

Abstract
Tumorigenesis requires the concerted action of multiple pathways, including pathways that stimulate proliferation and metabolism. Epidermal growth factor receptor (EGFR) is a transmembrane receptor-tyrosine kinase that is associated with cancer progression, and the EGFR inhibitors erlotinib/tarceva and tyrphostin/AG-1478 are potent anti-cancer therapeutics. Pgrmc1 (progesterone receptor membrane component 1) is a cytochrome b5-related protein that is up-regulated in tumors and promotes cancer growth. Pgrmc1 and its homologues have been implicated in cell signaling, and we show here that Pgrmc1 increases susceptibility to AG-1478 and erlotinib, increases plasma membrane EGFR levels, and co-precipitates with EGFR. Pgrmc1 co-localizes with EGFR in cytoplasmic vesicles and co-fractionates with EGFR in high density microsomes. The findings have therapeutic potential because a Pgrmc1 small molecule ligand, which inhibits growth in a variety of cancer cell types, de-stabilized EGFR in multiple tumor cell lines. EGFR is one of the most potent receptor-tyrosine kinases driving tumorigenesis, and our data support a role for Pgrmc1 in promoting several cancer phenotypes at least in part by binding EGFR and stabilizing plasma membrane pools of the receptor.
Keywords: Breast Cancer, Cancer Therapy, Drug Action, Lung, Receptor Regulation, Receptor-tyrosine Kinase, EGFR, Experimental Therapeutics, Lung Cancer
Introduction
The proliferation of a number of cancers is driven by receptor-tyrosine kinases, which span the cell membrane and transmit signals from polypeptide hormones. Activation of the epidermal growth factor receptor (EGFR)2-tyrosine kinase has been linked to increased proliferation, angiogenesis, metastasis, and decreased apoptosis (1). EGFR is up-regulated in a variety of tumors (2), and EGFR and HER2/neu overexpression are associated with a poor prognosis in multiple tumor types (3). EGFR is inhibited by a growing number of drugs, including antibodies such as cetuximab and small molecule inhibitors such as erlotinib (Tarceva/OSI-774) and gefitinib (3, 4). However, the degree to which patients respond favorably to these drugs varies markedly between those expressing wild-type EGFR and mutants (5–7). One of the emerging themes in targeting kinases is the importance of processing events, intracellular transport, and interaction partners among the kinases (8, 9). In the present study we demonstrate an association between EGFR and progesterone receptor membrane component 1 (Pgrmc1).
Pgrmc1 is related to cytochrome b5 and has binding sites for Src homology 2 (SH2) and SH3 domain-containing proteins and consensus phosphorylation sites for tyrosine kinases (10). Pgrmc1 is induced in a variety of cancers, including breast, thyroid, colon, ovary, and lung cancer (11–16). Furthermore, Pgrmc1 expression increases with tumor stage in ovarian cancer (11), and Pgrmc1 is elevated in estrogen receptor-negative breast tumors (17, 18). Pgrmc1 was originally identified by its induction by dioxin during liver tumorigenesis (19) and is one of six genes in a signature predicting non-genotoxic carcinogens (20). Pgrmc1 is required for key functions in tumor growth, promoting survival in cancer cells (21) particularly after damage from chemotherapeutic drugs (11, 22). The damage resistance function of Pgrmc1 is conserved with its yeast homologues (23–25).
Pgrmc1 promotes multiple phenotypes in cancer cells, including apoptotic resistance, anchorage-independent growth, invasion, tumor growth, and metastasis (21, 26, 27). In some cases progesterone signaling promotes the anti-apoptotic activity of Pgrmc1 (21), and Pgrmc1 was originally named Hpr6.6 (human membrane progesterone receptor) and mPR (membrane progesterone receptor) (29). Pgrmc1 does not share any apparent homology to hormone receptors (30, 31), and recombinant Pgrmc1 does not have direct progesterone binding activity (30). Furthermore, in lung cancer cells progesterone inhibits growth, and Pgrmc1 antagonizes this activity(27). However, Pgrmc1 is required for some aspects of progesterone signaling through an unidentified mechanism, which may include binding to the RNA-binding protein PAIR-BP1 (plasminogen activator inhibitor 1 mRNA-binding protein (32)].
Pgrmc1 is related to cytochrome b5 and binds to heme (23, 25, 33, 34). Pgrmc1 is highly expressed in the liver, where it likely binds to P450 proteins (34) and proteins that regulate cholesterol synthesis (35). However, Pgrmc1 binding partners in cancer cells are largely unknown. One intriguing possibility is that Pgrmc1 promotes cancer cell proliferation via altered signaling. In support of this model, Pgrmc1 increases cell signaling in breast cancer cells (17, 36), elevating the phosphorylation of the serine-threonine kinase Akt after damage and the apoptosis inhibitor IκB basally (36). The Caenorhabditis elegans homologue of Pgrmc1, VEM-1, is also implicated in cell signaling during development (37).
In the present study we provide a new mechanism through which Pgrmc1 promotes tumor growth. We show that Pgrmc1 binds to EGFR and stabilizes EGFR at the plasma membrane. We have found that EGFR and Pgrmc1 co-localize in a microsomal fraction, where Pgrmc1 is found in the lumen. Pgrmc1 increases susceptibility to EGFR inhibitors, likely because it increases EGFR levels at the plasma membrane. Finally, we have shown that a Pgrmc1 ligand induces EGFR degradation and antagonizes the activity of EGFR inhibitors. The results suggest that Pgrmc1 acts, at least in part, by regulating EGFR.
