

Abstract
The role of adenylate kinase (AK) as a determinant of K-ATP channel activity in human pancreatic β-cells was investigated. We have identified that two cytosolic isoforms of AK, AK1 and AK5 are expressed in human islets and INS-1 cells. Elevated concentrations of glucose inhibit AK1 expression and AK1 immunoprecipitates with the Kir6.2 subunit of K-ATP. AK activation by ATP + AMP stimulates K-ATP channel activity and this stimulation is abolished by AK inhibitors. We propose that glucose stimulation of β-cells inhibits AK through glycolysis and also through the elevation of diadenosine polyphosphate levels. Glucose-dependent inhibition of AK increases the ATP/ADP ratio in the microenvironment of the K-ATP channel promoting channel closure and insulin secretion. The down-regulation of AK1 expression by hyperglycemia may contribute to the defective coupling of glucose metabolism to K-ATP channel activity in type 2 diabetes.
Keywords: Adenylate kinase, K-ATP channel, Diadenosine pentaphosphate, Expression, Glucose, β-Cell, Islet
It is widely accepted that a critical early step for glucose-induced insulin secretion is the closure of ATP-sensitive potassium (K-ATP) channels in the plasma membrane of pancreatic β-cells. The closure of K-ATP channels results in β-cell depolarization, activation of voltage-sensitive calcium channels and calcium influx that stimulates insulin secretion [1].
K-ATP channels in pancreatic β-cells are hetero-octomers comprised of four type-1 sulfonylurea receptors (SUR1) and four pore-forming Kir6.2 subunits [2]. Binding of ATP to Kir6.2 inhibits channel activity whereas the SUR1 subunits have two nucleotide binding folds that interact to form nucleotide binding sites. K-ATP channel activity is inhibited when ATP is bound but increases when MgADP is bound [3]. Thus the level of K-ATP channel activity is regulated by the ratio of ATP to MgADP bound to SUR1 subunits and glucose-induced inhibition of AK activity as a regulator of nucleotide binding and channel activity is the focus of this study.
AK reversibly catalyzes the reaction ATP + AMP ↔ 2 ADP and is regulated by the availability of AMP or ADP [4]. A role for AK in regulating β-cell K-ATP channels was suggested by Larsson et al. [5] who observed that AMP increased channel activity in the presence of ATP, a condition in which AK mediated ADP production is expected. It was subsequently shown that AK activity in β-cells is inhibited by glucose and that this inhibition precedes insulin secretion [6]. These observations led to the suggestion that AK activity might determine ATP and ADP levels in the microenvironment of K-ATP channels [6].
A study using mice with targeted gene deletion of cytosolic AK1 supports a role for AK in the regulation of K-ATP channel activity in β-cells [7]. In these AK1 knockout mice, the activation of K-ATP channels by AMP in the presence of ATP is reduced, but not abolished [7]. The residual, AK1-independent, K-ATP activation could be explained by the expression of a second cytosolic AK isoform in β-cells. Herein, we present data showing that human islets express both AK1 and AK5, the two cytosolic isoforms of AK. Furthermore, we show that the expression of AK1 is decreased by high glucose, coordinate with changes in expression of K-ATP channel subunits [8]. The expression of both AK1 and AK5 in β-cells raises the possibility that both isoforms have roles in regulating the ATP/ADP ratio controlling K-ATP channel activity.
We suggest that glucose metabolism inhibits AK through two mechanisms: (1) ATP production and the depletion of AMP and (2) generation of diadenosine polyphosphates. We propose that glucose regulates both the activity and expression of cytosolic AK isoforms that control the activity of K-ATP channels and thereby control insulin secretion.
