

Abstract
This unit details the materials and methods required for both active induction and adoptive transfer of experimental autoimmune encephalomyelitis (EAE) in the SJL mouse strain using intact proteins or peptides from the two major myelin proteins: proteolipid protein (PLP) and myelin basic protein (MBP). Detailed materials and methods required for the purification of both PLP and MBP are also described. Modifications of the specified protocols may be necessary for efficient induction of active or adoptive EAE in other mouse strains.
Keywords: CFA • complete Freund's adjuvant, CNS • central nervous system, EAE • experimental autoimmune encephalomyelitis, MBP • myelin basic protein, MOG • myelin oligodendrocyte glycoprotein, PLP • proteolipid protein
Introduction
Experimental autoimmune encephalomyelitis (EAE) is a CD4+ T cell–mediated autoimmune disease characterized by perivascular CD4+ T cell and mononuclear cell inflammation and subsequent primary demyelination of axonal tracks in the central nervous system (CNS), leading to progressive hind-limb paralysis. EAE provides a powerful model for the study of the pathogenesis and immune regulation of CD4+ TH1/TH17–mediated tissue damage and is generally considered to be a relevant model for the human immune-mediated demyelinating disease multiple sclerosis. In the SJL (H-2s) mouse, the disease is characterized by a relapsing-remitting course of paralysis, which allows assessment of the efficacy of various immunoregulatory strategies in a progressive autoimmune disease setting. In C57BL/6 (H-2b) mice, the disease displays a chronic-progressive clinical course, while in other mouse strains, such as PL/J (H-2u) and B10.PL (H-2u), the disease is normally acute and self-limiting, and is not characterized by clinical relapses.
Actively induced EAE consists of an induction phase and an effector phase. The induction phase of the disease involves the priming of myelin epitope–specific CD4+ T cells following immunization with myelin proteins or peptides in complete Freund's adjuvant (CFA). The effector phase consists of multiple stages: (1) migration of activated myelin-specific T cells to the CNS, which involves extravasation of the T cells across the tight endothelial junctions comprising the blood-brain barrier; (2) elaboration of chemokines and cytokines by the myelin-specific T cells, which induce the influx of peripheral mononuclear phagocytes into the CNS parenchyma; (3) activation of peripheral monocytes/macrophages and CNS-resident microglial cells by TH cell–derived cytokines; and (4) demyelination of CNS axonal tracts by the phagocytic activity of activated mononuclear cells and by the inflammatory and cytotoxic effects of cytokines (e.g., IFN- γ, LT/TNF-β, IL-17, TNF- α, and NO) released from activated CD4+ T cells and monocytes. The effector phase of the disease is modeled by the adoptive-transfer model of EAE, in which disease is induced by the peripheral introduction of a preactivated population of myelin epitope–specific CD4+ T cells to a naive mouse.
This unit details the materials and methods required for both active induction (see basic Protocol) and adoptive transfer (see Alternate Protocol) of EAE in the SJL and C57BL/6 mouse strains using intact proteins or peptides from three myelin proteins—proteolipid protein (PLP), myelin oligodendrocyte glycoprotein (MOG), and myelin basic protein (MBP). Detailed materials and methods required for the purification of both PLP and MBP are also described (see Support Protocols 1 and 2). It should be cautioned that modifications of the specified protocols may be necessary for efficient induction of active or adoptive EAE in other mouse strains, and the reader is urged to consult the literature for details (also see Commentsry for more discussion).
Basic Prtocol
Active Induction of EAE with PLP and MBP Protein or Peptide
Experimental autoimmune encephalomyelitis (EAE) can be induced in SJL mice by immunization with proteolipid protein (PLP), myelin basic protein (MBP), or peptides corresponding to the immunodominant epitopes of MBP (MBP84-104), MOG (MOG92-106), or PLP (PLP139-151 and PLP178-191). Similarly, disease can be induced in C57BL/6 mice by immunization with the peptide corresponding to the immunodominant epitope of MOG (MOG35-55). Peptides can either be purchased from commercial vendors or synthesized according to established methods (see Chapter 9). Bovine MBP can be purified in the laboratory (see Support Protocol 2) and is available commercially from Sigma (see APPENDIX 5). Although either intact MBP or its immunodominant epitope MBP84-104 peptide can be used to actively induce EAE in SJL mice, pertussis toxin must be administered in conjunction with either of these proteins. Likewise, disease induction with MOG35-55 in the C57BL/6 mouse requires the administration of pertussis toxin. In the following Basic Protocol, the term neuroantigen will be used to denote intact PLP, intact MBP, PLP139-151, PLP178-191, MBP84-104, MOG92-106, and MOG35-55.
Materials
Incomplete Freund's adjuvant (IFA; Difco)
Mycobacterium tuberculosis H37Ra (killed and desiccated; Difco)
Solutions of MBP or PLP (see Support Protocols 1 and 2); PLP139-151, PLP178-191, MOG92-106, MBP84-104, or MOG35-55.
Female SJL or C57BL/6 mice, 5 to 8 weeks old (e.g., Jackson Labs or Harlan Labs)
Pertussis toxin (List Biologicals)
Carbol fuchsin solution (optional; see recipe)
13-ml polystyrene test tube
18- and 25-G needles (Becton Dickinson)
1-ml glass tuberculin syringe with Luer-lok (e.g., VWR)
Oster small-animal clipper (model A2)
-
Prepare complete Freund's adjuvant (CFA) by mixing 10 ml IFA with 40 mg M. tuberculosis H37Ra (final concentration 4 mg/ml M. tuberculosis).
Commercial preparations of CFA contain only 1 mg/ml M. tuberculosis. The 4 mg/ml concentration is critical.
-
Make an emulsion of neuroantigen and CFA by mixing 1 ml of a 2 mg/ml neuroantigen solution with 1 ml of the CFA, then repeatedly drawing up and expelling the mixture into a 13-ml polystyrene test tube using an 18-G blunt-end needle attached to a 1-ml glass tuberculin syringe (alternatively, prepare emulsion by sonication).
The lowest amount of a given neuroantigen and M. tuberculosis concentration that can induce reliable EAE varies by antigen/peptide lot and by the source and age of the SJL mice, and thus should be determined empirically by the individual investigator. As a guideline, the authors have found that a single immunization with 50 nmol (87 μg)/mouse of PLP139-151 in CFA with a final concentration of 2 mg/ml M. tuberculosis is sufficient to induce severe EAE in >90% of immunized SJL mice. In contrast, active disease induction with the immunodominant MBP84-104 peptide requires two immunizations with 100 nmol of peptide spaced a week apart. Active induction with MBP or PLP is usually achieved by a single immunization with 50 to 100 μg of the intact myelin protein. Active induction of EAE in C57BL/6 mice is achieved by a single immunization of 200 μg MOG35-55.
Draw the emulsion into the 1-ml glass tuberculin syringe with 18-G needle, taking care not to introduce air bubbles, and then replace the 18-G needle with a 25-G needle for immunization.
Shave the backs of mice using small-animal clipper.
-
Inject 0.1 ml emulsion subcutaneously in the shaved backs of the mice, distributing it over three sites (i.e., ∼0.033 ml per site): one along the midline of the back between the shoulders, and two on either side of the midline on the lower back.
When inducing EAE with MBP or MBP84-104 in SJL mice also inject 400 ng pertussis toxin i.p. or i.v. on the day of and 2 days following the first immunization. i.p. injection of 200 ng pertussis toxin on the day of and two days following immunization is also required to induce reliable disease in C57BL/6 mice.
Pertussis toxin is required to produce severe and reliable EAE with MBP (but not PLP) in SJL mice and with MOG35-55 in C57BL/6 mice. There is no apparent difference between the two delivery routes in terms of EAE development.
-
As needed, separate mice into groups and mark them by painting numbers on their backs or marking their tails with carbol fuchsin solution.
Separation of neuroantigen-primed mice into experimental groups is totally dependent on the design of the experiment. For example, the primed mice can be separated into a control and treatment group for studies designed to test the effects of particular therapeutic agents on disease progression.
Monitor mice every other day for the development of clinical symptoms (Tables 15.1.3. and 15.1.4; also see Anticipated Results), which will become evident between 12 and 28 days after immunization. Once disease has developed, monitor mice daily for changes in disease severity.
Table 15.1.3.
Grading System for Clinical Assessment of EAE
| Score | Clinical signs |
|---|---|
| 0 | Normal mouse; no overt signs of disease |
| 1 | Limp taila or hind limb weaknessb but not both |
| 2 | Limp taila and hind limb weaknessb |
| 3 | Partial hind limb paralysisc |
| 4 | Complete hind limb paralysisd |
| 5 | Moribund state; death by EAE: sacrifice for humane reasons |
Limp tail: complete flaccidity of the tail, and absence of curling at the tip of the tail when mouse is picked up.
Hind limb weakness: observed as a waddling gait, the objective sign being that, in walking, mouse's hind limbs fall through the wire cage tops.
