

Abstract
The keratin IF network of epidermal keratinocytes provides a protective barrier against mechanical insult, it is also a major player in absorbing stress in these cells. The human papilloma virus (HPV) type 16 E1^E4 protein accumulates in the upper layers of HPV16-infected epithelium and is known to associate with and reorganise the keratin IF network in cells in culture. Here, we show that this function is conserved amongst a number of HPV alpha-group E1^E4 proteins and that the differentiation-dependent keratins are also targeted. Using time-lapse microscopy, HPV16 E1^E4 was found to effect a dramatic cessation of keratin IF network dynamics by associating with both soluble and insoluble keratin. Network disruption was accompanied by keratin hyperphosphorylation at several sites, including K8 S73, which is typically phosphorylated in response to stress stimuli. Keratin immunoprecipitated from E1^E4-expressing cells was also found to be ubiquitylated, indicating that it is targeted for proteasomal degradation. Interestingly, the accumulation of hyperphosphorylated, ubiquitylated E1^E4-keratin structures was found to result in an impairment of proteasomal function. These observations shed new light on the mechanism of keratin IF network reorganisation mediated by HPV16 E1^E4 and provide an insight into the depletion of keratin co-incident with E1^E4 accumulation observed in HPV-infected epithelium.
Keywords: E1^E4, Human papilloma virus, Keratin
Introduction
There are over 100 different HPV types, which infect stratified epithelium and cause lesions that range in severity from benign warts and verucccas to invasive carcinomas (Doorbar, 2005). HPV DNA has been detected in 99.7% of cervical cancers, with alpha-group types HPV16 and HPV18 occurring most frequently (Doorbar, 2006; Walboomers et al., 1999). The productive HPV life cycle is closely linked to the differentiation state of the epithelium. The viral E1^E4 protein (an E4 splice variant of replication protein E1) is expressed throughout the epithelium, but accumulates to high levels in the upper layers. Accumulation of E1^E4 coincides with the onset of viral-genome amplification, but precedes the expression of the viral structural proteins (L1 and L2) (Doorbar et al., 1997; Middleton et al., 2003; Peh et al., 2004). The E1^E4 proteins of HPV1 and HPV16 have been shown to associate with the keratin intermediate filament (IF) network in cultured cells (Doorbar et al., 1991; Roberts et al., 1993). The binding of HPV16 E1^E4 (16E1^E4), via an N-terminal leucine-rich motif (‘LLKLL’), to keratin and subsequent E1^E4 multimerisation, mediated by a C-terminal multimerisation domain, has been shown to result in reorganisation of keratin IF networks (Roberts et al., 1994; Roberts et al., 1997; Wang et al., 2004). Keratin association has not been demonstrated for any of the other alpha-group HPV E1^E4 proteins.
Keratins are encoded from more than 50 different genes and form obligate non-covalent heteropolymers of one type I and one type II keratin, with unique complements serving as markers that distinguish different epithelial cell types (Fuchs and Weber, 1994; Moll et al., 1982). In the stratified epithelium of the cervix and vulva, where the HPV E1^E4 protein is expressed, K5-K14 polymers predominate in the basal keratinocytes, whereas the differentiation-dependent keratins K4-K13 and K1-K10 are expressed in the suprabasal keratinocytes of the upper layers.
In epithelial cells, the keratin IF network is a highly dynamic structure, which can be completely reorganised during cellular events such as mitosis and apoptosis (Toivola et al., 2002; Fausther et al., 2004; Ridge et al., 2005). Keratin solubility and organisation of keratin IF networks, is regulated by phosphorylation, with keratin sequence diversity providing unique target sites for different kinases and phosphatases (Izawa and Inagaki, 2006). Keratin phosphorylation is also known to modulate its ubiquitylation and hence turnover (Ku and Omary, 2000). In stressed cells, the keratin IF network acts as a sink for stress-activated protein kinase (SAPK) activity, resulting in keratin hyperphosphorylation, ubiquitylation and proteasomal degradation, which leads to network disassembly (Fausther et al., 2004; Ridge et al., 2005; Toivola et al., 2002).
The keratin IF network is fundamental, not only to cellular integrity, but also to epithelial stability through association with intercellular desmosomal junctions (Getsios et al., 2004; Green and Gaudry, 2000). In a number of keratin-associated disorders, specific keratin mutations are known to render the cell more susceptible to cellular stress, whereas others compromise polymerisation, resulting in epidermal fragility (Lane and McLean, 2004; Rugg and Leigh, 2004). It is thought that keratin disruption mediated by 16E1^E4 might compromise epithelial integrity, thus facilitating virion release (Bryan and Brown, 2000).
