

Abstract
miRNAs have been shown to be essential for normal cartilage development in the mouse. However, the role of specific miRNAs in cartilage function is unknown. Using rarely available healthy human chondrocytes (obtained from 8 to 50 year old patients), we detected a most highly abundant primary miRNA H19, whose expression was heavily dependent on cartilage master regulator SOX9. Across a range of murine tissues, expression of both H19- and H19-derived miR-675 mirrored that of cartilage-specific SOX9. miR-675 was shown to up-regulate the essential cartilage matrix component COL2A1, and overexpression of miR-675 rescued COL2A1 levels in H19- or SOX9-depleted cells. We thus provide evidence that SOX9 positively regulates COL2A1 in human articular chondrocytes via a previously unreported miR-675-dependent mechanism. This represents a novel pathway regulating cartilage matrix production and identifies miR-675 as a promising new target for cartilage repair.
Keywords: Collagen, Connective Tissue, Differentiation, Hypoxia, MicroRNA, SOX9, Tissue-specific MicroRNA, Type II Collagen
Introduction
MicroRNAs (miRNAs)3 are a recently discovered class of endogenous regulators of gene expression and, together with transcription factors, are the most numerous regulators defining cell-specific gene expression (1). Specifically, miRNAs are small non-protein coding RNA species that bind to complementary sequences in the 3′-UTR of target mRNA and inhibit their translation (by blocking translation or inducing transcript decay/degradation) (2). Thus, miRNAs affect cellular phenotype in a direct manner. Deletion studies in mice have shown that individual miRNAs can strongly influence specific mammalian differentiation processes, e.g. B cell differentiation (3), cardiomyocyte hypertrophy (4). In vitro studies using human preadipocytes showed that a single miRNA was critical for adipogenesis (5). However, at present, almost nothing is known about the function of individual miRNAs in cartilage or chondrocytes, with only one published study reporting detection of a tissue-specific microRNA (miR-140) in murine embryonic cartilage (6). However, the importance of miRNAs in normal skeletal development has been demonstrated recently by Kobayashi and colleagues (7), who generated cartilage-specific Dicer-null mice. (Dicer is a critical enzyme in the miRNA biogenesis pathway.) Dicer-null growth plates showed both greatly decreased chondrocyte proliferation and accelerated terminal differentiation (hypertrophy), leading to severe growth defects. Hence, miRNAs are essential for normal chondrocyte differentiation in the murine growth plate.
The chondrocyte is the sole cell type in cartilage, and it alone produces and maintains the tough extracellular matrix that gives the tissue its load-bearing function. The chondrocyte phenotype is unstable in vitro (8) and also is altered in disease states (9). Uncovering mechanisms that promote the differentiated phenotype has implications for cartilage repair and restoration of tissue function as it enables identification of potential therapeutic intervention points.
In the current study, from genome-wide profiling experiments using healthy human chondrocytes, we unexpectedly detected a most highly abundant noncoding RNA transcript, H19, whose expression correlates highly with the differentiated phenotype. Recent work has shown that H19 is a primary miRNA transcript, which is processed to the mature form, which is miR-675 (10). The role of miR-675 itself is completely unexplored; however, the pre-miR-675 region of H19 is highly conserved in therian mammals implying this miRNA plays a key role in H19 function (11). All miRNAs are processed initially from much longer transcripts known as primary miRNA to short stem-loop structures called pre-miRNA and finally to mature, functional miRNA, which are only 21–23 nucleotides in length (12). Although little is known about their regulation, it is believed that miRNA levels are dependent heavily on transcription (i.e. of the primary transcript) (12). Here, we report that expression of primary miRNA H19, like the main cartilage matrix genes, is dependent on master chondrocyte regulator transcription factor SOX9. In addition, inhibition of H19 and/or miR-675 significantly down-regulates COL2A1, the most abundant and functionally important cartilage matrix protein; whereas overexpression increases COL2A1 levels. Finally, overexpression of miR-675 rescues COL2A1 levels in H19- or SOX9-depleted cells, suggesting that regulation of COL2A1 by SOX9/H19 is mediated specifically by the mature microRNA. Hence, we propose that miR-675 lies at the core of a new mechanism promoting the differentiated chondrocyte phenotype and matrix production and thus represents a potential new target for cartilage repair.