EXPERIMENTAL PROCEDURES
Tissue Culture and RNAi
Cells were grown in Dulbecco's modified Eagle's medium with 10% serum supreme (Lonza, Basel, Switzerland) and antibiotics and were maintained at 37 °C in 5% CO2 in air. A549, MDA-MB-231, and HCC827 cells were purchased from the American Type Culture Collection. MDA-MB-468 and H1650 cells were generously provided by Drs. Rina Plattner and Heinz Kohler (University of Kentucky Markey Cancer Center). H157 and H358 cells were provided by Dr. Hsin-Hsiung Tai (University of Kentucky College of Pharmacy). For growth curves, cells were plated in 24-well dishes, harvested, and counted using a hemocytometer. The A549 derivatives infected with lentiviruses expressing control and Pgrmc1-knockdown short hairpin RNAs have been described previously (27). RNA inhibition by siRNA transfected was performed as described (22, 38). The Ad-LacZ and Ad-Pgr-hbd (previously called Ad-Hprhbd) adenoviruses have been described (22). The EGFR inhibitor AG1478 was from Biomol, Inc. (Plymouth Meeting, PA), and erlotinib was from LC Laboratories (Woburn, MA). AG-205 was purchased from Timtec, Inc. (Newark, DE). To construct the green fluorescent protein-Pgrmc1-encoding plasmid, the 5′ BamHI-ApaI fragment of Pgrmc1 was subcloned from the plasmid pRC40 into the BglII and ApaI sites of pEGFP-N1, forming the plasmid pRC76. The 3′ end of Pgrmc1 was then subcloned into the ApaI and AgeI sites of pRC76 by PCR using the primers Pgr+309F (TACGGGCCCGAGGGGCCGTATGG) and Pgr+563R-Age (CCTACCGGTCCATCATTTTTCCGGGCACA). The resulting plasmid fused the entire Pgrmc1 open reading frame to green fluorescent protein and was called pRC77. The identity of both plasmids was verified by sequencing. Plasmids were transfected using the Transpass reagent (New England Biolabs) following the manufacturer's instructions. Cells were visualized using a Leica DM IRBE inverted microscope at the University of Kentucky Imaging Facility.
Immunological Techniques
Protein levels were analyzed by Western blot using previously described techniques (38). The antibodies used in the study were anti-cadherin (Cell Signaling, Danvers, MA), anti-calnexin (C-20, Santa Cruz Biologicals, Santa Cruz, CA), anti-calnexin (Genscript, Scotch Plains, NJ), anti-caveolin (N-20, Santa Cruz), anti-EGFR (1005, Santa Cruz), CD9 (MEM-61, Santa Cruz), IMC-C225 (erbitux, ImClone Sytems, Branchburg, NJ), anti-fusin (H-118, Santa Cruz), anti-IGF1-R (Genscript), anti-Kit (Genscript), anti-ku70 (A-9, Santa Cruz), anti-Met (C-12, Santa Cruz), anti-poly(ADP-ribose) polymerase (H-250, Santa Cruz), anti-proliferating cell nuclear antigen (PCNA; PC-10, Santa Cruz), anti-phospho-EGFR-Tyr-992 (Upstate Biologicals, Lake Placid, NY), anti-phospho-EGFR-Tyr-1173 (Santa Cruz), anti-phosphotyrosine (Tyr(P)-100, Cell Signaling), anti-Rab5 (Genscript), anti-Shp1 (Upstate Biologicals) and anti-transferrin receptor (TfR; Biolegend, San Diego, CA, clone MEM-75). Western blots for Pgrmc1 were performed with the PGR-UK1 polyclonal antibody (27). Western blots were performed at least in duplicate. Proteins were immunoprecipitated in Nonidet P-40 buffer as described previously (39). For immunofluorescence, cells were fixed in 3.7% formaldehyde and permeabilized with 1% Triton X-100 before staining, as described previously (12). EGFR was detected with the Ab-13 antibody (Thermo Scientific, Fremont, CA).
For cell surface biotinylation, A549 cells were labeled with sulfo-NHS-SS-biotin (sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate), and the labeled proteins were purified with avidin-agarose using the Cell Surface Protein Isolation kit (Thermo Scientific). For comparison, the intracellular protein pool that did not bind avidin-agarose was also collected and stored as the “unbound” fraction. Cell surface labeling reactions were performed in triplicate, and 20 μl of the biotin-labeled eluate or unbound fraction were analyzed by SDS-PAGE or Western blot.
Density Gradient Centrifugation
All steps were performed according to Macdonald and Pike (40). Briefly, cells were scraped into cold TSCM buffer (20 mm Tris-HCl, pH 7.4, 250 mm sucrose, 1 mm CaCl2, and 1 mm MgCl2) and centrifuged at 250 × g for 2 min at 4 °C. The cells were then resuspended in 1 ml of TSCM buffer with protease inhibitors (0.1 m phenylmethylsulfonyl fluoride, aprotinin, and leupeptin) and 0.1 m Na3VO4, lysed by passage through 18-gauge needle 20 times, and centrifuged at 3900 rpm for 10 min at 4 °C. The supernatant was transferred into a clean tube, and the pellets were resuspended in 1 ml of TSCM buffer, homogenized, and centrifuged as described above. The two supernatants were combined and mixed with an equal volume of TS buffer (20 mm Tris-HCl, pH 7.4, and 250 mm sucrose) containing 50% OptiPrep (Sigma). The mixture was then overlaid by a step gradient of 2 ml each of 20, 15, 10, and 5% OptiPrep in TS buffer. The gradient was centrifuged in a SW-41 Ti rotor at 52,000 × g for 90 min at 4 °C. Fractions (1 ml) were collected from the bottom of the tube, and the distribution of proteins was analyzed by Western blot.
Proteolytic Microsome Digestion
2 × 108 cells were washed once with phosphate-buffered saline and re-suspended in 6 ml of TS buffer (with protease inhibitors) and homogenized with 30 strokes from a Dounce homogenizer. The lysates were then centrifuged at 12,000 × g for 20 min at 4 °C, and the supernatants were centrifuged again at 100,000 × g for 45 min at 4 °C. The pellets were re-suspended in TS buffer and incubated with 0.01–5 μg/ml proteinase K (Sigma) with or without 1% Triton X-100 for 40 min at 32 °C. The reaction was stopped by adding protease inhibitors and incubating the reactions on ice for 10 min.
RESULTS
In A549 non-small cell lung cancer cells, Pgrmc1 promotes proliferation in the absence of serum. To test the model that Pgrmc1 elevates growth factor receptor function, we treated A549 cells with the EGFR inhibitors AG1478/tyrphostin and erlotinib. Pgrmc1 knockdown suppressed growth (Fig. 1A, dashed line, A549/RNAi cells), as expected, and the difference in viability between A549/con and A549/RNAi cells was no longer significant after treatment with AG1478 (Fig. 1A) or erlotinib (data not shown). A549 cells expressing the dominant-negative Pgrmc1 mutant, Pgr-hbd (22), also have diminished proliferation (Fig. 1B, zero point), and the difference between A549 cells expressing a control protein or Pgr-hbd was eliminated by AG1478 treatment (Fig. 1B).
FIGURE 1.