Although the regions of 16E1^E4 responsible for keratin reorganisation have been mapped, little is known regarding the mechanism of E1^E4-mediated keratin IF network reorganisation or indeed whether the differentiation-dependent keratins are affected in the same way. This study is focused on the effects of E1^E4 on the keratin IF network. We have observed, using time-lapse imaging, that the keratin IF network has a significantly altered dynamic state when associated with 16E1^E4. This was accompanied by both hyperphosphorylation and ubiquitylation of keratin.
Together, our data present a model of HPV16 E1^E4-mediated keratin reorganisation, in which E1^E4 targets both soluble and filamentous keratin, disrupting network dynamics and resulting in keratin phosphorylation and ubiquitylation. This model offers an insight into the mechanism of keratin depletion, co-incident with E1^E4 accumulation, which we have observed in HPV16-infected cervical epithelium.
Results
E1^E4 proteins of a number of alpha-group HPV types all have the ability to associate with the keratin IF network
Association of 16E1^E4 with keratin has been shown to be fundamental to its ability to disrupt the keratin IF network (Wang et al., 2004). Amino acid sequences key to 16E1^E4-mediated keratin reorganisation have been mapped (Roberts et al., 1997; Wang et al., 2004) and interestingly, these sequences are highly conserved amongst E1^E4 proteins of other alpha-group viruses (Fig. 1A). We have looked at a number of E1^E4 proteins from both high-risk (types 16, 18, 31, 35 and 45) and low risk (types 2 and 11) alpha-group HPVs, to assess their keratin-binding capability. SiHa cells were transfected with pcDNA vectors to express the various E1^E4 proteins. Cells were fixed and stained with antibodies against E1^E4 (types 16, 18, 31, 35 and 45; TVG405, types 11 and 2 rabbit polyclonal) and keratin (C2562) 24 hours after transfection. All of the E1^E4 proteins were found to co-localise with the keratin IF network (Fig. 1B) and, as with 16E1^E4, filamentous, bundle and aggregate E1^E4-keratin co-localisation patterns were observed.
Fig. 1.

Keratin association is a conserved feature of alpha-group HPV E1^E4 proteins. (A) Amino acid sequence alignment for the E1^E4 proteins indicated. Conserved amino acids are highlighted in red and lower consensus sequences in blue. Sequences important in 16E1^E4 keratin reorganisation are indicated below. (B) SiHa cells, transfected with E1^E4 from a number of different virus types as indicated, were triple stained to visualise E1^E4, keratin and nuclei. The merged images demonstrate clear co-localisation of all E1^E4 proteins with the keratin IF network. Scale bars: 5 μm.
It appears from these data that the ability of the E1^E4 protein to associate with the keratin IF network is conserved amongst the diverse HPV types found in the alpha group. This function is most probably facilitated by a high degree of sequence conservation.
HPV16 E1^E4 associates with and causes disruption of several keratin networks
Previous studies have shown that 16E1^E4 reorganises the keratin IF networks of both SiHa and HaCaT cells (Doorbar et al., 1991; Wang et al., 2004). Work thus far has, however, focused on the association of E1^E4 to the well-characterised K8-K18 pair, which dominate the SiHa keratin IF network, K7-K16 is also present in these cells as a minor constituent (Bowden et al., 1992). In HPV16-infected cervical tissue E1^E4 has been shown to associate with K13 (the predominant keratin type in the upper layers of the epithelium) (Wang et al., 2004). Here, we found that in the differentiated cells of HPV16 genome-containing organotypic rafts, E1^E4 co-localised with the K10 filament network (Fig. 2A). K10 network reorganisation was also apparent in E1^E4-positive cells (Fig. 2A). It is however unclear how E1^E4-mediated reorganisation of these differentiation-dependent keratin networks, which predominate in the upper layers of the cervix and vulva where 16E1^E4 is expressed (Esquius et al., 1991; Kuppers et al., 1998; Smedts et al., 1990), compares with its reorganisation of the simple epithelial keratins.
Fig. 2.