EXPERIMENTAL PROCEDURES
Cell Culture
Healthy articular cartilage was obtained from patients after informed consent and following local ethics committee guidelines. Cartilage was harvested from the femoral condyle and tibial plateau following amputation due to sarcomas not involving the joint. Tissue was obtained from nine donors (six male and three female; age range of 8–50 years, average age of 30 years). Cartilage specimens were collected on the day of surgery and cut into small pieces (1–2 mm3). Diced cartilage was placed in 1.5 mg/ml collagenase type 2 (Worthington®) with Dulbecco's modified Eagle's medium containing 10% fetal calf serum (Biosera) and incubated at 37 °C for 18 h with shaking. Isolated human articular chondrocytes (HACs)3 were then passed through a cell strainer, pelleted, and washed twice with medium. Cells were seeded at a density of 8 × 103 cells/cm2 in Dulbecco's modified Eagle's medium with 10% fetal calf serum. Cultures were passaged at confluency (∼7 days) and subsequently seeded at 5 × 103 cells/cm2. Both primary (unpassaged) and passaged cells (up to passage 2) were used in experiments.
Transient Transfections
HACs were transfected as described previously (13). Briefly, HACs were seeded at 15 × 104 cells/well in 6-cm dishes. The following day, transfection with siRNA was carried out at a final concentration of 10 nm using Lipofectamine 2000 (Invitrogen) for 4 h in serum-free OptiMEM I. Gene-specific siRNAs against H19 and SOX9 were used (Eurofins MWG Operon, Ebersberg, Germany). As a negative control, siRNA against luciferase (Dharmacon) was transfected in parallel. Following transfection, medium was changed with pre-equilibrated Dulbecco's modified Eagle's medium (in 20% or 1% O2) containing 10% fetal calf serum, and HACs were incubated in each oxygen environment in a Galaxy® triple gas incubator for 3 days. The same procedure was used to transfect pre-miR-675 (Ambion, Inc./Applied Biosystems, Warrington, UK), antisense oligonucleotides (anti-miR-675) (Exiqon, Vedbaek, Denmark) or a negative control anti-miR microRNA inhibitor (catalog no. 199002-00, Exiqon).
RNA Extraction, Reverse Transcription, and Real-time PCR
Total RNA, including small RNA, was extracted and prepared using the Qiagen miRNeasy kit for both HACs and mouse tissue. RNA from mouse tissue was pooled from 10 mice (Balb/B, male, 5–6-weeks-old), and the procedure was repeated for a further 10 mice.
COL2A1, SOX9, or H19 and the housekeeping gene RPLP0 were analyzed using TaqMan technology. cDNA was generated using the Promega kit and random hexamers from 0.5 μg of total RNA. For these genes, predeveloped primer/probe sets were used (Applied Biosystems).
miR-675 PCR amplifications were performed using SYBR Green. 0.1 μg of total RNA (including small RNA) were first polyadenylated and then reverse-transcribed following the manufacturer's instructions (NCode miRNA kit, Invitrogen). miR-675 also was measured using miRNA-specific TaqMan-based stem loop primers. In both cases, the ΔΔCt method of relative quantitation was used to calculate relative transcript levels.
Protein Extraction and Western Blotting
For type II collagen-secreted protein analysis, HACs were serum-starved for 2 days, 24 h after transfection. Approximately 1 ml of conditioned medium was precipitated with trichloroacetic acid, loaded on a 6% PAGE gel and blotted with anti COL2A1 antibody (Chemicon MAB8887, 1:1000). The exact volume of medium used was adjusted after standardization according to total protein content (measured using the Bradford assay) from the cell layer.
