

Abstract
Centrosomes are the major microtubule-organizing centers in animal cells and regulate formation of a bipolar mitotic spindle. Aberrant centrosome number causes chromosome mis-segregation, and has been implicated in genomic instability and tumor development. Previous studies have demonstrated a role for the DNA replication factors MCM5 and Orc1 in preventing centrosome reduplication. Cyclin A-Cdk2 localizes on centrosomes by means of a modular centrosomal localization sequence (CLS) that is distinct from that of cyclin E. Here, we show that cyclin A interacts with both MCM5 and Orc1 in a CLS-dependent but Cdk-independent manner. Although the MRAIL hydrophobic patch is contained within the cyclin A CLS, binding of both MCM5 and Orc1 to cyclin A does not require a wild-type hydrophobic patch. The same domain in MCM5 that mediates interaction with cyclin E also binds cyclin A, resulting in centrosomal localization of MCM5. Finally, unlike its function in DNA synthesis, MCM5-mediated inhibition of centrosome reduplication in S-phase-arrested CHO cells does not require binding to other MCM family members. These results suggest that cyclins E and A sequentially prevent centrosome reduplication throughout interphase by recruitment of DNA replication factors such as MCM5 and Orc1.
Keywords: Cyclin A, Centrosomes, MCM5, Orc1
Introduction
As the major microtubule-organizing centers in animal cells, centrosomes dictate the bipolarity of the mitotic spindle that is essential for ensuring equal segregation of chromosomal DNA between two daughter cells. Centrosome amplification has been reported in virtually all types of solid tumors and is an underlying cause of aneuploidy, probably contributing to the development and progression of human cancers (Fukasawa, 2005; Ganem et al., 2009; Lingle et al., 2005). Centrosome duplication and DNA replication are fundamentally similar in that both undergo once-and-only-once semi-conservative duplication during every cell cycle. Both duplication events are initiated near the G1-S boundary and are coupled by increased cyclin-dependent kinase 2 (Cdk2) activity. Many groups have provided evidence that increased Cdk2 activity in late G1 is essential for these events, as inhibition of Cdk activity in either Xenopus egg extracts or mammalian cells prevents both DNA replication and centrosome duplication (Coverley et al., 2002; Hinchcliffe et al., 1999; Lacey et al., 1999; Matsumoto et al., 1999). In Xenopus egg extracts and certain mammalian cell lines, prolonged S-phase arrest allows multiple rounds of centrosome reduplication to occur, but only when Cdk2 is active (Balczon et al., 1995; Hinchcliffe et al., 1999; Lacey et al., 1999; Matsumoto et al., 1999). A crucial role for Cdk2 in this process has been supported by the identification of specific substrates directly involved in centrosome duplication, such as nucleophosmin (also known as B23) and CP110 (Chen et al., 2002; Okuda et al., 2000; Tokuyama et al., 2001).
Although nearly all centrosome duplication studies have investigated the regulatory role of Cdk2 coupled to cyclin E, recent studies suggest the involvement of other cyclin-Cdk complexes. Indeed, knockout mice lacking Cdk2 or either cyclins E1 or E2 are viable, and fibroblasts isolated from cyclin E double-knockout embryos show no mitotic defects when allowed to cycle continuously (Berthet et al., 2003; Geng et al., 2003; Ortega et al., 2003; Parisi et al., 2003). Thus, it appears that another Cdk2 activator, presumably cyclin A, can fulfill all essential cyclin E functions. A recent study confirmed this hypothesis by demonstrating that ablation of both cyclin E and cyclin A prevents cell division (Kalaszczynska et al., 2009).
Previously, our laboratory identified a 20 amino acid domain in cyclin E as a modular centrosomal localization sequence (CLS) necessary for the centrosomal localization of cyclin E. We showed that expression of the CLS domain displaces both endogenous cyclin E and cyclin A from centrosomes, leading to cell-cycle arrest (Matsumoto and Maller, 2004). Using a bacterial two-hybrid screen of a HeLa cDNA library, we showed that direct interaction of cyclin E with the DNA replication factor MCM5 is dependent on an intact CLS but independent of Cdk2 binding (Ferguson and Maller, 2008). Furthermore, this interaction recruits MCM5 to centrosomes and expression of MCM5 inhibits centrosome amplification in S-phase-arrested CHO cells. Recently, we reported the characterization of the cyclin A CLS, which has many structural and biochemical features in common with the cyclin E CLS and is also crucial for cell-cycle progression (Pascreau et al., 2010). We were therefore interested in determining whether cyclin A has a role in preventing centrosome reduplication.
Here, we show that cyclin A directly binds MCM5 and recruits it to centrosomes. More importantly, although the CLS of cyclin A resides in a different region of the cyclin molecule than cyclin E, we demonstrate that interaction between cyclin A and MCM5 still depends on an intact CLS and is independent of Cdk binding. Furthermore, MCM5 interaction with cyclin A or cyclin E, and inhibition of centrosome reduplication does not require binding to other MCM family members. It was recently reported that Orc1, an essential component in DNA replication licensing, is also recruited to centrosomes through interaction with cyclins A and E, and represses centrosome amplification (Hemerly et al., 2009). We report that, like MCM5, interaction between cyclin A and Orc1 is dependent on an intact CLS and independent of Cdk2. These studies suggest that, in a normal cell cycle, cyclin A can prolong inhibition of centrosome reduplication throughout S phase and G2 by CLS-dependent centrosomal recruitment of MCM5 and Orc1.
Results
Immunoprecipitates of endogenous cyclin A from asynchronous HeLa cells readily co-precipitated MCM5, with no significant binding to an IgG control (Fig. 1A). To confirm whether MCM5 directly binds cyclin A, GST or GST-tagged cyclin A was purified from baculovirus-infected Sf9 cells and incubated with [35S]-methionine-labeled MCM5 protein. The GST pulldown was separated by SDS-PAGE and analyzed by autoradiography. GST-cyclin A displayed significant binding to MCM5 compared with GST alone (Fig. 1B). Altogether, these data indicate that cyclin A directly interacts with MCM5 in vivo.
