

Abstract
We investigated the ability of AMP-activated protein kinase (AMPK) to activate PPARγ coactivator-1α (PGC-1α) in the brain, liver and brown adipose tissue (BAT) of the NLS-N171-82Q transgenic mouse model of Huntington's disease (HD). In the striatum of the HD mice, the baseline levels of PGC-1α, NRF1, NRF2, Tfam, COX-II, PPARdelta, CREB and ERRα mRNA and mitochondrial DNA (mtDNA), were significantly reduced. Administration of the creatine analog beta guanidinopropionic acid (GPA) reduced ATP and PCr levels and increased AMPK mRNA in both the cerebral cortex and striatum. Treatment with GPA significantly increased expression of PGC-1α, NRF1, Tfam and downstream genes in the striatum and cerebral cortex of wild-type (WT) mice, but there was no effect on these genes in the HD mice. The striatum of the untreated HD mice showed microvacuolation in the neuropil, as well as gliosis and huntingtin aggregates, which were exacerbated by treatment with GPA. GPA treatment produced a significant increase in mtDNA in the cerebral cortex and striatum of WT mice, but not in HD mice. The HD mice treated with GPA had impaired activation of liver PGC-1α and developed hepatic steatosis with accumulation of lipids, degeneration of hepatocytes and impaired activation of gluconeogenesis. The BAT in the HD mice showed vacuolation due to accumulation of neutral lipids, and age-dependent impairment of UCP-1 activation and temperature regulation. Impaired activation of PGC-1α, therefore, plays an important role in the behavioral phenotype, metabolic disturbances and pathology of HD, which suggests the possibility that agents that enhance PGC-1α function will exert therapeutic benefits in HD patients.
INTRODUCTION
A characteristic feature of Huntington's disease (HD) is weight loss despite increased caloric intake, which occurs early in the course of the illness. A number of studies have shown that HD patients are in negative energy balance (1–3). In transgenic mouse models of HD, similar observations have been made, and weight loss occurs progressively with a loss of muscle bulk (4,5). In addition, well-recognized metabolic deficits occur in the brain and muscle in HD. There is glucose hypometabolism on positron emission tomography imaging, even in presymptomatic gene carriers (6–9). NMR spectroscopy reveals increased lactate in the cerebral cortex and basal ganglia, and impaired phosphocreatine and ATP production in muscle, in both HD patients and in presymptomatic gene carriers (10–13). Biochemical studies show reduced activities of complexes II–III and aconitase in the striatum of human HD brain tissue (14–16).
In human HD lymphoblastoid cell lines, ATP to ADP ratios are reduced, and the decreases correlate with increases in the CAG repeat length (17). In the striatal cells, which were obtained from mutant huntingtin knock-in mouse embryos, mitochondrial respiration and ATP production were significantly impaired (18). The mitochondrial toxins 3-nitropropionic acid and malonate, which selectively inhibit succinate dehydrogenase and complex II of the electron transport chain, produce a clinical and pathologic phenotypes in rodents, primates and humans that closely resemble HD (19–22).
Mutant huntingtin (htt) causes impairment of mitochondrial function and trafficking by several different mechanisms (23,24). First, huntingtin may interact directly with mitochondria. Lymphoblast mitochondria from HD patients and brain mitochondria from HD transgenic mice depolarize at lower calcium loads than controls, and mutant htt is localized to mitochondria by electron microscopy (23,25,26). The N-terminus of htt was recently reported to be associated with mitochondria (23), and the N-terminal 17 amino acids are essential for this interaction (27). Phosphorylation of the N-terminal serines at positions 13 and 16 blocks the phenotype induced by mutant htt in HD transgenic mice (15).
Another mechanism by which mutant htt may affect mitochondrial function is by altering transcription (28,29). Huntington interacts with a number of transcription factors, including p53, cyclic AMP response element binding protein (CREB), TAFII-130 and SP1 (30–32). A link to the transcriptional coactivator PGC-1α was first suggested by observations that PGC-1α-deficient mice show striatal degeneration and a hyperkinetic movement disorder (33,34). PGC-1α is a transcriptional coactivator, which plays a key role in energy homeostasis, adaptive thermogenesis, alpha-oxidation of fatty acids and glucose metabolism (35,36). PGC-1α was originally identified as a PPARgamma-interacting protein in BAT (36), PGC-1α and a close homolog, PGC-1β, are highly expressed in BAT, and slow-twitch skeletal muscle, tissues known for their high mitochondrial content and energy demands (37). PGC-1α's ability to activate a diverse set of metabolic programs in different tissues depends on its ability to form heteromeric complexes with a variety of transcription factors, including nuclear respiratory factors, NRF1 and NRF2, and the nuclear hormone receptors PPARα, PPARdelta, PPARgamma, estrogen-related receptor alpha (ERRα) and thyroid receptor (37). Many nuclear encoded mitochondrial genes are modulated by PGC-1α, including those encoding cytochrome c, complexes I through V and the mitochondrial transcription factor A (Tfam) (38).
PGC-1α plays a critical role in mitochondrial biogenesis and in studies of cortical, midbrain and cerebellar granule neurons, both PGC-1α and PGC-1β control mitochondrial density (39). Overexpression of PGC-1β or PGC-1α, or activation of the latter by SIRT1, protects neurons from mutant htt-induced loss of mitochondria and cell death (39). The SIRT1 activator, resveratrol, increases the activity of PGC-1α and improves mitochondrial activity as a consequence of its deacetylation of PGC-1α, which increases its effects on liver, fat and muscle metabolism (40). Activation of PGC-1α by resveratrol protects against the adverse effects of cell stress, metabolic disturbances and high caloric intake (41,42). We found that resveratrol treatment of the N171-82Q transgenic mouse model produced increased PGC-1α and reduced vacuolation in BAT and reduced glucose levels, but there were no beneficial effects in the striatum (unpublished observation).
PGC-1α knock-out mice exhibit impaired mitochondrial function, a hyperkinetic movement disorder and striatal degeneration, all features also observed in HD (33,34). Furthermore, impaired PGC-1α function and levels occur in striatal cell lines, transgenic mouse models of HD and in postmortem brain tissue from HD patients (43,44). We recently showed that PGC-1α is reduced in muscle from HD transgenic mice and in muscle biopsies and myoblasts from HD patients (45).
There is reduced PGC-1α mRNA in adipose tissue from R6/2 mice at 9 weeks of age, and in cultured adipocytes, mutant Htt interferes with PGC-1α coactivation of PPARgamma and RxRα reporter constructs (46). A study of Htt transfected neurons showed impairment of the CREB-PGC-1α cascade, which was restored by blocking extrasynaptic NMDA receptors (47). Furthermore, transplantation of adipose-derived stem cells produced increased PGC-1α levels in R6/2 mice, and resulted in improved motor function, survival and reduced loss of striatal neurons (48). Administration of guanidinopropionic acid (GPA), a creatine analog, results in reduced creatine transport, and reduced creatine kinase activity, leading to depletion of creatine and PCr and increased AMP kinase (AMPK), followed by phosphorylation of PGC-1α leading to its activation and mitochondrial biogenesis (49–52). We previously showed that following GPA treatment, there was impaired mitochondrial biogenesis in the muscle of NLS-N171-82Q HD mice (45). In the present study, we extended these results by determining the effects of energy deprivation, and its ability to activate PGC-1α, in the brain tissue, liver and BAT of HD transgenic mice. We examined the effects of treatment of HD transgenic mice with the catabolic stressor beta GPA, which depletes PCr and ATP, leading to activation of AMPK and PGC-1α.
RESULTS
Decreased expression of PGC-1α and downstream genes in the striatum of HD mouse
In these studies, we utilized NLS-N171-82Q transgenic mouse model of HD herein referred to as HD mice (53). These mice have the same N-terminal construct as N171-82Q mice fused to a nuclear localization signal. They develop progressive neurologic deficits, weight loss and impaired survival; however, the phenotype is not as severe as that in the N171-82Q mice lacking an NLS (53). We initially examined PGC-1α mRNA levels in the striatum and the cerebral cortex of 26-week-old wild-type (WT) and NLS-N171-82Q HD mice. There was a significant decrease in PGC-1α mRNA in the striatum of the HD mice when compared with WT, but no significant change in the cerebral cortex (Fig. 1A). Using western blots, we confirmed a reduction in striatal PGC-1α protein levels, but no alteration in the cerebral cortex (Fig. 1B).
Figure 1.

Levels of PGC-1α and energy metabolites in HD and WT mice. PGC-1α mRNA levels in the striatum and the cerebral cortex of WT and HD mice (A). Western blots of PGC-1a immunoreactivity in the striatum and the cerebral cortex of WT and HD mice (B). Both the PGC-1α mRNA and protein levels are significantly reduced in the striatum. The effects of treatment with GPA on ATP, Cr and PCr in the striatum and the cerebral cortex of WT and HD mice (C). The AMPK mRNA levels in the striatum and the cerebral cortex in WT and HD mice at baseline and following GPA (D).
We measured ATP, Cr and PCr levels in both the striatum and the cerebral cortex at baseline and following chronic energy deprivation. We created artificial chronic energy deprivation in the NLS-N171-82Q HD mice by injecting the creatine analogue GPA for 10 weeks. GPA is a catabolic stressor, which competes for creatine transport and depletes intracellular PCr and creatine levels, and reduces creatine kinase activity (54,55). Chronic GPA treatment creates a chronic energy stress condition in brain and muscle, which resembles changes that occur during endurance exercise training.