Statistical Analysis
Data were compared using one-way analysis of variance with Bonferroni's post hoc test using Prism 4 software (GraphPad). Results are presented as mean ± S.E. p < 0.05 was considered statistically significant. n values refer to data obtained from individual donor chondrocyte cultures.
RESULTS
Noncoding RNA H19 Is Highly Expressed in Human Articular Chondrocytes, Correlates with the Differentiated Phenotype, Is SOX9-dependent, and Regulates COL2A1
H19 gene expression levels were measured in primary HACs and were found to be as great as the most abundant cartilage matrix genes, COL2A1 and aggrecan (Fig. 1A, AGC). H19 levels correlated with expression of the differentiated HAC phenotype, i.e. levels were high in primary cells, significantly decreased with passage of the cells, which leads to their dedifferentiation, and subsequently increased with hypoxia-induced redifferentiation of the chondrocytes (p < 0.01) (Fig. 1B) (14).
FIGURE 1.

Noncoding RNA H19 is highly expressed in normal HACs and correlates with the differentiated phenotype. A, H19 gene expression levels were comparable to the most abundant cartilage matrix genes aggrecan (AGC) and COL2A1 as measured in primary HACs. (n = 7, i.e. cultures from seven different patients). B, H19 expression levels in HACs significantly decreased with passage and subsequently increased upon redifferentiation in hypoxia (**, p < 0.01; n = 7). C, H19 clusters directly with SOX9 in HACs. Unbiased, hierarchical clustering analysis of genome-wide array data were performed on an entire data set of 35 arrays from HACs (n = 7, i.e. separate cultures derived from seven individual donors) undergoing dedifferentiation and redifferentiation in culture. The scale refers to Z-scores as it reduces the multiplicative error effect of the expression intensity measurements. Gene expression is relative to that of ribosomal protein RPLP0.
Unbiased, hierarchical clustering analysis of genome-wide microarray data obtained previously (15) revealed H19 clusters with the master chondrocyte regulator, transcription factor SOX9 (Fig. 1C). Such an extremely tight clustering of both genes through the dedifferentiation/redifferentiation process suggested their possible be co-regulation. We therefore investigated whether H19 is regulated by SOX9 by depletion of the latter using RNA interference. As shown in Fig. 2A, depletion of SOX9 in HACs greatly decreased H19 levels (p < 0.01). Depletion of H19 had no effect on SOX9 levels (data not shown).
FIGURE 2.

H19 is SOX9-dependent and positively regulates COL2A1 in HACs. A, H19 levels are significantly reduced by RNA interference-mediated depletion of SOX9 in HACs. (TaqMan PCR analysis, **, p < 0.01). B, depletion of H19 in HACs significantly reduced COL2A1 mRNA levels (**, p < 0.01). C, depletion of H19 greatly reduced levels of COL2A1 detectable in the medium of cultured HACs. All experiments were performed in 1% oxygen over a period of 3 days. Gene expression is relative to that of ribosomal protein RPLP0; Ctrl, negative control siRNA-targeting luciferase; si, small interfering.
Having established its SOX9 dependence, we next investigated the effect of H19 on the chondrocyte phenotype. H19 depletion in HACs significantly reduced COL2A1 mRNA levels (p < 0.01) (Fig. 2B). In addition, levels of COL2A1 secreted into the culture medium were reduced greatly in H19-depleted cells (Fig. 2C). Two different siRNA oligonucleotides targeting different sequences in the H19 transcript were used, and both showed a similar effect on COL2A1, suggesting the results are not due to any possible off-target effects of the siRNAs (Fig. 2C).