Fig. 1.
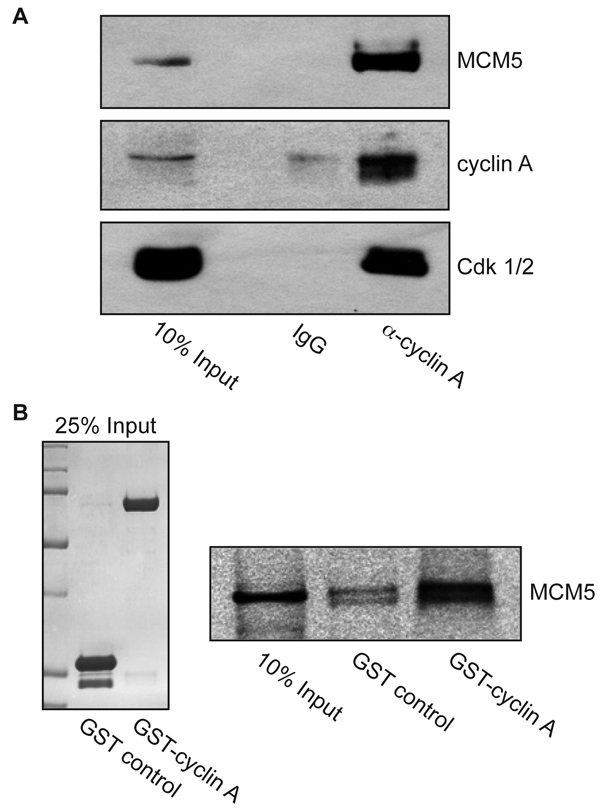
Cyclin A interacts with MCM5. (A) Endogenous cyclin A co-immunoprecipitates MCM5. Lysates from asynchronous HeLa S3 cells were subjected to immunoprecipitation with antibodies to either cyclin A or control IgG, as indicated. Immunoprecipitates were analyzed by western blot using antibodies to MCM5 (upper), cyclin A (middle) and PSTAIR (lower), which recognizes both Cdk2 and Cdk1. (B) Cyclin A directly interacts with MCM5. MCM5 protein was produced and radiolabeled with [35S]-methionine in an in vitro TNT-coupled system, and incubated with glutathione-agarose beads bound to GST or to GST-cyclin A. Beads were washed, eluted with SDS-PAGE sample buffer, electrophoresed on 10% gels and analyzed by autoradiography. Left: Coomassie-stained gel of 25% input of GST and GST-cyclin A. Right: autoradiograph of MCM5.
Because interaction between MCM5 and cyclin E is dependent on a wild-type CLS but independent of Cdk2 (Ferguson and Maller, 2008), we sought to determine whether interaction with cyclin A has the same requirements. Recent work from this laboratory showed that amino acids 201-255 of cyclin A are sufficient for centrosomal localization and that this cyclin A CLS resides in a different region of the cyclin molecule compared with cyclin E (Pascreau et al., 2010). To identify the region of cyclin A responsible for MCM5 binding, CHO-K1 cells were transiently co-transfected with a construct encoding full-length, wild-type MCM5 and various 6-Myc-tagged constructs of cyclin A (Fig. 2A). After transfection, CHO cell lysates were subjected to Myc immunoprecipitation and analyzed for MCM5 binding by immunoblotting. Initial analysis of two fragments of cyclin A, the N-terminal domain (amino acids 1-200) and the C-terminal domain (amino acids 201-432), demonstrated that MCM5 binding could be fully attributed to the C-terminal half of the protein, which contains the cyclin box and includes the CLS (Fig. 2B). Further subdivision of the C-terminus of cyclin A showed that two fragments of cyclin A containing the CLS, amino acids 201-255 and amino acids 201-301, clearly bound MCM5, whereas two fragments that do not contain the CLS, amino acids 256-301 and amino acids 302-432, showed no significant MCM5 binding (Fig. 2C). It is important to note that the two cyclin A fragments comprising amino acids 201-255 and 201-301 have previously been shown to not bind Cdks (Pascreau et al., 2010), indicating that both centrosomal localization and MCM5 binding are Cdk-independent properties of cyclin A. It was previously reported that mutation to arginine of four conserved surface residues within the cyclin A CLS, I213, E220, E224 and K226 (IEEK-R), disrupts localization to centrosomes (Pascreau et al., 2010). A cyclin A CLS construct containing these mutations was subjected to immunoprecipitation to determine whether MCM5 binding is likewise disrupted (Fig. 2D). Whereas the CLS-containing fragment comprising amino acids 201-301 displayed significant MCM5 binding, mutation of these four conserved residues dramatically decreased MCM5 association. These data indicate that the CLS of cyclin A is an essential domain for binding of MCM5 and that this interaction is independent of Cdk binding.
Fig. 2.