Disrupting Pgrmc1 function suppresses sensitivity to EGFR inhibitors. A, A549/con (solid line) or A549/RNAi (dashed line) cells were maintained in media lacking serum and treated with 2.5–10 μm EGFR inhibitor AG1478 for 96 h. Percent viability was determined by cell counting, and for all of the panels % viability refers to the cell density relative to untreated cells. B, AG1478 susceptibility in Ad-LacZ and Ad-Pgr-hbd-infected A549 cells, were measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay 4 days after infection in serum-free media containing increasing doses of AG1478. Solid lines represent cells infected with the control Ad-LacZ, whereas Ad-Pgr-hbd-infected cells are indicated by a dashed line. C, A549 cells were treated with vehicle (solid line) or 10 μm AG-205 (dashed line) plus increasing doses of erlotinib and counted. D, MDA-MB-231 breast cancer cells were treated with AG-205 and erlotinib as described in panel C. Each of the experiments is representative of experiments performed at least in triplicate. The results indicate that increases in proliferation in Pgrmc1-expressing cells are reversed by EGFR inhibitors.
Pgrmc1 is inhibited by the compound AG-205 (28), which arrests growth in lung cancer cells. Although erlotinib suppressed growth in vehicle-treated cells (Fig. 1C, solid line), it had no effect on viability in combination with AG-205 (Fig. 1C, dashed line). We then tested the same drug combinations in the breast cancer cell line MDA-MB-231 and found that AG-205 antagonized the anti-neoplastic activity of erlotinib, as it did in lung cancer cells (Fig. 1D). One explanation for these findings is that EGFR activity is diminished basally in A549/RNAi cells.
Next we determined the extent to which Pgrmc1 alters EGFR plasma membrane levels. Cell surface proteins were labeled with biotin, purified with avidin-agarose, and analyzed by Western blot. Pgrmc1 knockdown decreased plasma membrane EGFR levels by 7.4-fold (Fig. 2A, lane 4, A549/RNAi cells) compared with control cells (Fig. 2A, lane 3), a significant result (p = 0.01, t test) in triplicate labeling reactions. In contrast, both of the plasma membrane proteins E-cadherin and CXCR4 were not significantly changed in A549/RNAi cells (Fig. 2, C and D), and SDS-PAGE of the avidin-bound proteins revealed few changes in band intensity (Fig. 2E). The nuclear protein PCNA served as a control for intracellular proteins, and PCNA was present primarily in the protein fraction that did not bind avidin (Fig. 2B, lanes 1 and 2). Although PCNA bound slightly to avidin (Fig. 2B, lanes 3 and 4), 36-fold more PCNA was in the unbound fraction when taking into account the relative dilution of the bound and unbound samples. When Pgrmc1 was inhibited by siRNA at a different site in the Pgrmc1 transcript, EGFR membrane levels were similarly decreased (Fig. 2, F and G). Immunofluorescence for EGFR confirmed increased plasma membrane staining in A549/con compared with A549/RNAi cells (Fig. 2, H and I). The results indicate that Pgrmc1 stabilizes EGFR pools at the plasma membrane.
FIGURE 2.

Pgrmc1 increases plasma membrane-associated EGFR levels. In panels A–E, the cell surface proteins of A549/con and A549/RNAi cells were biotin-labeled with sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate and purified using avidin-agarose columns. Lanes 1 and 2 of panels A and B are Western blots of the proteins that failed to bind to the avidin-agarose columns (unbtn), whereas lanes 3 and 4 are Western blots of the avidin-bound proteins (biotin). The Western blots were probed for EGFR (A), PCNA (B), E-cadherin (C), and CXCR4/fusin (D). In panel E, one-tenth of the total pool of avidin-bound proteins was separated by SDS-PAGE and stained with Coomassie Blue. The 7.4-fold decrease in EGFR at the plasma membrane was repeated in triplicate biotinylation reactions and was statistically significant (p = 0.01). In panels F and G, Pgrmc1 expression was inhibited by siRNA transfection, and the cells were labeled and analyzed similarly to panels A–D for EGFR (F) and E-cadherin (G). In H and I, EGFR was stained by immunofluorescence (IF) in A549/con and A549/RNAi cells, respectively, indicating an increased intracellular pool of the receptor.
We then tested the extent to which Pgrmc1 regulates EGFR directly. In A549 lung cancer cells, Pgrmc1 was detected in EGFR precipitates (Fig. 3A, lower panel, lane 2) but not in precipitates using an unrelated antibody (Fig. 3A, lane 1). In the inverse experiment, EGFR co-precipitated with Pgrmc1 (Fig. 3A, lane 4). As a control, the same lysates were precipitated with preimmune serum from the same animal in which the Pgrmc1 antibody was raised (Fig. 3A, lane 3). The EGFR-Pgrmc1 association was not altered after EGF stimulation (Fig. 3B, lanes 1 and 2), suggesting that the proteins are constitutively associated. As an additional control, EGFR was precipitated from A549/con and A549/RNAi cells, and Pgrmc1 co-precipitating with EGFR decreased 5-fold in A549/RNAi cells compared with A549/con cells (Fig. 3C, lower panel). Similar to A549 cells, Pgrmc1 co-precipitated with EGFR in MDA-MB-231 breast cancer cells (Fig. 3D, lower panel).
FIGURE 3.

EGFR and Pgrmc1 co-precipitate and co-localize. A, EGFR was precipitated with the antibody (Ab) IMC-C225 from serum-starved A549 cells and probed for EGFR (top panel) or Pgrmc1 (second panel). Lane 1 is a control precipitation with an irrelevant antibody. For the inverse experiment, Pgrmc1 was precipitated from A549 cells with pre-immune serum (PIS, lane 3) or an anti-Pgrmc1 antibody (α-Pgr, lane 4). EGFR was detected in the latter reaction (upper panel, lane 4). WB, Western blot. B, Pgrmc1 was precipitated from A549 cells before (lane 1) and after (lane 2) stimulation with 50 ng/ml EGF for 15 min, and precipitates were probed for Pgrmc1 (upper panel) or EGFR (lower panel). C, EGFR was immunoprecipitated (IP) from serum-starved A549/con (lane 1) or A549/RNAi (lane 2) cells. Immunoprecipitation reactions were probed for EGFR (top) or Pgrmc1 (bottom). D, EGFR was immunoprecipitated with IMC-C225 from MDA-MB-231 breast cancer cells, and precipitation reactions were analyzed by Western blot for EGFR (upper panel) or Pgrmc1 (lower panel). E, the upper panels show fluorescence of Pgrmc1- green fluorescent protein expressed in A549 cells and immunofluorescence (IF) for EGFR, which was detected with a rhodamine-labeled secondary antibody. The lower panel shows a merged image, indicating that Pgrmc1 and EGFR co-localize to an intracellular region adjacent to the nuclear membrane. The bar indicates 25 μm.
