

Abstract
Glycogen synthase kinase 3 (GSK3) is a highly conserved serine/threonine protein kinase that plays important roles in a variety of physiological and developmental processes in animals. It is well known that the GSK3 kinase-catalyzed protein phosphorylation often requires a stable kinase-substrate docking interaction, which is achieved mainly by two mechanisms as follows: priming phosphorylation of a substrate by a distinct kinase to create a docking phosphate group and scaffold protein-mediated protein complex formation. Brassinosteroid-INsensitive 2 (BIN2) is an Arabidopsis GSK3-like kinase that negatively regulates brassinosteroid (BR) signaling by phosphorylating BES1 (bri1 EMS suppressor 1) and BZR1 (brassinazole-resistant 1), two highly similar transcription factors critical for bringing about characteristic BR responses. However, little is known about the biochemical mechanism by which BIN2 phosphorylates its substrates. Here, we show that BIN2 interacts directly with BZR1 through a 12-amino acid BIN2-docking motif adjacent to the C terminus of BZR1. Interestingly, this 12-amino acid motif is sufficient to allow a Drosophila GSK3 substrate Armadillo to be phosphorylated by BIN2 in vitro. Deletion of this motif inhibits the phosphorylation and subsequent degradation of BZR1 in vivo, resulting in phenotypic suppression of a hypermorphic bin2 mutation and enhanced resistance to a BR biosynthesis inhibitor. We thus concluded that BIN2 utilizes a direct kinase-substrate docking mechanism to phosphorylate BZR1 and regulate its protein stability.
Keywords: Enzyme Mechanisms, Glycogen Synthase Kinase 3, Protein Phosphorylation, Protein-Protein Interactions, Steroid Hormone, Brassinosteroid Signaling
Introduction
GSK3 kinases are a group of highly conserved serine/threonine kinases that play key regulatory roles in many signaling pathways (1, 2). It is well known that GSK3 kinases are constitutively active to phosphorylate a variety of protein substrates but become inactivated in response to biological signals (1, 3). The majority of known GSK3 substrates contain repeats of a short consensus phosphorylation motif, (S/T)XXX(S/T) (S/T corresponds to serine/threonine, and X represents any amino acid), but their phosphorylation by GSK3 kinases often requires a tight kinase-substrate docking interaction (3). Two mechanisms are utilized by animal GSK3 kinases to achieve such an interaction (4). The first mechanism requires a priming phosphorylation of the C-terminal Ser/Thr residue of the (S/T)XXX(S/T) motif by a different kinase to generate a primed phosphorylation site on a substrate, which can then interact with a specific phosphate-binding pocket comprising three highly conserved basic residues (Arg96, Arg180, and Lys205 in the human GSK3β kinase) within the catalytic core of GSK3 (5–7). The second mechanism involves a scaffold protein that simultaneously binds GSK3 and its substrate to significantly increase the local substrate concentration for GSK3, thus facilitating phosphorylation (8).
Brassinosteroid-INsensitive 2 (BIN2) is an Arabidopsis GSK3-like kinase that functions together with several of its nine homologs to negatively regulate the intracellular signal transduction of brassinosteroids (BRs)3 (9–14), a unique class of plant polyhydroxysteroids critical for normal plant growth and development (15). Unlike the animal steroid hormones that initiate their signaling by binding to intracellular steroid receptors, BRs are perceived by a cell surface receptor complex that contains two similar yet distinct leucine-rich repeat receptor-like kinases, BRI1 (BRassinosteroid-Insensitive 1) and BAK1 (BRI1-associated receptor Kinase 1), that heterodimerize and transphosphorylate to initiate BR signaling (16–19). In the absence of BR, BIN2 is a constitutively active kinase that phosphorylates downstream signaling proteins to block the BR signaling (20–22). Gain-of-function mutations or overexpression of the Arabidopsis BIN2 gene inhibit BR signaling (10, 23), whereas simultaneous elimination of BIN2 and its two closest homologs or chemical inhibition of multiple Arabidopsis GSK3-like kinases results in constitutive activation of the BR signal transduction pathway (13, 14, 24).
Using a yeast two-hybrid approach, we previously identified two highly similar Arabidopsis proteins, initially named as BIN2 SUBSTRATE 1 and 2 (BIS1 and BIS2) but later changed to BES1 and BZR1, as potential BIN2 substrates (20), which were independently discovered by genetic approaches as positive BR signaling components capable of directly binding to promoters of known BR-responsive genes (21, 25–29) or interacting with other nuclear proteins to regulate gene expression (30, 31). BES1 and BZR1 contain many copies of the conserved GSK3 phosphorylation motif, are efficiently phosphorylated by BIN2 in vitro, and exist as both hyper- and hypophosphorylated forms in vivo (20–22). Treatment of plants with brassinolide (BL), the most active member of the BR family, led to rapid dephosphorylation of both BZR1 and BES1 (21, 22, 32). It was generally believed that BIN2 phosphorylates BZR1 and BES1 in vivo to target them for degradation, to prevent their nuclear localization, and/or to inhibit their DNA binding activities (14, 21, 22, 27, 33–35). Despite all these exciting discoveries, little is known about the biochemical mechanism by which BIN2 phosphorylates its substrates.
Although BIN2 contains a conserved primed phosphate-binding site (10), its phosphorylation activity of BZR1 and BES1 in vitro does not require a priming phosphorylation or a scaffold protein but involves a direct kinase-substrate interaction (20). Here, we report that the Arabidopsis GSK3 kinase interacts with BZR1 via a 12-amino acid BIN2-docking motif (DM) near the BZR1 C terminus. We show that deletion of this motif inhibited the phosphorylation and subsequent degradation of BZR1 in vivo and that expression of a mutant BZR1 protein lacking the DM was able to promote hypocotyl elongation and rescue the short hypocotyl phenotype of the dark-grown bin2-1 mutant. Our study thus revealed a direct docking mechanism for a plant GSK3-like kinase to phosphorylate its substrate, which is different from the two common docking mechanisms of mammalian GSK3 kinases requiring a third protein.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
Construction of pACT2-BZR1 and pGEX-KG-BZR1 plasmids was described previously (20). Convenient restriction sites were used to remove different portions from the full-length BZR1 in pACT2 (Clontech) or pGEX-KG vector (36) to make the following truncated variants of BZR1 fusion proteins for yeast two-hybrid and in vitro pulldown and in vitro phosphorylation assays: Nh, the N-terminal half containing amino acids Met1 to Gly159; Ch, the C-terminal half containing Pro162 to Gly336; ΔN, containing Ser102 to Gly336; ΔM, deleting Ser103 to Met202; ΔC, carrying amino acids Met1 to Ala203; N, Met1 to Ser102; M, Ser102 to Ala203; C, Ala203 to Gly336; ΔC82, Met1 to Ser255; C82, Ser255 to Gly336; and M:C82, the M fragment fused with the C82 fragment. Site-directed mutagenesis using the QuikChange II site-directed mutagenesis kit (Stratagene) was performed on the C82 portion of M:C82 to introduce new restriction sites, which were used to delete varying numbers of amino acids from the N- or C-terminal end of the C82 fragment. These truncated C82 fragments were then religated with the M fragment to generate serial deletion M:C82 constructs in pACT2 or pGEX-KG vector.