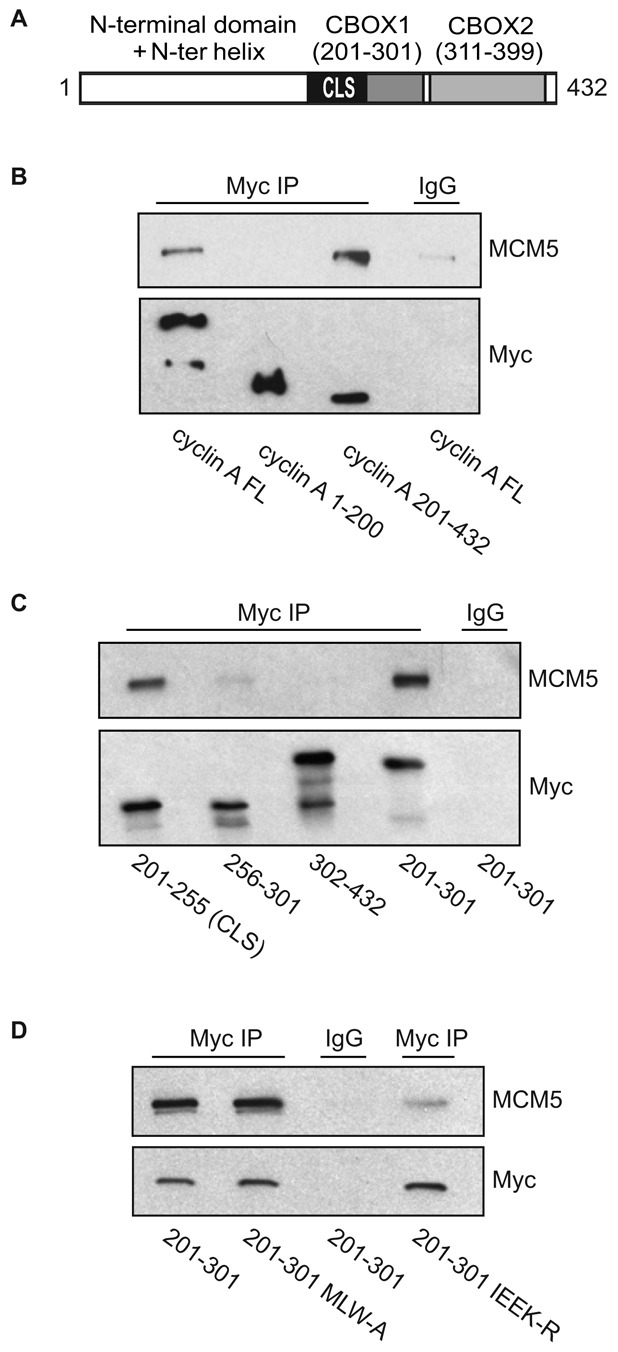
MCM5 interaction with cyclin A is CLS dependent. (A) Schematic representation of human cyclin A. The N-terminal domain (amino acids 1-200) is shown in white. The C-terminal region (amino acids 201-432) contains the cyclin domain, which can be divided into two cyclin box folds (CBOX1 and CBOX2), represented in gray. The CLS motif is highlighted in black. (B) MCM5 interacts with the C-terminus of cyclin A. CHO-K1 cells were transiently co-transfected with the indicated constructs of Myc-tagged cyclin A (FL, full length) and wild-type MCM5. Approximately 20 hours after transfection, cell lysates were prepared and subjected to immunoprecipitation (IP) with control IgG or antibody to Myc. The immunoprecipitates were separated on 10% SDS-PAGE gels and analyzed by western blotting for MCM5 and Myc-cyclin A. (C) MCM5 interacts with the CLS region of cyclin A. CHO-K1 cells were transiently co-transfected with the indicated constructs of Myc-tagged cyclin A. Analysis of MCM5 binding was carried out as in B. (D) Mutation of the cyclin A CLS disrupts MCM5 interaction. CHO-K1 cells were transiently co-transfected with wild-type MCM5 and Myc-tagged cyclin A amino acids 201-301 containing a wild-type CLS, the hydrophobic patch mutant CLS (MLW-A) or mutant CLS (IEEK-R), as indicated. Analysis of MCM5 co-immunoprecipitation was carried out as in B.
The CLS domain of cyclin A includes within it the conserved MRAIL hydrophobic patch that is involved in substrate recognition and the binding of p27Kip1. Mutation of three residues in the patch, M210, L214 and W217, to alanine (MLW-A) has been reported to impair binding of substrates and p27Kip1 without affecting binding to Cdk2 (Furstenthal et al., 2001; Russo et al., 1996; Schulman et al., 1998). Therefore, we analyzed the ability of this mutant to bind MCM5. Surprisingly, MCM5 bound the MLW-A mutant of cyclin A to the same extent as the wild-type protein (Fig. 2D).
Previous evidence indicates that the cyclin E CLS interacts with a specific domain of MCM5 (amino acids 533-569) that is distinct from the domains involved in DNA replication (Ferguson and Maller, 2008). The two-hybrid screen that identified the interaction of the cyclin E CLS and MCM5 resulted in isolation of multiple partial cDNA constructs encoding MCM5. Alignment of these sequences revealed a minimal 74 amino acid region of MCM5, amino acids 496-569, shared among all interacting constructs. It was important to determine the domain of MCM5 that mediates interaction with cyclin A. In an in vitro GST pulldown assay, the C-terminus of MCM5 (amino acids 496-734) and the minimal 74-residue fragment (amino acids 496-569) specifically bound to cyclin A (Fig. 3). The 74 amino acid fragment of MCM5 was then subdivided into two equal segments, tagged with GFP and analyzed in another GST pulldown assay. Whereas neither GFP alone nor GFP-MCM5 amino acids 496-532 displayed any appreciable binding to cyclin A, GFP-MCM5 amino acids 533-569 specifically bound cyclin A, indicating that the same region of MCM5 mediates binding to both cyclin E and cyclin A.
Fig. 3.

Identification of the cyclin A-binding domain of MCM5. HA-tagged MCM5 (top panels) and GFP-tagged MCM5 (bottom panel) constructs were radiolabeled with [35S]-methionine in an in vitro TNT reaction and subjected to pulldown analysis with GST or GST-tagged cyclin A as in Fig. 1B. Right: autoradiographs of MCM5. Left: Coomassie-stained gel of 25% input of GST and GST-tagged cyclin A.