H19 and H19-derived miR-675, Like SOX9, Are Differentially Expressed in Murine Articular Cartilage
The noncoding RNA H19 is a primary miRNA giving rise to a 23-nucleotide mature miRNA, miR-675, which, before final Dicer cleavage, forms one arm of a hairpin structure (Fig. 3A). The so-called “passenger” strand, designated miR-675*, is three nucleotides shorter than miR-675 and contains four mismatches.
FIGURE 3.

H19 and H19-derived miR-675 are, like SOX9, differentially expressed in articular cartilage. A, miR-675 containing hairpin structure of human H19. B, real-time PCR data of miR-675, H19 and Sox9 levels in mouse spleen, lungs, kidney, heart, liver, and articular cartilage. Data represents two pooled RNA samples, each obtained from ten BalbB mice. Note: Expression levels were arbitrarily set to a value of 1 in cartilage. C, during serial passage of primary HACs (which causes dedifferentiation) expression of miR-675 and H19 followed that of SOX9 and COL2A1, which all showed decreased expression. (Results are the average from three independent experiments.) P, passage number. Gene expression is relative to that of ribosomal protein RPLP0.
To investigate the tissue distribution of miRNA-675, its expression was assessed across a range of murine tissues (male BalbB, aged 5–6 weeks). Its primary transcript H19 and cartilage-specific Sox9 also were measured. As can be seen in Fig. 3B, miR-675 and H19 are highly expressed in articular cartilage compared with all other tissue investigated. Their expression pattern also closely follows Sox9, which complements our data from human chondrocytes showing a SOX9 dependence on H19 expression (Fig. 2, B and C). In addition, expression of miR-675 and H19 in HACs mirrored that of SOX9 and COL2A1 during dedifferentiation of the cells, all showing decreased expression upon serial passage (Fig. 3C).
miR-675 Positively Regulates COL2A1 Expression in Human Articular Chondrocytes
The next step was to assess whether H19-derived miR-675 itself was regulating COL2A1. To investigate this, the mature 23-nucleotide miRNA was inhibited (with antisense oligonucleotides) or overexpressed in transient transfection experiments. Transfection efficiency in HACs was high, with no negative effect on cell viability as shown by the use of fluorescently labeled anti-miR-675 sequences (Fig. 4A). Like H19 depletion, miR-675 inhibition significantly reduced COL2A1 gene expression (p < 0.01) (Fig. 4B) and secretion of COL2A1 protein into the culture medium (Fig. 4C). These effects were detected in both primary and passaged HACs. To minimize the number of cultures needed, the negative control used in these experiments was an siRNA-targeting luciferase because preliminary experiments showed similar results to the negative control anti-miR microRNA inhibitor (Exiqon).
FIGURE 4.

Inhibition of miR-675 decreases COL2A1 mRNA and secreted protein levels in HACs. A, HACs were successfully transfected with fluorescently labeled anti-miR-675 at 40 nm. B, Lipofectamine-mediated transient transfection of anti-miR-675 (40 nm) significantly reduced COL2A1 mRNA levels in HACs (**, p < 0.01), n = 7, i.e. data were obtained from cultures derived from seven different donors and expressed relative to ribosomal protein RPLP0. C, levels of COL2A1 secreted into the culture medium were similarly reduced upon inhibition of miR-675. p, passage number. In all experiments, HACs were incubated in 1% oxygen for 3 days before analysis. Ctrl, negative control siRNA-targeting luciferase; ns, not significant.
Conversely, overexpression of miR-675 significantly increased COL2A1 mRNA and secreted protein in HACs compared with negative control levels (Fig. 5, A and B). SOX9 depletion resulted in greatly reduced levels of secreted COL2A1 levels, whereas H19 depletion similarly reduced COL2A1 levels without altering endogenous SOX9 levels (Fig. 5C). This reduction in COL2A1 levels by either SOX9 or H19 depletion could be rescued completely by concurrent overexpression of miR-675 (Fig. 5D). This suggests that miR-675 is mediating the effect of SOX9 and SOX9-regulated H19 on COL2A1 levels in HACs. Unlike, COL2A1, expression of SOX9-dependent COL9A1 was not affected by depletion of H19 or overexpression of miR-675 (Fig. 5E).