Methods
Cell culture
The culture medium for INS-1 cells was RPMI 1640 containing 10 mM HEPES, 11.1 mM glucose, 10% fetal bovine serum, 100 U/ml penicillin G, 100 μg/ml streptomycin, 2.0 mM L-glutamine, 1.0 mM sodium pyruvate, and 50 μM 2-mercaptoethanol. Human islets (obtained from the Islet Cell Resource Centers) were maintained in CMRL with 10% fetal bovine serum, 100 U/ml penicillin G and 100 μg/ml streptomycin. Prior to recording, INS-1 cells or human islets were trypsinized and plated onto Concanavalin A (1 mg/ml) coated glass coverslips. Reagents for cell culture were obtained from Invitrogen LifeTechnologies (Rockville, MD).
Measurement of K-ATP channel currents
K-ATP channel currents were measured using inside-out patches with patch pipettes pulled from borosilicate glass. Pipettes were fire-polished and filled with a solution containing (in mM): 140 KCl, 1.0 MgCl2, 2.0 CaCl2, 5 HEPES (pH 7.3). Filled pipettes had resistances of 3.5–8 MΩ. Patches were excised into a solution containing (in mM): 70 K2SO4, 2.0 MgCl2, 0.1 CaCl2, 1.1 EGTA, 0.2 GTP, 5 HEPES (pH 7.3). Currents were acquired using a HEKA EPC-9 amplifier under the control of Pulse v.8.31 software (Instrutech Crop., Mineola, NY, USA) and recorded using a Digidata 1440A data acquisition system with pClamp10 software, also used for data analysis (Molecular Devices, Sunnyvale, CA) All experiments were performed at room temperature (22–26 °C).
Cytosolic solutions for inside-out patches were rapidly exchanged using Dynaflow Pro II chambers (Cellectricon Inc., Gaithersburg, MD) under computer control. Chemicals were from Sigma–Aldrich.
Polymerase cyclase reactions (PCR) and quantitative PCR (QPCR)
PCR primers were designed against published sequences using DNA-STAR software (DNASTAR Inc., Madison, WI). The identity of PCR products was confirmed by sequencing at an MGH core facility. Primers and probes for QPCR were designed using Beacon Designer (Premier Biosoft, Palo Alto, CA) software and reactions performed using Qiagen OneStep kits with 100 ng of RNA in a Cepheid SmartCycler. RNA was isolated from cells using RNeasy kits (Qiagen Inc., Valencia, CA). Changes in expression levels were calculated using standard ΔΔCt methods relative to GAPDH as previously described [9,10].
Labeling AK1 protein and immunoprecipitation
A cDNA clone of human AK1 (MGC-1808 obtained from ATCC, Manassas, VA) was subcloned into the pcDNA3.1/V5-His vector (Invitrogen Corp., Carlsbad, CA). This DNA template was used for in vitro transcription/translation with 35S-methionine using a TNT T7 reticulocyte lysate system (Promega, Madison, WI) for 90 min at 30 °C. This 35S-labeled AK1 protein was added to either cell-free RIPA lysis buffer (RIPA) or to whole-cell extracts (in RIPA) from mouse brain or membrane preparations from MIN6 cells and resuspended in RIPA. An antibody to Kir6.2 (αKir6.2, gift from Dr. A. Kuniyasu) was then added to each of these solutions for immunoprecipitation (IP). The antibody-Kir6.2 complex was pulled down using protein A-Sepharose, washed three times with RIPA buffer and run on pre-cast 4–12% NuPAGE gels (Invitrogen). Gels were soaked in 10% acetic acid and 30% methanol, then enlightning (NEN Life sciences), dried and exposed to film at −70 °C to detect 35S-labelled AK1.
Graphing and statistical analysis was performed using Origin software (OriginLab Corp., Northampton, MA). Values shown are means ± SEM and significance was assessed by one-way ANOVA.
Results
Expression of adenylate kinase isoforms in β-cells
There are six known isoforms of human AK [11]. AK2 expression has been reported in pancreas but is localized to the mitochondrial intermembrane space [12] and is unlikely to play a role in regulating K-ATP channels. Of the six known isoforms, AK1 and AK5 are cytosolic. We therefore examined the expression of AK isoforms in islets and insulinoma cells.