Partial hind limb paralysis: mouse can no longer use hind limbs to maintain rump posture or walk but can still move one or both limbs to some extent.
Complete hind limb paralysis: total loss of movement in hind limbs; mouse drags itself only on its forelimbs. Mice at this stage are given food on the cage floor, long sipper tubes, and daily subcutaneous saline injections to prevent death by dehydration.
Table 15.1.4.
Clinical Stages of Disease
| Disease stage | Definition |
|---|---|
| Acute phase | First clinical episode |
| Remission | Phase of clinical improvement following a clinical episode; characterized by a reduction in clinical score for at least 2 days after the peak score of the acute phase or a disease relapse has been reached |
| Relapse | Increase of at least one grade in clinical score for at least 2 days after remission has been attained |
Alternate Protocol
Adoptive Induction of EAE with PLP-, MBP-, or MOG-Specific Lymphocytes
As an alternative to direct induction with PLP, MBP, or MOG (see Basic Protocol), EAE can also be induced in SJL or C57BL/6 mice by adoptive transfer of in vitro neuroantigen-activated lymphocytes from mice immunized with these encephalitogenic neuroantigens. Induction of EAE by this method usually results in more severe disease, with higher incidence and a more accelerated and synchronous disease course. The clinical and histological features of EAE induced by adoptive transfer of neuroantigen-activated lymphocytes are identical to those seen after active disease induction. EAE can also be reproducibly induced by the adoptive transfer of long-term T cell lines or clones specific for PLP139-151, PLP178-191, MBP84-104, or MOG35-55 (see UNIT 313 for the generation of murine T cell clones).
Additional Materials (also see Basic Protocol)
Mishell-Dutton balanced salt solution (BSS; see recipe)
Complete DMEM-5 (APPENDIX 2A)
Recombinant IL-12 (R&D Systems)
100-mesh stainless steel screen (e.g., Fisher)
50-ml conical polypropylene centrifuge tubes
37°C, 7.5% CO2 tissue culture incubator
Additional reagents and equipment for active induction of EAE with PLP, MBP, or MOG (see Basic Protocol), euthanasia (UNIT 1.8), removal of lymphoid organs (UNIT 1.9), and trypan blue exclusion (APPENDIX 3B)
Immunize SJL or C57BL/6 mice with PLP, MBP, or MOG peptides (see Basic Protocol, steps 1 to 5).
-
Seven to fourteen days following immunization, euthanize the mice and remove the draining lymph nodes (inguinal, brachial, and axillary; see UNIT 1.9). Place the lymph nodes in Mishell-Dutton BSS.
HBSS (APPENDIX 2A) can be substituted for Mishell-Dutton BSS; however, the authors have observed less cell clumping with Mishell-Dutton BSS.
Generate a single cell suspension by pressing the lymph nodes through 100-mesh screen using the plunger from a disposable 3-ml syringe. Wash the screen with BSS and add wash to single-cell suspension.
-
Prepare a culture of the neuroantigen-primed lymph node cells at a concentration of 6 × 106/ml in complete DMEM-5 containing 50 μg/ml neuroantigen. Incubate 72 to 96 hr in a 37°C, 7.5% CO2 tissue culture incubator.
The authors routinely reactivate neuroantigen-primed lymph node cells in vitro with homologous antigen (the optimal concentration should be determined for each batch of protein or synthetic peptide); however, the draining lymph node cells can also be reactivated with 1 μg/ml Con A for 48 hr. Con A activation will reduce the frequency of neuroantigen-specific cells in the culture, thereby necessitating injection of larger numbers of total cells to achieve reliable and severe EAE expression. The numbers of Con A–activated lymph node cells that are injected into normal recipient mice will have to be determined empirically by the investigator. Adoptive transfer of EAE to C56Bl/6 mice requires an in vitro reactivation culture that contains 20 to 25 ng/ml recombinant IL-12.
Harvest the neuroantigen-activated lymphocytes by centrifuging the cultures in 50-ml conical polypropylene test tubes for 15 min at 300 × g. Wash the cell pellet with BSS and centrifuge again as before.
Resuspend the cells in BSS and determine cell count and viability using trypan blue exclusion (APPENDIX 3B).
-
Inject 1 × 107 viable lymphocytes in 0.3 to 0.5 ml BSS i.p. or i.v. into normal recipient SJL or C57BL/6 mice, then divide mice into experimental groups and continue monitoring them for development of EAE (see Basic Protocol, steps 6 and 7).
Pertussis toxin is not required in either the lymphocyte donor or the recipient SJL mouse when inducing EAE by the adoptive transfer of PLP-, PLP139-151-, PLP178-191-, MBP-, or MBP84-104-specific lymphocytes. However, i.p. injection of a 200 ng pertussis toxin on the day of and two days following adoptive transfer of MOG35-55-specific lymphocytes is required for adoptive disease induction in C57BL/6 recipients.
The authors have observed little or no difference between the two routes of administration in terms of disease incidence or severity. However, onset of EAE in mice receiving an i.v. injection of cells may be 1 to 2 days faster than for i.p. injected animals.
Support Protocol 1
Purification of Proteolipid Protein
PLP is an integral membrane protein and the major protein constituent of CNS myelin. PLP is highly conserved among species, thereby making bovine PLP suitable for use in the induction of murine EAE. PLP is not commercially available and must be purified. Intact PLP can be purified from bovine white matter (spinal cord or brain) using the following protocol, which involves extracting the protein from myelin using a series of organic solvents. The protein itself is very hydrophobic and is only converted into an aqueous phase at the end of the procedure, immediately prior to in vivo or in vitro use. Aqueous PLP is unstable and can be stored for only 1 month.
Materials
Bovine brain (Pel-Freez)
Chloroform/methanol (CM; see RECIPE), 4°C
Methanol, 4°C
Acetone (Aldrich), 4°C
CM/acetic acid (see recipe)
Diethyl ether (Aldrich)
2-chloroethanol (Aldrich)
Stainless steel Waring blender
Whatman no. 1 filter paper (Whatman or Fisher)
Corex centrifuge bottles (Corning)
Buchner funnel (Fisher)
Separatory funnel
Nitrogen gas flow
Lipophilic Sephadex LH-60-120 packed in 1 m × 2.5 cm, Teflon-fitted column (both from Pharmacia Biotech or Sigma), equilibrated with CM/acetic acid
Spectrapor 12- to 14-kD-MWCO dialysis tubing (Spectrum or Fisher)
BCA assay kit (Pierce)
Additional reagents and equipment for dialysis (APPENDIX 3H) and SDS-PAGE (UNIT 8.4)
Day 1: Prepare total lipid extract and precipitate
-
1
Mix 100 g (wet weight) bovine brain with 1900 ml 4°C CM.
Care should be taken when using CM, because it is a health hazard as well as a solvent that will dissolve most materials except glass, stainless steel, and Teflon.
-
2
Homogenize the mixture in a Waring blender and vacuum filter through Whatman no. 1 filter paper using a Buchner funnel. Save the filtrate, called the total lipid extract (TLE), for PLP purification.
If desired, TLE can be stored 6 to 12 months at 4°C before proceeding with the experiment.
-
3
Mix 200 ml TLE with 40 ml water, transfer into Corex centrifuge bottles, and centrifuge the mixture 30 min at 1000 × g.
-
4
Discard the upper phase (the lower phase contains PLP). Dissolve the white precipitate at the interface by adding 4°C methanol dropwise while gently mixing.
This step will take some time and the amount of methanol needed can vary. Take note of the total final volume to facilitate step 5.
-
5
Add 4 vol 4°C acetone, mix well, and transfer to a separatory funnel. Let stand at least 1 hr (but no longer than overnight) at 4°C.
Day 2: Remove solvents and purify PLP through lipophilic Sephadex column
-
6
Collect the precipitate through the funnel bottom into Corex centrifuge bottles, leaving most of the acetone behind in the funnel. Centrifuge 30 min at 3000 × g.
-
7
Remove the solvents and dry the precipitate ∼10 min under nitrogen flow. Dissolve the dried precipitate in 10 ml CM/acetic acid.
The nitrogen provides an inert gas flow that is necessary to limit oxidation and dry off the solvents.
At this step of the purification process the PLP preparation can be stored up to 8 weeks at 4°C before proceeding.
-
8
Apply the 10-ml PLP preparation to a lipophilic Sephadex LH-60-120 column (1 m × 2.5 cm, Teflon-fitted) that has been equilibrated with 500 ml CM/acetic acid.
-
9
Elute the PLP from the column by applying 500 ml CM/acetic acid. Collect 2-ml fractions in glass tubes. Measure OD280 of the fractions, and pool and save those with OD280 ≥0.5 (usually ten to fifteen fractions).
The pooled fractions can be stored up to 4 weeks at 4°C before proceeding.