The Met receptor-tyrosine kinase is a binding partner for EGFR, and Met was less abundant in EGFR precipitation reactions from Pgrmc1-knockdown cells than control cells (supplemental Fig. 1A, middle panel). In contrast, the Shp1 protein-tyrosine phosphatase preferentially precipitated with EGFR from A549/RNAi cells compared with A549/con cells (supplemental Fig. 1A, lower panel). EGFR Tyr-1173 is a binding site for Shp1 (41), and we found that basal EGFR Tyr1173 phosphorylation was elevated in A549/RNAi cells (supplemental Fig. 1B). The results suggest that Pgrmc1 suppresses basal EGFR-Tyr1173 phosphorylation, promotes an association between EGFR and Met at the plasma membrane, and inhibits the stability of the EGFR-Shp1 complex.
Pgrmc1 has been previously reported as a phosphoprotein (17), and immunoprecipitated Pgrmc1 reacted with an anti-phosphotyrosine antibody (supplemental Fig. 1C). After EGF stimulation, Pgrmc1 phosphorylation decreased slightly and was returned to its previous levels after 15 min of EGF treatment (supplemental Fig. 1C). To test whether EGFR phosphorylated Pgrmc1, we immunoprecipitated EGFR and performed an in vitro kinase assay with recombinant Pgrmc1 expressed in bacteria. We detected only a weakly phosphorylated band co-migrating with Pgrmc1 (data not shown), suggesting that Pgrmc1 is not a direct EGFR substrate.
To determine the subcellular localization in which Pgrmc1 interacts with EGFR, A549 cells were transfected with a Pgrmc1-green fluorescent protein fusion vector, stained for EGFR, and analyzed by fluorescence microscopy. As expected, Pgrmc1 localized to a perinuclear ring and to punctate sites that were broadly distributed in the cytoplasm (Fig. 3E, upper left), although in some views, more pronounced vesicles were detected in the cytoplasm. EGFR also localized to punctate cytoplasmic sites and to the cell periphery (Fig. 3E, upper right) and the two proteins co-localized in the punctate cytoplasmic sites (Fig. 3E, lower panel). To further characterize the region in which Pgrmc1 and EGFR co-localized, we fractionated cells by density gradient centrifugation. EGFR was concentrated in both high and low density fractions (Fig. 4A, lanes 1 and 5–8, respectively), whereas Pgrmc1was concentrated in the highest density fraction (Fig. 4B, lane 1). This high density fraction includes the endoplasmic reticulum marker calnexin (Fig. 4C, lane 1) and the secretory vesicle marker Rab5 (Fig. 4D). The plasma membrane localized to fractions 3–8, using the plasma membrane protein CD9 as a marker (Fig. 4E). Caveolin was detected throughout the gradient (Fig. 4F), suggesting that proteins were present in each fraction.
FIGURE 4.
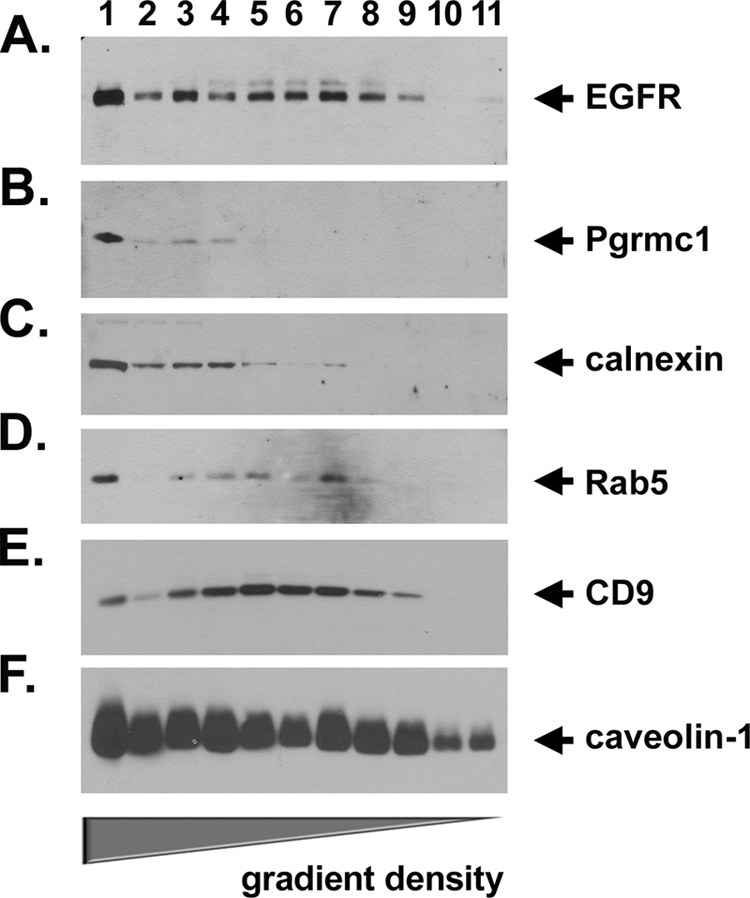
EGFR and Pgrmc1 co-fractionate in a high density vesicle fraction. A549 cells were lysed and fractionated on an Opti-prep density gradient then analyzed by Western blot for EGFR (A), Pgrmc1 (B), calnexin (C, a marker for the endoplasmic reticulum), Rab5 (D, a marker for secretory vesicles), CD9 (E, a plasma membrane protein), and caveolin (lower panel). The results suggest that Pgrmc1 and EGFR co-fractionate primarily in intracellular microsomes.
To map the topology of Pgrmc1, we isolated a microsomal fraction from A549 lysates by centrifugation at 12,000 × g and 100,000 × g. The final pellet was then digested with proteinase K with and without Triton X-100 to determine the extent to which proteins are protected from digestion within the microsomal lumen (diagrammed in Fig. 5A). Pgrmc1 was cleaved to a lower molecular weight form and then completely degraded when detergent was present (Fig. 5B, lanes 8–10). In contrast, Pgrmc1 was protected from degradation at the 0.1 μg/ml dose of proteinase K when the microsomal membranes were intact (Fig. 5B, lane 3). The protease was active in both sets of samples because the cytoplasmic terminus of calnexin was efficiently digested in the absence and presence of detergent (Fig. 5C, lanes 3–5 and 8–10). The amino terminus of calnexin, which faces the endoplasmic reticulum lumen, was protected from digestion, although it was digested to a lower molecular weight form (Fig. 5D, lanes 2–5). Protein disulfide isomerase, an endoplasmic reticulum lumen protein, was digested in the presence of detergent but protected in its absence (Fig. 5C, lanes 8–10 and 3–5, respectively). In contrast, Pgrmc1 was digested at the 1 and 5 μg/ml doses of proteinase K, whereas protein disulfide isomerase and calnexin were not (Fig. 5D-E, lanes 4–5), suggesting that Pgrmc1 localizes to a type of microsome that is sensitive to digestion with high levels of proteinase K.