Centrosomal localization of MCM5 dependent on cyclin E binding inhibits centrosome reduplication in S-phase-arrested CHO cells. We have proposed that the recruitment of MCM5 to centrosomes functions as a potential feedback mechanism to prevent cells from reduplicating centrosomes after initiation of DNA synthesis (Ferguson and Maller, 2008). It is evident that sustaining this inhibition throughout S phase and G2 after cyclin E levels decline would be crucial for ensuring the once-and-only-once duplication of centrosomes that is essential for maintaining genomic stability. Expression of the CLS of either cyclin A or cyclin E displaces both endogenous cyclins, indicating homologous functionality (Matsumoto and Maller, 2004; Pascreau et al., 2010). Displacement of these cyclins also results in the displacement of endogenous MCM5, which can be restored by exogenous cyclin E that is centrosomally localized by means of a different targeting motif (Ferguson and Maller, 2008) – the pericentrin-AKAP450 centrosomal targeting (PACT) domain (Gillingham and Munro, 2000). Fusion of the PACT domain to cyclin A causes centrosomal localization even in cells expressing the CLS domain (Pascreau et al., 2010). It was first determined whether expression of the CLS of cyclin A displaces endogenous MCM5 from centrosomes. Cells were transiently transfected with GFP-tagged cyclin A amino acids 1-200, the GFP-tagged wild-type cyclin A CLS or the GFP-tagged IEEK-R mutant CLS, followed by immunofluorescent analysis of endogenous MCM5 localization (Fig. 4A,B). Whereas expression of GFP-tagged cyclin A 1-200 or the GFP-tagged IEEK-R mutant CLS did not alter MCM5 localization, expression of the wild-type GFP-tagged CLS resulted in the striking displacement of MCM5 from centrosomes. Cells were then co-transfected with the wild-type GFP-tagged CLS and either PACT-fused 6-Myc-tagged full-length cyclin A or the PACT-fused 6-Myc-tagged amino acids 1-200 of cyclin A, a fragment that does not contain the CLS and is unable to bind MCM5 (Fig. 2B). Analysis of endogenous MCM5 localization in cells coexpressing GFP-tagged CLS and these constructs revealed that PACT-fused full-length cyclin A restores MCM5 localization to centrosomes, whereas the fragment unable to bind MCM5 does not. This indicates that cyclin A, like cyclin E, directly recruits MCM5 to centrosomes and suggests that MCM5-mediated inhibition of centrosome reduplication persists throughout interphase.
Fig. 4.

Centrosomal localization of MCM5 is dependent on cyclin A. (A) Localization of endogenous MCM5 was analyzed in CHO-K1 cells expressing either wild-type or mutant GFP-CLS, or co-expressing wild-type GFP-CLS and the indicated PACT-fused cyclin A constructs. Cells were stained with antibody to γ-tubulin (blue) and MCM5 (red). Expression and localization of GFP-tagged constructs were observed by direct fluorescence (green). Line scans measuring centrosome-associated relative fluorescence intensity are shown on the right, with the green, blue and red lines representing the GFP-, γ-tubulin- and MCM5-associated fluorescence, respectively. Arrows indicate the position of centrosomes. Insets: magnified image of the centrosomal region. Scale bars: 10 μm. (B) Graphical analysis of the centrosomal localization of MCM5. Several experiments similar to the one in A were performed with the indicated constructs. Over 100 cells were analyzed for each condition in each experiment. Error bars indicate mean ± s.d. (n=3).
We next sought to determine whether MCM5-mediated inhibition of centrosome reduplication requires binding to other MCM family members. Previous evidence indicates that expression of MCM5 but not MCM2, a related member of the MCM family, inhibits centrosome reduplication in S-phase-arrested CHO cells (Ferguson and Maller, 2008), suggesting a function specific to MCM5. Binding of MCM5 to other MCM family members is required for DNA replication functions of MCM5. However, the necessity for MCM5 to bind other family members in order to inhibit centrosome reduplication has not been determined.
In Saccharomyces cerevisiae, mutation of highly conserved cysteine residue 183 to tyrosine (C183Y) in MCM5 disrupts binding to family members (Dalton and Hopwood, 1997). The equivalent cysteine residue in human MCM5, C172, was therefore mutated to tyrosine (C172Y) and examined for binding to MCM3. Multiple studies show that MCM5 and MCM3 exist as a tightly associated dimer that can easily be isolated from other MCMs (Bochman et al., 2008; DaFonseca et al., 2001). Therefore, binding to MCM3 reflects binding to all family members. Either wild-type HA-tagged MCM5 or HA-tagged MCM5 C172Y was transiently transfected into CHO cells, immunoprecipitated with an antibody to HA and analyzed by immunoblotting (Fig. 5A). Whereas wild-type MCM5 clearly co-immunoprecipitated MCM3, no detectable MCM3 was associated with MCM5 C172Y, indicating a loss of binding to family members. To determine whether mutation of C172 had any effect on cyclin binding to MCM5, radiolabeled MCM5 C172Y was incubated with GST-cyclin A and GST-cyclin E. The resultant pulldown indicates that the MCM5 C172Y mutant binds efficiently to both cyclins (Fig. 5B). The potential for centrosomal localization of this MCM5 mutant was then examined by immunostaining with antibodies to HA and γ-tubulin (Fig. 5C). Nearly all cells expressing HA-tagged MCM5 C172Y showed co-localization with γ-tubulin, indicating that this protein still localizes to centrosomes.
Fig. 5.