We measured levels of high-energy phosphate metabolites such as ATP and PCr by HPLC in WT and HD mice treated with either GPA or normal saline (NS) (Fig. 1C). We found significantly reduced levels of ATP in the striatum and the cerebral cortex of HD mice, yet significant increases in PCr, which we observed previously, and which are associated with reduced creatine kinase mRNA (56) and with reduced cystolic creatine kinase activity (unpublished observations). Levels of Cr were significantly reduced in HD mouse striatum; however, decreases in the cerebral cortex were not significant. Following administration of GPA, there was a significant reduction of ATP, creatine and PCr in the striatum and cerebral cortex of both HD and WT mice, with greater reductions in the striatum than those seen in the cerebral cortex. In the striatum of the HD mice, AMPK mRNA was reduced when compared with WT; however, there was no significant difference in the cerebral cortex. Following treatment with GPA, there was a significant increase in AMPK mRNA in WT striatum and cerebral cortex; however, no significant change in the HD mice (Fig. 1D).
We examined WT and NLS-N171-82Q HD mice mRNA at 26 weeks of age (53). At baseline, there was a significant reduction in PGC-1α, NRF1, NRF2, Tfam, COX2, PPARdelta, CREB and ERRα mRNA in the striatum of the HD mice when compared with WT (Fig. 2A). In the cerebral cortex, although levels of many mRNAs trended lower in the HD tissue, only COX IV, PPARdelta and ERRα were significantly reduced (Fig. 2B). Following GPA treatment, AMPK mRNA and protein levels were significantly increased in striatum and cortex of WT mice but were unchanged in HD mice (Fig. 1D). GPA administration resulted in a significant increase in PGC-1α mRNA in the striatum and cerebral cortex of WT mice, which did not occur in the HD mice (Fig. 2A and B). Similarly, GPA treatment of the WT mice resulted in an increase in NRF1, NRF2, Tfam, COX2, PPARα, PPARdelta, cytochrome c and ERRα in the striatum of WT mice, which did not occur in the HD mice. In the cerebral cortex, GPA treatment of WT mice resulted in significant increases of NRF1, NRF2, COX IV, PPARα, PPARdelta, cytochrome c and ERRα. These results indicate impaired PGC-1α transcription at baseline in HD mice, and an impaired ability to activate AMPK and the PGC-1α pathway following energy deprivation.
Figure 2.

Relative mRNA expression of PGC-1α and related genes in the HD and WT mice. Relative mRNA of PGC-1α, PGC-1beta and downstream genes in the striatum (A) and the cerebral cortex (B) of WT and HD mice at baseline and following GPA treatment.
Brain PGC-1α immunoreactivity
PGC-1α immunoreactivity in both the striatum and cerebral cortex is shown at baseline and following chronic energy deprivation with GPA (Fig. 3). Small differences in the intensity of PGC-1α immunolabeling were observed. At baseline, there was reduced PGC-1α immunoreactivity in the cerebral cortex, and particularly, in the striatum of the HD mice (Fig. 3D–F). Following treatment with GPA, there was increased PGC-1α immunoreactivity in the cortex and striatum of WT mice (Fig. 3G–I); however, there was no discernable increase in PGC-1α immunoreactivity following GPA treatment of the HD mice (Fig. 3J–L).
Figure 3.

PGC-1α immunoreactivity in the brain. PGC-1α immunoreactivity in the cortex and the striatum of untreated WT (A–C), untreated HD (D–F), GPA-treated WT (G–I) and GPA-treated HD (J–L) mice. The HD mouse brain shows reduced immunoreactivity compared with WT, particularly in the striatum. GPA treatment of WT mice enhanced PGC-1 α immunoreactivity in the cortex and the striatum. GPA treatment of HD mice also increased PGC-1α immunoreactivity in the cortex and the striatum but to a lesser extent than WT mice. Scale bar = 150 µm.
Neuropathology and striatal vacuolation in HD mice under chronic energy deprivation conditions
Ablation of PGC-1α causes defects in energy metabolism and striatal degeneration in PGC-1α knock-out mice (33,34). We assessed neuropathology and striatal degeneration in the brains of the HD mice during lowered energy conditions, by histological analysis using hematoxylin and eosin (H&E staining) and Oil red O staining. We observed no structural abnormalities in the frontal cortex of the HD mice under basal conditions (Fig. 4B) when compared with WT mice (Fig. 4A). Light microscopic examination of H&E stained brain sections revealed the presence of spongiform lesions (microvacuolation) in the striatum of HD mice under basal conditions, when compared with WT mice (Fig. 4E and F). Following GPA administration, during energy-stressed conditions, we found neuronal microvacuolation in the striatum (Fig. 4H), and to a lesser extent in cortex of the HD mice (Fig. 4D), which were absent in WT mice (Fig. 4C and G). The number and size of the spongiform lesions was increased in both the cerebral cortex and striatum of HD mice under chronic energy-stressed conditions (Fig. 4D and H). The patchy areas of spongiform lesions include microvacuolation of neurons and neuropil in the striatum, and to a lesser extent in the cerebral cortex.
Figure 4.

Histopathology of WT and HD striatum and cerebral cortex. Hematoxylin-and-eosin and Oil red O staining of the cortex and striatum of WT and HD mice with or without GPA treatment. Note vacuolation (arrows) in the HD striatum which is more severe following GPA treatment. The GPA-treated HD sriatum showed more severe vacuolation than the GPA-treated HD cortex. Oil red O staining did not reveal any significant lipid accumulation in any of the sections. Insets are higher magnification of the respective boxed areas showing the vacuolation. Scale bar = 100 µm.
PGC-1α controls several metabolic functions including fatty acid oxidation and gluconeogenesis. PGC-1α knock-out mice exhibit impaired fatty acid oxidation and an inability to maintain cellular lipid balance during fasting conditions (33,34). Therefore, we examined whether the spongiform lesions observed in the striatum in chronic energy deprived conditions are associated with impaired lipid metabolism. We examined whether there was accumulation of neutral lipids, using Oil red O staining. We observed no Oil red O staining in the cerebral cortex or striatum of either WT or HD mice under basal conditions (Fig. 4I, M, J and N) or following GPA administration (Fig. 4K, O, L and P). The spongiform areas of vacuolation in the cerebral cortex and striatum are, therefore, not due to accumulation of lipids.
Astrogliosis, huntingtin aggregates and neurofilament immunostaining, and amounts of mitochondrial DNA
Immunostaining for glial fibrillary acidic protein (GFAP) showed mild astrogliosis in the HD mouse striatum at baseline, which was markedly exacerbated by treatment with GPA (Fig. 5B and D). There was mild astrogliosis in the WT striatum after treatment with GPA (Fig. 5C), but it was not as severe as that in the HD mouse striatum (Fig. 5D). The number of Htt aggregates was examined using the EM48 antibody. The HD striatum showed EM48 immunoreactive aggregates at baseline which became much more prevalent following GPA treatment (Fig. 5F and H). Quantification of the number of aggregates showed that they were significantly increased in the HD striatum, and that the GPA treatment produced a further significant increase in the number of aggregates (Fig. 5B).
Figure 5.
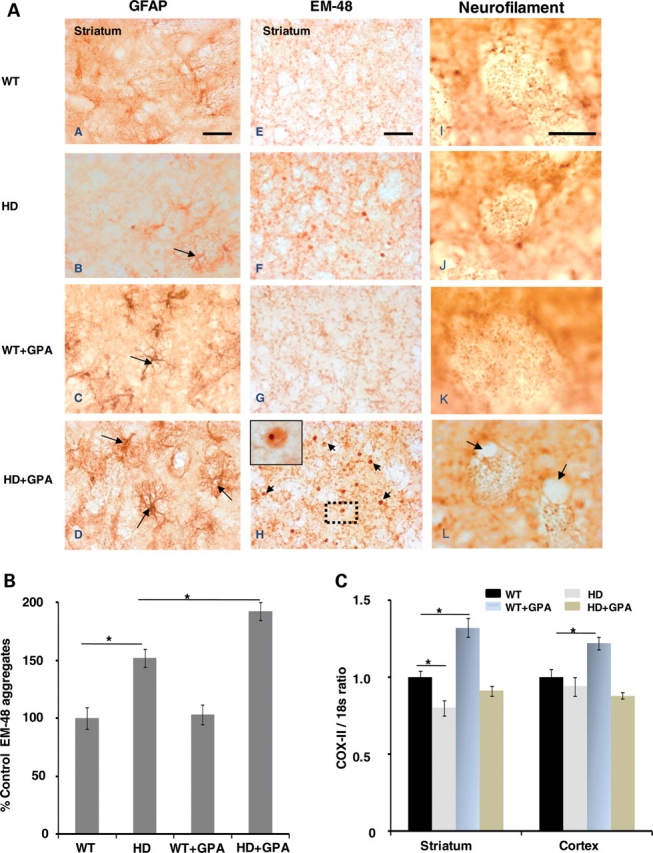
Gliosis, huntingtin aggregates formation, neurofilaments immunostaining and measurement of mt DNA. Photomicrographs (A) showing glial fibrillary acidic protein (GFAP), EM-48 and neurofilament immunoreactivity in the striatum of WT and HD mice with or without GPA treatment. GFAP-labeled reactive astrocytes (long arrows) are evident in the HD striatum (B). Astrocytosis in the HD striatum is exacerbated by GPA treatment (D). GPA treatment of WT mice also resulted in astrocytosis, (C) but not as severe as HD mice. The HD striatum exhibits EM-48 immunoreactive aggregates (F) that become more abundant (short arrows) after GPA treatment (H). Inset shows a high magnification of the boxed area showing an intranuclear aggregate. Neurofilament staining of axons in cross-section revealed atrophy of striatal neurites in HD mice (J, L). Vacuoles associated with axons are also evident in the GPA-treated HD striatum (L). Bar graphs show the quantification of EM-48 aggregates in the striatum (B) and COX-II/18s ratio (C). Scale bar = 100 µm.