FIGURE 5.

Pgrmc1 localizes to the microsomal lumen. A, shown is a diagram of the protease digestion experiments used in panels B–E. An isolated microsomal fraction was incubated with proteinase K (dark gray circles) in the absence (left) and presence (right) of detergents to distinguish proteins that were protected by the microsomal membranes from cytoplasmic proteins. B–E, Western blots of microsomal fractions incubated without protease (lanes 1 and 6) or with 0.01 (lanes 2 and 7), 0.1 (lanes 3 and 8), 1 (lanes 4 and 9), or 5 μg/ml (lanes 5 and 10) proteinase K in the absence (lanes 1–5) or presence (lanes 6–10) of 1% Triton X-100 are shown. Blots were probed for Pgrmc1 (B), calnexin (cytoplasmic epitope (C) and lumen epitope (D)) and protein disulfide isomerase (PDI) (E, which localizes to the endoplasmic reticulum lumen). The results indicate that Pgrmc1 is protected from digestion by the microsomal membrane (compare lanes 3 and 8) but localizes to microsomes that are sensitive to high levels of proteinase K.
MDA-MB-468 human breast cancer cells are a model system for EGFR regulation because they express high levels of the receptor. Pgrmc1 expression was inhibited with two separate siRNA oligonucleotide duplexes to distinct regions of the Pgrmc1 coding sequence (diagrammed in Fig. 6A). We will refer to the siRNAs as siPGR and siPGR2. As a control, parallel cultures were transfected with a control siRNA called siCON. Transfection with siPGR caused a nearly complete inhibition of EGFR levels (Fig. 6B, lane 2). This was reflected in a 28-fold decrease in the predominant 180-kDa tyrosine-phosphorylated band (Fig. 6C, lane 2). As expected, Pgrmc1 levels were almost completely inhibited (Fig. 6D, lane 2). We utilized the DNA end-binding protein ku70 as a control for equal protein loading (Fig. 6E) because we did not detect any ku70 alterations in these experiments. Pgrmc1 did not affect EGFR transcription because EGFR transcript levels were unchanged in siPGR-transfected cells (supplemental Fig. 2A), whereas Pgrmc1 levels decreased (supplemental Fig. 2B). Pgrmc1 demonstrated a degree of specificity for EGFR, because levels of the receptor-tyrosine kinases insulin-like growth factor 1 receptor (IGF-1R) and c-Kit and the membrane-associated transferrin receptor (tfR) were unaffected or modestly induced by the loss of Pgrmc1 (Fig. 6, F–H). A second siRNA targeting Pgrmc1 (which we will refer to as siPGR2, Fig. 6A) attenuated EGFR levels to a lesser extent (Fig. 6B, lanes 3 and 4). Using the siPGR2 siRNA, the levels of the 180-kDa tyrosine-phosphorylated band were diminished (Fig. 6C, lanes 3 and 4), whereas ku70 was unaffected (Fig. 6E, lanes 3 and 4).
FIGURE 6.

Pgrmc1 increases EGFR levels in MDA-MB-468 breast cancer cells. Panel A is a diagram indicating the positions of the siRNA molecules targeting Pgrmc1. Exons 1, 2, and 3 are indicated by ex. MDA-MB-468 cells were transfected with a control siRNA (siCON, lanes 1 and 3) or two separate siRNAs targeting Pgrmc1 (siPGR and siPGR2, lanes 2 and 4, respectively). In panels B–E, protein levels were analyzed by Western blot for EGFR (B), phosphotyrosine (C), Pgrmc1 (D), and ku70 (E) as a control for protein loading. Panels F–H show Western blot analyses for insulin-like growth factor 1 receptor (IGF-1R; F), c-Kit (G), and transferrin receptor (tfR) (H). I, MDA-MB-468 cells transfected with siCON (solid line) or siPGR (dashed line) were incubated with increasing doses of the EGFR inhibitor AG1478, and viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. % viability refers to absorbance after treatment relative to untreated control cells. In panels J-K, MDA-MB-468 cells were treated with increasing doses of the Pgrmc1 ligand AG-205. Protein expression was analyzed by Western blot for EGFR (J) and ku70 (K). Cells were treated with 0 (lanes 1), 2 (lanes 2), 10 (lanes 3), or 50 (lanes 4) μm AG-205 for 24 h in serum-free medium. In panels L–M, A549 cells were treated with the same doses and analyzed for EGFR (L) and ku70 (M). The results show that the Pgrmc1 ligand AG-205 decreases EGFR protein levels. Expression analyses were repeated at least in duplicate throughout.
MDA-MB-468 cells transfected with siPGR proliferated at a slower rate than control cells (Fig. 6I, 0 dose), and as for A549 and MDA-MB-231 cells, Pgrmc1-knockdown cells were less susceptible than control cells to the EGFR inhibitor AG-1478 (Fig. 6I, dashed line). The Pgrmc1 inhibitor AG-205 reduced EGFR levels 8.7 + 2.5-fold in MDA-MB-468 cells (Fig. 6J, p = 0.04, t test). ku70 levels were not affected (Fig. 6K). Similarly, AG-205 treatment reduced EGFR levels in A549 cells (Fig. 6L) by 4.5 ± 1.6-fold (p = 0.004, t test), whereas ku70 levels (Fig. 6 m) were unchanged, and EGFR transcription was not altered (data not shown).
Because Pgrmc1 regulates EGFR levels, we then tested the extent to which cells expressing different EGFR variants respond to AG-205. A549, H157, and H358 lung cancer cells express the wild-type form of EGFR, and all have similar levels of susceptibility to AG-205 (Table 1). MDA-MB-231 and MDA-MB-468 breast cancer cells express wild-type EGFR, although EGFR expression in the latter is greatly elevated, and both lines were sensitive to AG-205. Like MDA-MB-468 cells, A431 cells overexpress EGFR, and A431 cells were the most AG-205-sensitive cell line tested (Table 1). Indeed, A431 cells rounded and demonstrated markers of apoptosis after AG-205 treatment (data not shown). In contrast, HCC827 and H1650 cells, which express a ΔE746-A750 deletion mutant of EGFR, were relatively insensitive to AG-205 inhibition (Table 1). The results suggest that in the cell lines tested structural features of EGFR are important for the growth inhibiting activity of AG-205.