To identify amino acids critical for the BZR1-BIN2 interaction, the region between Gln294 and Val320 of M:C82 was screened by alanine-scanning mutagenesis (37), in which three adjacent amino acids were simultaneously mutated to alanine by PCR-based site-directed mutagenesis. Site-directed mutagenesis was also performed on the full-length glutathione S-transferase (GST)-BZR1 fusion protein by simultaneously deleting Val309–Lys310–Pro311 (ΔVKP), Trp312–Glu315 (ΔWEGE), Arg316–Ile317–His318 (ΔRIH), or Val309 to Val320 (ΔDM). To determine the sufficiency of the C82 fragment or the 12-amino acid DM for BIN2 binding, their coding sequences (an annealed double-stranded oligonucleotide was used for the 12-amino acid motif) were translationally fused at their N termini to a 2.3-kb EcoRI/NcoI fragment of the Drosophila Armadillo (Arm) cDNA (38) and cloned into the pGEX-KG vector to create Arm:C82 and Arm:DM fusion constructs. The DM was also fused to the M fragment in the pGEX-KG vector to create the M:DM plasmid.
Protein Expression and in Vitro Kinase Assays
The entire BIN2 open reading frame was cloned into the pMAL-C2 vector (New England Biolabs) to create a maltose-binding protein (MBP) BIN2 fusion kinase, and various truncated/mutated BZR1 proteins were expressed as GST-tagged recombinant proteins using the pGEX-KG plasmids described above. Protein induction and purification were carried out according to the manufacturers' recommended protocols or the method described previously (36). A small peptide (BZRtide) corresponding to Asn306–Val320 of the BZR1 protein was synthesized at the Protein Structure Facility, University of Michigan, Ann Arbor, and used for the kinase competition assay with the full-length GST-BZR1 protein. The in vitro BIN2 kinase assay was performed as described previously (20).
Yeast Two-hybrid and GST Pulldown Assays
The wild-type and various truncated pACT2-BZR1 constructs described above were individually transformed into yeast Y187 cells, and the transformed yeast cells were then mated with Y190 cells containing the pAS2-BIN2 plasmid. The BIN2-BZR1 interaction in the resulting diploid cells was assayed using a method described previously (20). For the GST pulldown assays, MBP-BIN2 was labeled with [γ-32P]ATP by autophosphorylation (20). Glutathione-Sepharose beads (Amersham Biosciences) containing ∼2 μg of the wild-type or a truncated GST-BZR1 protein were incubated with 20 μl of γ-32P-labeled MBP-BIN2 in the pulldown buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.1% Nonidet P-40 (v/v), and 1 mm dithiothreitol) at 4 °C for 2 h and washed five times with the pulldown buffer. The bound proteins were removed from the beads by boiling in 20 μl of 2× SDS sample buffer (0.10 m Tris-HCl (pH 6.8), 10% glycerol (v/v), 3% SDS (w/v), 0.2 m dithiothreitol, and 0.004% bromphenol blue (w/v)), separated by 7.5% SDS-PAGE, and analyzed by autoradiography for the presence of γ-32P-labeled MBP-BIN2.
Generation of Transgenes and Transgenic Plants
The full-length BZR1 cDNA was used to replace the BRI1 open reading frame of the pBRI1:BRI1-GFP plasmid (39), generating the pBRI1:BZR1-GFP construct to produce a BZR1 fusion protein with green fluorescent protein (GFP) in transgenic Arabidopsis plants. The QuikChange site-directed mutagenesis kit (Stratagene) was used to create mutated BZR1 genes containing the P234L mutation (P234L) or to delete the BIN2-DM (ΔDM). Both the wild-type and bin2-1-mutated BIN2 genomic transgenes (gBIN2 and gBIN2-1) were previously described (10), and a site-directed mutagenesis experiment was performed to introduce the R80A mutation into the gBIN2-1 transgene to generate a double mutation gBIN2-1(R80A) transgene. These transgenes were mobilized into the Agrobacterium tumefaciens GV3101 strain and transformed into the wild-type Arabidopsis plants using a vacuum transformation protocol (40). The resulting transgenic lines were screened by RNA blot analysis to identify lines with similar transgene expression levels. The pBRI1:BZR1(ΔDM)-GFP transgene from a selected line was crossed into the homozygous bin2-1 mutant (10) for phenotypic analysis.
Protein Blot Analysis
Seeds of Arabidopsis wild-type (ecotype Columbia) and various transgenic lines were germinated on half-strength Murashige and Skoog (½ MS) agar medium containing 0.5% (w/v) sucrose and grown under a long day photoperiodic condition (16- h light/8-h dark) at 22 °C for 3 weeks. The seedlings were removed from agar plates and incubated in liquid ½ MS medium supplemented with 1 μm brassinolide (BL, Chemiclones, Inc., Canada) or 10 μm proteasome inhibitor MG132 (Sigma) for 2 h. Immediately after treatments, equal amounts of seedlings were homogenized and boiled in 60 μl of 2× SDS sample buffer, and 20 μl of each sample was loaded onto a 7.5% SDS-polyacrylamide gel. Separated proteins were transferred onto Immobilon-P membrane (Millipore) and analyzed with a polyclonal anti-GFP antibody (Torrey Pines Biolabs, Houston, TX). The developed film was digitized, and the signals were quantified using the ImageJ software (rsbweb.nih.gov).
Hypocotyl Measurement
Seeds of the wild-type, bin2-1 (10), bzr1-D (26), and selected BZR1-GFP transgenic lines were germinated and grown on ½ MS medium supplemented with or without 2 μm brassinazole (Brz) (41) in the dark for 5 days. The hypocotyl lengths of the Brz-treated seedlings of different genotypes were manually measured for comparison. A total of ∼60 seedlings for each genotype were analyzed from three independent experiments to quantify relative resistance to the BR biosynthesis inhibitor Brz.