FIGURE 5.

Overexpression of miR-675 up-regulates COL2A1 expression and rescues COL2A1 levels in H19- and SOX9-depleted HACs. Overexpression of miR-675 (10 nm) significantly up-regulates COL2A1 mRNA (A) (*, p < 0.05) and secreted protein levels (B) in HACs (1 nm and 10 nm of miR-675 used). H19 depletion resulted in reduced levels of secreted COL2A1, whereas SOX9 was unaffected (C). In addition, overexpression of miR-675 (10 nm) rescues COL2A1 levels in H19- and SOX9-depleted HACs (D). Expression of COL9A1 was not affected by depletion of H19 or by overexpression of H19-derived miR-675 (E). In all experiments, HACs were incubated in 1% oxygen for 3 days before analysis. p, passage number. Gene expression is relative to that of ribosomal protein RPLP0. Ctrl, negative control siRNA-targeting luciferase; si, small interfering.
DISCUSSION
In the present study, we have identified H19-derived miR-675 as a highly expressed, cartilage-specific, SOX9-dependent positive regulator of COL2A1, the most abundant and functionally important cartilage matrix protein. Thus, this represents a new mechanism regulating cartilage matrix expression and identifies miR-675 as a potential new target for cartilage repair. H19 is a non-protein coding, imprinted, maternally expressed gene whose expression is abundant in embryonic tissue of endodermal and mesodermal origin but is thought to be repressed after birth (16, 17). Germ line deletion in mice is not lethal to embryos, suggesting it does not play a key role in development (18). Sequence analysis of H19 suggests this gene is functional, since it appears the RNA is subject to a stabilizing selection (19). Nevertheless, despite extensive study, its role remains unclear. However, important recent work by Cullen and colleagues (10) has shed new light on H19 and has shown it can function as a primary miRNA transcript, giving rise to the mature miRNA, miR-675. Although the function of miR-675 is unknown, the miRNA-containing region of H19 is highly conserved, and H19 is reported to have been a primary miRNA gene for 148 million years (11), suggesting the mature miRNA may play a fundamental role in H19 function. In comparing a range of tissues, our data show H19 is, in fact, differentially expressed in cartilage in young adult mice. Indeed, we also found H19 was extremely highly expressed in healthy primary human articular chondrocytes (average age of 30 years), surprisingly at comparable levels to that of the most abundant matrix genes COL2A1 and aggrecan core protein. It is possible that H19 has a function in chondrocytes in addition to generation of miR-675. However, the rescue of COL2A1 levels in H19-depleted HACs by overexpression of miR-675 suggests that regulation of COL2A1 by H19 is mediated by the mature microRNA, rather than the full-length H19.
Transcription factor SOX9 is essential for cartilage formation (20). Here, we provide the first evidence of the SOX9 regulation of a primary microRNA transcript, H19. We hypothesize that H19 is targeted directly by SOX9 because analysis of the human H19 promoter has revealed two putative SOX9 binding sites in the proximal promoter. Most interestingly, the two sites are paired, i.e. staggered by two nucleotides, and arranged in opposite orientation to each other; such paired SOX9 binding sites have been shown in SOX9 target genes (21). Future work will investigate SOX9 recruitment to these putative binding sites in the H19 promoter in HACs.