TABLE 1.
IC50 values for AG-205 in various cell lines
| Cell line | Tissue of origin | Serum-starved | EGFR |
|---|---|---|---|
| μm | |||
| A431 | Epitheloid | 8 | Wild type |
| MDA-MB-231 | Breast | 18 | Wild type |
| MDA-MB-468 | Breast | 12 | Wild type |
| A549 | Lung | 15 | Wild type |
| H157 | Lung | 10 | Wild type |
| H358 | Lung | 12 | Wild type |
| H1650 | Lung | >100 | ΔE746-A750 |
| HCC827 | Lung | >100 | ΔE746-A750 |
DISCUSSION
Pgrmc1 has been linked to cancer through its expression and biological activities. Pgrmc1 is induced by carcinogens (19) and is part of a six-gene signature that predicts non-genotoxic carcinogens (20). In addition, Pgrmc1 is overexpressed in tumors (11, 12, 16), suppresses apoptosis (11, 21, 43), and promotes anchorage-independent growth, invasion, and in vivo tumor growth (26, 27). In the present study we provide a novel activity of Pgrmc1 in cancer, increasing the cell surface stability of EGFR.
EGFR promotes proliferation and invasion in A549 cells (44–46), and Pgrmc1 promotes the same phenotypes. Both Pgrmc1-knockdown and EGFR inhibition suppress growth, but inhibition of both proteins does not have an additive effect. Indeed, Pgrmc1-knockdown cells had the same level of viability with or without EGFR inhibitors, as control cells treated with EGFR inhibitors. Pgrmc1 was inhibited by three independent approaches; that is, short hairpin RNA, expression of a heme binding-deficient mutant, and pharmacological inhibition with a Pgrmc1 ligand. The results are consistent with a model in which Pgrmc1 and EGFR function through a common pathway.
We have found that Pgrmc1 associates with EGFR and maintains EGFR at the plasma membrane. Pgrmc1 and EGFR co-localize within the same microsomal fraction, and the microsomal localization for Pgrmc1 agrees with previous reports (12, 30, 47). In contrast, exogenous forms of Pgrmc1 are sometimes localized to the nucleus in ovarian cells (11), and Pgrmc1 is also found at the plasma membrane in ovarian and neuronal cell types (43, 48) but not in A549 cells (49). Indeed, we did not detect Pgrmc1 in Western blots of plasma membrane fractions or of biotinylated proteins at the cell surface (Fig. 2 and data not shown). We have mapped the topology of Pgrmc1 to the lumen of microsomes, but Pgrmc1 is more labile to proteases than classical endoplasmic reticulum lumen proteins, calnexin, and protein disulfide isomerase. An alternate interpretation of the data is that subpopulations of Pgrmc1 localize to the cytoplasmic or lumenal surface of microsomes, but the mechanism driving this localization is unclear. Nölte et al. also concluded that Pgrmc1 localized to the microsomal lumen in rat liver extracts (47), but their analysis utilized an antibody with an amino-terminal (cytoplasmic) epitope, and the ability of the microsomal membrane to protect Pgrmc1 from degradation was untested. In the lumen, Pgrmc1 likely interacts with the EGFR extracellular, ligand binding domain. The function of Pgrmc1 in the microsomal lumen is unknown, but numerous redox associated pathways localize to the lumen, including protein folding and glycosylation pathways. Notably, Pgrmc1 homologues have reducing activity (33), and Pgrmc1 and its homologues regulate P450-mediated redox reactions (23, 30, 34). We also note that the predicted isoelectric point for Pgrmc1 is 4.56, consistent with the acidic nature of the microsomal lumen. The target of Pgrmc1 in the microsomal lumen is currently under investigation.
The ability of Pgrmc1 to stabilize EGFR at the plasma membrane follows our observation that Pgrmc1 promotes proliferation in the absence of serum but has little effect when serum is present (27). A549 cells express a mutated form of K-ras, which drives constitutive EGFR ligand secretion (50) and forms an autocrine loop promoting cell survival. Our results are consistent with a model in which, in the absence of serum growth factors, Pgrmc1 supports autocrine signaling, at least in part, through EGFR. Such autocrine signaling would not be required in serum-rich media, and we found that A549 cells are sensitive to erlotinib and tyrphostin in the absence of serum, where autocrine signaling is essential. An alternate model is that Pgrmc1 could sustain autocrine signaling through other routes, such as the release of growth factors into the media.
We also tested the hypothesis that Pgrmc1 stabilizes EGFR plasma membrane pools through increased cholesterol metabolism. Pgrmc1 elevates cholesterol synthesis under specific conditions (34), and EGFR is regulated by cholesterol levels (51). However, we were unable to detect any changes in cholesterol synthesis in Pgrmc1-knockdown lung cancer cells using two different assays (27). Our results do not exclude the possibility that Pgrmc1 may regulate signaling via cholesterol in other cell types.
In contrast to A549 cells, Pgrmc1 promoted growth in MDA-MB-468 cells even in the presence of 10% serum (27). We resorted to siRNA in MDA-MB-468 cells because short hairpin RNA resulted in abundant 3′ transcripts and the expression of a variant 24-kDa protein (data not shown). EGFR protein levels were dependent on Pgrmc1 in MDA-MB-468 cells (Fig. 6) after inhibition with two separate siRNA molecules or with the Pgrmc1 inhibitor AG-205. The dependence of EGFR levels in MDA-MB-468 cells compared with A549 cells probably reflects the more modest expression levels of wild-type EGFR in A549 cells (52).
EGFR is one of the most prominent therapeutic targets in cancer and is inhibited clinically by the antibody fragment erbitux/cetuximab and by the small molecule inhibitors erlotinib and gefitinib. These inhibitors prevent ligand binding and kinase activation, respectively, but the fraction of patients that responds to erlotinib is somewhat limited. Patients with the EGFR ΔE746-A750 mutation have an improved response to erlotinib (5, 53), perhaps because the EGFR ΔE746-A750 mutant is constitutively active and inefficiently down-regulated (54). Cell lines expressing these constitutively active EGFR mutants are not inhibited by AG-205, and RNAi for Pgrmc1 did not inhibit EGFR levels in HCC827 cells (data not shown). In contrast to the wild-type EGFR, we propose that ΔE746-A750 mutants do not require Pgrmc1 for their membrane localization.
