized with MYHC-α peptide as in A and i.p. injected with hamster IgG (100 μg) or anti-CTLA-4 (100 μg). C, The prevalence and severity of MYHC-α–induced myocarditis.
To determine whether there is a heightened Th17 response in MYHC-α–immunized CD28−/− mice treated with hCTLA-4-Ig, we analyzed ex vivo IL-17–producing CD4+ T cells in draining lymph nodes and heart lymphocyte infiltrates in these mice. We found that there was a significant increase in IL-17–producing CD4+ T cells in draining lymph nodes in MYHC-α–immunized CD28−/− mice receiving hCTLA-4 Ig compared with those receiving rhIgG1-Fc (Supplemental Fig. 6A). hCTLA-4-Ig treatment resulted in severe lymphocyte infiltration in the heart with a significant increase in the proportion of CD3+ T cells within the CD45+ population (Fig. 4A) and in the in vivo proliferation of infiltrating CD3+ T cells in the heart (Supplemental Fig. 6B). Consistent with this observation, treating WT mice with anti–CTLA-4 Ab induced extremely severe myocarditis and resulted in severe infiltration of CD3+Tcells in these hearts, which were mostly IL-17 positive (Fig. 4B, 4C). The above data suggest that blocking CTLA-4–B7 interaction potentiates Th17-mediated autoimmune responses during induction of EAM. To further confirm that the suppressive role of CTLA-4 in autoimmune myocarditis is mediated by Th17, we treated MYHC-α–immunized CD28−/− mice injected with hCTLA4-Ig with a neutralizing anti–IL-17 Ab. As shown in Fig. 3A, neutralization of IL-17 significantly suppressed the development of EAM and diminished the IL-17–producing CD4+ T cells in draining lymph nodes (Supplemental Fig. 6A) and the heart-infiltrating CD3+ T cells in CD28−/− mice that received hCTLA-4-Ig treatment (Fig. 4A). Taken together, our data indicate that CTLA-4–B7 interaction regulates the susceptibility of CD28−/− mice to EAM by controlling Th17 responses.
FIGURE 4.
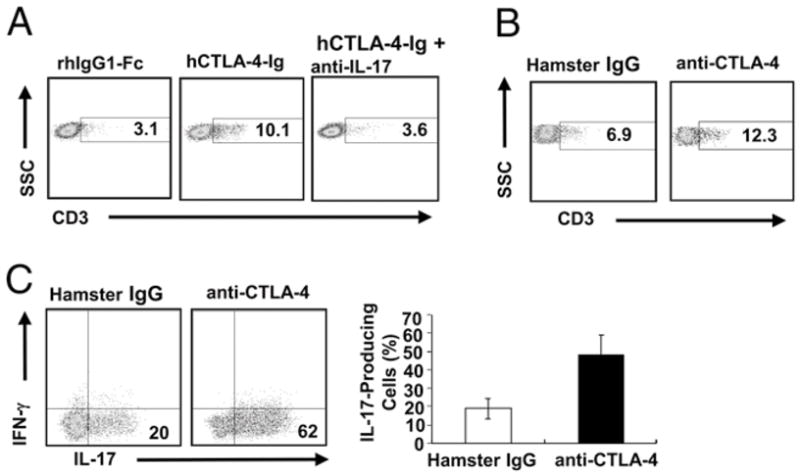
CTLA-4 inhibits autoimmune Th17 responses during EAM induction. Lymphocytes were isolated from the hearts of MYHC-α–immunized CD28−/− mice (A) or WT mice (B) with indicated treatment and stained with anti-CD3 and anti-CD45. The percentages of CD3+ T cells in gated CD45+ cells in the hearts were shown. C, The infiltrating lymphocytes in B were subjected to PMA plus ionomycin restimulation. Numbers in the gate indicate the percentage of IL-17–producing cells in the gated CD4+ T cells, and values in the column represent mean ± SEM. The data shown are representative of a group of 10 mice in two independent experiments.