We also examined neurofilament staining of axons. The neurofilament staining showed atrophy of axons in the HD mouse striatum both at baseline and following administration of GPA (Figs 5J and L). The neurofilament staining showed the presence of vacuoles, which were more prevalent after treatment with GPA. We also examined whether there was evidence for reduced numbers of mitochondria, and mitochondrial DNA (mtDNA), in the HD mouse striatum. We examined the ratio of mRNA for COX-II, a mtDNA encoded gene, to the mRNA for 18s rRNA, a nuclear-encoded transcript. This has been shown to be a reliable index of mtDNA content (57). There was a significant reduction in the COX II/18s rRNA ratio in the striatum of HD mice at baseline, when compared with WT mice. Following administration of GPA, there was a significant increase in the COXII/18s rRNA ratio in the striatum of the WT mice, but no increase in the HD mice. In the cerebral cortex, there was no alteration in the COXII/18s rRNA ratio at baseline, but there was a significant increase after GPA treatment of WT mice, which was not observed in the HD mice.
Morphological alteration in hepatocytes and hepatic steatosis during energy-stressed conditions in HD mice
We observed no significant gross morphological difference between H&E stained liver sections from WT and HD mice under basal conditions (Fig. 6). The HD mice livers showed well-preserved morphology of hepatocytes with intact round nuclei (Fig. 6B). Under energy-deprived conditions, the HD mice exhibited marked morphological alterations, with hepatic steatosis and hepatocyte degeneration, with reduced numbers of intact nuclei, and accumulation of neutral lipids as detected with Oil red O (Fig. 6D). All of these morphological abnormalities observed in HD mouse liver under energy-deprived conditions were not evident in WT mice under energy-deprived conditions (Fig. 6C). In addition, we examined blood glucose levels following the administration of pyruvate. The HD mice are known to develop impaired glucose tolerance, which occurred as shown in Figure 6 in which mean glucose levels were increased in HD mice under both free and fasting conditions. Following the administration of GPA, there was a significant reduction in blood glucose in the WT mice and to a lesser extent in the HD mice. Following GPA treatment, the rate of increase of pyruvate-induced glucose increases was reduced in the WT mice, most likely due to increased glucose uptake in muscle. The rate of increase of glucose in the GPA-treated HD mice was not affected following pyruvate administration. We also examined mRNA levels in the liver of WT and HD mice at baseline and following GPA treatment. At baseline, PGC-1α, Tfam and hepatocyte nuclear factor alpha (HNFα mRNA) were significantly decreased in the HD mice. Following GPA treatment PGC-1, Tfam and HNFα mRNA were significantly increased in WT mice; however, there were no significant increases in the HD mice, except for an increase in medium chain acyl-CoA dehydrogenase (MCAD) mRNA. In the HD mice, PGC-1α, Tfam, G6Pase, HNFα and phosphoenolpyruvate carboxykinase (PEPCK) mRNA levels were significantly lower than those in WT mice after GPA. In PGC-1α-deficient mice, the induction of PEPCK and G6Pase mRNA is impaired in response to fasting, similar to our observations in the HD mice (34).
Figure 6.
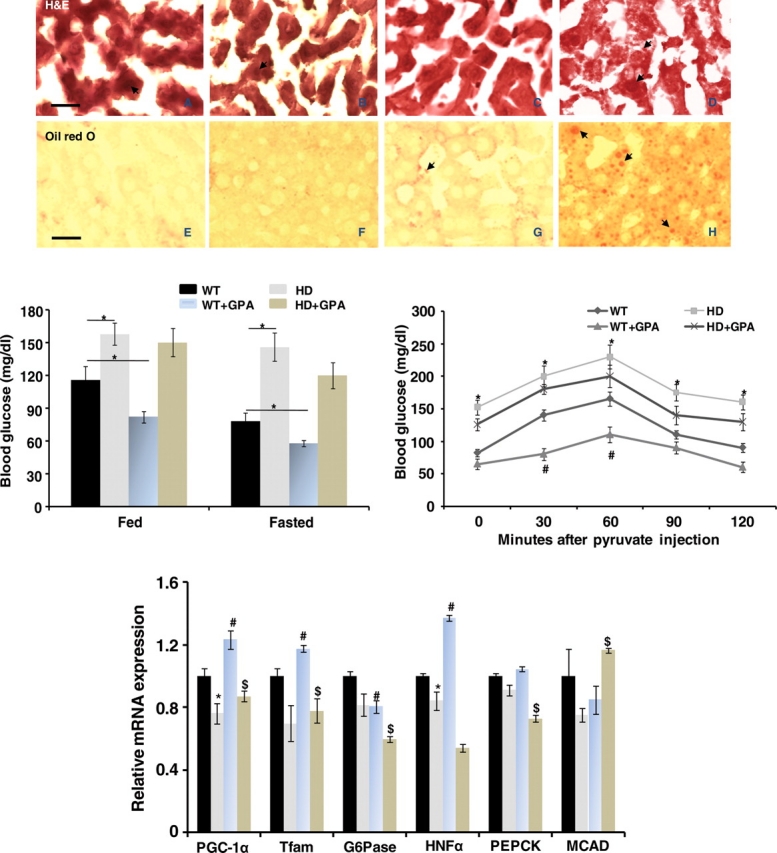
Histopathology of the liver, measurement of glucose and liver mRNA. Hematoxylin-and-eosin and Oil red O staining of the liver of WT and HD mice with or without GPA treatment. Vacuolation of hepatocytes is present in the liver of HD mice treated with GPA. Oil red O staining revealed neutral lipid accumulation (red staining, arrows) in the liver of GPA-treated HD mice. Graphs show blood glucose levels (mg/dl). Following administration of pyruvate, the blood glucose was significantly increased in the HD mice when compared with WT, and administration of GPA reduced the blood glucose in both WT and HD mice. The mRNA for liver PGC-1α and downstream genes both before and following GPA is shown in the bottom graph. Before GPA both PGC-1α and Tfam were reduced in HD mouse liver, and they significantly increased in WT after treatment with GPA but not in HD. G6Pase, HNFα and PEPCK are involved in gluconeogenesis, and they showed a pattern similar to PGC-1α, whereas MCAD, which is involved in fatty acid metabolism was reduced in HD mouse liver at baseline, but showed a significant increase after GPA administration. Scale bar = 50 µm.
Brown adipose tissue and thermoregulation
The WT and HD mouse BAT was examined for mRNA of PGC-1α and UCP-1 both at baseline and after exposure to 4°C temperature for 3 h. The PGC-1α levels were decreased slightly at baseline, but they increased in both the WT and HD mice in response to cold (Fig. 7A), similar to previous observations in N171-82Q transgenic HD mice (44). The BAT of the HD mice was markedly vacuolated when compared with WT mice, and Oil red O staining showed that this was due to accumulation of neutral lipids (Fig. 7C). The response of WT and HD mice to cold exposure at 4°C for 3 h is shown in Figure 7B. The HD mice showed age-dependent impairment of temperature regulation. There was no impairment at 90, 120 and 180 days of age (data not shown), but increasing impairment starting at 210 days and 240 days of age, with the temperature dropping 2–35.5°C following cold exposure at 4°C (Fig. 7B).
Figure 7.

PGC-1α and UCP1 mRNA levels, temperature regulation and BAT vacuolation in WT and HD mice. PGC-1α and UCP1 mRNA levels in BAT of control and HD transgenic mice at baseline and following exposure to 4°C for3 h (A). Exposure to cold for 3 h resulted in progressive hypothermia in the HD mice at 210 and 240 days of age (B). Brown adipose tissues of WT and NLS N171-82Q mice (120 days) stained with hematoxylin-and-eosin showing increased vacuolation in the NLS N171-82Q mouse. Oil red O staining (inset) revealed abundant accumulation of larger lipid droplet arrows and (red staining) in the NLS N171-82Q mouse compared with WT (C).
DISCUSSION
Increasing evidence implicates mitochondrial dysfunction and metabolic impairment in the pathogenesis of HD. In particular, PGC-1α is a key transcriptional co-regulator that is important in protecting neurons against oxidative damage, and which appears to be involved in HD pathogenesis (43–45,58). PGC-1α induces the transcription of cellular programs regulating mitochondrial respiration, oxidative stress defence and adaptive thermogenesis (35,36). PGC-1α mRNA is reduced in the striata from postmortem HD patient brains, striata from an HD knock-in mouse model and in the STHdhQ111 cultured HD striatal cell line (43). It was suggested that mutant Htt interferes with the formation of the CREB/TAF4 complex that regulates transcription of the gene encoding PGC-1α.