Targeting Pgrmc1 as a therapeutic approach for cancer offers several advantages. One of the potential problems with the current strategies for inhibiting EGFR is that they target the activated state of the protein. Unfortunately, EGFR promotes growth in part in a kinase-independent manner. As a result, RNAi for EGFR is toxic to cells (55, 56), even where EGFR inhibitors are not, and deletion of the mouse EGFR gene is lethal (42), whereas loss of kinase activity is not (28). Thus, one appealing feature of Pgrmc1 inhibitors, such as AG-205, is that they potentially target both kinase-dependent and -independent EGFR functions. Furthermore, Pgrmc1 targeting may be useful for tumors that express wild-type forms of EGFR, which respond poorly to current EGFR inhibitors. Although many aspects of the delivery and toxicity of AG-205 remain to be tested, the existence of a novel pathway regulating EGFR stability suggests new avenues for research and therapeutic development, some of which may be useful for inhibiting cancer growth.
Acknowledgments
We are grateful to Drs. Graham Carpenter, David Kaetzel, Michael Kilgore, Rina Plattner, Hollie Swanson, and Xuwei Yang for helpful suggestions and to Mary Gail Engle and Jim Begley for help with imaging.
This work was supported in part by American Cancer Society Grant 85-001-19-IRG and by the Kentucky Lung Cancer Research Program.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
- EGFR
- epidermal growth factor receptor
- Pgrmc1
- progesterone receptor membrane component 1
- PCNA
- proliferating cell nuclear antigen
- siRNA
- small interfering RNA
- PBS
- phosphate-buffered saline.
REFERENCES
- 1.Ritter C. A., Arteaga C. L. (2003) Semin. Oncol. 30, 3–11 [DOI] [PubMed] [Google Scholar]
- 2.Salomon D. S., Brandt R., Ciardiello F., Normanno N. (1995) Crit. Rev. Oncol. Hematol. 19, 183–232 [DOI] [PubMed] [Google Scholar]
- 3.Ono M., Kuwano M. (2006) Clin. Cancer Res. 12, 7242–7251 [DOI] [PubMed] [Google Scholar]
- 4.Bareschino M. A., Schettino C., Troiani T., Martinelli E., Morgillo F., Ciardiello F. (2007) Ann. Oncol. 18, vi35–41 [DOI] [PubMed] [Google Scholar]
- 5.Lynch T. J., Bell D. W., Sordella R., Gurubhagavatula S., Okimoto R. A., Brannigan B. W., Harris P. L., Haserlat S. M., Supko J. G., Haluska F. G., Louis D. N., Christiani D. C., Settleman J., Haber D. A. (2004) N. Engl. J. Med. 350, 2129–2139 [DOI] [PubMed] [Google Scholar]
- 6.Paez J. G., Jänne P. A., Lee J. C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F. J., Lindeman N., Boggon T. J., Naoki K., Sasaki H., Fujii Y., Eck M. J., Sellers W. R., Johnson B. E., Meyerson M. (2004) Science 304, 1497–1500 [DOI] [PubMed] [Google Scholar]
- 7.Sequist L. V., Lynch T. J. (2008) Annu. Rev. Med. 59, 429–442 [DOI] [PubMed] [Google Scholar]
- 8.Sebastian S., Settleman J., Reshkin S. J., Azzariti A., Bellizzi A., Paradiso A. (2006) Biochim. Biophys. Acta 1766, 120–139 [DOI] [PubMed] [Google Scholar]
- 9.Laurent-Puig P., Lievre A., Blons H. (2009) Clin. Cancer Res. 15, 1133–1139 [DOI] [PubMed] [Google Scholar]
- 10.Cahill M. A. (2007) J. Steroid Biochem. Mol. Biol. 105, 16–36 [DOI] [PubMed] [Google Scholar]
- 11.Peluso J. J., Liu X., Saunders M. M., Claffey K. P., Phoenix K. (2008) J. Clin. Endocrinol. Metab. 93, 1592–1599 [DOI] [PubMed] [Google Scholar]
- 12.Crudden G., Loesel R., Craven R. J. (2005) Tumour Biol. 26, 142–146 [DOI] [PubMed] [Google Scholar]
- 13.Dressman H. K., Hans C., Bild A., Olson J. A., Rosen E., Marcom P. K., Liotcheva V. B., Jones E. L., Vujaskovic Z., Marks J., Dewhirst M. W., West M., Nevins J. R., Blackwell K. (2006) Clin. Cancer Res. 12, 819–826 [DOI] [PubMed] [Google Scholar]
- 14.Irby R. B., Malek R. L., Bloom G., Tsai J., Letwin N., Frank B. C., Verratti K., Yeatman T. J., Lee N. H. (2005) Cancer Res. 65, 1814–1821 [DOI] [PubMed] [Google Scholar]
- 15.Difilippantonio S., Chen Y., Pietas A., Schlüns K., Pacyna-Gengelbach M., Deutschmann N., Padilla-Nash H. M., Ried T., Petersen I. (2003) Eur. J. Cancer 39, 1936–1947 [DOI] [PubMed] [Google Scholar]
- 16.Rohe H. J., Ahmed I. S., Twist K. E., Craven R. J. (2009) Pharmacol. Ther. 121, 14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neubauer H., Clare S. E., Wozny W., Schwall G. P., Poznanovic S., Stegmann W., Vogel U., Sotlar K., Wallwiener D., Kurek R., Fehm T., Cahill M. A. (2008) Breast Cancer Res. 10, R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Craven R. J. (2008) Breast Cancer Res. 10, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Selmin O., Lucier G. W., Clark G. C., Tritscher A. M., Vanden Heuvel J. P., Gastel J. A., Walker N. J., Sutter T. R., Bell D. A. (1996) Carcinogenesis 17, 2609–2615 [DOI] [PubMed] [Google Scholar]
- 20.Nie A. Y., McMillian M., Parker J. B., Leone A., Bryant S., Yieh L., Bittner A., Nelson J., Carmen A., Wan J., Lord P. G. (2006) Mol. Carcinog. 45, 914–933 [DOI] [PubMed] [Google Scholar]
- 21.Peluso J. J., Romak J., Liu X. (2008) Endocrinology 149, 534–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crudden G., Chitti R. E., Craven R. J. (2006) J Pharmacol. Exp. Ther. 316, 448–455 [DOI] [PubMed] [Google Scholar]