MCM5 centrosomal function is independent of other MCMs. (A) MCM5 C172Y does not interact with MCM3. CHO-K1 cells were transiently transfected with either HA-tagged wild-type MCM5 or HA-tagged MCM5 C172Y mutant. Approximately 20 hours after transfection, cell lysates were subjected to immunoprecipitation with antibody to the HA tag. The immunoprecipitates were separated on 10% SDS-PAGE gels and analyzed by western blotting with antibodies to MCM5 and MCM3. (B) MCM5 C172Y interacts with both cyclin A and cyclin E. HA-tagged MCM5 C172Y protein was produced and radiolabeled with [35S]-methionine in an in vitro TNT-coupled system. The MCM5 C172Y protein was then incubated with glutathione-agarose beads bound to GST, GST-tagged cyclin A or GST-tagged cyclin E. Beads were washed, eluted with SDS-PAGE sample buffer, electrophoresed on 10% gels and analyzed by autoradiography. Left: Coomassie-stained gel of 25% input of GST, GST-cyclin A and GST-cyclin E. Right: autoradiograph of MCM5 C172Y. (C) MCM5 C172Y is centrosomally localized. CHO-K1 cells were transiently transfected with HA-tagged MCM5 C172Y. Approximately 20 hours after transfection, cells were methanol fixed and stained with antibodies to γ-tubulin and HA, as indicated. Arrowheads mark the centrosome. Inset: magnified image of the centrosomal region of the merged image. Scale bar: 10 μm. (D) Expression of MCM5 C172Y inhibits centrosome reduplication. CHO-K1 cells arrested in S phase by HU were transiently transfected with either HA-tagged wild-type MCM5 or HA-tagged MCM5 C172Y. The number of γ-tubulin-staining foci (centrosomes) was monitored by immunofluorescence in HA-expressing cells and compared with control non-expressing cells. The error bar indicates mean ± s.d. (n=3). Over 150 cells were counted for each condition in each experiment.
To determine whether this MCM5 mutant inhibits centrosome reduplication, we analyzed centrosome duplication in hydroxyurea (HU)-arrested CHO-K1 cells expressing either HA-tagged wild-type MCM5 or HA-tagged MCM5 C172Y. After 48 hours of S-phase arrest, control cells contained an average of seven γ-tubulin foci, whereas cells expressing either HA-tagged wild-type MCM5 or HA-tagged MCM5 C172Y contained an average of only four (Fig. 5D). These data show that MCM5-mediated inhibition of centrosome reduplication does not require the formation of a heterohexameric complex with the other MCM family members.
During the course of these studies, it was reported that cyclin A also directly binds Orc1, an essential component of the pre-replication complex. It was suggested that this interaction supports Orc1 localization to centrosomes and inhibits centrosome amplification (Hemerly et al., 2009). We therefore sought to determine whether the region of cyclin A containing the CLS that mediates MCM5 binding is also essential for Orc1 association. Myc immunoprecipitates of the same cyclin A constructs described in Fig. 2 clearly demonstrated that binding of Orc1 is mediated by the C-terminal cyclin-box-containing region, amino acids 201-432, of cyclin A (Fig. 6A). Further analysis showed that the CLS region of cyclin A, amino acids 201-255, mediates interaction with Orc1 (Fig. 6B). Moreover, like the association of cyclin A with MCM5, mutation of CLS surface residues (IEEK-R), but not the MRAIL hydrophobic patch subdomain within the CLS (MLW-A), disrupted the interaction of Orc1 with cyclin A (Fig. 6C).
Fig. 6.
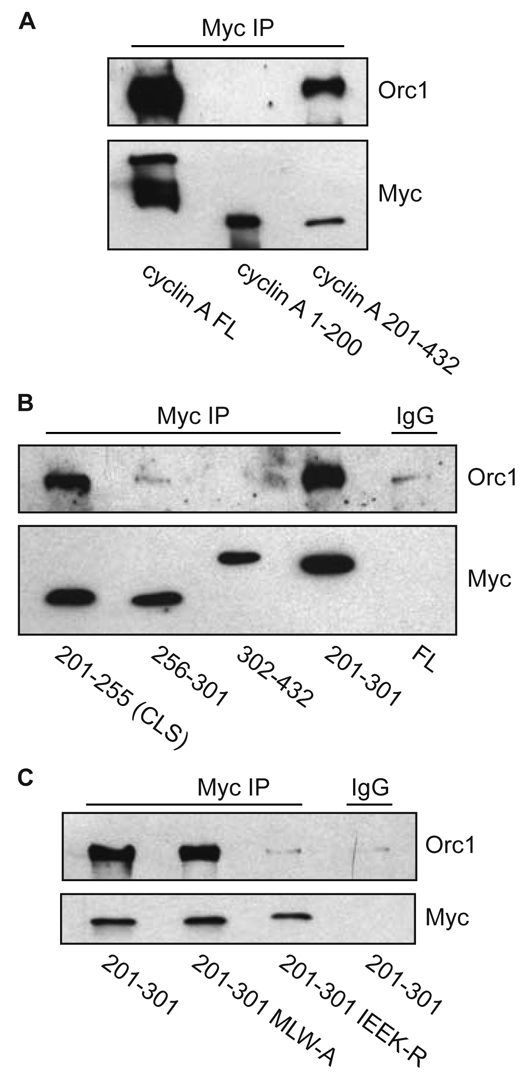
Cyclin A interaction with Orc1 is CLS dependent, but MRAIL independent. (A) Orc1 interacts with the C-terminus of cyclin A. CHO-K1 cells were transiently co-transfected with wild-type Orc1 and full-length (FL) Myc-tagged cyclin A, Myc-tagged cyclin A N-terminus (amino acids 1-200) or Myc-tagged cyclin A C-terminus (amino acids 201-432), as indicated. Approximately 20 hours after transfection, cell lysates were prepared and subjected to immunoprecipitation with anti-Myc antibody. The immunoprecipitates were separated on 10% SDS-PAGE gels and analyzed by western blotting for Orc1 and Myc. Non-immune rabbit IgG was used as a control. (B) Orc1 specifically interacts with the CLS of cyclin A. CHO-K1 cells were transiently co-transfected with indicated constructs of Myc-tagged cyclin A and wild-type Orc1. Analysis of Orc1 binding to cyclin A was carried out by immunoprecipitation analysis as in A. Amino acids 201-255 form the cyclin A CLS. (C) Mutation of the cyclin A CLS but not of the MRAIL hydrophobic pocket (MLW-A) disrupts Orc1 interaction with cyclin A. CHO-K1 cells were transiently co-transfected with indicated constructs of Myc-tagged cyclin A and wild-type Orc1. Analysis of Orc1 binding to cyclin A was carried out by immunoprecipitation analysis as in A.