Days 3 and 4: Precipitate PLP with ether and dialyze
-
10
Precipitate 20 ml of the pooled fractions with 80 ml (4 vol) of 4°C diethyl ether for 1 hr in a 4°C cold room. Centrifuge 30 min at 3000 × g.
This converts PLP from the liquid to the aqueous phase.
-
11
Decant solvents and dry precipitate thoroughly under nitrogen (this usually takes ∼15 min) to ensure that there is no water or ether in the precipitate.
-
12
Dissolve precipitate in 5 ml 2-chloroethanol.
CAUTION: 2-chloroethanol is extremely toxic upon inhalation and contact. Be sure to stir until entirely dissolved, at least 1 hr.
-
13
Transfer to 12- to 14-kD-MWCO Spectrapor dialysis tubing and dialyze (APPENDIX 3H) against 6 liters deionized water for three changes (a minimum of ½ day/change).
Expect the volume in the dialysis bag to increase by 25%.
Day 5: Determine protein concentration and purity
-
14
Determine protein concentration using BCA assay kit and analyze purity by SDS-PAGE (UNIT 8.4) using 12% acrylamide in the gel.
Aqueous PLP should be stored no longer than 1 month at 4°C.
Support Protocol 2
Purification of Myelin Basic Protein
MBP is the second most abundant protein in CNS myelin, with a high basic amino acid content. Bovine MBP is purified from the water-soluble fraction of myelin, does not require an aqueous conversion step, and can be lyophilized and stored for long periods of time. Guinea pig MBP can also be used for the induction of EAE in SJL mice. The relative ease of purification along with its commercial availability make MBP a more desirable encephalitogenic protein for the study of EAE.
Materials
Bovine brain or spinal cord (Pel-Freez)
Chloroform/methanol (CM; see recipe), 4°C
0.01 N HCl (Aldrich)
Saturated (NH4)2SO4 (ammonium sulfate; UNIT 2.7)
0.25 N NaOH
Buchner funnel (Fisher)
Stainless steel Waring blender
Whatman no. 90 and no. 4 filter papers (Whatman or Fisher)
Corex centrifuge bottles (Corning)
Spectrapor 12- to 14-kD-MWCO dialysis tubing (Spectrum or Fisher)
Additional reagents and equipment for dialysis (APPENDIX 3H)
Day 1: Perform initial extraction with chloroform/methanol
-
1
Homogenize 100 g (wet weight) bovine brain or spinal cord with 500 ml 4°C CM using a Waring blender. Add 2.5 liters CM to homogenate and stir overnight at 4°C.
Day 2: Separate aqueous and lipid fractions and acid precipitate MBP
-
2
Using a Buchner funnel, vacuum filter the 3 liters brain homogenate/CM through Whatman no. 90 filter paper.
-
3
Save the residue on the filter paper. Using a Waring blender, homogenize in 3 liters CM.
-
4
Using a Buchner funnel, vacuum filter the 3 liters brain homogenate/CM through Whatman no. 90 filter paper.
-
5
Save the residue on the filter paper and resuspend in 1 liter water. Stir mixture 1 hr at 4°C.
-
6
Using a Buchner funnel, vacuum filter the 1-liter mixture through Whatman no. 90 filter paper.
-
7
Resuspend residue on the filter paper in 300 ml of 0.01 N HCl. Monitoring pH with a pH meter, maintain the pH of the mixture between 3.0 and 3.2 by adding 0.01 N HCl dropwise until reading is stabilized (usually 1 hr).
-
8
Centrifuge the solution in Corex bottles for 10 min at 47,000 × g.
-
9
Save the supernatant and filter through Whatman no. 4 filter paper.
-
10
Add 1 vol saturated (NH4)2SO4 dropwise to the supernatant with stirring. Leave the precipitate overnight at 4°C with stirring.
Day 3: Dialyze
-
11
Centrifuge the precipitate 10 min at 47,000 × g.
-
12
Discard the supernatant and dissolve the precipitate in 50 ml of 0.01 N HCl. Transfer to Spectrapor 12- to 14-kD-MWCO dialysis tubing and dialyze (APPENDIX 3H) overnight against 6 liters deionized water. Change the dialysis bath once after the first 6 hr.
Day 4: Adjust pH
-
13
Centrifuge the contents of the dialysis bag 10 min at 47,000 × g. Save supernatant and discard precipitate.
-
14
Adjust the pH of the supernatant to 7.0 using 0.25 N NaOH while stirring the mixture.
The pH of the supernatant prior to adjustment will be ∼4.5.
-
15
Centrifuge the solution 10 min at 47,000 × g. Discard the precipitate and lyophilize the supernatant using standard Virtis lyophilizer. Store the lyophilized product (MBP) desiccated at 4°C.
The amount of protein in a given MBP preparation should be determined prior to in vivo use. Salt concentration can alter the weight of a preparation and dramatically affect the ability of a given preparation to induce reliable and severe EAE.
Support Protocol 3
Isolation of CNS-Infiltrating Lymphocytes
Upon induction of EAE, inflammatory cells infiltrate the CNS, resulting in ascending paralysis of the animal. The nature of the ascending paralysis is due to the fact that the cells begin infiltrating the CNS in the lumbar region of the spinal cord. The disease progresses in severity as more of the spinal cord becomes inflamed. Thus, care should be taken when harvesting spinal cords such that as much of the lumbar region is obtained as possible, because the majority of the infiltrates are located within this area. In addition, the CNS is highly vascularized; therefore, an important and critical parameter is to completely perfuse the animal with PBS prior to removal of the cord, in order to ensure that the cells ultimately obtained were those residing within the cord parenchyma and not merely circulating within the vasculature. Single-cell suspensions obtained from inflamed spinal cord can be used for a number of assays, e.g., analysis of cellular infiltrates via flow cytometry, or isolation of specific cellular subsets via cell sorting or magnetic beads.
Materials
Mouse (?additional specifications?)
Phosphate-buffered saline (PBS; APPENDIX 2A)
50 mM 2-mercaptoethanol (2-ME)
Neural Tissue Dissociation Kit (Miltenyi Biotec, cat. no. 130-092-628)
0.9 M sucrose in HBSS (see APPENDIX 2A for HBSS)
150-mm petri dish
-
Surgical instruments:
Hemostats
Scissors
Forceps
Scalpel
25-G butterfly
12-cc syringe
19-G needle
6-cc syringe
60-mm petri dishes
2-ml pipets
15-ml conical centrifuge tubes
Refrigerated centrifuge
Additional reagents and equipment for euthanasia of the mouse by CO2 asphyxiation (UNIT 1.8)
Prepare the animal
-
1
Euthanize the animal via CO2 asphyxiation (UNIT 1.8).
Cervical dislocation is not recommended as this damages the spinal cord.
-
2
Place the animal in a 150-mm petri dish and expose the heart by reflecting the chest wall.
The chest wall does not need to be completely removed, and can be held out of the way with a hemostat.
Perfuse the animal
-
3
Attach a 25-G butterfly to a 12-cc syringe that has been filled with PBS. Carefully insert the needle into the left ventricle of the heart and secure it in place by crossclamping the heart with a second hemostat.
-
4
Clip the right atrium with scissors. Apply a small amount of pressure to the syringe and check for correct placement of the needle by observing an efflux of blood from the right atrium.
If there has been correct placement of the needle, there will be minimal resistance and dark red blood will begin to flow from the right atrium. If the needle has been incorrectly placed, blood will not flow from the atrium and the lungs will inflate. If this occurs, carefully reposition the needle.
-
5
Once correct placement has been confirmed, perfuse the animal with the entire 12 cc of PBS.
By the end of the perfusion, the effluent from the atrium should be clear and the liver should have lost its red color. If this does not occur, leave the needle in place, detach the syringe from the butterfly and refill with PBS, then reattach and continue to perfuse.
Isolate the spinal cord
-
6
Expose the spinal column by peeling back the skin from the base of the tail to the head of the animal. Identify where the base of the spinal column attaches to the pelvis.
-
7
Using a pair of scissors, make a perpendicular cut through the column at this point. Then, cut along each side of the column to the base of the skull, and make another perpendicular cut at this point to remove the column.
-
8
Attach a 19-G needle to a 6-cc syringe that has been filled with PBS. Fill a 60-mm petri dish with PBS. Hold the column with forceps and identify the spinal canal at the caudal end of the column. Carefully insert the 19-G needle at this point.
Resistance will be encountered as the needle enters the canal.
-
9
Slowly push past the resistance in the canal until the point where the resistance is suddenly lost. At this point, slowly pull back on the needle until it feels as if the needle is “set” in the canal. Hold the column over the 60-mm petri dish (that was prepared with PBS in step 7) and depress the plunger to flush the cord from the column.
Dissociate spinal cord tissue
-
10
Transfer the cord to a second 60-mm petri dish (?containing PBS?). Using scalpel and a pair of forceps, cut the cord into very small pieces.