AK expression was tested using PCR with RNA isolated from human or mouse islets and insulinoma (rat-derived INS-1 and mouse-derived MIN6) cells. Amplified bands were isolated and their identity confirmed by sequencing. AK1 and AK5 were detected in human islet, consistent with the hypothesis that these two cytosolic isoforms play a role in regulating K-ATP channel activity. A weak band for AK5 was also detected in mouse islet.
The glucose-dependence of AK1 expression was examined by QPCR using INS-1 cells (Fig. 1A). INS-1 cells were grown in normal culture medium (11 mM glucose), or in culture medium containing 2 mM glucose for 3d or 25 mM for 5d. Cells grown in normal medium were also washed with 2 mM or 25 mM glucose for 4 h to examine short term expression changes. RNA was extracted from each batch of cells and used in QPCR experiments to detect changes in expression. Incubation in 25 mM glucose produces about a fourfold decrease in AK1 message whereas 2 mM glucose produces about a threefold increase relative to cells grown in 11 mM glucose (Fig. 1A).
Fig. 1.
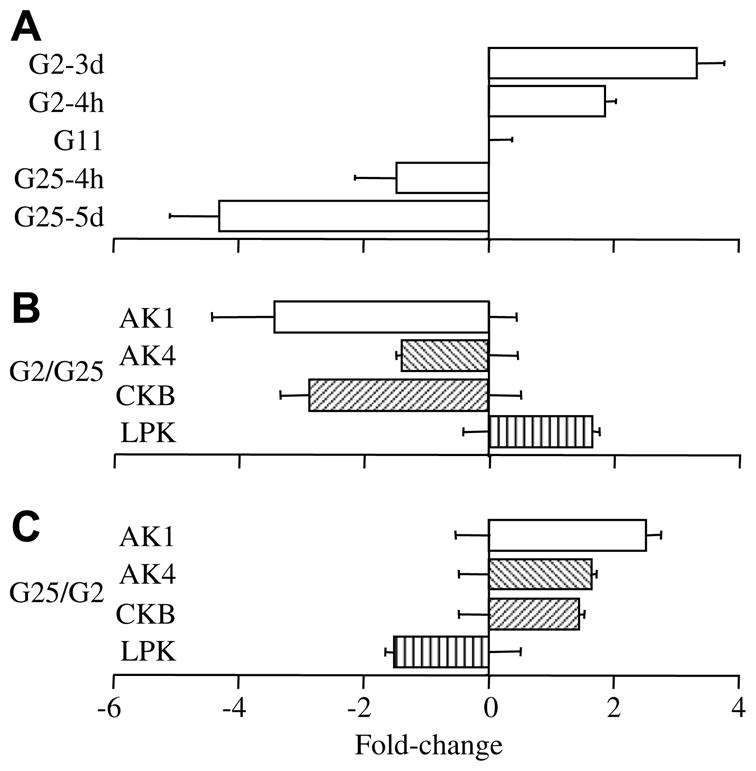
Glucose-regulation of AK expression in INS-1 cells measured by QPCR. (A) AK1 expression changes induced by glucose. Fold changes in expression level are plotted relative to the level found in INS-1 cells grown in normal RPMI culture medium (G11). Transferring cells to 2 mM glucose RPMI (G2) for 4 h or 3d increased AK1 expression by 1.9 ± 0.2 (n = 9)- and 3.3 ± 0.4 (n = 10)-fold, respectively, overcontrol levels. Increasing glucose to 25 mM (G25) decreased AK1 expression by 1.5 ± 0.7-fold (n = 12) at 4 h and by 4.3 ± 0.8-fold (n = 12) at 5d. (B) Glucose-mediated changes in expression are reversible. INS-1 cells grown in 2 mM glucose for 3d were transferred into fresh 2 mM glucose or 25 mM glucose for 4 h and fold changes in expression are plotted for G25 relative to values for cells in fresh G2. Error bars from the zero-fold change axis represent SEM for values in G2. Increasing glucose to 25 mM reversed the G2-mediated increase and decreased AK1 expression 3.4 ± 1.0-fold, AK4 decreased by 1.4 ± 0.1-fold, CKB decreased 2.9 ± 0.5-fold and LPK increased 1.7 ± 0.1-fold (all values n = 11 from the same RNA samples). (C) INS-1 cells were also transferred from 25 mM glucose to either 2 mM or fresh 25 mM glucose overnight. These changes produced opposite effects to those shown in (B). AK1 increased 2.5 ± 0.2-fold, AK4 increased 1.7 ± 0.1-fold, CKB increased 1.4 ± 0.1-fold and LPK decreased 1.5 ± 0.1-fold (n = 6 for each value).