The STHdhQ111 striatal neuronal line exhibited reduced expression of nuclear encoded mitochondrial genes, which are regulated by PGC-1α including cytochrome c and cytochrome oxidase IV. In knock-in HD transgenic mice (with 140 CAG repeats inserted into the murine Htt gene), the expression of PGC-1α was reduced several fold in medium spiny neurons, but increased almost 50-fold in nNOS interneurons, consistent with a role of PGC-1α in the selective vulnerability of medium spiny neurons, and the resistance of interneurons which are spared in HD. Expression of PGC-1α is rapidly induced in response to cold, and it regulates expression of key components of adaptive thermogenesis including UCP1, which uncouples respiration resulting in heat production in BAT (44). There is a marked hypothermia at baseline and following cold exposure in several HD transgenic mouse models (44). Following cold exposure, UCP1 expression did not increase in BAT from N171-82Q HD mice relative to WT animals, implicating impairment of PGC-1α induction of UCP1 and active thermogenesis in HD mice. In the primary brown adipocytes of N171-82Q HD mice, there is a reduced ATP/ADP ratio and reduced numbers of mitochondria (44). These mice show vacuolated BAT, similar to our present results and findings in PGC-1α-deficient mice (44).
Microarray expression data from the caudate nucleus of HD patient postmortem brain tissue showed that there was reduced expression of 24 out of 26 PGC-1α target genes (44). Consistent with impaired PGC-1α function in HD, there is evidence for oxidative stress in HD plasma, postmortem brain tissue and transgenic mice (14,59,60). The increase in oxidative damage may be a consequence of impaired expression PGC-1α and PGC-1beta, since they are required for induction of many ROS detoxifying enzymes including Cu/Zn superoxide dismutase, manganese superoxide dismutase and glutathione peroxidase (58).
We found that an impairment of PGC-1α regulation contributes to muscle dysfunction in HD (45). There is a loss of muscle bulk in HD patients, as well as in HD transgenic mouse models, which is due to a myopathy, perhaps due to metabolic dysfunction (61). Polyglutamine inclusions, as well as changes in gene expression and calcium regulation, are observed in muscle from both HD transgenic mice and HD patients (62,63). Cardiac dysfunction with abnormal mitochondria occurs in R6/2 HD transgenic mice, similar to findings in PGC-1α-deficient mice (4,64). In R6/2 HD transgenic mice, the quadriceps and gastrocnemius muscles develop normally until 6 weeks of age, after which they severely atrophy, and there is supersensitivity to acetylcholine and morphological abnormalities of the neuromuscular junctions (5).
We recently investigated whether PGC-1α dysfunction plays a role in muscle abnormalities in the NLS-N171-82Q transgenic mouse model of HD, in which an N-terminal huntingtin gene is fused to a nuclear localization signal (53). We found significant reductions of PGC-1α and Tfam in the muscle of untreated HD mice. We then treated the HD mice with the creatine analog GPA for 10 weeks, which depletes intramuscular ATP/AMP ratios, resulting in activation of muscle AMPK, increased PGC-1α and mitochondrial biogenesis (49,54,55). AMPK directly phosphorylates PGC-1α and increases its expression through a feedback loop (52).
Following treatment with GPA, AMPK mRNA and protein levels, and PGC-1α mRNA levels were significantly increased in the muscle of WT mice, but there was no effect in the HD mice (45). In WT mice, there was an induction of genes of oxidative phosphorylation, as well as mitochondrial biogenesis, and increased numbers of mitochondria, an increase in numbers of oxidative muscle fibers and motor performance, which did not occur in HD transgenic mice (65). Furthermore, in muscle biopsies from HD patients, PGC-1α, PGC-1β and oxidative fibers were decreased. Oxygen consumption, PGC-1α and NRF1 in response to GPA were significantly reduced in myoblasts from HD patients, and this was reversed by a knockdown of mutant huntingtin with siRNA.
These results are consistent with an effect of mutant Htt in blocking PGC-1α expression, either due to direct binding of Htt to the PGC-1α promoter impairing its expression, or by indirectly modulating its expression by interfering with the ability of other transcription factors, such as CREB, to activate the PGC-1α promoter. Recent work has shown that Htt binds to DNA (66). Several studies have shown that there is reduced CREB and CBP in Htt transvected cell lines and transgenic mice, which can be reversed by increasing cAMP which activates CREB (67,68). The phosphodiesterase type IV inhibitor rolipram increases CREB phosphorylation, neuropathologic behavior and abnormalities in the R6/2 transgenic mouse model of HD (67,68). Depletion of CREB binding protein has been linked to cell death mediated by mutant huntingtin in vitro (31,69,70). CREB plays an important role in the regulation of PGC-1α, and its ability to induce gluconeogenesis and to increase in response to H2O2 (71,72). The regulation of PGC-1α by CREB/TAF4-dependent transcription was shown to be impaired in HD (43).
In the present study, we expanded these observations into brain tissue from the GPA-treated NLS-N171-82Q transgenic mice. We found that AMPK mRNA and protein expression were decreased in the striatum of our HD transgenic mice, when compared with WT mice. Following GPA treatment, AMPK mRNA and protein levels were significantly increased in the striatum and cerebral cortex of WT mice, but were unchanged in the HD mice, consistent with an inability to activate PGC-1α expression. Furthermore, following GPA, there was an increase in PGC-1α, NRF1, Tfam, COX2, PPARalpha, PPARdelta, cytochrome c and ERRα expression in the striatum of WT mice, but there was an inability to activate these genes in the HD mice. Similarly, GPA treatment increased expression of PGC-1α, NRF1, NRF2, COX IV, PPARα, PPARdelta, cytochrome c and ERRα in the cerebral cortex of WT mice, but these effects, except for an increase in PPARdelta and ERRα, were absent in the HD mice.
Interestingly, we also observed that following GPA administration, there was neuronal vacuolation in the HD mice which was absent in control mice. This is of interest since similar microvacuolation is seen in the striatum of PGC-1α-deficient mice (33,34). The PGC-1α-deficient mice developed a hyperkinetic movement disorder as well as microvacuolation within the striatum, similar to that which we observed in the HD mice treated with GPA (33,34). The microvacuolation that we observed was present in HD mice on a normal diet, but was significantly worsened by the administration of GPA, showing that a metabolic stressor exacerbated the pathology. The microvacuolation in the striatum was not due to accumulation of neutral lipids as assessed using Oil red O staining.
There was astrogliosis in the striatum, which was worsened following administration of GPA. Interestingly, the number of EM-48 aggregates was also increased after GPA treatment. The vacuoles in the striatum are found both in neurons and in the neuropil associated with axons as shown by neurofilament staining. This is similar to observations made in PGC-1α-deficient mice (33,34). In these mice, spongiform lesions in the striatum were associated with gliosis, and the spongiform lesions appeared to arise from the loss of axons in the striatum (34). In the PGC-1α-deficient mice, there were normal numbers of cortical neurons but neurons from the deep layers showed small vacuoles in both the neurons and neuropil (33). The neuropathologic observation of spongiform degeneration is of interest, since similar lesions occur in MnSOD null mice (73), and PGC-1α plays an important role in controlling expression of MnSOD (71). In addition, there was a reduction in the COX II/18s rRNA ratio in the HD striatum, consistent with reduced mtDNA and numbers of mitochondria. In WT mice, GPA treatment activated AMPK, which increased PGC-1α, NRF1 and Tfam, and this was accompanied by an increase in COX II/18s rRNA, consistent with mitochondrial biogenesis, increased mtDNA and increased numbers of mitochondria. This pathway, which leads to an increase in mitochondria in response to an energetic stress, was blocked in the HD mice.
We also observed that there was an increase in steatosis as well as degeneration of hepatocytes in the livers of HD mice following treatment with GPA. This is of interest since it was shown that after a 24 h fast, PGC-1α-deficient mice develop hepatic steatosis, with increased neutral lipids as assessed using Oil red O stains (33). This appears to be related to impaired mitochondrial respiration as well as increased synthesis and impaired metabolism of lipids (33). A reduction of hepatic PGC-1α impairs fatty acid oxidation by medium chain acyl-CoA dehydrogenase (MCAD), which contributes to the hepatic steatosis. There was impaired ability to activate genes involved in gluconeogenesis, similar to observations in PGC-1α-deficient mice, but blood glucose levels were not reduced since there are alternate pathways to activate gluconeogenesis (34,74). We also observed that in BAT of the HD mice, there is a marked vacuolation, which is due to accumulation of neutral lipids. The HD mice also show age-dependent impaired temperature regulation, consistent with abnormal BAT, and an inability to upregulate uncoupling protein 1 (UCP1). PGC-1α is known to control the expression of UCP-1 in response to cold (33,34). These findings are similar to those that were observed in PGC-1α-deficient mice (33). The PGC-1α-deficient mice also develop abnormal vacuolation of BAT and inability to upregulate UCP1, and marked hypothermia in response to a cold challenge (33,34). Other authors recently showed reduced PGC-1α levels in BAT of 9-week-old R6/2 mice and interference with PGC-1α coactivation in adipocytes by mutant Htt (46). Furthermore, transplanted adipose-derived stem cells, which increase PGC-1α levels, improve motor behavior, survival and loss of striatal neurons in the R6/2 transgenic mouse model of HD (48).