-
11
Add 13.5 μl of 50 mM 2-mercaptoethanol to 10 ml of Solution 2 from the Neural Tissue Dissociation Kit. Mix 1900 μl of Solution 2 (containing the 2-ME) with 50 μl of Solution 1 from the kit. Using a 2-ml pipet, collect the minced pieces of spinal cord from the petri dish with the solution mixture and transfer the suspension to a 15-ml tube. Incubate in a 37.C water bath for 20 min, vortexing every 5 min.
-
12
Resuspend Solution 4 from the kit with 1 ml of Storage Buffer (also from kit). Add 10 μl of Solution 4 to 20 μl of Solution 3 from the kit, then add to the sample. Pipet the sample up and down with a 2-ml pipet until the tissue appears completely dissociated. Incubate the sample for an additional 15 min in the 37.C water bath, vortexing every 5 min. Dissociate any remaining pieces of tissue by pipetting up and down once again with a 2-ml pipet.
Wash cells
-
13
Wash the cells once in PBS by centrifuging ?? min at ?? × g, ??°C, aspirating the supernatant, adding ?? ml PBS, then centrifuging again as before and removing the supernatant.
Removal of the supernatant by aspiration is preferable to decanting, as the pellet can be quite loose.
-
14
Resuspend the pellet in 10 ml ice cold 0.9 M sucrose in HBSS. Centrifuge 10 min at 850 × g, 4.C.
The myelin will form a compact layer at the top, while the cells will pellet at the bottom of the tube.
-
15
Remove the myelin and supernatant by aspiration.
-
16
Wash the cells once more in PBS by centrifuging ?? min at ?? × g, ??.C, aspirating the supernatant, adding ?? ml PBS, then centrifuging again as before and removing the supernatant. Proceed with any further processing.
Reagents and Solutions
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see APPENDIX 5.
Carbol fuchsin solution
3 ml 88% phenol
5 ml 100% ethanol
0.5 g pararosaniline
H2O to 50 ml total
Store indefinitely at room temperature in sealed glass container
Use carbol fuchsin with care, as it stains permanently.
Chloroform/methanol (CM)
Mix 2 vol chloroform with 1 vol methanol and stir ≥1 hr at room temperature. Store indefinitely in sealed glass container at room temperature in a chemical fume hood.
Chloroform/methanol/1% acetic acid (CM/acetic acid)
Mix 2 vol chloroform with 1 vol methanol and add glacial acetic acid to 1% (v/v). Stir ≥1 hr at room temperature. Store indefinitely in sealed glass container at room temperature in a chemical fume hood.
Mishell-Dutton balanced salt solution (BSS)
| Stock solution 1: | Stock solution 2: |
| 10 g glucose | 1.86 g CaCl2·2H2O |
| 0.6 g KH2PO4 | 4.0 g KCl |
| 3.58 g Na2HPO4·7H2O | 80.0 g NaCl |
| 20 ml 0.5% phenol red | 1.04 g anhydrous MgCl2 |
| H2O to 1 liter | 2.0 g MgSO4·7H2O |
| H2O to 1 liter |
Prepare stock solutions 1 and 2 and filter sterilize through separate 0.22-μm-pore-size filters. Store ≤6 months at 4°C.
Working solution (1× BSS): Add 100 ml stock solution 1 to 700 ml water and then add 100 ml stock solution 2. Dilute to 1 liter total with water (do not add stock solution 1 directly to stock solution 2). Add 100 μl of 2 N NaOH to maintain pH at 7.4. Store ≤1 month at 4°C.
Commentary
Background Information
Experimental autoimmune encephalomyelitis (EAE) in mice was first described and established as an important model of CNS inflammatory autoimmune disease over 50 years ago (Olitsky and Yager, 1949). Mouse models include acute, chronic, and relapsing-remitting EAE. EAE in the H-2u strains (PL/J and B10.PL) is usually characterized by an acute paralytic episode from which the mice recover either partially or totally, while EAE in the C57BL/6 mouse is usually manifest as a chronic disease (Tompkins et al., 2002). Relapsing EAE (R-EAE) is normally seen in the SJL mouse or F1 hybrids of SJL mice and offers the advantage that it can be used to study the pathogenesis and immunoregulation of T cell–mediated demyelination (McRae et al., 1992). R-EAE has histopathological and clinical similarities to the relapsing-remitting form of human multiple sclerosis, and thereby serves as an ideal system in which to examine the molecular basis of disease induction and progression.
R-EAE in the SJL mouse is a CD4+ T cell–mediated autoimmune disease directed against protein components of CNS myelin (Paterson and Swanborg, 1988) resulting in a relapsing-remitting clinical course of paralysis (Brown and McFarlin, 1981). R-EAE can be actively induced by sensitization with mouse spinal cord homogenate (MSCH; Brown and McFarlin, 1981), MBP (Fritz et al., 1983), PLP (Trotter et al., 1987), or synthetic peptides corresponding to the major encephalitogenic regions of PLP, MBP, or myelin oligodendrocyte protein (MOG; see Table 15.1.1). Alternatively, R-EAE can be induced by the adoptive transfer of neuroantigen/epitope-specific in vitro–activated CD4+ T cells (Pettinelli and McFarlin, 1981; McRae et al., 1992; see Table 15.1.2). The disease is characterized clinically by transient ascending hind limb paralysis and histologically by perivascular mononuclear-cell infiltration and fibrin deposition in the brain and spinal cord with adjacent areas of acute and chronic demyelination (Brown et al., 1982; Paterson and Swanborg, 1988). CNS damage apparently results from the direct and indirect effects of chemokines (Karpus et al., 1995) and pro-inflammatory cytokines such as IFN-γ, LT/TNF, and IL-17 (Powell et al., 1990; Ruddle et al., 1990; Selmaj et al., 1991a; Langrish et al., 2005; Park et al., 2005; Chen et al., 2006), which cause the chemoattraction and activation of additional monocytes and macrophages. Activated macrophages, in turn, mediate myelin damage mainly by a terminal, nonspecific bystander response (Cammer et al., 1978; Selmaj et al., 1991b).
Table 15.1.1.
Encephalitogenic Peptides of MBP, PLP, and MOG in Various Inbred Mouse Strains
| Mouse strain | H-2 type | Peptide | Sequencea | Reference |
|---|---|---|---|---|
| PL/J, B10.PL | H-2u | MBPAc1-11 | Ac-ASQKRPQRHG | Zamvil et al. (1986) |
| PLP178-191 | NTWTTCQSIAFPSK | Unpublished (B10.PL) | ||
| MBP35-47 | TGILDSIGRFFSG | Zamvil et al. (1988) | ||
| PLP43-64 | EKLIETYFSKNYQDYEYLINVI | Whitham et al. (1991) | ||
| SJL | H-2s | MBP89-101 | VHFFKNIVTPRTP | Sakai et al. (1988) |
| MBP84-104 | VHFFKNIVTPRTPPPSQGKGR | Tan et al. (1992) | ||
| PLP139-151b | HSLGKWLGHPDKF | Tuohy et al. (1993) | ||
| PLP104-117 | KTTICGKGLSATVT | Tuohy and Thomas (1993) | ||
| PLP178-191 | NTWTTCQSIAFPSK | Greer et al. (1992) | ||
| PLP57-70 | YEYLINVIHAFQYV | Greer et al. (1996) | ||
| MOG92-106 | DEGGYTCFFRDHSYQ | Amor et al. (1994) | ||
| (PL/J X SJL) F1 | H-2s/u | MBPAc1-11 | Ac-ASQKRPQRHG | Zamvil et al. (1986) |
| PLP43-64 | EKLIETYFSKNYQDYEYLINVI | Whitham et al. (1991) | ||
| PLP139-151 | HSLGKWLGHPDKF | Whitham et al. (1991) | ||
| C57BL/6 | H-2b | MOG35-55 | MEVGWYRSPFSRVVHLYRNGK | Mendel et al. (1995) |
| PLP178-191 | NTWTTCQSIAFPSK | Tompkins et al. (2002) | ||
| C3H | H-2k | PLP103-116 | YKTTICGKGLSATV | Tuohy et al. (1988a) |
| SWR | H-2q | PLP215-232 | PGKVCGSNLLSICKTAEF | Endoh et al. (1990) |
| (SJL X B10.PL) F1 | H-2s/q | PLP139-151 | HSLGKWLGHPDKF | Unpublished |
| PLP178-191 | NTWTTCQSIAFPSK | Unpublished | ||
| MBPAc1-11 | Ac-ASQKRPQRHG | Unpublished | ||
| (SJLX C3H/HeJ)F1c | H-2s/k | PLP190-209 | SKTSASIGSLCADARMYGVL | Muller et al. (2000) |
| PLP215-232 | PGKVCGSNLLSICKTAEFQ | Greer et al. (1996) | ||
| BALB/cPtc | H-2d | PLP178-191 | NTWTTCQSIAFPSK | Greer et al. (1992) |
| NOD | H-2g7 | PLP56-70 | DYEYLINVIHAFQYV | Girvin et al. (2000) |
| MOG35-55 | MEVGWYRSPFSRVVHLYRNGK | Slavin et al. (1998) |
Sequences for MBP peptides are based on different species variants of MBP, which have different numbering systems; sequences for PLP and MOG peptides are based on the mouse sequence. The reader is urged to consult the indicated references for more detailed information.