It has been reported that CTLA-4−/− T cells display a predominantly Th2 phenotype (19), suggesting that CTLA-4 engagement may inhibit Th2 cell differentiation. However, this conclusion was not exclusive because the dominant effect observed in CTLA-4−/− T cells in this study is mediated by CD28, which has been shown to be essential for Th2 differentiation (20). Using in vitro and in vivo Th17 differentiation assays, we found that the CTLA-4–B7 engagement has no significant effect on Th1 or Th2 differentiation but significantly suppresses Th17 responses and Th17-mediated autoimmunity, further supporting a crucial role for CTLA-4 in regulating Th17 cell differentiation. Therefore, we have discovered a previously unknown function for CTLA-4 in Th cell differentiation.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant R01 AI090901 (to J.Z.) and American Heart Association Grant 09GRNT2010084 (to J.Z.). J.Z. is an American Lung Association Career Investigator.
We thank Dr. Jeffrey Bluestone for providing the CTLA-4−/−B7-1−/−/B7-2−/− mice.
Abbreviations used in this paper
- B6
C57BL/6
- EAM
experimental autoimmune myocarditis
- h
human
- rh
recombinant human
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 2.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 3.Salomon B, Bluestone JA. Complexities ofCD28/B7:CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- 4.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6–dependent Th17 cell development and survival. J Exp Med. 2008;205:2281–2294. doi: 10.1084/jem.20071119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valaperti A, Marty RR, Kania G, Germano D, Mauermann N, Dirnhofer S, Leimenstoll B, Blyszczuk P, Dong C, Mueller C, et al. CD11b+ monocytes abrogate Th17 CD4+ T cell-mediated experimental autoimmune myocarditis. J Immunol. 2008;180:2686–2695. doi: 10.4049/jimmunol.180.4.2686. [DOI] [PubMed] [Google Scholar]
- 7.Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carreno BM, Bennett F, Chau TA, Ling V, Luxenberg D, Jussif J, Baroja ML, Madrenas J. CTLA-4 (CD152) can inhibit T cell activation by two different mechanisms depending on its level of cell surface expression. J Immunol. 2000;165:1352–1356. doi: 10.4049/jimmunol.165.3.1352. [DOI] [PubMed] [Google Scholar]
- 9.Li D, Gál I, Vermes C, Alegre ML, Chong AS, Chen L, Shao Q, Adarichev V, Xu X, Koreny T, et al. Cutting edge: Cbl-b: one of the key molecules tuning CD28- and CTLA-4–mediated T cell costimulation. J Immunol. 2004;173:7135–7139. doi: 10.4049/jimmunol.173.12.7135. [DOI] [PubMed] [Google Scholar]
- 10.Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4–deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity. 1997;7:885–895. doi: 10.1016/s1074-7613(00)80406-9. [DOI] [PubMed] [Google Scholar]
- 11.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 12.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 13.Mandelbrot DA, McAdam AJ, Sharpe AH. B7-1 or B7-2 is required to produce the lymphoproliferative phenotype in mice lacking cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) J Exp Med. 1999;189:435–440. doi: 10.1084/jem.189.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, Candeloro P, Belladonna ML, Bianchi R, Fioretti MC, Puccetti P. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3:1097–1101. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- 15.Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol. 2002;3:611–618. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- 16.Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol. 1997;15:297–322. doi: 10.1146/annurev.immunol.15.1.297. [DOI] [PubMed] [Google Scholar]
- 17.Hirota K, Hashimoto M, Yoshitomi H, Tanaka S, Nomura T, Yamaguchi T, Iwakura Y, Sakaguchi N, Sakaguchi S. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J Exp Med. 2007;204:41–47. doi: 10.1084/jem.20062259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rangachari M, Mauermann N, Marty RR, Dirnhofer S, Kurrer MO, Komnenovic V, Penninger JM, Eriksson U. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203:2009–2019. doi: 10.1084/jem.20052222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ubaldi V, Gatta L, Pace L, Doria G, Pioli C. CTLA-4 engagement inhibits Th2 but not Th1 cell polarisation. Clin Dev Immunol. 2003;10:13–17. doi: 10.1080/10446670310001598519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McArthur JG, Raulet DH. CD28-induced costimulation of T helper type 2 cells mediated by induction of responsiveness to interleukin 4. J Exp Med. 1993;178:1645–1653. doi: 10.1084/jem.178.5.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.