Discussion
During interphase of the cell cycle, Cdk2 is positively regulated through association with either cyclin E or cyclin A. These cyclins have different functions during the cell cycle, with cyclin E regulating the G1-S transition, and cyclin A controlling late S-phase progression and the G2-M transition. However, the recent isolation of cyclin E-null mouse embryonic fibroblasts that undergo continuous normal mitotic cycles indicates that a certain redundancy exists between these cyclins (Geng et al., 2003). Presumably, cyclin A coupled to Cdk2 can readily compensate for the loss of cyclin E and promote the G1-S transition. Similarly, DNA synthesis in Xenopus egg extracts can be supported by Cdk2 binding to either cyclin E or cyclin A (Strausfeld et al., 1996; Woo and Poon, 2003).
We recently identified a modular domain encompassing amino acids 201-255 of cyclin A as a CLS motif responsible for centrosomal localization. Although the cyclin A CLS resides in a different molecular region than the cyclin E CLS, they are structurally very similar and also functionally homologous, as expression of either displaces endogenous cyclins A and E from centrosomes and inhibits S-phase progression (Pascreau et al., 2010). Here, we advance previous findings of CLS-dependent, direct interaction between MCM5 and cyclin E to show that cyclin A also directly binds MCM5 in a CLS-dependent, Cdk-independent manner and that this interaction recruits MCM5 to centrosomes.
Significantly, the cyclin A CLS includes the highly conserved hydrophobic patch containing the MRAIL amino acid sequence. This region has been reported to mediate binding of the cyclin-Cdk complex to substrates and regulatory proteins that contain a complementary hydrophobic sequence known as the K/RXL or Cy motif (Schulman et al., 1998; Takeda et al., 2001; Wilmes et al., 2004). It was previously reported that the binding of Orc1 to cyclin A is dependent on a K/RXL motif within Orc1 (Hemerly et al., 2009). Although these results suggest that the interaction of cyclin A with Orc1 might be MRAIL dependent, our studies with the MLW-A hydrophobic patch mutant of the cyclin A CLS indicate that a wild-type MRAIL sequence is not required. Instead, mutation of four downstream residues essential for CLS function results in reduced binding of both Orc1 and MCM5 to cyclin A. The ability of a CLS with hydrophobic patch mutations to still bind Orc1 and MCM5 probably reflects the contributions of other subdomains within the CLS to mediate binding.
Interestingly, although it has been reported that interaction between cyclin E and Orc1 is indirect (Hemerly et al., 2009), we previously described a direct interaction between cyclin E and MCM5 (Ferguson and Maller, 2008). Using MCM5 truncation mutants, we have shown here that the same region of MCM5 mediates direct binding to both cyclins A and E. Additionally, this region, amino acids 533-569, does not contain a K/RXL motif, indicating that the mechanism of binding to MCM5 does not require a wild-type hydrophobic patch; this was confirmed in our experiments (Fig. 2D). Although the CLS domains of cyclin A and cyclin E reside in distinct regions of the molecules, the fact that both CLS domains have a similar sequence and three-dimensional structure and that mutation of four CLS residues disrupts binding to both MCM5 (Fig. 2) and Orc1 (Fig. 6) indicates that interaction occurs through conserved CLS mechanisms. Thus, the CLS is not only a localization motif, but also an interaction domain for specifically targeting Cdk2 complexes to centrosomes.
It was previously shown that the expression of MCM5 but not MCM2, a related MCM family member, inhibits centrosome duplication in S-phase-arrested CHO-K1 cells (Ferguson and Maller, 2008). The work shown here suggests that recruitment of MCM5 and Orc1 to centrosomes by interaction with cyclin A contributes to inhibition of centrosome reduplication throughout late S phase and G2. Because centrosomes are still able to duplicate in cells arrested during these later interphase stages (Hanashiro et al., 2008), it is crucial that mechanisms preventing reduplication are actively maintained. Importantly, we have shown here that MCM5-mediated inhibition of centrosome reduplication does not require binding to any other MCM proteins. Thus, MCM5 regulation of centrosome number does not require assembly of the heterohexameric MCM complex and is distinct from its role in DNA replication.
Our data show that cyclin E and cyclin A function similarly in binding MCM5 and Orc1, controlling initiation of S phase and regulating centrosome duplication. With evidence that loss of cyclin E can be readily compensated by cyclin A, we propose that cyclin A plays a central role in regulating and maintaining centrosome number throughout interphase. Additionally, these studies support an apparent trend whereby proteins involved in DNA replication have dual functions in coordinating and regulating both DNA replication and centrosome duplication (Bettencourt-Dias and Glover, 2007; Ferguson and Maller, 2008; Hemerly et al., 2009; Tachibana et al., 2005).
Materials and Methods
Cell culture and transfection
CHO-K1 and HeLa S3 cells were grown as described previously (Ferguson and Maller, 2008). All transfections were carried out with Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's directions.
Immunoprecipitation
For endogenous cyclin A immunoprecipitation (IP), HeLa S3 cells were lysed in NP-40 lysis buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% NP-40) supplemented with 1× complete protease inhibitor (Roche), 1 mM AEBSF (Sigma), and phosphatase inhibitor cocktails 1 and 2 (Sigma). Agarose protein A beads (Sigma) were incubated with either anti-cyclin A antibody (Santa Cruz, sc-751) or control rabbit IgG (Santa Cruz) for 2 hours at 4°C, followed by incubation overnight at 4°C with 1 mg of lysate protein. Immunoprecipitates were separated on 10% SDS-PAGE, transferred to PDVF membranes and immunoblotted for proteins of interest. For blotting, the following primary antibodies were used against cyclin A (Cell Signaling, BF683) and MCM5 (Abcam, 17967). Coimmunoprecipitation of Cdk2 and Cdk1 was detected with a monoclonal antibody to PSTAIR (Sigma, P7962).