In mouse fetal cartilage, Sox9 is thought to directly regulate Col2a1 through binding to specific enhancer elements in the first intron because mutation of these sites abolishes expression of a COL2A1-driven reporter gene in transgenic mice (20, 22). Binding of SOX9 to similar COL2A1 enhancer elements in vitro also has been demonstrated in human fetal chondrocytes (23). More recently, endogenous binding of Sox9 to Col2a1 was demonstrated in mouse growth plate chondrocytes (24). We have shown previously that COL2A1 expression is highly dependent on SOX9 in normal HACs (13). While not discounting the possibility of direct binding, in the present study, we provide evidence (from juvenile and adult HAC experiments) that SOX9 can activate COL2A1 indirectly via a miR-675 mediated mechanism, because COL2A1 levels can be rescued in SOX9-depleted cells specifically by overexpression of miR-675. Our data in human articular chondrocytes raise the possibility of differences in transcriptional control of cartilage genes between developing cartilage and the permanent articular cartilage. However, it remains to be seen whether miR-675 is specific to articular cartilage or whether it plays a similar role in COL2A1 regulation in growth plate chondrocytes.
Expression profiling studies by ourselves and others have recently identified altered expression of specific microRNAs in cartilage pathologies such as osteoarthritis (OA) (25, 26). However, the microRNA expression profile is likely to change with disease progression. For example, expression of miR-146 was found to be down-regulated in late stage human OA cartilage compared with normal controls (25) but increased in low-grade (presumably early stage) OA cartilage (27). The specific role and phenotypic effects of these differentially expressed miRNAs remain to be elucidated. Due to its relatively recent discovery (10), miR-675 was not analyzed in these profiling studies; however, it would be most interesting to investigate whether its expression is altered in OA. For example, could aberrant expression of miR-675 explain the reported lack of correlation between SOX9 and COL2A1 expression in human OA cartilage (28)?
Our results suggest that, in addition to the action of transcription factors and of growth factors and their downstream signaling pathways, miRNAs also exert significant control over the chondrocyte phenotype. In addition, there are most likely complex interactions between these different classes of regulators. For example, miR-199a has been shown to inhibit transcription factor Smad1 by directly targeting it, and growth factor BMP2 can transiently decrease miR-199a levels in chondrocytic cell lines leading to the release of Smad1 inhibition, which, because Smad1 is a key mediator of BMP signaling, allows BMP2-induced chondrocytic differentiation to occur (29). In the present study, we have shown that the master cartilage-specific transcription SOX9, which is thought to drive cartilage matrix gene expression (20), also drives expression of H19, a highly abundant primary microRNA transcript, which gives rise to mature miR-675. This miRNA, in turn, positively regulates COL2A1 expression in human articular chondrocytes. Interestingly, transcription of both H19 and Col2a1 were reported to be coordinately regulated in an immortalized hamster cell line study (30). Similarly, as HACs are dedifferentiated through serial passage, we observed that H19 and COL2A1 levels decrease in an almost identical manner. From our data, we propose a new miRNA-dependent mechanism by which key transcription factor SOX9 can activate COL2A1 transcription. In silico analysis reveals COL2A1 is not a predicted target of miR-675, and because miRNAs most commonly inhibit expression of their direct targets, it may be that H19-derived miR-675 is up-regulating COL2A1 by a derepression mechanism, i.e. by targeting a COL2A1 transcriptional repressor (Fig. 6). In fact, it is thought that derepression may be an important mechanism by which miRNAs influence cell-specific gene expression (1). Further work is now needed to test this hypothesis, i.e. to identify the direct miR-675 targets, and in particular those regulating COL2A1. Interestingly, we have found that miR-675 does not appear to regulate the SOX9-dependent gene COL9A1, indicating that miR-675 does not mediate all of the effects of SOX9. The full phenotypic effects of miR-675 in HACs remain to be elucidated. A growing body of evidence suggests that miRNAs are essential for both normal development and tissue and cellular homeostasis (31). In a recent study, miRNA delivery conferred dramatic tumor suppression without toxicity in a hepatic tumor mouse model, thus highlighting the potential therapeutic value of miRNAs (32). Based on our findings, miR-675, by up-regulating the most important cartilage matrix protein COL2A1, offers a means to induce cartilage repair. This exciting possibility first must be tested in an appropriate animal model.