Confocal Microscopy
Transgenic plants expressing a GFP-tagged BZR1 protein were grown on ½ MS agar medium in dark at 22 °C for 5 days. Seedlings were mounted on glass slides with water. The green fluorescent signals of transgenic seedlings were examined using an LSM 510 confocal microscope (Zeiss) filtered with FITC10 set (excitation 488 nm with emission 505–530 and 530–560 nm) (20).
RESULTS
BR Regulatory Activity of BIN2 Does Not Require a Priming Phosphorylation Activity
We previously showed that the in vitro BZR1/BES1 phosphorylation activity of BIN2 does not require a priming phosphorylation or need a scaffold protein (20). Sequence comparison between BIN2 and animal GSK3 kinases indicated that the three basic residues (Arg96, Arg180, and Lys205 in the human GSK3β kinase) comprising the binding site for a priming phosphate (6) are conserved in BIN2 (Arg80, Arg164, and Lys189) (10). To determine whether the in vivo function of BIN2 to block BR signaling in Arabidopsis requires priming phosphorylation of its physiological substrates, we conducted PCR-based site-directed mutagenesis to replace the conserved Arg80 with an Ala residue in a genomic BIN2 transgene that also carries the gain-of-function bin2-1 mutation (E263K). Previous experiments demonstrated that the corresponding Arg-Ala mutation in the mammalian GSK3β destroys the phosphate-binding pocket and prevents the phosphorylation of GSK3 substrates that require priming phosphorylation (6), while our earlier transgenic studies showed that expression of a bin2-1-mutated BIN2 genomic transgene (gBIN2-1) was able to block BR signaling in Arabidopsis, generating strong dwarf transgenic plants resembling the homozygous bin2-1 mutant (10). We reasoned that if the BR regulatory activity of BIN2 requires priming phosphorylation of its substrates, the R80A mutation would nullify the ability of the gBIN2-1 transgene to cause the dwarf phenotype. Fig. 1 showed that when transformed into the wild-type Arabidopsis plants, the gBIN2-1(R80A) transgene was still capable of creating severe dwarf transgenic lines, indicating that the R80A mutation had no detectable effect on the biological activity of BIN2 to inhibit BR signaling. We thus concluded that the BIN2 function in BR signaling does not involve priming phosphorylation of its physiological substrates in Arabidopsis.
FIGURE 1.

R80A mutation has no effect on the in vivo BIN2 activity. Shown here (from left to right) are 5-week-old plants of a representative gBIN2 line, a representative gBIN2-1 transgenic line, and a representative gBIN2-1(R80A) transgenic line grown in soil at 22 °C under a 16-h light/8-h dark photoperiodic growth condition.
C-terminal End of BZR1 Is Critical for Its Phosphorylation by BIN2
Because both BZR1 and BES1 were discovered as BIN2-interacting proteins by a yeast two-hybrid screen and our previous phosphorylation assays showed that both proteins could be efficiently phosphorylated by BIN2 in vitro (20), we suspected that the two BIN2 substrates might carry a dedicated BIN2-binding site that is functionally equivalent to a scaffold protein that binds both GSK3 and its substrates to facilitate phosphorylation. To discover such a BIN2-binding site, we performed both yeast two-hybrid and in vitro pulldown assays with various truncated BZR1 proteins (see “Experimental Procedures”). As shown in Fig. 2, A–C, all BZR1 fragments containing the C-terminal 135 amino acids (C fragment) were able to bind BIN2, whereas all BZR1 proteins lacking the C fragment failed to interact with the Arabidopsis GSK3-like kinase, indicating that the C fragment is essential for the BZR1-BIN2 interaction. These truncated BZR1 proteins were also tested for in vitro phosphorylation by BIN2. As shown in Fig. 2D, only those BZR1 fusion proteins capable of interaction with BIN2 were phosphorylated by the BIN2 kinase, demonstrating that a stable BIN2-BZR1 interaction is required for BIN2 to phosphorylate BZR1 in vitro.
FIGURE 2.

Kinase-substrate interaction is essential for BIN2 to phosphorylate BZR1. A, schematic structures of various BZR1 proteins. The patterned boxes represent the regions containing the predicted GSK3 phosphorylation ((S/T)XXX(S/T)) motifs. The amino acids contained in various BZR1 proteins (with their names listed on the left) are the following: FL (Met1–Gly336), Nh (Met1–Gly159), Ch (Pro162–Gly336), ΔN (Ser102–Gly336), ΔM (Met1–Ser102 plus Ala203–Gly336), ΔC (Met1–Ala203), N (Met1–Ser102), M (Ser102–Ala203), and C (Ala203–Gly336). B, BIN2-BZR1 interaction in yeast was assayed by growth of yeast cells on medium lacking His (the −His column) and blue color on 5-bromo-4-chloro-3-indolyl-β-d-galactoside-containing medium (the 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) column). C, GST pulldown assay testing the in vitro BIN2-BZR1 interaction. Equal amounts of various GST-BZR1 proteins bound to glutathione-Sepharose beads were incubated with equal amounts of 32P-labeled MBP-BIN2 fusion protein. After washing, proteins remaining on the beads were separated by SDS-PAGE. The upper panel shows 20% of the 32P-labeled MBP-BIN2 input and the lower panel reveals (by autoradiography) the amount of 32P-MBP-BIN2 proteins brought down by various GST-BZR1 proteins. D, in vitro phosphorylation assays. The same GST-BZR1 fusion proteins used in C were incubated with the MBP-BIN2 kinase in a GSK3 assay buffer (20) at 25 °C for 30 min. The reaction mixtures were separated by SDS-PAGE. Gels were stained with Coomassie Blue (the upper panel), and phosphorylated proteins were visualized by autoradiography (the lower panel). GST-BZR1* indicates the full-length GST-BZR1 fusion protein or its truncated/mutated variants.
A further deletion of N-terminal 53 amino acids from the C fragment, removing 3 of its 4 putative GSK3 phosphorylation motifs, eliminated its phosphorylation by BIN2 while having little effect on its interaction with BIN2 (Fig. 3, A–C). By contrast, deletion of the C82 fragment from BZR1 prevented both BZR1-BIN2 interaction and BIN2-catalyzed BZR1 phosphorylation (Fig. 3, A–C). The C82 fragment, when fused to the M fragment containing multiple GSK3 phosphorylation motifs, was able to allow BIN2 to phosphorylate the M fragment that itself could not be phosphorylated by the Arabidopsis GSK3 kinase (Figs. 2D and 3, A and C). Interestingly, the C82 fragment can also allow BIN2 to phosphorylate Armadillo (Arm), a known substrate of the Drosophila GSK3 kinase Shaggy (Fig. 3D) (38). It was known that efficient phosphorylation of Arm by Shaggy requires both priming phosphorylation and a scaffold protein (42). Simply mixing the Escherichia coli-expressed GST-Arm fusion protein and MBP-BIN2 in the GSK3 phosphorylation buffer did not lead to any detectable phosphorylation of the Arm protein (Fig. 3D). These results indicated that the C82 fragment of BZR1 contains a portable BIN2-binding motif that functions similarly to a scaffold protein.