The PLP139-151 sequence has a serine (S) for cysteine (C) substitution at position 140 to enhance solubility.
The EAE observed in these mice is nonclassical. In (SJL X C3H/HeJ)F1 mice, the disease causes imbalance and axial rotary movement (rotary EAE). In BALB/cPt, mice show lack of balance and forelimb paralysis in the absence of hindlimb paralysis.
Table 15.1.2.
Summary of General Parameters for Various EAE Adoptive Transfer Modelsa
| Mouse strain | Antigen specificity | Donor immunization period (days) | In vitro antigen concentration (mg/ml) | In vitro IL-12 (ng/ml) | In vitro culture Time (hr) | Number of blasts transferred (×106) | Disease type | Disease severity |
|---|---|---|---|---|---|---|---|---|
| SJL | PLP | 7-14 | 50-100 | — | 72-96 | 5-10 | Relapsing-remitting | Severe |
| PLP139-151 | 7-14 | 20 | — | 72-96 | 1-5 | Relapsing-remitting | Severe | |
| MBP | 7-14 | 50-100 | — | 72-96 | 40-60 | Relapsing-remitting | Moderate | |
| MBP84-104 | 7-14 | 50 | — | 72-96 | 10-20 | Relapsing-remitting | Moderate | |
| C57BL/6 | MBP | 10-14 | 50-100 | — | 72-96 | 50 | Monophasic/chronic | Mild |
| MBP84-104 | 10-14 | 50 | — | 72-96 | 50 | Monophasic/chronic | Mild | |
| MOG | 10-14 | 50 | 25 | 72-96 | 20 | Chronic | Moderate | |
| MOG35-55 | 10-14 | 10 | 25 | 72-96 | 20 | Chronic | Moderate | |
| B10.PLb | MBPAc1-11 | — | 50 | 10 | 72 | 1 | Monophasic | Moderate |
| B10.S | MBP | 10-11 | 25 | 20 | 96 | 35 | Monophasic | Moderate |
| MBP87-106 | 10-11 | 50 | 20 | 96 | 35 | Monophasic | Moderate |
These parameters have been optimized in different laboratories as previously described. SJL protocol parameters are from Pettinelli and McFarlin (1981), Pettinelli et al. (1982), Miller et al. (1991), McRae et al. (1992), Fritz and Zhao (1994), Segal et al. (1994), Skundric et al. (1993, 1994), and Tuohy and Thomas (1995). C57BL/6 protocol parameters are from Mendel et al. (1995), Shaw et al. (1995), Clark et al. (1997), Segal et al. (1998), Mendel and Shevach (2002), and Tompkins et al. (2002). B10.PL protocol parameters are from Walker and Mannie (2002). B10.S protocol parameters are from Segal and Shevach (1996).
Note that the B10.PL system employs T cell receptor transgenic donors which do not require in vivo priming, only in vitro culture with MBPAc1-11 peptide and rIL-12.
The mechanisms responsible for the clinical remission following the acute paralytic episode are not well understood. However, it has been demonstrated that the primary disease relapse is mediated predominantly by T cells specific for endogenous myelin epitopes which are activated as a result of presentation of myelin debris released during acute disease, a phenomenon known as epitope spreading (McRae et al., 1995; Miller et al., 1995; McMahon et al., 2005). The relapsing-remitting clinical disease thus makes it an ideal system for studying the evolution of T cell responses during the chronic disease and for determining the efficacy of various immunoregulatory strategies for treatment of an ongoing autoimmune disease (Vanderlugt and Miller, 2002). Acute, chronic, and relapsing models have been employed to study afferent regulation of CD4+ T cell-mediated inflammatory responses. The effects of various immunotherapeutic regimens administered either prior to disease induction, or after disease induction but prior to the onset of the acute clinical phase of paralysis, can thereby be determined. The relapsing-remitting disease in the SJL mouse is ideal for studying efferent regulation of disease progression, as various immunotherapies can be administered during the remission phase following acute disease and the effects of the therapy on subsequent disease relapses can be investigated.
Critical Parameters
Genetic control of susceptibility to murine EAE is complex and involves genes located both within and outside of the major histocompatibility complex (MHC) and differs with the different myelin proteins used for disease induction (Andersson and Karlsson, 2004). In mouse spinal cord homogenate (MSCH)–induced EAE, genes governing sensitization to histamine and other vasoactive amines (Linthicum and Frelinger, 1982; Lublin, 1982; Gonatas et al., 1986) and other unmapped genes (Montgomery and Rauch, 1982; Munoz and Mackay, 1984; Knobler et al., 1985) influence susceptibility. Susceptibility to acute EAE induced by intact MBP maps to the I-A subregion of the murine H-2 complex (Fritz et al., 1984). In contrast, acute EAE induced with intact PLP is controlled primarily by non-MHC genes (Tuohy et al., 1988b,c, Andersson and Karlsson, 2004). Obviously, the induction of EAE with defined peptides of myelin proteins in various mouse strains (see Table 15.1.1) depends on the ability of the various peptides to form a stable association with the particular MHC class II molecules present. The complex genetic control reflects the influences of genes acting at different stages of the disease process and varies with the etiology of each disease model.
The requirement for pertussis toxin for efficient induction of active disease in certain protocols is thought to relate to its ability to increase permeability of the blood-brain barrier. The authors have not had to use pertussis toxin with PLP or the immunodominant peptides of PLP to induce reliable EAE. However, others have reported that induction of EAE with PLP and PLP139-151 requires the use of pertussis toxin. In general, R-EAE is more easily induced in female than in male SJL mice. In addition, female mice are preferred due to the fact that male SJL mice are quite aggressive and should be housed one mouse per cage to avoid fighting and reduce stress, which can negatively affect the induction of disease. It should be noted that increased stress and production of corticosteroids have been shown to inhibit EAE (Mason, 1991). The reader is encouraged to consult the relevant literature for details pertaining to disease induction in different mouse strains using the various myelin proteins and peptides.
Anticipated Results
R-EAE is characterized clinically by the development of ascending flaccid hind limb paralysis. Each mouse is graded daily and assigned a score ranging from 0 to 5 as defined by the clinical criteria outlined in Table 15.1.3. The clinical stages of the disease are summarized in Table 15.1.4 and defined as follows. Acute phase: first clinical sign of disease, in which mice show ascending paralysis following active disease induction or adoptive transfer of encephalitogenic CD4+ T cells. In PLP139-151-induced disease, most mice reach a clinical score of 3 or 4 during this phase. Remission: the phase of clinical improvement that follows a clinical episode. A remission is defined as a reduction in clinical score for at least 2 days after the peak score of the acute phase or of a disease relapse has been reached. Most mice retain some neurological deficit, although occasionally a mouse may recover completely. Relapse: the phase of increasing clinical disease seen after remission. This is normally defined as an increase of at least one grade in clinical score maintained for at least 2 days after remission has occurred. A typical relapsing disease course in a group of 10 naive SJL recipients of 3 × 107 PLP139-151-specific lymph node T cells is illustrated in Figure 15.1.1 (Panel A). Note the more severe acute disease phase followed by the appearance of a primary and secondary relapse interspersed with periods of remission. This data is presented as the mean clinical score of all the mice in the group versus the time post T cell transfer. As such, the clinical severity of the relapses in the group as a whole tends to be underestimated since as the relapses are not synchronous (i.e., individual mice relapse on different days after remitting from the acute phase of paralysis). A typical chronic disease course in a group of 5 naive C57BL/6 recipients of 1.5 × 107 MOG35-55-specific lymph node T cells is shown in Figure 15.1.1 (Panel B). Note the chronic course of the disease and the lack of remission or appreciable recovery, however this can vary depending on the number of encephalitogenic cells transferred.
Figure 15.1.1.