Taken together, these findings provide further evidence for an impairment of PGC-1α activity and function in HD transgenic mice. Polymorphisms in the PGC-1α gene alter the age of onset of HD, providing further genetic evidence that PGC-1α plays a role in HD pathogenesis (75,76). The abnormality that we have observed is both a decrease in the levels of PGC-1α at baseline, as well as an inability to upregulate it in response to physiologic stimuli, such as cold exposure or a depletion of high-energy phosphates. This impairment correlates with worsened pathology in both the striatum and the liver, which show increased vacuolation, and in the striatum with an increase in astrogliosis and Htt aggregates.
An impairment of PGC-1α transcriptional activation may, therefore, play a critical role in the pathogenesis of HD. This suggests that agents, which can increase the activity of PGC-1α or its upstream transcriptional activators, may be useful in the treatment of HD. There are several potential means to modulate PGC-1α expression and activity. PGC-1α expression is reduced by acetylation by GCN5 and increased by deacetylation by SIRT1, therefore, agents which either inhibit GCN5, or which increase the activity of SIRT1, may be useful (77). PGC-1α, SIRT1 and AMPK form an energy-sensing network which controls energy expenditure in various tissues (78,79). SIRT1 activity is increased by resveratrol or its cofactor NAD (41,42). Our studies of resveratrol in the N171-82Q HD mice showed improvement of vacuolation of brown adipose tissue and glucose levels, but no improvements of motor function, survival or striatal pathology, due to inadequate uptake into the central nervous system (Ho and Beal, unpublished findings). Another approach is to utilize PPAR-γ agonists such as rosiglitazone, which prevents mitochondrial dysfunction and oxidative stress in mutant Htt striatal cells (80). Another activator of PGC-1α is bezafibrate, which is a PPAR panagonists leading to increased PGC-1α levels and improvement in an experimental mitochondrial myopathy (81). AMPK can be activated with metformin, which was reported in one study to be beneficial in R6/2 HD mice (82). Increases in PGC-1α activity need to be precisely modulated since overexpression can lead to a myopathy or cardiomyopathy (83,84). A moderate increase in PGC-1α, however, produces beneficial metabolic effects (85). An increase in PGC-1α expression or activity may, therefore, be beneficial in ameliorating both the phenotype and the pathology that occur in HD.
MATERIALS AND METHODS
Transgenic animals and treatments
The NLS-N171-82Q mice with a C3H/C57Bl6 background were a kind gift from Dr David Borchelt, Chris Ross and Gabrielle Schilling. These mice have the human prion promoter driving expression of an 82-CAG repeat. In NLS-N171-82Q HD mice, a nuclear localization signal (NLS) derived from atrophin-1 was fused to the N-terminus of an N171-82Q construct (53). These mice develop phenotypes identical to N171-82Q mice, although less severe. HD offspring were genotyped by PCR assay of DNA obtained from tail tissue. The animals were housed at Weill Medical College of Cornell University animal house and were kept on a 12 h light/dark cycle, with food and water continuously available. Experiments were carried out using procedures that minimized pain and discomfort. All experiments were conducted within National Institutes of Health guidelines for animal research and were approved by the Weill Cornell Medical College Animal Care and Use Committee.
WT littermates and HD male and female mice were divided equally in different treatment groups. WT and HD mice were treated with GPA, which is a creatine analog that competes for the transport of creatine in the skeletal muscle and inhibits creatine kinase activity. Chronic GPA treatment causes chronic energy depletion by depleting intracellular PCr and ATP concentrations and mimics endurance exercise training conditions (54,55). Thus, chronic GPA treatment is a method to pharmacologically create chronic energy deprivation conditions. WT and HD mice at the age of 16 weeks received GPA by single i.p. injection daily for 10 weeks (0.2 ml/mouse; 0.5 M). WT and HD mice, which received a daily single injection of saline (0.2 ml/mouse) for the same duration, served as controls as described earlier (45).
Gene expression analysis by RT–PCR
After indicated treatments, tissues were harvested and frozen immediately. Total RNA was isolated from frozen WT and HD mice striatum and cortex using the TriZol Reagent. Genomic DNA was removed using RNase-free DNase (Ambion). RNA pellets were resuspended in DEPC-treated water (Ambion). Total RNA purity and integrity was confirmed by ND-1000 NanoDrop (NanoDrop Technologies) and 2100 Bioanalyzer (Agilent), respectively, with an average 260/280 ratio for all study samples ranging from 1.9 to 2.1 and average RIN numbers ranging from 5.0 to 7.5. All qPCR plating was performed on ice. Real-time RT–PCR was performed using the ABI prism 7900 HT sequence detection system (Applied Biosystems, Foster City, CA, USA) based on the 5′-nuclease assay. Serial dilutions of 320 ng/well, 32 ng and 3.2 ng of total RNA were plated with two representative genes and housekeeping gene β-actin or 18s rRNA. Probe cleavage was consistently observed with 320 ng/well, with average Ct value of 30, for the two representative genes. Consistent amplification was observed in all three-dilution points for β-actin or 18 s rRNA. One-step, singleplex and triplicates of each sample were plated. A working solution of 160 ng/µl was prepared for each sample and 2.5 µl of this solution, making 400 ng/qPCR well, was distributed to each triplicate qPCR well. A master-mix, comprising one-step RT qPCR MasterMix Plus reagents (no AmpErase UNG, RTQPRT- 032X, EuroGentec, San Diego, CA, USA), 20X TaqMan Gene Expression Assay (Applied Biosystems) and Molecular Biology Grade H2O (HyClone) was prepared for each target gene and 5.5 µl of this Primer-Probe master-mix was added to the PCR well totaling 8 µl qPCR reaction volume. Thermal cycling conditions were 48°C for 30 min, 95°C for 10 min and then 45 cycles of 95°C for 15 s and 60°C for 1 min. Relative expression was calculated using the ΔΔCt method. TaqMan-based gene expression assays were used for RT–PCR experiments (Table 1).
Table 1.
Primer sequences used for quantitative real-time PCR
| Mouse Sequence (ABI TaqMan Gene expression assays) | |
|---|---|
| Gene name | TaqMan probe set ID |
| PGC-1α | Mm00447183_m1 |
| PGC-1β | Mm00504720_m1 |
| NRF-1 | Mm00447996_m1 |
| NRF-2 | Mm00477784_m1 |
| TFAM | Mm00447485_m1 |
| AMPK | Mm01264787_m1 |
| CREB | Mm00501607_m1 |
| ERR-α | Mm00433143_m1 |
| COX-IV | Mm00446387_m1 |
| PPARα | Mm00440939_m1 |
| PPARδ | Mm00803186_m1 |
| CYTC | Mm01621044_g1 |
HPLC assay
Briefly, mice were sacrificed by cervical dislocation and hind limbs were immediately (within 1–2 s) dropped into the liquid nitrogen for a snap freezing. Striatum and cortex were quickly dissected on a mechanically refrigerated (−40°C) thermal plate (Sigma, San Diego, CA, USA). Two hundred microliter ice-cold acetonitrile was added to frozen tissues and tissues were homogenized with a pellet pestle while adding 100 µl of ice-cold ddH2O. The homogenate was centrifuged at 14 000 rpm, 4°C for 15 min and 150 µl of supernatant was added with 150 µl of ddH2O, centrifuged again and made ready for the HPLC assay. Standards were prepared exactly in the same way. Fifty microliter of the preparation was injected to a HPLC system which comprises a Perkin Elmer M-250 binary LC pump (Norwalk, CT, USA), a Waters 717 plus autosampler, a Waters 490 programmable multi-wavelength UV detector (Milford, MA, USA) and an ESA 501 chromatography data process system (Chelmsford, MA, USA). The gradient elution was performed on a TSK-GEL HPLC column (Tosoh Biosep, Japan) with a mobile phase rate of 1 ml/min by two buffers. Buffer A contained 25 mm NaH2PO4, 100 mg/l tetrabutylammonium with pH5.5, whereas organic buffer B was 10% (v/v) acetonitrile made in a buffer of 200 mm NaH2PO4, 100 mg/l tetrabutylammonium with pH 4.0. The UV detector was programmed 210 nm from 0 to14 min for detecting creatine and phosphocreatine which appeared at about 3.7 and 10 min, respectively, and 260 nm from 10 to 40 min for AMP, ADP and ATP which were eluted out at about 18, 22 and 25 min, respectively. The standard curve of each compound was constructed by plotting peak heights (mV) versus concentrations, ranging from 10 to 1000 µM for creatine and phosphocreatine and 5 to 500 µM for ATP and its metabolites. The quantification was carried out using the external standard calibration. Protein concentrations were measured by using BCA protein assay kit (Pierce, USA). Data were expressed as nmol/mg protein.