FIGURE 3.
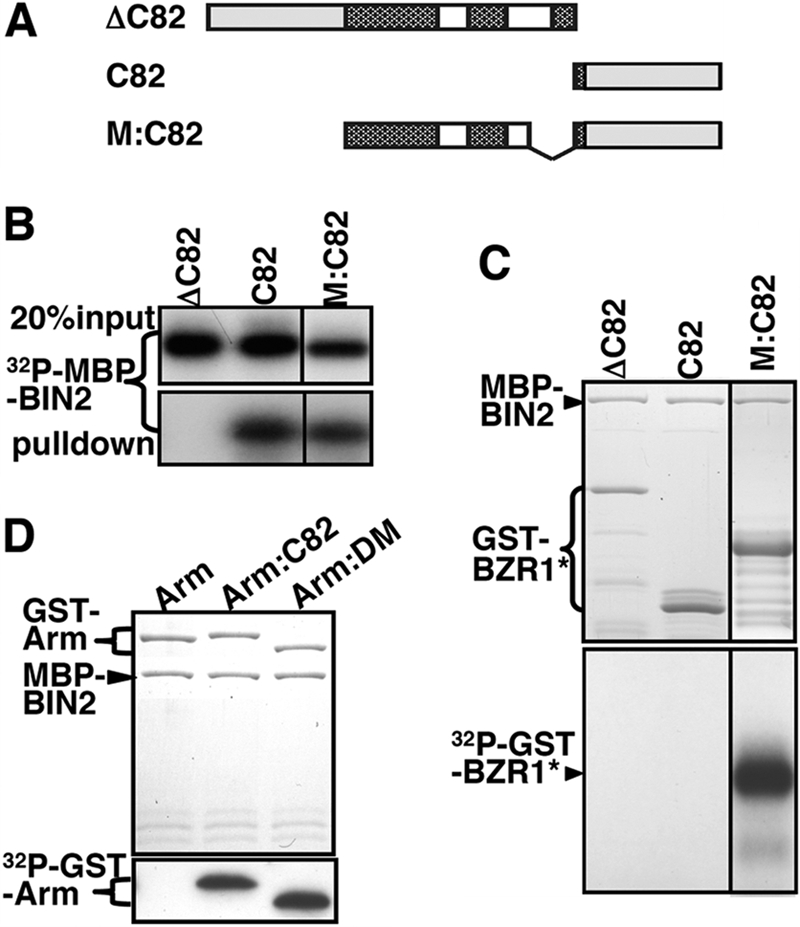
C-terminal 82 amino acids carry a portable BIN2-docking motif critical for the BIN2-BZR1 interaction. A, schematic structures of three truncated/fused BZR1 proteins as follows: ΔC82 and C82 lacking and containing the C-terminal 82 amino acids, respectively, whereas M:C82 is a fusion protein between the M fragment (Ser102–Ala203) and the C82 fragment (Ser255–Gly336). B, similar to Fig. 2C but using different GST-BZR1 fusion proteins. C and D, testing in vitro phosphorylation of different GST fusion proteins by MBP-BIN2. The listed GST fusion proteins, including fusion proteins between a Drosophila GSK3 substrate Arm with C82 or our identified BIN2-docking motif (DM, see Fig. 5A), were incubated with MBP-BIN2 in 30 μl of GSK3 assay buffer at 25 °C for 30 min, and the reactions were stopped by adding SDS sample buffer and 5 min of boiling at 95 °C. The denatured samples were separated by SDS-PAGE and analyzed by Coomassie Blue staining (upper panels) and autoradiography (lower panels).
Identification of a 12-Amino Acid Motif as the Minimal BIN2-docking Motif
To define a minimal region responsible for BIN2 binding, we took advantage of the newly generated M:C82 fusion protein that consists of a BIN2-binding module (C82) and a BIN2 phosphorylation module (the M fragment). Because a direct docking interaction is required for BIN2 to phosphorylate BZR1, any mutation in C82 that inhibits the BIN2-BZR1 interaction would prevent the BIN2-catalyzed BZR1 phosphorylation in vitro. We thus generated serial deletion mutants of the C82 fragment (from both ends), expressed each of the mutated M:C82 proteins as GST fusion proteins in E. coli, and tested their phosphorylation by the MBP-tagged BIN2 in vitro. As shown in Fig. 4, A and C, N-terminal deletion of 15 (255–269) or 32 (255–286) amino acids had little effect; elimination of 43 (255–297) amino acids slightly reduced and removing 60 (255–314) or 66 (255–320) amino acids completely inhibited the in vitro BIN2-catalyzed BZR1 phosphorylation. We also found that C-terminal deletion of 16 (321–336) amino acids had no effect on phosphorylation, but further deletion of 7 additional (314–320) amino acids significantly reduced phosphorylation, and removing the C-terminal 40 (297–336) amino acids completely prevented the BIN2-catalyzed BZR1 phosphorylation (Fig. 4, B and D). Combining the results of the two sets of serial deletion experiments, we concluded that amino acids of Gly297–Val320 are important for the BZR1-BIN2 interaction.
FIGURE 4.

Identification of the 12-amino acid BIN2-DM. A and B, convenient restriction sites were introduced into the C82 fragment of the GST-M:C82 fusion protein by PCR-based site-directed mutagenesis at indicated positions. Two series of deletion constructs were created from the N terminus (ΔN) or the C terminus (ΔC) of the C82 domain by double enzyme digestion (with one enzyme cutting a terminal end and the other cutting a newly created restriction site at the indicated positions) followed by re-ligation. Pattern bars represent regions carrying predicted GSK3 phosphorylation motifs, and the names of serially deleted GST-BZR1 proteins are shown on the left. C and D, similar to Fig. 2D, testing the in vitro phosphorylation of the serially deleted GST-BZR1 proteins by the MBP-tagged BIN2. The GST-M:C82 (M:C82) and GST-M (M) fusion proteins were used as the positive and negative control, respectively. E, three consecutive amino acids were simultaneously changed to three alanine residues for the 24-amino acid (Gly297–Val320) segment, and a total eight mutated forms (m1 to m8) of the GST-M:C82 fusion protein were created (left panel). Asterisks indicate nonmutated amino acids. The mutated proteins were subject to the in vitro phosphorylation assay described in Fig. 2D. C–E, the upper panels show Coomassie Blue staining of the fusion proteins used in the kinase assays, and the lower panels are autoradiographs showing the intensity of 32P-labeled GST-BZR1 fusions.