FIGURE 6.
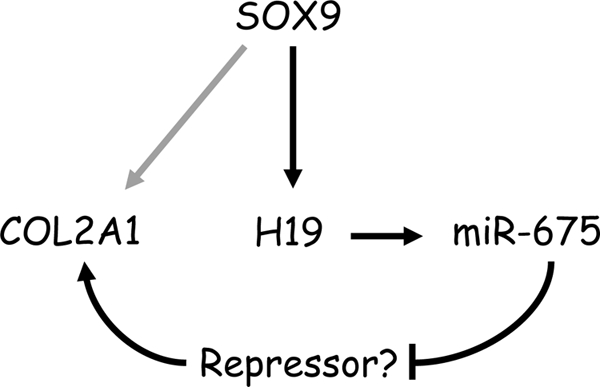
Proposed new mechanism of SOX9 activation of COL2A1 in HACs. Although evidence exists for direct activation of Col2a1 by Sox9 in murine growth plate chondrocytes (gray arrow), in the present study, we provide evidence for an additional pathway where master cartilage regulator transcription factor SOX9 acts through H19-derived miR-675, which up-regulates COL2A1 in human articular chondrocytes. We postulate this may occur by a derepression mechanism, i.e. by miR-675 targeting a COL2A1 transcriptional repressor.
This work was supported by Arthritis Research UK and Biotechnology and Biological Sciences Research Council (United Kingdom) Grant BB/G006555/1.
- miRNA
- microRNA
- HAC
- human articular chondrocyte
- siRNA
- small interfering RNA
- OA
- osteoarthritis.
REFERENCES
- 1.Hobert O. (2008) Science 319, 1785–1786 [DOI] [PubMed] [Google Scholar]
- 2.Bartel D. P. (2004) Cell 116, 281–297 [DOI] [PubMed] [Google Scholar]
- 3.Xiao C., Calado D. P., Galler G., Thai T. H., Patterson H. C., Wang J., Rajewsky N., Bender T. P., Rajewsky K. (2007) Cell 131, 146–159 [DOI] [PubMed] [Google Scholar]
- 4.van Rooij E., Sutherland L. B., Qi X., Richardson J. A., Hill J., Olson E. N. (2007) Science 316, 575–579 [DOI] [PubMed] [Google Scholar]
- 5.Esau C., Kang X., Peralta E., Hanson E., Marcusson E. G., Ravichandran L. V., Sun Y., Koo S., Perera R. J., Jain R., Dean N. M., Freier S. M., Bennett C. F., Lollo B., Griffey R. (2004) J. Biol. Chem. 279, 52361–52365 [DOI] [PubMed] [Google Scholar]
- 6.Tuddenham L., Wheeler G., Ntounia-Fousara S., Waters J., Hajihosseini M. K., Clark I., Dalmay T. (2006) FEBS Lett. 580, 4214–4217 [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi T., Lu J., Cobb B. S., Rodda S. J., McMahon A. P., Schipani E., Merkenschlager M., Kronenberg H. M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 1949–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von der Mark K., Gauss V., von der Mark H., Müller P. (1977) Nature 267, 531–532 [DOI] [PubMed] [Google Scholar]
- 9.Aigner T., Söder S., Gebhard P. M., McAlinden A., Haag J. (2007) Nat Clin Pract. Rheumatol 3, 391–399 [DOI] [PubMed] [Google Scholar]
- 10.Cai X., Cullen B. R. (2007) RNA 13, 313–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smits G., Mungall A. J., Griffiths-Jones S., Smith P., Beury D., Matthews L., Rogers J., Pask A. J., Shaw G., VandeBerg J. L., McCarrey J. R., Renfree M. B., Reik W., Dunham I. (2008) Nat. Genet. 40, 971–976 [DOI] [PubMed] [Google Scholar]
- 12.Cullen B. R. (2004) Mol Cell 16, 861–865 [DOI] [PubMed] [Google Scholar]
- 13.Lafont J. E., Talma S., Murphy C. L. (2007) Arthritis Rheum. 56, 3297–3306 [DOI] [PubMed] [Google Scholar]