To examine the reversibility of these glucose-induced changes, plates of cells grown in 2 mM (3d) glucose were washed with media containing either 2 mM or 25 mM glucose and incubated for 4 h (Fig. 1B). The change in expression in 25 mM glucose relative to 2 mM glucose is shown for AK1, AK4, CKB, and hepatic (L-type) pyruvate kinase (LPK). Glucose increases LPK gene transcription [13,14] and was used as a positive control. Increasing glucose reduced expression of AK1, AK4, and CKB but increased LPK. Incubation for 4 h in 25 mM glucose significantly decreased the expression of AK1 (p < 0.01), AK4 (p < 0.05) and CKB (p = 0.001), and increased the expression of LPK (p < 0.001). Similarly, cells maintained in 25 mM glucose for 5d were transferred to fresh medium with either 2 mM or 25 mM glucose overnight. Reduction of the glucose level in the culture medium significantly increased AK1, AK4, and CKB (p < 0.01) expression and decreased LPK (p < 0.001) expression (Fig. 1C).
AK1 associates with Kir6.2
Experiments were performed using in vitro transcribed and translated AK1 labeled with 35S-methionine (Fig. 2). A labeled band of the predicted size for AK1 (ca. 22 kDa) was observed only in reactions with the AK1 expression vector added and not with the empty vector (EV) or reticulocyte lysate alone (RL). This 35S-labeled AK1 was added to either cell-free RIPA lysis buffer (RIPA) or to cell membrane extracts (in RIPA) from mouse brain or MIN6 cells (Fig. 2, right panel). Immunoprecipitation (IP) using a Kir6.2 antibody was performed to test for association of Kir6.2 with AK1. This test was prompted by a report that the stimulatory effect of AK on K-ATP channel activity was not observed in isolated patches from β-cells [7], suggesting that AK might not directly couple to the channel. This report conflicts with previous studies in cardiac muscle and our own studies, shown below, demonstrating K-ATP activation by AK in inside-out patches. A weak band corresponding to the size of AK1 was observed from cell-free RIPA extracts and stronger bands were observed when either brain or MIN6 membrane extracts that contain Kir6.2 protein were added. Co-immunoprecipitation of AK1 with Kir6.2 in β-cells and mouse brain supports previous reports using cardiac muscle [15].
Fig. 2.