To test whether one or more amino acids in the 297–320 segment are crucial for the BIN2-BZR1 docking interaction, we performed an alanine-scanning mutagenesis experiment (37) by simultaneously mutating three consecutive amino acids to an alanine triplet, and we tested the in vitro phosphorylation of the mutated M:C82 fusion proteins by BIN2. As shown in Fig. 4E, simultaneously mutating three consecutive amino acids to alanine in the 1st half of the 297–320 segment had little effect on the BIN2-catalyzed BZR1 phosphorylation, whereas AAA mutations in the 2nd half reduced the BZR1 phosphorylation by ∼50%, suggesting that no individual amino acid is essential for the BZR1-BIN2 interaction and that the second half of the 24-amino acid segment is important for BIN2 binding.
To verify the role of amino acids 309–320 in mediating the BZR1-BIN2 interaction, we performed site-directed mutagenesis to simultaneously delete 3 or 4 consecutive amino acids within this motif from the full-length GST-BZR1 fusion protein, and we tested their in vitro phosphorylation by BIN2. As shown in Fig. 5A, deletion of 3 or 4 amino acids significantly inhibited phosphorylation, and deletion of the entire 12-amino acid motif completely eliminated the BZR1 phosphorylation. We named this 12-amino acid motif BIN2-docking motif or BIN2-DM. Further support for the critical role of the BIN2-DM in the phosphorylation of BZR1 by BIN2 was provided by our finding that a 15-amino acid oligopeptide (NSQVKPWEGERIHDV, named as BZRtide), containing the entire BIN2-DM plus 3 additional amino acids at its N-terminal end, prevented the BIN2-BZR1 interaction (Fig. 5B) and inhibited the BIN2-catalyzed BZR1 phosphorylation in a dose-dependent manner (Fig. 5C). Interestingly, this 12-amino acid BIN2-DM was sufficient to allow the Arabidopsis GSK3 kinase to phosphorylate not only the M fragment of BZR1 (Fig. 5D) but also the Drosophila Arm protein (Fig. 3D). These results thus revealed that BZR1 carries a portable docking motif to facilitate its phosphorylation by BIN2, which represents a novel docking mechanism for a GSK3 kinase to phosphorylate its substrates with no help from a third protein.
FIGURE 5.

12-Amino acid BIN2-DM is necessary and sufficient for BIN2 to bind and phosphorylate BZR1. A, in vitro phosphorylation assay of the full-length wild-type (FL) or a mutated GST-BZR1 fusion protein lacking 3 or 4 amino acids (ΔVKP, ΔWEGE, or ΔRIH) or the entire BIN2-DM (ΔDM). The amino acid sequence of the BIN2-DM is shown on the left, and asterisks indicate deleted amino acids. B, GST pulldown assay was performed to test if a BIN2-DM (underlined amino acids)-containing peptide (306NSQVKPWEGERIHDV320, named as BZRtide) inhibits the BIN2/BZR1 interaction. Equal amounts of 32P-labeled MBP-BIN2 (the upper panel) were incubated with equal amounts of the full-length GST-BZR1 fusion protein bound to glutathione-Sepharose beads and different concentrations of BZRtide, and the amounts of pulled down MBP-BIN2 were revealed by autoradiography (the lower panel). C, BZRtide competitively inhibits the BIN2-catalyzed BZR1 phosphorylation by preventing the BIN2-BZR1 interaction. Equal amounts of GST-BZR1 were incubated with equal amounts of MBP-BIN2 but different concentrations of BZRtide in a 30-μl GSK3 kinase assay solution (20), and the levels of phosphorylation were revealed by autoradiography of an SDS-polyacrylamide gel that separates the reaction mixtures. D, GST-tagged M, M:C82, or the M:DM (the M fragment fused to the BIN2-DM) fusion protein was tested for phosphorylation by MBP-BIN2. The upper panel shows the amount of proteins used in the kinase assay by Coomassie Blue staining, and the lower panel indicates the levels of phosphorylation by autoradiography.
Deletion of the BIN2-DM Prevents the BZR1 Phosphorylation in Vivo
To determine whether such a direct GSK3-substrate docking mechanism is responsible for the in vivo phosphorylation of BZR1, we expressed the wild-type and two mutated BZR1-GFP fusion proteins, one containing the bzr1-D mutation (BZR1(P234L)) that changed Pro234 to Leu (P234L) and significantly increased BZR1 stability (20, 26), and the other lacking the entire BIN2-DM (BZR1(ΔDM)), in transgenic Arabidopsis plants (Fig. 6A). One representative transgenic line for each construct with similar transgene expression levels (Fig. 6B) was selected to compare the protein abundance and phosphorylation status of the three BZR1-GFP fusion proteins by immunoblotting analysis with anti-GFP antibodies. Because of the presence of more than 25 predicted GSK3 phosphorylation motifs, the phosphorylation status of BZR1 can be examined by a significant mobility change on protein gel (22). As expected, the BZR1(P234L)-GFP protein accumulated at high levels as both phosphorylated and nonphosphorylated forms (Fig. 6C, lane 4), whereas the wild-type BZR1-GFP proteins accumulated at a much lower level predominantly as the phosphorylated form (Fig. 6C, lane 1). By contrast, the BZR1(ΔDM)-GFP fusion proteins accumulated at a higher level as the nonphosphorylated form (Fig. 6C, lane 7), indicating that the DM deletion prevented the in vivo phosphorylation of BZR1. Our data thus demonstrated that the in vivo phosphorylation of BZR1 by BIN2 is likely mediated through a direct GSK3-substrate docking mechanism.
FIGURE 6.

Direct kinase-substrate docking mechanism is responsible for the in vivo phosphorylation of BZR1 by BIN2. A, schematic presentation of three pBRI1:BZR1-GFP fusion genes driven by a BRI1 promoter, including the wild-type and two mutated variants carrying the P234L mutation or lacking the BIN2-DM (ΔDM). B, RNA blot analysis with a GFP probe showing similar levels of transgene expression in three representative pBRI1:BZR1-GFP transgenic lines expressing one of the three transgenes shown in A. The lower panel shows equal loading of total RNAs by ethidium bromide staining. C, deletion of the BIN2-DM prevents BZR1 phosphorylation and inhibits BZR1 degradation in vivo. 3-Week-old seedlings were treated with 1 μm BL or 10 μm MG132 for 2 h in liquid ½ MS medium, and the amount of BZR1-GFP fusion proteins was analyzed by immunoblotting using an anti-GFP antibody. The resulting signals were digitized and quantified by ImageJ (rsbweb.nih.gov). The numbers shown above and below the protein bands in the top panel are relative abundance (%) of phosphorylated (pBZR1-GFP) and nonphosphorylated bands (BZR1-GFP), respectively, to that of the phosphorylated wild-type BZR1-GFP after normalization against the signal intensity of the nonspecific band (indicated by an asterisk). D, deletion of the BIN2-DM enhances the nuclear accumulation of BZR1-GFP in dark-grown hypocotyls. The intensity of GFP fluorescence signal was monitored at different regions of elongating hypocotyls: 1, the region just below the apical hook; 2, the upper middle region; 3, the lower middle region; and 4, the bottom region.