- 14.Murphy C. L., Polak J. M. (2004) J. Cell. Physiol. 199, 451–459 [DOI] [PubMed] [Google Scholar]
- 15.Lafont J. E., Talma S., Hopfgarten C., Murphy C. L. (2008) J. Biol. Chem. 283, 4778–4786 [DOI] [PubMed] [Google Scholar]
- 16.Goshen R., Rachmilewitz J., Schneider T., de-Groot N., Ariel I., Palti Z., Hochberg A. A. (1993) Mol. Reprod. Dev. 34, 374–379 [DOI] [PubMed] [Google Scholar]
- 17.Lustig O., Ariel I., Ilan J., Lev-Lehman E., De-Groot N., Hochberg A. (1994) Mol. Reprod. Dev. 38, 239–246 [DOI] [PubMed] [Google Scholar]
- 18.Rachmilewitz J., Goshen R., Ariel I., Schneider T., de Groot N., Hochberg A. (1992) FEBS Lett. 309, 25–28 [DOI] [PubMed] [Google Scholar]
- 19.Hurst L. D., Smith N. G. (1999) Trends Genet. 15, 134–135 [DOI] [PubMed] [Google Scholar]
- 20.Lefebvre V., Huang W., Harley V. R., Goodfellow P. N., de Crombrugghe B. (1997) Mol. Cell. Biol. 17, 2336–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenkins E., Moss J. B., Pace J. M., Bridgewater L. C. (2005) Matrix Biol. 24, 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bell D. M., Leung K. K., Wheatley S. C., Ng L. J., Zhou S., Ling K. W., Sham M. H., Koopman P., Tam P. P., Cheah K. S. (1997) Nat. Genet. 16, 174–178 [DOI] [PubMed] [Google Scholar]
- 23.Stokes D. G., Liu G., Dharmavaram R., Hawkins D., Piera-Velazquez S., Jimenez S. A. (2001) Biochem. J. 360, 461–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han Y., Lefebvre V. (2008) Mol. Cell Biol. 28, 4999–5013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones S. W., Watkins G., Le Good N., Roberts S., Murphy C. L., Brockbank S. M., Needham M. R., Read S. J., Newham P. (2009) Osteoarthritis Cartilage 17, 464–472 [DOI] [PubMed] [Google Scholar]
- 26.Iliopoulos D., Malizos K. N., Oikonomou P., Tsezou A. (2008) PLoS One 3, e3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamasaki K., Nakasa T., Miyaki S., Ishikawa M., Deie M., Adachi N., Yasunaga Y., Asahara H., Ochi M. (2009) Arthritis Rheum. 60, 1035–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aigner T., Gebhard P. M., Schmid E., Bau B., Harley V., Pöschl E. (2003) Matrix Biol. 22, 363–372 [DOI] [PubMed] [Google Scholar]
- 29.Lin E. A., Kong L., Bai X. H., Luan Y., Liu C. J. (2009) J. Biol. Chem. 284, 11326–11335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owen R. D., Hosoi J., Montgomery J. C., Wiseman R., Barrett J. C. (1993) Cell Growth & Differ. 4, 1013–1021 [PubMed] [Google Scholar]
- 31.Kloosterman W. P., Plasterk R. H. (2006) Dev. Cell 11, 441–450 [DOI] [PubMed] [Google Scholar]
- 32.Kota J., Chivukula R. R., O'Donnell K. A., Wentzel E. A., Montgomery C. L., Hwang H. W., Chang T. C., Vivekanandan P., Torbenson M., Clark K. R., Mendell J. R., Mendell J. T. (2009) Cell 137, 1005–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]