Western blot
The Striatum and the cortex of WT and HD mice were homogenized in cell extraction buffer containing 50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% SDS, 0.5% NP-40, 0.5% deoxycholate supplemented with protease and phosphatase inhibitors (Sigma). Samples were homogenized, sonicated for 5 s and centrifuged at 4000 g for 30 min. Protein concentrations of the supernatant were determined using the BCA protein assay, as per the manufacturer's recommended protocol. Equal amounts of protein (45 µg) were loaded on to a 4–20% Tris-glycine gel (Invitrogen). Membranes were then blocked for 1 h at room temperature in Tris-buffered saline/Tween-20 (TBST) (50 mm Tris–HCl, 150 mm NaCl, pH 7.4, 1% Tween-20) containing 5% non-fat dried milk. The membranes were incubated overnight at 4°C for PGC-1α (1:2,000, a kind gift of Dr B.M. Spiegelman), and β-actin (1:10 000, Chemicon). Membranes were then washed three times with TBST and incubated for 1 h with HRP-conjugated secondary antibody and the immunoreactive proteins detected using a chemiluminescent substrate (Pierce) according to the manufacturer's instructions. Films were scanned by film processor (Konica Minolta & Graphic Inc., SRX-101A, USA). Protein expression was quantified using Scion Image for Windows (NIH, USA).
Mitochondrial DNA (mtDNA) content
Total DNA was isolated by a standard phenol-chloroform extraction procedure. Fragments of the mitochondrial COX-II gene and the nuclear 18S gene were amplified in duplicate by quantitative real-time PCR. The absolute content of mtDNA was expressed as the mtDNA-to-nuclear-DNA ratio (COX-II mtDNA/18S nuclear DNA) (57).
Histological analysis
Brain and liver tissues were dissected and overnight post-fixed in 4% paraformaldehyde followed by washing with phosphate-buffered saline (PBS). Twenty micrometer thin liver and brain sections were cut for H&E and Oil red O staining.
Immunohistochemistry
Rats from each group were perfused with 0.1 M phosphate-buffered saline (PBS; pH 7.2) followed by chilled 4% paraformaldehyde for fixation of tissues. Brains were removed and post fixed in the same fixative overnight, followed by cryopreservation in 10, 20 and 30% sucrose in PBS (W/V). Serial coronal sections were cut on a freezing microtome at 20 µm thickness and collected in PBS. Endogenous peroxidase activity was inhibited by incubation with freshly prepared 0.5% H2O2 in methanol for 15 min. Non-specific binding sites were blocked by incubating in PBS containing 1.5% NGS, 0.5% BSA and 0.1% Triton X-100. The tissue sections/cultures were then incubated overnight in primary antibodies raised against PGC-1α, EM-48 (Santacruz, USA), Neurofilament (Santacruz) and GFAP (Sigma). Sections were then incubated in anti-mouse secondary antibody conjugated to peroxidase (1:150) for 2 h at room temperature followed by three washes with PBS. Color was developed for peroxidase-linked antibody with 3-3′ diaminobenzidine (DAB) as chromogen. Sections were transferred onto slides, dehydrated, cleared, mounted in DPX, cover slipped and then visualized under microscope.
Quantitative analysis of huntingtin aggregates
For each animal, five serial coronal EM48-immunostained sections through the striatum (300 µm apart) from the level of interaural 4.9/bregma 1.1 to interaural 3.7/bregma −0.1 were analyzed. Unbiased stereological counts of striatal neurons containing EM48-immunoreactive intranuclear aggregates were obtained using the Stereo Investigator software (Microbrightfield, Colchester, VT, USA). The optical fractionator probe was used to quantify the number of neurons with EM48-immunoreactive aggregates in a defined volume of the striatum. The size of the x–y sampling grid was 600 µm × 600 µm, and the counting frame thickness was 14 µm with 3 µm guard zones. Twenty to 24 fields (three to five cells per field) per section were counted. The software program was used to estimate the total number of huntingtin aggregates in the striatum across all the sections analyzed.
Cold challenge
NLS-N171-82Q mice (n = 6 total per group) from different age groups (60, 90, 120, 180, 210 and 240 days) and age-matched WT were subjected to an energetic stress in the form of a cold challenge at 4°C for 3 h and rectal temperature recorded hourly using a rectal probe (Physitemp instruments).
Statistical analysis
Statistical analysis was performed using GraphPad InStat statistical analysis software version 3.05 for Windows (San Diego, CA, USA). The mean significant difference in the experimental groups was determined using one-way analysis of variance (ANOVA) followed by the Tukey–Kramer post hoc multiple comparisons test. Values of P < 0.05 were considered to be statistically significant.
FUNDING
This work was supported by the NS 39258 and Huntington's Disease Society of America Coalition for the Cure.
ACKNOWLEDGEMENTS
Dr David Borchett, Chris Ross and Gabrielle Schilling are thanked for providing us with NLS-N171-82Q transgenic HD mice. We are also thankful to Laragen Inc., Los Angels, CA for mice PCR genotyping. The secretarial assistance of Greta Strong is gratefully acknowledged.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Pratley R.E., Salbe A.D., Ravussin E., Caviness J.N. Higher sedentary energy expenditure in patients with Huntington's disease. Ann. Neurol. 2000;47:64–70. doi:10.1002/1531-8249(200001)47:1<64::AID-ANA11>3.0.CO;2-S. [PubMed] [Google Scholar]
- 2.Goodman A.O., Murgatroyd P.R., Medina-Gomez G., Wood N.I., Finer N., Vidal-Puig A.J., Morton A.J., Barker R.A. The metabolic profile of early Huntington's disease—a combined human and transgenic mouse study. Exp. Neurol. 2008;210:691–698. doi: 10.1016/j.expneurol.2007.12.026. doi:10.1016/j.expneurol.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 3.Djousse L., Knowlton B., Cupples L.A., Marder K., Shoulson I., Myers R.H. Weight loss in early stage of Huntington's disease. Neurology. 2002;59:1325–1330. doi: 10.1212/01.wnl.0000031791.10922.cf. [DOI] [PubMed] [Google Scholar]
- 4.Sathasivam K., Hobbs C., Turmaine M., Mangiarini L., Mahal A., Bertaux F., Wanker E.E., Doherty P., Davies S.W., Bates G.P. Formation of polyglutamine inclusions in non-CNS tissue. Hum. Mol. Genet. 1999;8:813–822. doi: 10.1093/hmg/8.5.813. doi:10.1093/hmg/8.5.813. [DOI] [PubMed] [Google Scholar]
- 5.Ribchester R.R., Thomson D., Wood N.I., Hinks T., Gillingwater T.H., Wishart T.M., Court F.A., Morton A.J. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington's disease mutation. Eur. J. Neurosci. 2004;20:3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x. doi:10.1111/j.1460-9568.2004.03783.x. [DOI] [PubMed] [Google Scholar]
- 6.Grafton S.T., Mazziotta J.C., Pahl J.J., St George-Hyslop P., Haines J.L., Gusella J., Hoffman J.M., Baxter L.R., Phelps M.E. Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington's disease. Arch. Neurol. 1992;49:1161–1167. doi: 10.1001/archneur.1992.00530350075022. [DOI] [PubMed] [Google Scholar]
- 7.Feigin A., Leenders K.L., Moeller J.R., Missimer J., Kuenig G., Spetsieris P., Antonini A., Eidelberg D. Metabolic network abnormalities in early Huntington's disease: an [(18)F]FDG PET study. J. Nucl. Med. 2001;42:1591–1595. [PubMed] [Google Scholar]
- 8.Antonini A., Leenders K.L., Spiegel R., Meier D., Vontobel P., Weigell-Weber M., Sanchez-Pernaute R., de Yebenez J.G., Boesiger P., Weindl A., et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington's disease. Brain. 1996;119:2085–2095. doi: 10.1093/brain/119.6.2085. [DOI] [PubMed] [Google Scholar]
- 9.Kuwert T., Lange H.W., Boecker H., Titz H., Herzog H., Aulich A., Wang B.C., Nayak U., Feinendegen L.E. Striatal glucose consumption in chorea-free subjects at risk of Huntington's disease. J. Neurol. 1993;241:31–36. doi: 10.1007/BF00870669. doi:10.1007/BF00870669. [DOI] [PubMed] [Google Scholar]
- 10.Jenkins B.G., Koroshetz W.J., Beal M.F., Rosen B.R. Evidence for impairment of energy metabolism in vivo in Huntington's disease using localized 1H NMR spectroscopy. Neurology. 1993;43:2689–2695. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- 11.Koroshetz W.J., Jenkins B.G., Rosen B.R., Beal M.F. Energy metabolism defects in Huntington's disease and effects of coenzyme Q10. Ann. Neurol. 1997;41:160–165. doi: 10.1002/ana.410410206. doi:10.1002/ana.410410206. [DOI] [PubMed] [Google Scholar]
- 12.Lodi R., Schapira A.H., Manners D., Styles P., Wood N.W., Taylor D.J., Warner T.T. Abnormal in vivo skeletal muscle energy metabolism in Huntington's disease and dentatorubropallidoluysian atrophy. Ann. Neurol. 2000;48:72–76. doi:10.1002/1531-8249(200007)48:1<72::AID-ANA11>3.0.CO;2-I. [PubMed] [Google Scholar]
- 13.Saft C., Zange J., Andrich J., Muller K., Lindenberg K., Landwehrmeyer B., Vorgerd M., Kraus P.H., Przuntek H., Schols L. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington's disease. Mov. Disord. 2005;20:674–679. doi: 10.1002/mds.20373. doi:10.1002/mds.20373. [DOI] [PubMed] [Google Scholar]