Deletion of the BIN2-DM Increases the Protein Abundance of BZR1
As shown in Fig. 6C, lane 7, inhibition of in vivo BZR1 phosphorylation by BIN2-DM deletion resulted in a more than 4-fold increase in the abundance of BZR1-GFP compared with its wild-type counterpart. Treatment with either BL or MG132 failed to increase but instead slightly reduced the level of BZR1(ΔDM)-GFP (Fig. 6C, lanes 8 and 9), although similar treatments of BL and MG132 did lead to an ∼19- and ∼4-fold increase in the abundance of nonphosphorylated and phosphorylated wild-type BZR1-GFP, respectively (Fig. 6C, lanes 2 and 3). The increased protein abundance of BZR1(ΔDM)-GFP was also observed through confocal microscopic analyses of the BZR1-GFP transgenic seedlings. Consistent with a previous report (26), the wild-type BZR1-GFP signal was mainly detected in the region right below the apical hook of the dark-grown seedlings where cells are rapidly elongating, and cells at the lower half of the etiolated hypocotyls showed very low green fluorescence signal (Fig. 6D, WT column). By contrast, the BZR1(ΔDM)-GFP was accumulated in the nuclei along the entire length of the hypocotyls (Fig. 6D, ΔDM column), a pattern similar to that of BZR1(P234L)-GFP protein (Fig. 6D, P234L column) (26).
The DM-deleted BZR1 Protein Enhances BR Signaling
To determine whether BZR1(ΔDM) is functional in promoting BR signaling, we germinated seeds and grew the seedlings of wild-type, bzr1-D, BZR1-GFP, BZR1(P234L)-GFP, and BZR1(ΔDM)-GFP transgenic lines in the dark on medium containing 2 μm Brz, a specific inhibitor of BR biosynthesis (41). Such a Brz resistance assay has been widely used to quantify relative strength of BR signaling. Consistent with previously published results (20, 26), both bzr1-D and the BZR1(P234L)-GFP transgenic seedlings were resistant to Brz, having longer hypocotyls than the wild-type and BZR1-GFP transgenic seedlings (Fig. 7A). The average hypocotyl lengths of the wild-type and pBRI1:BZR1-GFP transgenic seedlings are 3–4 mm, whereas the mean hypocotyl lengths of the bzr1-D and pBRI1:BZR1(P234L)-GFP transgenic seedlings are between 9 and 10 mm (Fig. 7B). Fig. 7, A and B, indicated that the pBRI1:BZR1(ΔDM)-GFP transgenic seedlings were also resistant to the Brz treatment with their average hypocotyl length similar to that of the bzr1-D mutant, indicating that the DM-deleted BZR1 protein was still active to enhance BR signaling. We also crossed the pBRI1:BZR1(ΔDM)-GFP transgene into the homozygous bin2-1 mutant that was known to accumulate BIN2 in the nucleus (14) and discovered that the hypocotyl length of the dark-grown seedlings of the resulting F1 seeds was significantly longer than the corresponding dark-grown bin2-1 heterozygous seedlings, indicating that the expression of the DM-deleted BZR1-GFP could at least partially suppress the bin2-1 mutation (Fig. 7C). Our results thus provided an unequivocal support for the nonphosphorylated BZR1 being the physiologically active form in regulating BR signaling.
FIGURE 7.

BIN2-DM-deleted BZR1 can stimulate BR signaling. A, shown here from left to right are 5-day-old Arabidopsis seedlings of wild type, the bzr1-D mutant, and three transgenic lines expressing the wild type, P234L-mutated, and DM-deleted BZR1-GFP fusion protein grown on ½ MS medium containing 2 μm Brz in the dark. B, quantitative analysis of Brz resistance by measuring average hypocotyl lengths of dark-grown seedlings of five genotypes shown in A. A total of ∼60 seedlings per genotype were manually measured from triplicate experiments, and error bars denote standard errors. C, shown here from left to right are 5-day-old Arabidopsis seedlings of the wild-type (WT), a heterozygous and a homozygous bin2-1 mutant (bin2-1/+ and bin2-1), and an F1 offspring from a cross between bin2-1 and the pBRI1:BZR1(ΔDM)-GFP transgenic line grown on ½ MS medium in the dark.
DISCUSSION
In this study, we demonstrated that an Arabidopsis GSK3-like kinase, BIN2, requires a direct kinase-substrate docking interaction to phosphorylate its substrate in vitro and in vivo. First, our yeast two-hybrid assays, in vitro protein binding experiments, and in vitro phosphorylation studies showed that a BIN2-BZR1 interaction was a prerequisite for the Arabidopsis GSK3 kinase to phosphorylate its substrate. Second, using various truncated or mutated BZR1 fusion proteins, we discovered that the BIN2-BZR1 interaction is largely mediated by a 12-amino acid DM located near the BZR1 C-terminal end. It should be interesting to mention that the C termini of a Maize BES1 homolog (accession number ACN27365.1) and a Sorghum BES1 homolog (accession number: XM_002468545) contain 3 and 4 copies of a 12-amino acid motif, F(N/S)AWEGEK(A/V)(T/S)GA, respectively, which is similar to the BIN2-DM with an identical WEGE core sequence. Third, we showed that deletion of the DM from the full-length BZR1 protein prevented its phosphorylation by BIN2 in vitro and in vivo. Importantly, we demonstrated that overexpression of the BZR1(ΔDM) protein in transgenic Arabidopsis plants gave rise to phenotypes similar to that of the bzr1-D mutant and suppressed the gain-of-function bin2-1 mutation, indicating that the 12-amino acid DM is not essential for its biological activity but serves an important regulatory function. Our study thus extended the kinase-substrate docking requirement to a plant GSK3-like kinase and uncovered a novel mechanism for a GSK3 kinase to dock its substrates carrying a portable docking motif.