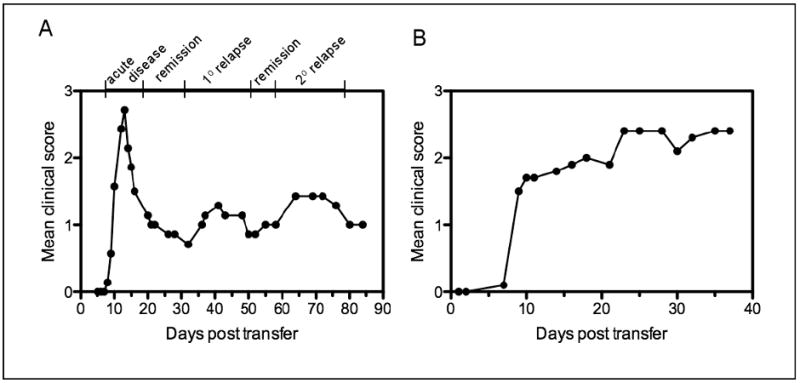
Typical course of relapsing and chronic EAE in SJL and C57BL/6 recipients of PLP139-151- or MOG35-55-specific LN Cells, respectively. (A) A group of 10 naive, female SJL mice was injected i.v. with 3 × 107 PLP139-151-specific LN cells on day 0 and scored for clinical signs of disease over the next 85 days. A typical relapsing-remitting disease pattern is seen, with disease onset beginning ∼7 to 10 days post-transfer and peak acute disease seen at 14 to 15 days. The mice then exhibited clinical remission followed by a primary relapse beginning ∼35 days post-transfer and peaking around ∼40 days. A second remission was followed by a secondary relapse which peaked 65 to 70 days post-transfer. (B) A group of 5 naive, female C57BL/6 mice was injected i.v. with 1.5 × 107 MOG35-55-specific LN cells on day 0 and scored for clinical signs of disease over the next 38 days. A typical chronic disease pattern is seen, with disease onset beginning ∼7 days post-transfer.
There are a variety of ways in which EAE clinical data can be presented; these are summarized in Table 15.1.5. The statistical significance of the percentage of disease incidence between experimental groups is assessed by χ2 test. Depending on the experimental design, statistically significant differences between mean clinical scores, mean day of onset, and mean peak disease severity between experimental groups may be assessed by ANOVA or Student's t-test (Steel and Torrie, 1960). Depending on the experimental design, it is also customary to examine the spinal cords of affected mice using a variety of histologic criteria such as extent and/or severity of inflammatory cell infiltration and the extent of demyelination (reviewed in Dal Canto et al., 1995).
Table 15.1.5.
Clinical Data Presentation
| Clinical data parameter | Definition |
|---|---|
| Mean clinical score | Mean clinical score of all the animals in an experimental group at a particular time post disease initiation |
| Percent incidence | Number of mice displaying acute and/or relapsing clinical signs divided by the total number of animals in an experimental group |
| Mean day of onset or relapse | Mean day of first appearance of acute and/or relapsing clinical disease of those mice within an experimental group that displayed clinical signs |
| Mean peak disease severity | Mean of the highest clinical score reached by those mice within an experimental group that displayed clinical signs |
Transgenic EAE Models
There are a variety of T cell receptor (TCR) transgenic models of EAE on a variety of inbred mouse strains, including humanized strains expressing human HLA class II molecules and human TCRs. These models are summarized in Table 15.1.6. Spontaneous models utilizing mouse MHC II restricting elements include the 19G B10.PL (H-2u)/MBPAc1-11 TCR transgenic (Lafaille et al., 1994), the 5B6 SJL (H-2s)/PLP139-151 TCR transgenic (Waldner et al., 2000), and the 2D2 C57BL/6 (H-2b)/MOG35-55 TCR transgenic (Bettelli et al., 2003) displaying a variety of symptoms ranging from conventional clinical and histological signs of EAE (the 5B6 TCR transgenic on the wild-type SJL background and the 19G TCR transgenic on the B10.PL RAG-1−/− background), to optic neuritis without conventional EAE symptoms (the 2D2 TCR transgenic on the C57BL/6 background). A variant of the 2D2 TCR transgenic system has also been reported, wherein crossing the 2D2 TCR transgenics to a MOG-specific Ig heavy chain knock-in mice (IgHMOG; Bettelli et al., 2006; Krishnamoorthy et al., 2006) resulted in a large percentage of the F1 mice developing a severe form of EAE characterized by selective demyelinating lesions in the spinal cord and optic nerve, a pattern resembling human Devic disease (neuromyelitis optica). There have been three EAE transgenic models described which employ humanized mice expressed HLA restricting elements and human MBP peptide-specific TCRs (Madsen et al., 1999; Ellmerich et al., 2004, 2005; Quandt et al., 2004). Interestingly, with one exception (Ellmerich et al., 2005) there is no significant spontaneous development of EAE in these mice and induction of disease requires either peptide priming and treatment with pertussis toxin (Madsen et al., 1999; Ellmerich et al., 1999), or transfer of activated transgenic T cells (Quandt et al., 2004). The reader is encouraged to consult the relevant literature in order to determine the usefulness of the various transgenic models.
Table 15.1.6.
Mouse TCR Transgenic Models of EAE
| Background (MHC restriction) | TCR Specificity | Characterization of disease parameters | Reference |
|---|---|---|---|
| 19G B10.PL (I-Au) | Mouse MBPAc1-11 | High spontaneous incidence (100%) of conventional EAE on RAG-1−/− background with onset at ∼7-8 weeks; low incidence (∼15%) on conventional B10.PL background | Lafaille et al. (1994) |
| 5B6 SJL (I-As) | Mouse PLP139-151 | Moderate spontaneous incidence (40%-60%) on conventional SJL background; more severe conventional EAE upon peptide immunization and/or treatment with pertussis toxin | Waldner et al. (2000) |
| 2D2 C57BL/6 (H-2b) | Mouse MOG35-55 | No spontaneous conventional EAE, but ∼30% incidence of spontaneous optic neuritis; more severe induction of conventional EAE upon peptide immunization and/or treatment with pertussis toxin | Bettelli et al. (2003) |
| (2D2 × IgHMOG)F1 | Mouse MOG35-55 | 60% incidence of spontaneous conventional EAE in F1 mice with demyelinating lesions restricted to optic nerve and spinal cord, sparring the cerebellum | Madsen et al. (1999); Ellmerich et al. (2005) |
| HLA-DR2 (DRA*0101/DRB1*1501) | Human MBP84-102 (Ob1.A12) | Only ∼4% spontaneous EAE in (HLA-DR2 × Ob1.A12) F1 mice; high disease incidence (∼85% to 90%) in F1 mice primed with peptide and treated with pertussis toxin with variable clinical symptoms | Madsen et al. (1999) |
| HLA-DR15 | Human MBP84-102 (Ob1.A12) | Line 7 transgenics develop spontaneous paralyses at 4 to 5 months of age at a level of 60% (wild-type background) and 100% (Rag-1−/− background) and responses spread to other DR15-restricted myelin epitopes; other lines develop disease only upon peptide priming and treatment with pertussis toxin | Ellmerich et al. (2004, 2005) |
| DRB1*0401 | Human MBP111-129 (MS2-3C8) | Disease incidence in HLA-DRB1*0401 transgenic recipients of transgenic T cells ranged from 60% to 100%; clinical signs consist of ascending hindlimb paralysis along with atypical signs consistent with demyelination within the brainstem and cranial nerve roots | Quandt et al. (2004) |
Time Considerations
Assessment of EAE is obviously a time- and mouse-intensive technique. Depending on the experimental design, the first observation of clinical disease can occur anywhere from 4 to >100 days post induction. In addition, induction of adoptive EAE takes an additional 10 to 14 days to allow time for in vivo priming of donor mice and for in vitro activation of the neuroantigen-specific T cells.
The time required to isolate cells from the CNSwill of course depend greatly on the number of animals to be processed. However, for a modest experiment of 5 or 6 animals, at least half a day will be required. This procedure not only isolates any infiltrating leukocytes, but also isolates neurons, glia, and other CNS resident populations. The number of leukocytes obtained is directly proportional to the disease severity of the animal. An animal with an EAE clinical score of 0 will typically yield almost no leukocytes, while an animal with a clinical score or 4 to 5 may yield several hundred thousand. The nature and requirements of the experiment will determine whether cells from multiple animals will need to be pooled to obtain sufficient cell numbers. However, for flow cytometric analysis, it is reasonable to expect that enough cells will be obtained such that animals can be analyzed individually if so desired.