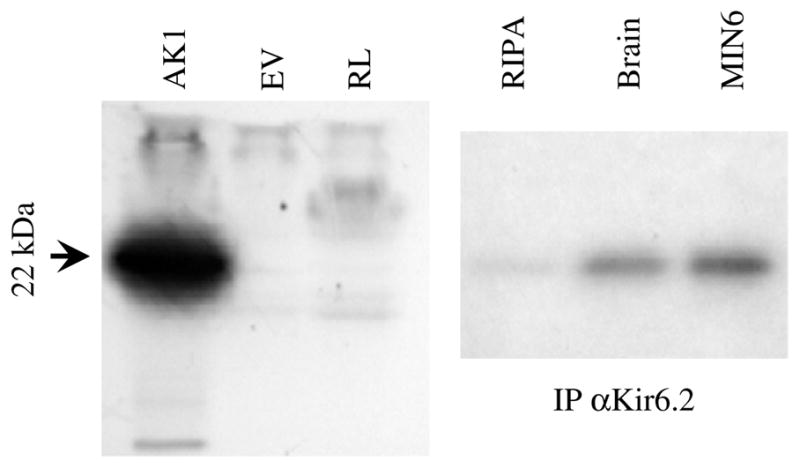
AK1 co-immunoprecipitates with Kir6.2. (Left panel) Specific 35S-incorporation into AK1 by in vitro transcription/translation was observed when an AK1 plasmid was added but not with the empty vector (EV) or the reticulocyte lysate (RL). (Right panel) Labeled AK1 was added to RIPA lysis alone or the lysis buffer with mouse brain or mouse-derived MIN6 membrane extracts. Immunoprecipitation of AK1 by αKir6.2 was observed with extracts of mouse brain and MIN6 cells but not with cell-free lysis buffer.
K-ATP channel currents are regulated by AK
The ability of AMP to increase K-ATP channel currents in the presence of ATP was investigated using inside-out patches from human islet (Fig. 3). We observed stimulation of K-ATP currents in response to AMP + ATP using both human islets and INS-1 cells consistent with AK generating ADP under these conditions and thus increasing channel activity (Fig. 3A). Segments of this recording (Fig. 3A) in the presence of ATP (Fig. 3B) and ATP + AMP (Fig. 3C) are shown on an expanded time scale. AMP + ATP stimulated channel activity 2.0 ± 0.3-fold (n = 4, p = 0.026) relative to ATP alone.
Fig. 3.

Addition of the AK substrates, ATP and AMP, stimulates K-ATP channel activity in human islets. (A) A continuous record of K-ATP channel currents in an inside-out patch held at −60 mV. Unitary current amplitudes are about 4 pA. The record starts with control (Cont.) solution (ATP- and AMP-free) being perfused onto the cytosolic face of the patch. This solution was then exchanged for one containing 30 μM AMP, then 30 μM ATP (zero AMP) then ATP + AMP (both at 30 μM) and finally washout with control solution. Solution applications are indicated by bars above the trace. Values for NPO were 4.40 (Cont.), 3.95 (AMP), 0.96 (ATP), 2.88 (ATP + AMP) and 4.53 (Wash) for this patch. (B) Currents in the presence of ATP are shown on expanded scales at the time indicated below trace A by the bar labeled B. (C) Currents are shown on expanded scales in the presence of ATP + AMP as indicated by bar C marked below trace A. Similar results were obtained in four patches from human β-cells.
The findings described above demonstrate that AK substrates can enhance K-ATP currents. The AK inhibitor diadenosine pentaphosphate (AP5A) was used to further test for a role of AK in regulating channel activity. The dose–response relation for the inhibition of K-ATP currents by AP5A was measured using AP5A at increasing concentrations (1–100 μM). These experiments indicate an IC50 of 14.9 ± 4.5 μM and a Hill coefficient of 1.22 ± 0.14 (n = 10). These values are similar to those previously reported for AP5A inhibition of K-ATP channels in cardiac muscle (16 μM and Hill coefficient of 1.6 [16]) and for the inhibition of islet K-ATP channels by AP4A (17 μM and Hill 1.2 [17]). The similarity of the IC50’s of AP4A and AP5A to inhibit K-ATP suggests a direct action on the channel as these two compounds have widely different binding affinities and inhibitory efficacy at AK [18,19].
AP5A can be used to demonstrate a role for AK in regulating K-ATP currents by its ability to block the stimulation of currents by the AK substrates ATP+AMP. In the absence of AP5A, ATP + AMP increased K-ATP currents 1.22 ± 0.06-fold (n = 6, p = 0.004). Addition of AP5A (20 μM) inhibited K-ATP currents by 45% (n = 6, p = 0.003) and in the presence of AP5A ATP + AMP did not significantly increase current amplitudes relative to ATP alone (1.07 ± 0.09). This block of the stimulation of K-ATP currents by ATP + AMP by the AK inhibitor AP5A supports a role for AK in regulating channel activity in β-cells.