Our discovery that deletion of the conserved BIN2-DM inhibited the in vivo BZR1 phosphorylation provided an opportunity to test a previous hypothesis that the BIN2-catalyzed phosphorylation of BZR1 and BES1 targets the two substrates for proteasome-mediated protein degradation (43). This hypothesis was based on several lines of indirect evidence: the BR-induced dephosphorylation and protein accumulation of BES1 and BZR1, increased protein stability of BES1 and BZR1 in the dominant bes1-D and bzr1-1D mutants with enhanced BR signaling, and increased accumulation of the phosphorylated BZR1 by treatment with MG132, a known proteasome inhibitor (21, 22, 26). However, this theory was challenged by recent reports suggesting that regulation of BR signaling by BIN2 does not involve protein degradation but is mainly mediated through inhibiting the DNA binding activity of the BIN2 substrates (14) or cytosolic localization of phosphorylated BZR1 (33–35). Our experiments using Arabidopsis transgenic lines expressing three different BZR1-GFP fusion transgenes, including the wild-type BZR1-GFP and its two mutant variants carrying the P234L mutation or lacking the BIN2-DM, revealed a likely causal relationship between BIN2-catalyzed phosphorylation and the protein stability of BZR1. We showed that deletion of the BIN2-DM from BZR1-GFP not only prevented its in vivo phosphorylation but also led to increased BZR1 abundance that could not be further increased by treatment with MG132 known to prevent degradation of phosphorylated BZR1 proteins (Fig. 5) (22). Because all three transgenes were driven by the same BRI1 promoter with similar transcript levels, the increased BZR1(ΔDM)-GFP abundance was likely caused by increased protein stability, although we could not completely eliminate the possibility that the deletion of the DM-encoding nucleotides might enhance the translational efficiency of the BZR1(ΔDM)-GFP transcripts. Thus, our study provided additional support for a role of BIN2 in regulating protein stability of its substrates, a major mechanism by which the mammalian GSK3 regulates multiple cellular processes (44).
It is thought that the kinase-substrate docking interaction is important for both substrate specificity and kinase regulation (4). The two known docking interactions of animal GSK3 kinases allow the same kinase to phosphorylate different substrates and be regulated by distinct mechanisms. Insulin signaling inhibits the kinase activity of GSK3 toward primed substrates by phosphorylating a conserved serine residue (Ser21 in GSK3α and Ser9 in GSK3β) at the N terminus of GSK3, which masks the priming phosphate binding pocket, with little impact on its phosphorylation activity toward nonprimed substrates (6). In the wingless signaling pathway, Wnt binding to its cell surface receptor Frizzled leads to recruitment of Axin, a crucial scaffolding protein for assembling the Axin-GSK3-β-catenin destruction complex, to the plasma membrane, thus preventing phosphorylation of β-catenin by GSK3 (8). Our previous study showed that one mechanism to inhibit BIN2 activity relies on proteasome-assisted protein degradation of BIN2 (45), whereas a recent report suggested that dephosphorylating an autophosphorylated tyrosine residue in the catalytic domain of BIN2 is critical for BR-mediated BIN2 inhibition (46). Our finding of a kinase-substrate docking requirement for BIN2 to phosphorylate its substrates strongly suggests that modulating the docking interaction between BIN2 and its substrates could be another important regulatory mechanism for BIN2 regulation. Such a modulation can be achieved by phosphorylation of a yet to be defined substrate-binding motif of BIN2, or the interaction between BIN2 and a regulatory BIN2-binding protein. Identification of such a substrate-binding motif and/or additional BIN2-binding proteins will lead to better knowledge of the regulatory mechanisms that inhibit the BIN2-catalyzed phosphorylation in response to the perception of steroid signals at the cell surface.
Unlike mammals that contain only two isoforms of GSK3 kinase (GSK3α and GSK3β) (47), Arabidopsis contains BIN2 and 9 other GSK3-like kinases that are thought to be involved in other physiological processes (48), including flower development (49, 50), stress, and wounding responses (51–53). Given the fact that the BIN2-docking motif exists in only six Arabidopsis proteins (20), all of which are regulated by BRs (25), it is quite possible that plant GSK3 kinases phosphorylate other substrates through different binding motifs. On the other hand, the three basic residues involved in binding primed phosphate within the catalytic core of GSK3 are conserved in the Arabidopsis GSK3-like kinases, leaving open the possibility that these Arabidopsis GSK3-like kinases might use priming phosphorylation to bind some of their substrates. Further studies on other GSK3-like kinases and identification of their possible substrates could shed additional light on the regulatory mechanisms of plant GSK3-like kinases.
Acknowledgments
We thank Drs. M. Fang and K. Cadigan (University of Michigan) for the plasmid of the Drosophila Arm cDNA and members of Li laboratory for stimulating discussions throughout this study.
This work was supported, in whole or in part, by National Institutes of Health Grant GM060519 (to J. L.).
- BR
- brassinosteroid
- BL
- brassinolide
- DM
- docking motif
- GST
- glutathione S-transferase
- GFP
- green fluorescent protein
- MS
- Murashige and Skoog
- Brz
- brassinazole
- MBP
- maltose-binding-protein.