Literature Cited
- Amor S, Groome N, Linington C, Morris MM, Dornmair K, Gardinier MV, Matthieu JM, Baker D. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J Immunol. 1994;153:4349–4356. [PubMed] [Google Scholar]
- Andersson A, Karlsson J. Genetics of experimental autoimmune encephalomyelitis in the mouse. Arch Immunol Ther Exp (Warsz) 2004;52:316–325. [PubMed] [Google Scholar]
- Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest. 2006;116:2393–2402. doi: 10.1172/JCI28334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, McFarlin DE. Relapsing experimental allergic encephalomyelitis in the SJL/J mouse. Lab Invest. 1981;45:278–284. [PubMed] [Google Scholar]
- Brown A, McFarlin DE, Raine CS. Chronologic neuropathology of relapsing experimental allergic encephalomyelitis in the mouse. Lab Invest. 1982;46:171–185. [PubMed] [Google Scholar]
- Cammer W, Bloom BR, Norton WT, Gordon S. Degradation of basic protein in myelin by neutral proteases secreted by stimulated macrophages: A possible mechanism of inflammatory demyelination. Proc Natl Acad Sci U S A. 1978;75:1554–1558. doi: 10.1073/pnas.75.3.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, Blumenschein W, Churakovsa T, Low J, Presta L, Hunter CA, Kastelein RA, Cua DJ. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RB, Grunnet M, Lingenheld EG. Adoptively transferred EAE in mice bearing the lpr mutation. Clin Immunol Immunopathol. 1997;85:315–319. doi: 10.1006/clin.1997.4450. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Melvold RW, Kim BS, Miller SD. Two models of multiple sclerosis: Experimental allergic encephalomyelitis (EAE) and Theiler's murine encephalomyelitis virus (TMEV) infection—a pathological and immunological comparison. Microsc Res Tech. 1995;32:215–229. doi: 10.1002/jemt.1070320305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellmerich S, Takacs K, Mycko M, Waldner H, Wahid F, Boyton RJ, Smith PA, Amor S, Baker D, Hafler DA, Kuchroo VK, Altmann DM. Disease-related epitope spread in a humanized T cell receptor transgenic model of multiple sclerosis. Eur J Immunol. 2004;34:1839–1848. doi: 10.1002/eji.200324044. [DOI] [PubMed] [Google Scholar]
- Ellmerich S, Mycko M, Takacs K, Waldner H, Wahid FN, Boyton RJ, King RH, Smith PA, Amor S, Herlihy AH, Hewitt RE, Jutton M, Price DA, Hafler DA, Kuchroo VK, Altmann DM. High incidence of spontaneous disease in an HLA-DR15 and TCR transgenic multiple sclerosis model. J Immunol. 2005;174:1938–1946. doi: 10.4049/jimmunol.174.4.1938. [DOI] [PubMed] [Google Scholar]
- Endoh M, Kunishita T, Nihei J, Nishizawa M, Tabira T. Susceptibility to proteolipid apoprotein and its encephalitogenic determinants in mice. Int Arch Allergy Appl Immunol. 1990;92:433–438. doi: 10.1159/000235176. [DOI] [PubMed] [Google Scholar]
- Fritz RB, Zhao ML. Encephalitogenicity of myelin basic protein exon-2 peptide in mice. J Neuroimmunol. 1994;51:1–6. doi: 10.1016/0165-5728(94)90122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz RB, Chou CH, McFarlin DE. Induction of experimental allergic encephalomyelitis in PL/J and (SJL/J × PL/J)F1 mice by myelin basic protein and its peptides: Localization of a second encephalitogenic determinant. J Immunol. 1983;130:191–194. [PubMed] [Google Scholar]
- Fritz RB, Perry LL, Chou CJ. Genetic control of myelin basic protein–induced experimental allergic encephalomyelitis in mice. In: Alvord EC Jr, Kies M, Suckling AJ, editors. Experimental Allergic Encephalomyelitis: A Useful Model for Multiple Sclerosis. Wiley-Liss; New York: 1984. pp. 235–242. [PubMed] [Google Scholar]
- Girvin AM, Dal Canto MC, Rhee L, Salomon B, Sharpe AH, Bluestone JA, Miller SD. A critical role for B7/CD28 costimulation in experimental autoimmune encephalomyelitis: A comparative study using costimulatory molecule-deficient mice and monoclonal antibody blockade. J Immunol. 2000;164:136–143. doi: 10.4049/jimmunol.164.1.136. [DOI] [PubMed] [Google Scholar]
- Gonatas NK, Greene MI, Waksman BH. Genetic and molecular aspects of demyelination. Immunol Today. 1986;7:121–126. doi: 10.1016/0167-5699(86)90072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer JM, Kuchroo VK, Sobel RA, Lees MJ. Identification and characterization of a second encephalitogenic determinant of myelin proteolipid protein (residues 178-191) for SJL mice. J Immunol. 1992;149:783–788. [PubMed] [Google Scholar]
- Greer JM, Sobel RA, Sette A, Southwood S, Lees MB, Kuchroo VK. Immunogenic and encephalitogenic epitope clusters of myelin proteolipid protein. J Immunol. 1996;156:371–379. [PubMed] [Google Scholar]
- Karpus WJ, Lukacs NW, McRae BL, Streiter RM, Kunkel SL, Miller SD. Prevention and treatment of experimental autoimmune encephalomyelitis with anti-MIP-1α. J Immunol. 1995;155:5003–5010. [PubMed] [Google Scholar]
- Knobler RL, Linthicum DS, Cohn M. Host genetic regulation of acute MHV-4 viral encephalomyelitis and acute experimental autoimmune encephalomyelitis in (BALB/cKe × SJL/J) ecombinant-inbred mice. J Neuroimmunol. 1985;8:15–28. doi: 10.1016/S0165-5728(85)80044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy G, Lassmann H, Wekerle H, Holz A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest. 2006;116:2385–2392. doi: 10.1172/JCI28330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 1994;78:399–408. doi: 10.1016/0092-8674(94)90419-7. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, Mc-Clanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linthicum DS, Frelinger JA. Acute autoimmune encephalomyelitis in mice. II. Susceptibility is controlled by the combination of H-2 and histamine sensitization genes. J Exp Med. 1982;156:31–40. doi: 10.1084/jem.156.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lublin FD. Delayed, relapsing experimental allergic encephalomyelitis in mice. Role of adjuvants and pertussis vaccine. J Neurol Sci. 1982;57:105–110. doi: 10.1016/0022-510x(82)90114-9. [DOI] [PubMed] [Google Scholar]
- Madsen LS, Andersson EC, Jansson L, Krogsgaard M, Andersen CB, Engberg J, Strominger JL, Svejgaard A, Hjorth JP, Holmdahl R, Wucherpfennig KW, Fugger L. A humanized model for multiple sclerosis using HLA-DR2 and a human T- cell receptor. Nat Genet. 1999;23:343–347. doi: 10.1038/15525. [DOI] [PubMed] [Google Scholar]
- Mason D. Genetic variation in the stress response: Suceptibility to experimental allergic encephalomyelitis and implications for human inflammatory disease. Immunol Today. 1991;12:57–60. doi: 10.1016/0167-5699(91)90158-P. [DOI] [PubMed] [Google Scholar]
- McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- McRae BL, Kennedy MK, Tan LJ, Dal Canto MC, Miller SD. Induction of active and adoptive chronic-relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol. 1992;38:229–240. doi: 10.1016/0165-5728(92)90016-e. [DOI] [PubMed] [Google Scholar]
- McRae BL, Vanderlugt CL, Dal Canto MC, Miller SD. Functional evidence for epitope spreading in the relapsing pathology of EAE in the SJL/J mouse. J Exp Med. 1995;182:75–85. doi: 10.1084/jem.182.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel I, Shevach EM. Differentiated Th1 autoreactive effector cells can induce experimental autoimmune encephalomyelitis in the absence of IL-12 and CD40/CD40L interactions. J Neuroimmunol. 2002;122:65–73. doi: 10.1016/s0165-5728(01)00465-9. [DOI] [PubMed] [Google Scholar]
- Mendel I, Kerlero DR, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: Fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. 1995;25:1951–1959. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- Miller SD, Tan LJ, Kennedy MK, Dal Canto MC. Specific immunoregulation of the induction and effector stages of relapsing EAE via neuroantigen-specific tolerance induction. Ann N Y Acad Sci. 1991;636:79–94. doi: 10.1111/j.1749-6632.1991.tb33440.x. [DOI] [PubMed] [Google Scholar]
- Miller SD, McRae BL, Vanderlugt CL, Nikcevich KM, Pope JG, Pope L, Karpus WJ. Evolution of the T cell repertoire during the course of experimental autoimmune encephalomyelitis. Immunol Rev. 1995;144:225–244. doi: 10.1111/j.1600-065x.1995.tb00071.x. [DOI] [PubMed] [Google Scholar]
- Montgomery IN, Rauch HC. Experimental allergic encephalomyelitis (EAE) in mice: Primary control of EAE susceptibility is outside the H-2 complex. J Immunol. 1982;128:421–425. [PubMed] [Google Scholar]