Discussion
The findings presented here support a role for adenylate kinases in controlling the ATP/ADP ratio in the microenvironment of K-ATP channels in human β-cells and regulating channel activity (Fig. 4). Under fasting conditions, AK generates ADP from ATP + AMP in the microenvironment of the K-ATP channel to maintain channel activity, hold the cell at a hyperpolarized membrane potential and prevent insulin secretion. Elevated blood glucose inhibits AK-mediated phosphoryl transfer through the metabolic synthesis of ATP from ADP and depletion of AMP. The depletion of AMP occurs as AK cycles ATP + AMP → 2ADP, this ADP is then phosphorylated to ATP and AK-mediated cycling continues with the net effect that AMP becomes depleted. This model is supported by the observation that AMP levels decline in glucose-stimulated β-cells in association with AK inhibition [6]. Glucose-induced inhibition of AK results in a localized increase in ATP and a decrease in ADP levels thereby promoting K-ATP channel closure and insulin secretion. Additionally, glucose metabolism generates diadenosine polyphosphates (APnA), compounds that inhibit AK and also directly inhibit K-ATP channel activity. We propose that these three effects, glycolytic generation of ATP and depletion of AMP to inhibit AK, APnA mediated inhibition of AK and direct inhibition of K-ATP by APnA, are all involved in the inhibition of K-ATP channels by glucose resulting in the stimulation of insulin secretion (Fig. 4).
Fig. 4.

A model for glucose-induced closure of K-ATP channels leading to depolarization, Ca2+-influx and insulin secretion. Glucose uptake through the Glut2 transporter enters the metabolic pathway and generates ATP from ADP. Metabolic synthesis of ATP from ADP depletes the local AMP concentration (through ATP + AMP → 2ADP and ADP then being converted to ATP) and inhibits the AK-mediated phosphoryl transfer cascade that under basal condition generates ADP from ATP in the microenvironment of the K-ATP channel. Inhibition of the AK cascade results in a localized increase in ATP and decrease of ADP causing channel closure. Additionally, glucose metabolism generates diadenosine polyphosphates (APnA) that further inhibit AK-mediated phosphotransfer and also directly interact with K-ATP to inhibit channel activity. We therefore propose that these three components are involved in the inhibition of K-ATP channels resulting in cell depolarization, activation of L-type voltage-dependent Ca2+ channels (L-VDCC) and the stimulation of insulin secretion.
Our studies extend previous reports on the role of AK in β-cells by identifying expression of the two cytosolic isoforms of AK, AK1, and AK5, and by showing that AK1 associates with Kir6.2 and regulates channel activity in isolated patches from human islets and INS-1 cells. Schulze et al. reported that AK1 regulates K-ATP currents in mouse islets and observed a reduction in the efficacy of AMP to stimulate activity of about 50% in AK1 knockout mice [7]. Our observation that the second cytosolic isoform of AK, AK5 is expressed in β-cells could explain this remaining stimulatory activity of AMP in AK1 knockout mice.
The reversible, decreased expression of AK1 with high glucose reflects changes in the expression of SUR1 and Kir6.2 [8]. This coordinate-regulation might play a role in the failing sensitivity of β-cell K-ATP channels to glucose in type 2 diabetes [20]. However, previous studies suggest that the overall level of AK activity does not decrease in islets from ob/ob diabetic mice with persistent hyperglycemia [21]. Our findings of decreased message levels for AK1 suggests that the effects of glucose slowly reverse or that expression of other AK isoforms increase to compensate for the decreased expression of AK1.