REFERENCES
- 1.Frame S., Cohen P. (2001) Biochem. J. 359, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doble B. W., Woodgett J. R. (2003) J. Cell Sci. 116, 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woodgett J. R. (2001) Sci. STKE 2001, re12. [DOI] [PubMed] [Google Scholar]
- 4.Harwood A. J. (2001) Cell 105, 821–824 [DOI] [PubMed] [Google Scholar]
- 5.Dajani R., Fraser E., Roe S. M., Young N., Good V., Dale T. C., Pearl L. H. (2001) Cell 105, 721–732 [DOI] [PubMed] [Google Scholar]
- 6.Frame S., Cohen P., Biondi R. M. (2001) Mol. Cell 7, 1321–1327 [DOI] [PubMed] [Google Scholar]
- 7.ter Haar E., Coll J. T., Austen D. A., Hsiao H. M., Swenson L., Jain J. (2001) Nat. Struct. Biol. 8, 593–596 [DOI] [PubMed] [Google Scholar]
- 8.Wu D., Pan W. (2010) Trends Biochem. Sci. 35, 161–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pérez-Pérez J. M., Ponce M. R., Micol J. L. (2002) Dev. Biol. 242, 161–173 [DOI] [PubMed] [Google Scholar]
- 10.Li J., Nam K. H. (2002) Science 295, 1299–1301 [DOI] [PubMed] [Google Scholar]
- 11.Choe S., Schmitz R. J., Fujioka S., Takatsuto S., Lee M. O., Yoshida S., Feldmann K. A., Tax F. E. (2002) Plant Physiol. 130, 1506–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rozhon W., Mayerhofer J., Petutschnig E., Fujioka S., Jonak C. (2010) Plant J. 62, 215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan Z., Zhao J., Peng P., Chihara R. K., Li J. (2009) Plant Physiol. 150, 710–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vert G., Chory J. (2006) Nature 441, 96–100 [DOI] [PubMed] [Google Scholar]
- 15.Clouse S. D., Sasse J. M. (1998) Annu Rev. Plant Physiol. Plant Mol. Biol. 49, 427–451 [DOI] [PubMed] [Google Scholar]
- 16.Li J., Chory J. (1997) Cell 90, 929–938 [DOI] [PubMed] [Google Scholar]
- 17.Li J., Wen J., Lease K. A., Doke J. T., Tax F. E., Walker J. C. (2002) Cell 110, 213–222 [DOI] [PubMed] [Google Scholar]
- 18.Nam K. H., Li J. (2002) Cell 110, 203–212 [DOI] [PubMed] [Google Scholar]
- 19.Wang X., Kota U., He K., Blackburn K., Li J., Goshe M. B., Huber S. C., Clouse S. D. (2008) Dev. Cell 15, 220–235 [DOI] [PubMed] [Google Scholar]
- 20.Zhao J., Peng P., Schmitz R. J., Decker A. D., Tax F. E., Li J. (2002) Plant Physiol. 130, 1221–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin Y., Wang Z. Y., Mora-Garcia S., Li J., Yoshida S., Asami T., Chory J. (2002) Cell 109, 181–191 [DOI] [PubMed] [Google Scholar]
- 22.He J. X., Gendron J. M., Yang Y., Li J., Wang Z. Y. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 10185–10190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J., Nam K. H., Vafeados D., Chory J. (2001) Plant Physiol. 127, 14–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Rybel B., Audenaert D., Vert G., Rozhon W., Mayerhofer J., Peelman F., Coutuer S., Denayer T., Jansen L., Nguyen L., Vanhoutte I., Beemster G. T., Vleminckx K., Jonak C., Chory J., Inzé D., Russinova E., Beeckman T. (2009) Chem. Biol. 16, 594–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin Y., Vafeados D., Tao Y., Yoshida S., Asami T., Chory J. (2005) Cell 120, 249–259 [DOI] [PubMed] [Google Scholar]
- 26.Wang Z. Y., Nakano T., Gendron J., He J., Chen M., Vafeados D., Yang Y., Fujioka S., Yoshida S., Asami T., Chory J. (2002) Dev. Cell 2, 505–513 [DOI] [PubMed] [Google Scholar]
- 27.He J. X., Gendron J. M., Sun Y., Gampala S. S., Gendron N., Sun C. Q., Wang Z. Y. (2005) Science 307, 1634–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li L., Yu X., Thompson A., Guo M., Yoshida S., Asami T., Chory J., Yin Y. (2009) Plant J. 58, 275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L. Y., Bai M. Y., Wu J., Zhu J. Y., Wang H., Zhang Z., Wang W., Sun Y., Zhao J., Sun X., Yang H., Xu Y., Kim S. H., Fujioka S., Lin W. H., Chong K., Lu T., Wang Z. Y. (2009) Plant Cell 21, 3767–3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L., Ye H., Guo H., Yin Y. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 3918–3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X., Li L., Li L., Guo M., Chory J., Yin Y. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 7618–7623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mora-García S., Vert G., Yin Y., Caño-Delgado A., Cheong H., Chory J. (2004) Genes Dev. 18, 448–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai M. Y., Zhang L. Y., Gampala S. S., Zhu S. W., Song W. Y., Chong K., Wang Z. Y. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 13839–13844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gampala S. S., Kim T. W., He J. X., Tang W., Deng Z., Bai M. Y., Guan S., Lalonde S., Sun Y., Gendron J. M., Chen H., Shibagaki N., Ferl R. J., Ehrhardt D., Chong K., Burlingame A. L., Wang Z. Y. (2007) Dev. Cell 13, 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryu H., Kim K., Cho H., Park J., Choe S., Hwang I. (2007) Plant Cell 19, 2749–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guan K. L., Dixon J. E. (1991) Anal. Biochem. 192, 262–267 [DOI] [PubMed] [Google Scholar]
- 37.Lefèvre F., Rémy M. H., Masson J. M. (1997) Nucleic Acids Res. 25, 447–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pai L. M., Orsulic S., Bejsovec A., Peifer M. (1997) Development 124, 2255–2266 [DOI] [PubMed] [Google Scholar]
- 39.Friedrichsen D. M., Joazeiro C. A., Li J., Hunter T., Chory J. (2000) Plant Physiol. 123, 1247–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bechtold N., Pelletier G. (1998) Methods Mol. Biol. 82, 259–266 [DOI] [PubMed] [Google Scholar]
- 41.Asami T., Min Y. K., Nagata N., Yamagishi K., Takatsuto S., Fujioka S., Murofushi N., Yamaguchi I., Yoshida S. (2000) Plant Physiol. 123, 93–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu C., Li Y., Semenov M., Han C., Baeg G. H., Tan Y., Zhang Z., Lin X., He X. (2002) Cell 108, 837–847 [DOI] [PubMed] [Google Scholar]
- 43.Li J. (2005) Curr. Opin. Plant Biol. 8, 526–531 [DOI] [PubMed] [Google Scholar]
- 44.Xu C., Kim N. G., Gumbiner B. M. (2009) Cell Cycle 8, 4032–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng P., Yan Z., Zhu Y., Li J. (2008) Mol. Plant 1, 338–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim T. W., Guan S., Sun Y., Deng Z., Tang W., Shang J. X., Burlingame A. L., Wang Z. Y. (2009) Nat. Cell Biol. 11, 1254–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woodgett J. R. (1990) EMBO J. 9, 2431–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jonak C., Hirt H. (2002) Trends Plant Sci. 7, 457–461 [DOI] [PubMed] [Google Scholar]
- 49.Dornelas M. C., Van Lammeren A. A., Kreis M. (2000) Plant J. 21, 419–429 [DOI] [PubMed] [Google Scholar]
- 50.Claisse G., Charrier B., Kreis M. (2007) Plant Mol. Biol. 64, 113–124 [DOI] [PubMed] [Google Scholar]
- 51.Piao H. L., Pih K. T., Lim J. H., Kang S. G., Jin J. B., Kim S. H., Hwang I. (1999) Plant Physiol. 119, 1527–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piao H. L., Lim J. H., Kim S. J., Cheong G. W., Hwang I. (2001) Plant J. 27, 305–314 [DOI] [PubMed] [Google Scholar]
- 53.Jonak C., Beisteiner D., Beyerly J., Hirt H. (2000) Plant Cell 12, 1467–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]