- 14.Browne S.E., Bowling A.C., MacGarvey U., Baik M.J., Berger S.C., Muqit M.M., Bird E.D., Beal M.F. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann. Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. doi:10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 15.Gu M., Gash M.T., Mann V.M., Javoy-Agid F., Cooper J.M., Schapira A.H. Mitochondrial defect in Huntington's disease caudate nucleus. Ann. Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. doi:10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 16.Tabrizi S.J., Cleeter M.W., Xuereb J., Taanman J.W., Cooper J.M., Schapira A.H. Biochemical abnormalities and excitotoxicity in Huntington's disease brain. Ann. Neurol. 1999;45:25–32. doi: 10.1002/1531-8249(199901)45:1<25::aid-art6>3.0.co;2-e. doi:10.1002/1531-8249(199901)45:1<25::AID-ART6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 17.Seong I.S., Ivanova E., Lee J.M., Choo Y.S., Fossale E., Anderson M., Gusella J.F., Laramie J.M., Myers R.H., Lesort M., et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 2005;14:2871–2880. doi: 10.1093/hmg/ddi319. doi:10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- 18.Milakovic T., Johnson G.V. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J. Biol. Chem. 2005;280:30773–30782. doi: 10.1074/jbc.M504749200. doi:10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- 19.Beal M.F., Brouillet E., Jenkins B., Henshaw R., Rosen B., Hyman B.T. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J. Neurochem. 1993;61:1147–1150. doi: 10.1111/j.1471-4159.1993.tb03633.x. doi:10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 20.Brouillet E., Hantraye P., Ferrante R.J., Dolan R., Leroy-Willig A., Kowall N.W., Beal M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl Acad. Sci. USA. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. doi:10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ludolph A.C., He F., Spencer P.S., Hammerstad J., Sabri M. 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can J. Neurol. Sci. 1991;18:492–498. doi: 10.1017/s0317167100032212. [DOI] [PubMed] [Google Scholar]
- 22.Brouillet E., Jenkins B.G., Hyman B.T., Ferrante R.J., Kowall N.W., Srivastava R., Roy D.S., Rosen B.R., Beal M.F. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J. Neurochem. 1993;60:356–359. doi: 10.1111/j.1471-4159.1993.tb05859.x. doi:10.1111/j.1471-4159.1993.tb05859.x. [DOI] [PubMed] [Google Scholar]
- 23.Orr A.L., Li S., Wang C.E., Li H., Wang J., Rong J., Xu X., Mastroberardino P.G., Greenamyre J.T., Li X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. doi:10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reddy P.H., Mao P., Manczak M. Mitochondrial structural and functional dynamics in Huntington's disease. Brain. Res. Rev. 2009;61:33–48. doi: 10.1016/j.brainresrev.2009.04.001. doi:10.1016/j.brainresrev.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choo Y.S., Johnson G.V., MacDonald M., Detloff P.J., Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. doi:10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 26.Panov A.V., Gutekunst C.A., Leavitt B.R., Hayden M.R., Burke J.R., Strittmatter W.J., Greenamyre J.T. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat. Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 27.Rockabrand E., Slepko N., Pantalone A., Nukala V.N., Kazantsev A., Marsh J.L., Sullivan P.G., Steffan J.S., Sensi S.L., Thompson L.M. The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 2007;16:61–77. doi: 10.1093/hmg/ddl440. doi:10.1093/hmg/ddl440. [DOI] [PubMed] [Google Scholar]
- 28.Cha J.H. Transcriptional signatures in Huntington's disease. Prog. Neurobiol. 2007;83:228–248. doi: 10.1016/j.pneurobio.2007.03.004. doi:10.1016/j.pneurobio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugars K.L., Rubinsztein D.C. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. doi:10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- 30.Dunah A.W., Jeong H., Griffin A., Kim Y.M., Standaert D.G., Hersch S.M., Mouradian M.M., Young A.B., Tanese N., Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. doi:10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 31.Nucifora F.C., Jr, Sasaki M., Peters M.F., Huang H., Cooper J.K., Yamada M., Takahashi H., Tsuji S., Troncoso J., Dawson V.L., et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 32.Steffan J.S., Kazantsev A., Spasic-Boskovic O., Greenwald M., Zhu Y.Z., Gohler H., Wanker E.E., Bates G.P., Housman D.E., Thompson L.M. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl Acad. Sci. USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. doi:10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leone T.C., Lehman J.J., Finck B.N., Schaeffer P.J., Wende A.R., Boudina S., Courtois M., Wozniak D.F., Sambandam N., Bernal-Mizrachi C., et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS. Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. doi:10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin J., Wu P.H., Tarr P.T., Lindenberg K.S., St-Pierre J., Zhang C.Y., Mootha V.K., Jager S., Vianna C.R., Reznick R.M., et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. doi:10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 35.Puigserver P., Spiegelman B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. doi:10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 36.Puigserver P., Wu Z., Park C.W., Graves R., Wright M., Spiegelman B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. doi:10.1016/S0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 37.Lin J., Handschin C., Spiegelman B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell. Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. doi:10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Kelly D.P., Scarpulla R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. doi:10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 39.Wareski P., Vaarmann A., Choubey V., Safiulina D., Liiv J., Kuum M., Kaasik A. PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J. Biol. Chem. 2009;284:21379–21385. doi: 10.1074/jbc.M109.018911. doi:10.1074/jbc.M109.018911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lagouge M., Argmann C., Gerhart-Hines Z., Meziane H., Lerin C., Daussin F., Messadeq N., Milne J., Lambert P., Elliott P., et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. doi:10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 41.Baur J.A., Pearson K.J., Price N.L., Jamieson H.A., Lerin C., Kalra A., Prabhu V.V., Allard J.S., Lopez-Lluch G., Lewis K., et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. doi:10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pearson K.J., Baur J.A., Lewis K.N., Peshkin L., Price N.L., Labinskyy N., Swindell W.R., Kamara D., Minor R.K., Perez E., et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. doi: 10.1016/j.cmet.2008.06.011. doi:10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cui L., Jeong H., Borovecki F., Parkhurst C.N., Tanese N., Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. doi:10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 44.Weydt P., Pineda V.V., Torrence A.E., Libby R.T., Satterfield T.F., Lazarowski E.R., Gilbert M.L., Morton G.J., Bammler T.K., Strand A.D., et al. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. doi:10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 45.Chaturvedi R.K., Adhihetty P., Shukla S., Hennessy T., Calingasan N., Yang L., Starkov A., Kiaei M., Cannella M., Sassone J., et al. Impaired PGC-1alpha function in muscle in Huntington's disease. Hum. Mol. Genet. 2009;18:3048–3065. doi: 10.1093/hmg/ddp243. doi:10.1093/hmg/ddp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phan J., Hickey M.A., Zhang P., Chesselet M.F., Reue K. Adipose tissue dysfunction tracks disease progression in two Huntington's disease mouse models. Hum. Mol. Genet. 2009;18:1006–1016. doi: 10.1093/hmg/ddn428. doi:10.1093/hmg/ddn428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okamoto S., Pouladi M.A., Talantova M., Yao D., Xia P., Ehrnhoefer D.E., Zaidi R., Clemente A., Kaul M., Graham R.K., et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. doi:10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee S.T., Chu K., Jung K.H., Im W.S., Park J.E., Lim H.C., Won C.H., Shin S.H., Lee S.K., Kim M., et al. Slowed progression in models of Huntington disease by adipose stem cell transplantation. Ann. Neurol. 2009;66:671–681. doi: 10.1002/ana.21788. doi:10.1002/ana.21788. [DOI] [PubMed] [Google Scholar]
- 49.Zong H., Ren J.M., Young L.H., Pypaert M., Mu J., Birnbaum M.J., Shulman G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl Acad. Sci. USA. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. doi:10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reznick R.M., Zong H., Li J., Morino K., Moore I.K., Yu H.J., Liu Z.X., Dong J., Mustard K.J., Hawley S.A., et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. doi:10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams D.B., Sutherland L.N., Bomhof M.R., Basaraba S.A., Thrush A.B., Dyck D.J., Field C.J., Wright D.C. Muscle-specific differences in the response of mitochondrial proteins to beta-GPA feeding: an evaluation of potential mechanisms. Am. J. Physiol. Endocrinol. Metab. 2009;296:E1400–E1408. doi: 10.1152/ajpendo.90913.2008. doi:10.1152/ajpendo.90913.2008. [DOI] [PubMed] [Google Scholar]