In summary, β-cells express two cytosolic isoforms of AK that play a role in regulating K-ATP channel activity. We propose that glucose-stimulation of β-cells inhibits AK through glycolysis and also through the generation of diadenosine polyphosphates. This dual inhibition of AK increases the ATP/ADP ratio in the microenvironment of the K-ATP channel and thereby induces channel closure, an effect that might be potentiated by direct interactions of diadenosine polyphosphates with channel subunits. The closure of K-ATP channels by these three mechanisms induces membrane depolarization, activation of voltage-dependent Ca2+ channels and the stimulation of insulin secretion. This sequence of events is consistent with a report that glucose-induced AK inhibition precedes Ca2+ influx and insulin secretion [6]. We also demonstrate that glucose regulates the expression of AK as well as its activity. High glucose down-regulates AK1 expression, an effect that parallels glucose-mediated effects on K-ATP channel subunits. We propose that this coordinate down-regulation of SUR1, Kir6.2 and AK1, a regulator of channel activity, may participate in the failure of β-cells to respond to glucose in type 2 diabetes.
Acknowledgments
A preliminary account of this information was presented at the 2005 American Diabetes Association annual meeting [22]. We thank Dr. A. Kuniyasu (Kumamoto University, Japan) for providing the antibody to Kir6.2. This work was supported by an American Diabetes Association research award (C.A.L.), by NIH awards R01 DK069575 and R01 DK045817 (G.G.H.).
References
- 1.Tarasov A, Dusonchet J, Ashcroft FM. Metabolic-regulation of the pancreatic beta-cell ATP-sensitive K+ channel: a pas de deux. Diabetes. 2004;53(Suppl 3):S113–S122. doi: 10.2337/diabetes.53.suppl_3.s113. [DOI] [PubMed] [Google Scholar]
- 2.Bryan J, Vila-Carriles WH, Zhao G, Babenko AP, Aguilar-Bryan L. Toward linking structure with function in ATP-sensitive K+ channels. Diabetes. 2004;53(Suppl 3):S104–S112. doi: 10.2337/diabetes.53.suppl_3.s104. [DOI] [PubMed] [Google Scholar]
- 3.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 4.Zeleznikar RJ, Heyman RA, Graeff RM, Walseth TF, Dawis SM, Butz EA, Goldberg ND. Evidence for compartmentalized adenylate kinase catalysis serving a high energy phosphoryl transfer function in rat skeletal muscle. J Biol Chem. 1990;265:300–311. [PubMed] [Google Scholar]
- 5.Larsson O, Ammala C, Bokvist K, Fredholm B, Rorsman P. Stimulation of the K-ATP channel by ADP and diazoxide requires nucleotide hydrolysis in mouse pancreatic beta-cells. J Physiol. 1993;463:349–365. doi: 10.1113/jphysiol.1993.sp019598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson LK, Schroeder W, Robertson RP, Goldberg ND, Walseth TF. Suppression of adenylate kinase catalyzed phosphotransfer precedes and is associated with glucose-induced insulin secretion in intact HIT-T15 cells. J Biol Chem. 1996;271:16544–16552. doi: 10.1074/jbc.271.28.16544. [DOI] [PubMed] [Google Scholar]
- 7.Schulze DU, Dufer M, Wieringa B, Krippeit-Drews P, Drews G. An adenylate kinase is involved in K(ATP) channel-regulation of mouse pancreatic beta-cells. Diabetologia. 2007;50:2126–2134. doi: 10.1007/s00125-007-0742-9. [DOI] [PubMed] [Google Scholar]
- 8.Moritz W, Leech CA, Ferrer J, Habener JF. Regulated expression of adenosine triphosphate-sensitive potassium channel subunits in pancreatic beta-cells. Endocrinology. 2001;142:129–138. doi: 10.1210/endo.142.1.7885. [DOI] [PubMed] [Google Scholar]