- Muller DM, Pender MP, Greer JM. A neuropathological analysis of experimental autoimmune encephalomyelitis with predominant brain stem and cerebellar involvement and differences between active and passive induction. Acta Neuropathol (Berl) 2000;100:174–182. doi: 10.1007/s004019900163. [DOI] [PubMed] [Google Scholar]
- Munoz JJ, Mackay IR. Production of experimental allergic encephalomyelitis with the aid of pertussigen in mouse strains considered genetically resistant. J Neuroimmunol. 1984;7:91–96. doi: 10.1016/s0165-5728(84)80009-0. [DOI] [PubMed] [Google Scholar]
- Olitsky PK, Yager RH. Experimental disseminated encephalomyelitis in white mice. J Exp Med. 1949;90:213–223. doi: 10.1084/jem.90.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson PY, Swanborg RH. Demyelinating diseases of the central and peripheral nervous systems. In: Sampter M, Talmage DW, Frank MM, Austen KF, Claman HN, editors. Immunological Diseases. Little, Brown; Boston: 1988. pp. 1877–1916. [Google Scholar]
- Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: Requirement for Lyt 1+ 2- T lymphocytes. J Immunol. 1981;127:1420–1423. [PubMed] [Google Scholar]
- Pettinelli CB, Fritz RB, Chou CHJ, Mc-Farlin DE. Encephalitogenic activity of guinea pig myelin basic protein in the SJL mouse. J Immunol. 1982;129:1209–1211. [PubMed] [Google Scholar]
- Powell MB, Mitchell D, Lederman J, Buckmeier J, Zamvil SS, Graham M, Ruddle NH, Steinman L. Lymphotoxin and tumor necrosis factor-α production by myelin basic protein-specific T cell clones correlates with encephalitogenicity. Int Immunol. 1990;2:539–544. doi: 10.1093/intimm/2.6.539. [DOI] [PubMed] [Google Scholar]
- Quandt JA, Baig M, Yao K, Kawamura K, Huh J, Ludwin SK, Bian HJ, Bryant M, Quigley L, Nagy ZA, McFarland HF, Muraro PA, Martin R, Ito K. Unique clinical and pathological features in HLA-DRB1*0401-restricted MBP 111-129-specific humanized TCR transgenic mice. J Exp Med. 2004;200:223–234. doi: 10.1084/jem.20030994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddle NH, Bergman CM, McGrath KM, Lingenheld EG, Grunnet ML, Padula SJ, Clark RB. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J Exp Med. 1990;172:1193–1200. doi: 10.1084/jem.172.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Zamvil SS, Mitchell DJ, Lim M, Rothbard JB, Steinman L. Characterization of a major encephalitogenic T cell epitope in SJL/J mice with synthetic oligopeptides of myelin basic protein. J Neuroimmunol. 1988;19:21–32. doi: 10.1016/0165-5728(88)90032-x. [DOI] [PubMed] [Google Scholar]
- Segal BM, Shevach EM. IL-12 unmasks latent autoimmune disease in resistant mice. J Exp Med. 1996;184:771–775. doi: 10.1084/jem.184.2.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal BM, Raine CS, McFarlin DE, Voskuhl RR, McFarland HF. Experimental allergic encephalomyelitis induced by the peptide encoded by exon 2 of the MBP gene, a peptide implicated in remyelination. J Neuroimmunol. 1994;51:7–19. doi: 10.1016/0165-5728(94)90123-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal BM, Dwyer BK, Shevach EM. An Interleukin (IL)-10/IL-12 immunoregulatory circuit controls susceptibility to autoimmune disease. J Exp Med. 1998;187:537–546. doi: 10.1084/jem.187.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmaj K, Raine CS, Farooq M, Norton WT, Brosnan CF. Cytokine cytotoxicity against oligodendrocytes. Apoptosis induced by lymphotoxin. J Immunol. 1991a;147:1522–1529. [PubMed] [Google Scholar]
- Selmaj K, Raine CS, Cross AH. Anti–tumor necrosis factor therapy abrogates autoimmune demyelination. Ann Neurol. 1991b;30:694–700. doi: 10.1002/ana.410300510. [DOI] [PubMed] [Google Scholar]
- Shaw MK, Kim C, Hao HW, Chen F, Tse HY. Induction of myelin basic protein-specific experimental autoimmune encephalomyelitis in C57BL/6 mice: Mapping of T cell epitopes and T cell receptor V beta gene segment usage. J Neurosci Res. 1996;45:690–699. doi: 10.1002/(SICI)1097-4547(19960915)45:6<690::AID-JNR5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Skundric DS, Kim C, Tse HY, Raine CS. Homing of T cells to the central nervous system throughout the course of relapsing experimental autoimmune encephalomyelitis in Thy-1 congenicmice. J Neuroimmunol. 1993;46:113–121. doi: 10.1016/0165-5728(93)90240-y. [DOI] [PubMed] [Google Scholar]
- Skundric DS, Huston K, Shaw M, Tse HY, Raine CS. Experimental allergic encephalomyelitis. T cell trafficking to the central nervous system in a resistant Thy-1 congenic mouse strain. Lab Invest. 1994;71:671–679. [PubMed] [Google Scholar]
- Slavin A, Ewing C, Liu J, Ichikawa M, Slavin J, Bernard CC. Induction of a multiple sclerosis-like disease in mice with an immunodominant epitope of myelin oligodendrocyte glycoprotein. Autoimmunity. 1998;28:109–120. doi: 10.3109/08916939809003872. [DOI] [PubMed] [Google Scholar]
- Steel RG, Torrie JH. Principals and Procedures of Statistics. McGraw-Hill; New York: 1960. [Google Scholar]
- Tan LJ, Kennedy MK, Miller SD. Regulation of the effector stages of experimental autoimmune encephalomyelitis via neuroantigen-specific tolerance induction. II. Fine specificity of effector T cell inhibition. J Immunol. 1992;148:2748–2755. [PubMed] [Google Scholar]
- Tompkins SM, Padilla J, Dal Canto MC, Ting JP, Van Kaer L, Miller SD. De novo central nervous system processing of myelin antigen is required for the initiation of experimental autoimmune encephalomyelitis. J Immunol. 2002;168:4173–4183. doi: 10.4049/jimmunol.168.8.4173. [DOI] [PubMed] [Google Scholar]
- Trotter JL, Clark HB, Collins KG, Wegeschiede CL, Scarpellini JD. Myelin proteolipid protein induces demyelinating disease in mice. J Neurol Sci. 1987;79:173–188. doi: 10.1016/0022-510x(87)90271-1. [DOI] [PubMed] [Google Scholar]
- Tuohy VK, Thomas DM. A third encephalitogenic determinant of myelin proteolipid protein (PLP) for SJL/J mice. J Immunol. 1993;150:194A. [PubMed] [Google Scholar]
- Tuohy VK, Thomas DM. Sequence 104-117 of myelin proteolipid protein is a cryptic encephalitogenic T cell determinant for SJL/J mice. J Neuroimmunol. 1995;56:161–170. doi: 10.1016/0165-5728(94)00143-c. [DOI] [PubMed] [Google Scholar]
- Tuohy VK, Lu ZJ, Sobel RA, Laursen RA, Lees MB. A synthetic peptide from myelin proteolipid protein induces experimental allergic encephalomyelitis. J Immunol. 1988a;141:1126–1130. [PubMed] [Google Scholar]
- Tuohy VK, Sobel RA, Lees MB. Susceptibility to PLP-induced EAE is regulated by non-H-2 genes. Ann N Y Acad Sci. 1988b;540:709–711. doi: 10.1111/j.1749-6632.1988.tb27221.x. [DOI] [PubMed] [Google Scholar]
- Tuohy VK, Sobel RA, Lees MB. Myelin proteolipid protein-induced experimental allergic encephalomyelitis. Variations of disease expression in different strains of mice. J Immunol. 1988c;140:1868–1873. [PubMed] [Google Scholar]
- Tuohy VK, Lu Z, Sobel RA, Laursen RA, Lees MB. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J Immunol. 1989;142:1523–1527. [PubMed] [Google Scholar]
- Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: Implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- Waldner H, Whitters MJ, Sobel RA, Collins M, Kuchroo VK. Fulminant spontaneous autoimmunity of the central nervous system in mice transgenic for the myelin proteolipid protein-specific T cell receptor. Proc Natl Acad Sci U S A. 2000;97:3412–3417. doi: 10.1073/pnas.97.7.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MR, Mannie MD. Acquisition of functional MHC class II/peptide complexes by T cells during thymic development and CNSdirected pathogenesis. Cell Immunol. 2002;218:13–25. doi: 10.1016/s0008-8749(02)00577-4. [DOI] [PubMed] [Google Scholar]
- Whitham RH, Jones RE, Hashim GA, Hoy CM, Wang RY, Vandenbark AA, Offner H. Location of a new encephalitogenic epitope (residues 43 to 64) in proteolipid protein that induces relapsing experimental autoimmune encephalomyelitis in PL/J and (SJL × PL)F1 mice. J Immunol. 1991;147:3803–3808. [PubMed] [Google Scholar]
- Zamvil SS, Mitchell DJ, Moore AC, Kitamura K, Steinman L, Rothbard JB. T-cell epitope of the autoantigen myelin basic protein that induces encephalomyelitis. Nature. 1986;324:258–260. doi: 10.1038/324258a0. [DOI] [PubMed] [Google Scholar]
- Zamvil SS, Mitchell DJ, Powell MB, Sakai K, Rothbard JB, Steinman L. Multiple discrete encephalitogenic epitopes of the autoantigen myelin basic protein include a determinant for I-E class II–restricted T cells. J Exp Med. 1988;168:1181–1186. doi: 10.1084/jem.168.3.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]