- 52.Jager S., Handschin C., St-Pierre J., Spiegelman B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl Acad. Sci. USA. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. doi:10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schilling G., Savonenko A.V., Klevytska A., Morton J.L., Tucker S.M., Poirier M., Gale A., Chan N., Gonzales V., Slunt H.H., et al. Nuclear-targeting of mutant huntingtin fragments produces Huntington's disease-like phenotypes in transgenic mice. Hum. Mol. Genet. 2004;13:1599–1610. doi: 10.1093/hmg/ddh175. doi:10.1093/hmg/ddh175. [DOI] [PubMed] [Google Scholar]
- 54.Fitch C.D., Jellinek M., Mueller E.J. Experimental depletion of creatine and phosphocreatine from skeletal muscle. J. Biol. Chem. 1974;249:1060–1063. [PubMed] [Google Scholar]
- 55.Shoubridge E.A., Challiss R.A., Hayes D.J., Radda G.K. Biochemical adaptation in the skeletal muscle of rats depleted of creatine with the substrate analogue beta-guanidinopropionic acid. Biochem. J. 1985;232:125–131. doi: 10.1042/bj2320125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luthi-Carter R., Hanson S.A., Strand A.D., Bergstrom D.A., Chun W., Peters N.L., Woods A.M., Chan E.Y., Kooperberg C., Krainc D., et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum. Mol. Genet. 2002;11:1911–1926. doi: 10.1093/hmg/11.17.1911. doi:10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- 57.D'Aurelio M., Vives-Bauza C., Davidson M.M., Manfredi G. Mitochondrial DNA background modifies the bioenergetics of NARP/MILS ATP6 mutant cells. Hum. Mol. Genet. 2010;19:374–386. doi: 10.1093/hmg/ddp503. doi:10.1093/hmg/ddp503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.St-Pierre J., Lin J., Krauss S., Tarr P.T., Yang R., Newgard C.B., Spiegelman B.M. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J. Biol. Chem. 2003;278:26597–26603. doi: 10.1074/jbc.M301850200. doi:10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- 59.Browne S.E., Beal M.F. Oxidative damage in Huntington's disease pathogenesis. Antioxid. Redox Signal. 2006;8:2061–2073. doi: 10.1089/ars.2006.8.2061. [DOI] [PubMed] [Google Scholar]
- 60.Hersch S.M., Gevorkian S., Marder K., Moskowitz C., Feigin A., Cox M., Como P., Zimmerman C., Lin M., Zhang L., et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2'dG. Neurology. 2006;66:250–252. doi: 10.1212/01.wnl.0000194318.74946.b6. doi:10.1212/01.wnl.0000194318.74946.b6. [DOI] [PubMed] [Google Scholar]
- 61.Kosinski C.M., Schlangen C., Gellerich F.N., Gizatullina Z., Deschauer M., Schiefer J., Young A.B., Landwehrmeyer G.B., Toyka K.V., Sellhaus B., et al. Myopathy as a first symptom of Huntington's disease in a Marathon runner. Mov. Disord. 2007;22:1637–1640. doi: 10.1002/mds.21550. doi:10.1002/mds.21550. [DOI] [PubMed] [Google Scholar]
- 62.Turner C., Cooper J.M., Schapira A.H. Clinical correlates of mitochondrial function in Huntington's disease muscle. Mov. Disord. 2007;22:1715–1721. doi: 10.1002/mds.21540. doi:10.1002/mds.21540. [DOI] [PubMed] [Google Scholar]
- 63.Gizatullina Z.Z., Lindenberg K.S., Harjes P., Chen Y., Kosinski C.M., Landwehrmeyer B.G., Ludolph A.C., Striggow F., Zierz S., Gellerich F.N. Low stability of Huntington muscle mitochondria against Ca2+ in R6/2 mice. Ann. Neurol. 2006;59:407–411. doi: 10.1002/ana.20754. doi:10.1002/ana.20754. [DOI] [PubMed] [Google Scholar]
- 64.Mihm M.J., Amann D.M., Schanbacher B.L., Altschuld R.A., Bauer J.A., Hoyt K.R. Cardiac dysfunction in the R6/2 mouse model of Huntington's disease. Neurobiol. Dis. 2007;25:297–308. doi: 10.1016/j.nbd.2006.09.016. doi:10.1016/j.nbd.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin J., Wu H., Tarr P.T., Zhang C.Y., Wu Z., Boss O., Michael L.F., Puigserver P., Isotani E., Olson E.N., et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. doi:10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 66.Benn C.L., Sun T., Sadri-Vakili G., McFarland K.N., DiRocco D.P., Yohrling G.J., Clark T.W., Bouzou B., Cha J.H. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 2008;28:10720–10733. doi: 10.1523/JNEUROSCI.2126-08.2008. doi:10.1523/JNEUROSCI.2126-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giampa C., Middei S., Patassini S., Borreca A., Marullo F., Laurenti D., Bernardi G., Ammassari-Teule M., Fusco F.R. Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington's disease. Eur. J. Neurosci. 2009;29:902–910. doi: 10.1111/j.1460-9568.2009.06649.x. doi:10.1111/j.1460-9568.2009.06649.x. [DOI] [PubMed] [Google Scholar]
- 68.DeMarch Z., Giampa C., Patassini S., Bernardi G., Fusco F.R. Beneficial effects of rolipram in the R6/2 mouse model of Huntington's disease. Neurobiol. Dis. 2008;30:375–387. doi: 10.1016/j.nbd.2008.02.010. doi:10.1016/j.nbd.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 69.Jiang H., Nucifora F.C., Jr, Ross C.A., DeFranco D.B. Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum. Mol. Genet. 2003;12:1–12. doi: 10.1093/hmg/ddg002. doi:10.1093/hmg/ddg002. [DOI] [PubMed] [Google Scholar]
- 70.Jiang H., Poirier M.A., Liang Y., Pei Z., Weiskittel C.E., Smith W.W., DeFranco D.B., Ross C.A. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 2006;23:543–551. doi: 10.1016/j.nbd.2006.04.011. doi:10.1016/j.nbd.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 71.St-Pierre J., Drori S., Uldry M., Silvaggi J.M., Rhee J., Jager S., Handschin C., Zheng K., Lin J., Yang W., et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. doi:10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 72.Herzig S., Long F., Jhala U.S., Hedrick S., Quinn R., Bauer A., Rudolph D., Schutz G., Yoon C., Puigserver P., et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. doi:10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 73.Hinerfeld D., Traini M.D., Weinberger R.P., Cochran B., Doctrow S.R., Harry J., Melov S. Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. J. Neurochem. 2004;88:657–667. doi: 10.1046/j.1471-4159.2003.02195.x. doi:10.1046/j.1471-4159.2003.02195.x. [DOI] [PubMed] [Google Scholar]
- 74.Estall J.L., Kahn M., Cooper M.P., Fisher F.M., Wu M.K., Laznik D., Qu L., Cohen D.E., Shulman G.I., Spiegelman B.M. Sensitivity of lipid metabolism and insulin signaling to genetic alterations in hepatic peroxisome proliferator-activated receptor-gamma coactivator-1alpha expression. Diabetes. 2009;58:1499–1508. doi: 10.2337/db08-1571. doi:10.2337/db08-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weydt P., Soyal S.M., Gellera C., Didonato S., Weidinger C., Oberkofler H., Landwehrmeyer G.B., Patsch W. The gene coding for PGC-1alpha modifies age at onset in Huntington's Disease. Mol. Neurodegener. 2009;4:3. doi: 10.1186/1750-1326-4-3. doi:10.1186/1750-1326-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taherzadeh-Fard E., Saft C., Andrich J., Wieczorek S., Arning L. PGC-1alpha as modifier of onset age in Huntington disease. Mol. Neurodegener. 2009;4:10. doi: 10.1186/1750-1326-4-10. doi:10.1186/1750-1326-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kelly T.J., Lerin C., Haas W., Gygi S.P., Puigserver P. GCN5-mediated transcriptional control of the metabolic coactivator PGC-1beta through lysine acetylation. J. Biol. Chem. 2009;284:19945–19952. doi: 10.1074/jbc.M109.015164. doi:10.1074/jbc.M109.015164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Canto C., Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. doi:10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dominy J.E., Jr, Lee Y., Gerhart-Hines Z., Puigserver P. Nutrient-dependent regulation of PGC-1alpha's acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim. Biophys. Acta. 2009 doi: 10.1016/j.bbapap.2009.11.023. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quintanilla R.A., Jin Y.N., Fuenzalida K., Bronfman M., Johnson G.V. Rosiglitazone treatment prevents mitochondrial dysfunction in mutant huntingtin-expressing cells: possible role of peroxisome proliferator-activated receptor-gamma (PPARgamma) in the pathogenesis of Huntington disease. J. Biol. Chem. 2008;283:25628–25637. doi: 10.1074/jbc.M804291200. doi:10.1074/jbc.M804291200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wenz T., Diaz F., Spiegelman B.M., Moraes C.T. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–256. doi: 10.1016/j.cmet.2008.07.006. doi:10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 82.Ma T.C., Buescher J.L., Oatis B., Funk J.A., Nash A.J., Carrier R.L., Hoyt K.R. Metformin therapy in a transgenic mouse model of Huntington's disease. Neurosci. Lett. 2007;411:98–103. doi: 10.1016/j.neulet.2006.10.039. doi:10.1016/j.neulet.2006.10.039. [DOI] [PubMed] [Google Scholar]
- 83.Miura S., Tomitsuka E., Kamei Y., Yamazaki T., Kai Y., Tamura M., Kita K., Nishino I., Ezaki O. Overexpression of peroxisome proliferator-activated receptor gamma co-activator-1alpha leads to muscle atrophy with depletion of ATP. Am. J. Pathol. 2006;169:1129–1139. doi: 10.2353/ajpath.2006.060034. doi:10.2353/ajpath.2006.060034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lehman J.J., Barger P.M., Kovacs A., Saffitz J.E., Medeiros D.M., Kelly D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 2000;106:847–856. doi: 10.1172/JCI10268. doi:10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liang H., Balas B., Tantiwong P., Dube J., Goodpaster B.H., O'Doherty R.M., DeFronzo R.A., Richardson A., Musi N., Ward W.F. Whole body overexpression of PGC-1alpha has opposite effects on hepatic and muscle insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2009;296:E945–E954. doi: 10.1152/ajpendo.90292.2008. doi:10.1152/ajpendo.90292.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]