- 9.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 10.Leech CA, Habener JF. Regulation of glucagon-like peptide-1 receptor and calcium-sensing receptor signaling by L-histidine. Endocrinology. 2003;144:4851–4858. doi: 10.1210/en.2003-0498. [DOI] [PubMed] [Google Scholar]
- 11.Ren H, Wang L, Bennett M, Liang Y, Zheng X, Lu F, Li L, Nan J, Luo M, Eriksson S, Zhang C, Su XD. The crystal structure of human adenylate kinase 6: An adenylate kinase localized to the cell nucleus. Proc Natl Acad Sci USA. 2005;102:303–308. doi: 10.1073/pnas.0407459102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Rompay AR, Johansson M, Karlsson A. Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol Ther. 2000;87:189–198. doi: 10.1016/s0163-7258(00)00048-6. [DOI] [PubMed] [Google Scholar]
- 13.Kawaguchi T, Takenoshita M, Kabashima T, Uyeda K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc Natl Acad Sci USA. 2001;98:13710–13715. doi: 10.1073/pnas.231370798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.da Silva Xavier G, Varadi A, Ainscow EK, Rutter GA. Regulation of gene expression by glucose in pancreatic beta -cells (MIN6) via insulin secretion and activation of phosphatidylinositol 3′-kinase. J Biol Chem. 2000;275:36269–36277. doi: 10.1074/jbc.M006597200. [DOI] [PubMed] [Google Scholar]
- 15.Carrasco AJ, Dzeja PP, Alekseev AE, Pucar D, Zingman LV, Abraham MR, Hodgson D, Bienengraeber M, Puceat M, Janssen E, Wieringa B, Terzic A. Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc Natl Acad Sci USA. 2001;98:7623–7628. doi: 10.1073/pnas.121038198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jovanovic A, Alekseev AE, Terzic A. Cardiac ATP-sensitive K+ channel: a target for diadenosine 5′,5″-P1,P5-pentaphosphate. Naunyn Schmiedebergs Arch Pharmacol. 1996;353:241–244. doi: 10.1007/BF00168763. [DOI] [PubMed] [Google Scholar]
- 17.Ripoll C, Martin F, Manuel Rovira J, Pintor J, Miras-Portugal MT, Soria B. Diadenosine polyphosphates. A novel class of glucose-induced intracellular messengers in the pancreatic beta-cell. Diabetes. 1996;45:1431–1434. doi: 10.2337/diab.45.10.1431. [DOI] [PubMed] [Google Scholar]
- 18.Lienhard GE, Secemski P1,P5-Di(adenosine-5′)pentaphosphate, a potent multisubstrate inhibitor of adenylate kinase. J Biol Chem. 1973;248:1121–1123. [PubMed] [Google Scholar]
- 19.Reinstein J, Vetter IR, Schlichting I, Rosch P, Wittinghofer A, Goody RS. Fluorescence and NMR investigations on the ligand binding properties of adenylate kinases. Biochemistry. 1990;29:7440–7450. doi: 10.1021/bi00484a013. [DOI] [PubMed] [Google Scholar]
- 20.Tsuura Y, Ishida H, Okamoto Y, Kato S, Sakamoto K, Horie M, Ikeda H, Okada Y, Seino Y. Glucose sensitivity of ATP-sensitive K+ channels is impaired in beta-cells of the GK rat. A new genetic model of NIDDM. Diabetes. 1993;42:1446–1453. doi: 10.2337/diab.42.10.1446. [DOI] [PubMed] [Google Scholar]
- 21.Borglund E, Brolin SE, Agren A. Adenylate kinase activity in various organs and tissues of mice with the obese-hyperglycemic syndrome (gene symbol Ob/Ob) J Histochem Cytochem. 1978;26:127–130. doi: 10.1177/26.2.203625. [DOI] [PubMed] [Google Scholar]
- 22.Leech CA, Stanojevic V, Habener JF. Phosphotransfer cascades and glucose sensing by ATP-sensitive potassium channels. Diabetes. 2005:A408. [Google Scholar]