- 23.Mallory J. C., Crudden G., Johnson B. L., Mo C., Pierson C. A., Bard M., Craven R. J. (2005) Mol. Cell. Biol. 25, 1669–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hand R. A., Jia N., Bard M., Craven R. J. (2003) Eukaryot. Cell 2, 306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson A. M., Reddi A. R., Shi X., Goldbeck R. A., Moënne-Loccoz P., Gibney B. R., Holman T. R. (2007) Biochemistry 46, 14629–14637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peluso J. J., Gawkowska A., Liu X., Shioda T., Pru J. K. (2009) Endocrinology 150, 4846–4854 [DOI] [PubMed] [Google Scholar]
- 27.Ahmed I. S., Rohe H. J., Twist K. E., Mattingly M. N., Craven R. J. (2010) J. Pharmacol. Exp. Ther. 333, 564–573 [DOI] [PubMed] [Google Scholar]
- 28.Luetteke N. C., Phillips H. K., Qiu T. H., Copeland N. G., Earp H. S., Jenkins N. A., Lee D. C. (1994) Genes Dev. 8, 399–413 [DOI] [PubMed] [Google Scholar]
- 29.Gerdes D., Wehling M., Leube B., Falkenstein E. (1998) Biol. Chem. 379, 907–911 [DOI] [PubMed] [Google Scholar]
- 30.Min L., Strushkevich N. V., Harnastai I. N., Iwamoto H., Gilep A. A., Takemori H., Usanov S. A., Nonaka Y., Hori H., Vinson G. P., Okamoto M. (2005) FEBS J. 272, 5832–5843 [DOI] [PubMed] [Google Scholar]
- 31.Mifsud W., Bateman A. (2002) Genome Biol. 3, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peluso J. J., Pappalardo A., Losel R., Wehling M. (2005) Biol. Reprod. 73, 261–270 [DOI] [PubMed] [Google Scholar]
- 33.Ghosh K., Thompson A. M., Goldbeck R. A., Shi X., Whitman S., Oh E., Zhiwu Z., Vulpe C., Holman T. R. (2005) Biochemistry 44, 16729–16736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes A. L., Powell D. W., Bard M., Eckstein J., Barbuch R., Link A. J., Espenshade P. J. (2007) Cell Metab. 5, 143–149 [DOI] [PubMed] [Google Scholar]
- 35.Suchanek M., Radzikowska A., Thiele C. (2005) Nat. Methods 2, 261–267 [DOI] [PubMed] [Google Scholar]
- 36.Hand R. A., Craven R. J. (2003) J. Cell. Biochem. 90, 534–547 [DOI] [PubMed] [Google Scholar]
- 37.Runko E., Kaprielian Z. (2004) J. Neurosci. 24, 9015–9026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mallory J. C., Crudden G., Oliva A., Saunders C., Stromberg A., Craven R. J. (2005) Mol. Pharmacol. 68, 1747–1756 [DOI] [PubMed] [Google Scholar]
- 39.Kurenova E., Xu L. H., Yang X., Baldwin A. S., Jr., Craven R. J., Hanks S. K., Liu Z. G., Cance W. G. (2004) Mol. Cell. Biol. 24, 4361–4371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macdonald J. L., Pike L. J. (2005) J. Lipid Res. 46, 1061–1067 [DOI] [PubMed] [Google Scholar]
- 41.Keilhack H., Tenev T., Nyakatura E., Godovac-Zimmermann J., Nielsen L., Seedorf K., Böhmer F. D. (1998) J. Biol. Chem. 273, 24839–24846 [DOI] [PubMed] [Google Scholar]
- 42.Miettinen P. J., Berger J. E., Meneses J., Phung Y., Pedersen R. A., Werb Z., Derynck R. (1995) Nature 376, 337–341 [DOI] [PubMed] [Google Scholar]
- 43.Peluso J. J., Pappalardo A., Losel R., Wehling M. (2006) Endocrinology 147, 3133–3140 [DOI] [PubMed] [Google Scholar]
- 44.Zhang M., Zhang X., Bai C. X., Song X. R., Chen J., Gao L., Hu J., Hong Q. Y., West M. J., Wei M. Q. (2005) Genet. Vaccines Ther. 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaramillo M. L., Banville M., Collins C., Paul-Roc B., Bourget L., O'Connor-McCourt M. (2008) Cancer Biol. Ther. 7, 557–568 [DOI] [PubMed] [Google Scholar]
- 46.Wang T., Niki T., Goto A., Ota S., Morikawa T., Nakamura Y., Ohara E., Ishikawa S., Aburatani H., Nakajima J., Fukayama M. (2007) Cancer Sci. 98, 506–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nölte I., Jeckel D., Wieland F. T., Sohn K. (2000) Biochim. Biophys. Acta 1543, 123–130 [DOI] [PubMed] [Google Scholar]
- 48.Krebs C. J., Jarvis E. D., Chan J., Lydon J. P., Ogawa S., Pfaff D. W. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 12816–12821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shin B. K., Wang H., Yim A. M., Le Naour F., Brichory F., Jang J. H., Zhao R., Puravs E., Tra J., Michael C. W., Misek D. E., Hanash S. M. (2003) J. Biol. Chem. 278, 7607–7616 [DOI] [PubMed] [Google Scholar]
- 50.Toulany M., Baumann M., Rodemann H. P. (2007) Mol. Cancer Res. 5, 863–872 [DOI] [PubMed] [Google Scholar]
- 51.Westover E. J., Covey D. F., Brockman H. L., Brown R. E., Pike L. J. (2003) J. Biol. Chem. 278, 51125–51133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldenberg A., Masui H., Divgi C., Kamrath H., Pentlow K., Mendelsohn J. (1989) J. Natl. Cancer Inst. 81, 1616–1625 [DOI] [PubMed] [Google Scholar]
- 53.Sordella R., Bell D. W., Haber D. A., Settleman J. (2004) Science 305, 1163–1167 [DOI] [PubMed] [Google Scholar]
- 54.Furukawa M., Nagatomo I., Kumagai T., Yamadori T., Takahashi R., Yoshimura M., Yoneda T., Takeda Y., Goya S., Matsuoka H., Kijima T., Yoshida M., Osaki T., Tachibana I., Greene M. I., Kawase I. (2007) DNA Cell Biol. 26, 178–185 [DOI] [PubMed] [Google Scholar]
- 55.Weihua Z., Tsan R., Huang W. C., Wu Q., Chiu C. H., Fidler I. J., Hung M. C. (2008) Cancer Cell 13, 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagy P., Arndt-Jovin D. J., Jovin T. M. (2003) Exp. Cell Res. 285, 39–49 [DOI] [PubMed] [Google Scholar]