For Myc IP, CHO-K1 cells were transfected with pcDNA5-FRT-Myc-tagged cyclin A constructs. Approximately 20 hours after transfection, cell lysates were incubated with either preconjugated Myc-antibody agarose beads (Santa Cruz, 9E10) or control preconjugated mouse IgG agarose beads (Santa Cruz) for 3 hours at 4°C. Immunoprecipitates were separated on 10% SDS-PAGE, transferred to nitrocellulose membranes and proteins of interest detected by immunoblot analysis. The following primary antibodies were used: Myc (Santa Cruz, 9E10-HRP conjugated); MCM5 (Abcam, 17967); and Orc1 (Bethyl).
For HA IP, HeLa S3 cells were transfected with pcDNA5-FRT-HA-tagged MCM5 constructs. Approximately 20 hours after transfection, cell lysates were incubated at 4°C with protein A agarose beads (Sigma) bound to either rabbit polyclonal antibody to HA tag (Abcam, 9110) or control normal rabbit IgG (Santa Cruz). Immunoprecipitates were separated on 10% SDS-PAGE and proteins of interest detected by immunoblot analysis with primary antibodies to MCM5 (Abcam, 17967) and MCM3 (Abcam, 60597).
GST pulldown
GST and GST-tagged human cyclin A were purified from baculovirus-infected Sf9 cells obtained from the University of Colorado Cancer Center Tissue Culture Core Facility as previously described (Ferguson and Maller, 2008). For pulldown analysis, GST and GST-tagged cyclin A or cyclin E bound to glutathione beads were incubated overnight at 4°C in TIF buffer (20 mM Tris pH 8.0, 150 mM NaCl, 1 mM MgCl2, 0.1% NP-40 and 10% glycerol), with MCM5 protein produced in an in vitro transcription-translation (TNT)-coupled reaction according to the manufacturer's protocol (Promega). MCM5 protein was radiolabeled with [35S]-methionine during the TNT reaction. Pulldowns were separated on a 10% SDS-PAGE. Gels were dried and exposed to Kodak Biomax MR film.
Microscopy
CHO-K1 cells were seeded onto collagen-coated glass coverslips (Iwaki, Japan) and fixed with –20°C methanol for 15 minutes. Cells were immunostained with indicated primary antibodies to MCM5 (Santa Cruz, 22780), γ-tubulin (Sigma, GTU088 or Abcam, ab27076), or HA–Alexa-Fluor-488 conjugate (Molecular Probes, 16B12). The GFP signal was visualized by direct fluorescence. All primary antibody incubations were for 1 hour at room temperature, followed by secondary antibody staining. Secondary antibodies, either goat anti-rabbit or anti-mouse conjugated to Alexa-Fluor-488 or Alexa-Fluor-594 (Molecular Probes), were incubated with cells for 45 minutes at room temperature. Microscopic observations were made on a Nikon Eclipse TE300 inverted microscope with a 100× oil-immersion objective (NA 1.4). Images were obtained with either a PCM2000 confocal laser scan or an air-cooled charge-coupled device (CCD) camera (SenSys Photometrics) attached to a 0.76× coupler (Diagnostic Instruments). Images and line scans measuring centrosome-associated integrated fluorescence intensity were generated using simple PCI imaging software (Compix).
Centrosome duplication
CHO-K1 cells were placed in media containing 4 mM HU for 24 hours, followed by transient transfection of either pcDNA5-FRT-HA-tagged wild-type MCM5 or HA-tagged MCM5 C172Y. 24 hours after transfection (for a total of 48 hours HU treatment), cells were methanol fixed and processed for immunofluorescent staining. Cells were stained for centrosomes using γ-tubulin (Sigma, AK-15) and HA (Molecular Probes, A-21287).
Acknowledgments
We thank Robert Sclafani and Frank Eckerdt for helpful discussions throughout the course of this work, and Sean Munro for providing the PACT cDNA plasmid. Baculovirus expression and DNA sequencing were performed by core facilities in the University of Colorado Cancer Center (CA46930). The Howard Hughes Medical Institute supported this work. R.L.F. and G.P. are Research Associates, and J.L.M. is an Investigator of the Howard Hughes Medical Institute. Deposited in PMC for release after 6 months.
References
- Balczon R., Bao L., Zimmer W. E., Brown K., Zinkowski R. P., Brinkley B. R. (1995). Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J. Cell Biol. 130, 105-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet C., Aleem E., Coppola V., Tessarollo L., Kaldis P. (2003). Cdk2 knockout mice are viable. Curr. Biol. 13, 1775-1785 [DOI] [PubMed] [Google Scholar]
- Bettencourt-Dias M., Glover D. M. (2007). Centrosome biogenesis and function: centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 8, 451-463 [DOI] [PubMed] [Google Scholar]
- Bochman M. L., Bell S. P., Schwacha A. (2008). Subunit organization of Mcm2-7 and the unequal role of active sites in ATP hydrolysis and viability. Mol. Cell. Biol. 28, 5865-5873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Indjeian V. B., McManus M., Wang L., Dynlacht B. D. (2002). CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev. Cell 3, 339-350 [DOI] [PubMed] [Google Scholar]
- Coverley D., Laman H., Laskey R. A. (2002). Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat. Cell Biol. 4, 523-528 [DOI] [PubMed] [Google Scholar]
- DaFonseca C. J., Shu F., Zhang J. J. (2001). Identification of two residues in MCM5 critical for the assembly of MCM complexes and Stat1-mediated transcription activation in response to IFN-gamma. Proc. Natl. Acad. Sci. USA 98, 3034-3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton S., Hopwood B. (1997). Characterization of Cdc47p-minichromosome maintenance complexes in Saccharomyces cerevisiae: identification of Cdc45p as a subunit. Mol. Cell. Biol. 17, 5867-5875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson R. L., Maller J. L. (2008). Cyclin E-dependent localization of MCM5 regulates centrosome duplication. J. Cell Sci. 121, 3224-3232 [DOI] [PubMed] [Google Scholar]
- Fukasawa K. (2005). Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 230, 6-19 [DOI] [PubMed] [Google Scholar]
- Furstenthal L., Kaiser B. K., Swanson C., Jackson P. K. (2001). Cyclin E uses Cdc6 as a chromatin-associated receptor required for DNA replication. J. Cell Biol. 152, 1267-1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem N. J., Godinho S. A., Pellman D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278-282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y., Yu Q., Sicinska E., Das M., Schneider J. E., Bhattacharya S., Rideout W. M., Bronson R. T., Gardner H., Sicinski P. (2003). Cyclin E ablation in the mouse. Cell 114, 431-443 [DOI] [PubMed] [Google Scholar]
- Gillingham A. K., Munro S. (2000). The PACT domain, a conserved centrosomal targeting motif in the coiled-coil proteins AKAP450 and pericentrin. EMBO Rep. 1, 524-529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanashiro K., Kanai M., Geng Y., Sicinski P., Fukasawa K. (2008). Roles of cyclins A and E in induction of centrosome amplification in p53-compromised cells. Oncogene 27, 5288-5302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemerly A. S., Prasanth S. G., Siddiqui K., Stillman B. (2009). Orc1 controls centriole and centrosome copy number in human cells. Science 323, 789-793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchcliffe E. H., Li C., Thompson E. A., Maller J. L., Sluder G. (1999). Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science 283, 851-854 [DOI] [PubMed] [Google Scholar]
- Kalaszczynska I., Geng Y., Iino T., Mizuno S., Choi Y., Kondratiuk I., Silver D. P., Wolgemuth D. J., Akashi K., Sicinski P. (2009). Cyclin A is redundant in fibroblasts but essential in hematopoietic and embryonic stem cells. Cell 138, 352-365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey K. R., Jackson P. K., Stearns T. (1999). Cyclin-dependent kinase control of centrosome duplication. Proc. Natl. Acad. Sci. USA 96, 2817-2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle W. L., Lukasiewicz K., Salisbury J. L. (2005). Deregulation of the centrosome cycle and the origin of chromosomal instability in cancer. Adv. Exp. Med. Biol. 570, 393-421 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y., Maller J. L. (2004). A centrosomal localization signal in cyclin E required for Cdk2-independent S phase entry. Science 306, 885-888 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y., Hayashi K., Nishida E. (1999). Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr. Biol. 9, 429-432 [DOI] [PubMed] [Google Scholar]
- Okuda M., Horn H. F., Tarapore P., Tokuyama Y., Smulian A. G., Chan P. K., Knudsen E. S., Hofmann I. A., Snyder J. D., Bove K. E., et al. (2000). Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 103, 127-140 [DOI] [PubMed] [Google Scholar]
- Ortega S., Prieto I., Odajima J., Martin A., Dubus P., Sotillo R., Barbero J. L., Malumbres M., Barbacid M. (2003). Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet. 35, 25-31 [DOI] [PubMed] [Google Scholar]
- Parisi T., Beck A. R., Rougier N., McNeil T., Lucian L., Werb Z., Amati B. (2003). Cyclins E1 and E2 are required for endoreplication in placental trophoblast giant cells. EMBO J. 22, 4794-4803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascreau G., Eckerdt F., Churchill M. E., Maller J. L. (2010). Discovery of a distinct domain in cyclin A sufficient for centrosomal localization independently of Cdk binding. Proc. Natl. Acad. Sci. USA 107, 2932-2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A. A., Jeffrey P. D., Patten A. K., Massague J., Pavletich N. P. (1996). Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 382, 325-331 [DOI] [PubMed] [Google Scholar]
- Schulman B. A., Lindstrom D. L., Harlow E. (1998). Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc. Natl. Acad. Sci. USA 95, 10453-10458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strausfeld U. P., Howell M., Descombes P., Chevalier S., Rempel R. E., Adamczewski J., Maller J. L., Hunt T., Blow J. J. (1996). Both cyclin A and cyclin E have S-phase promoting (SPF) activity in Xenopus egg extracts. J. Cell Sci. 109, 1555-1563 [DOI] [PubMed] [Google Scholar]
- Tachibana K. E., Gonzalez M. A., Guarguaglini G., Nigg E. A., Laskey R. A. (2005). Depletion of licensing inhibitor geminin causes centrosome overduplication and mitotic defects. EMBO Rep. 6, 1052-1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda D. Y., Wohlschlegel J. A., Dutta A. (2001). A bipartite substrate recognition motif for cyclin-dependent kinases. J. Biol. Chem. 276, 1993-1997 [DOI] [PubMed] [Google Scholar]
- Tokuyama Y., Horn H. F., Kawamura K., Tarapore P., Fukasawa K. (2001). Specific phosphorylation of nucleophosmin on Thr(199) by cyclin-dependent kinase 2-cyclin E and its role in centrosome duplication. J. Biol. Chem. 276, 21529-21537 [DOI] [PubMed] [Google Scholar]
- Wilmes G. M., Archambault V., Austin R. J., Jacobson M. D., Bell S. P., Cross F. R. (2004). Interaction of the S-phase cyclin Clb5 with an “RXL” docking sequence in the initiator protein Orc6 provides an origin-localized replication control switch. Genes Dev. 18, 981-991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo R. A., Poon R. Y. (2003). Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle 2, 316-324 [PubMed] [Google Scholar]