HPV16 E1^E4 associates with and reorganises both differentiation-dependent and simple epithelial keratin IF networks. (A) HPV16 organotypic raft tissues triple stained for 16E1 E4 (green), K10 (red) and DNA (blue). K10 network reorganisation is observed in many cells (arrows) and co-localisation of 16E1^E4 with the K10 network is highlighted at higher magnification in the lower panel. Scale bars: 5 μm. (B) HaCaT cells infected with rAd16E1^E4 triple stained to visualise keratin (K1-K10, K4-K13, K5-K14 and K8-K18) (red), 16E1^E4 (green) and nuclei (blue). The merged images clearly show co-localisation of 16E1^E4 with all of these keratin networks. Scale bars: 5 μm. (C) Immunofluorescence images of 16E1^E4-transfected SiHa and HaCaT cells, stained as above, illustrating the co-localisation of 16E1^E4 and keratin in filamentous, bundle and aggregate structures. Scale bars: 5 μm. Graphs show the percentage of cells displaying filamentous, bundled or aggregate 16E1^E4-keratin co-localisation patterns, during a 72 hour period, in both SiHa and HaCaT cells.
Next, we examined the localisation of the differentiation-dependent keratins and 16E1^E4 by immunofluorescence microscopy in HaCaT cells. HaCaT cells, which are a human keratinocyte cell line, express a complex array of keratins, including the differentiation-dependent keratins, K1-K10 and K4-K13 and the primary keratins, K8-K18 and K5-K14 (Breitkreutz et al., 1991; Breitkreutz et al., 1993). Cells infected with recombinant adenovirus (rAd) 16E1^E4 were fixed 24 hours after infection and stained for both keratin, using type-specific keratin antibodies as indicated, and 16E1^E4 (TVG405). As illustrated in Fig. 2B, E1^E4 co-localised with all of the keratin networks examined. At this time point, the 16E1^E4-keratin network had a filamentous staining pattern, which was similar to that observed previously in SiHa cells.
Having established that 16E1^E4 could associate with keratin IF networks containing both differentiation-dependent and primary keratins, we compared the effects of this association on the different keratin networks. HaCaT cells infected with rAd16E1^E4 were stained for 16E1^E4 (TVG405) and keratin (C2562) 24 hours after transfection. 16E1^E4-mediated keratin re-organisation in HaCaT cells, as with SiHa cells, resulted in the observation of a number of different immunofluorescence staining patterns (Fig. 2C). Here, we classified the different 16E1^E4-keratin structures as filamentous, bundled or aggregated, depending on the extent of re-organisation. At early time points, the 16E1^E4-keratin network had a filamentous staining pattern with the appearance of thickened keratin filaments. At later time points, the keratin network was observed to re-organise into perinuclear bundles, which wrapped around the nucleus. Perinuclear aggregates were also observed in some cells, and in these cells, the keratin IF network was often found to be intact. We suspected that this might be due to the transient expression of 16E1^E4 in this system, which allows reformation of the network at later time points.
To compare the effect of 16E1^E4 on the differentiation-dependent keratins in HaCaT cells with its effect on the simple keratins in SiHa cells, the immunofluorescence distribution of 16E1^E4 in both SiHa and HaCaT cells was recorded at 12, 24, 48 and 72 hours after transfection. At each time point, a total of 100 cells were categorised as having either a filamentous, bundled or aggregated 16E1^E4 staining pattern. As shown in Fig. 2C, each staining pattern had a very different, time-dependent distribution.
The HaCaT keratin IF network was more resistant to network re-organisation, because 30% of HaCaT cells, compared with 2% of SiHa cells, retained a filamentous 16E1^E4 staining pattern up to 72 hours. However, in both cell lines, the ratio of cells containing filamentous 16E1^E4-keratin, to those having either bundled or aggregated structures, decreased over the time period shown. These data clearly illustrate that 16E1^E4-mediated keratin IF reorganisation is not keratin-type specific, and that the expression of 16E1^E4 results in a progressive re-organisation of both simple epithelial and differentiation-dependent keratin networks.
Association of HPV16 E1^E4 with keratin results in the inhibition of keratin dynamics
Although 16E1^E4-mediated disruption of the keratin IF network has been well documented by immunofluorescence, the events that initiate this process are still not fully understood. We thus used time-lapse imaging to probe keratin dynamics in the time period before network reorganisation is observed by immunofluorescence. In epithelial cells, GFP-K13 has been used to monitor keratin dynamics (Windoffer and Leube, 1999). Since keratin dynamics has not previously been characterised in SiHa cells, cells were transfected with the pHK13ΔP.EGFP plasmid and GFP-K13 was used to establish a picture of basic keratin dynamics. GFP-K13, which was easily visualised by immunofluorescence, became incorporated into filamentous structures that were indistinguishable from the endogenous keratin networks (supplementary material Fig. S1A). Time-lapse images of these cells recorded over a 2 hour period revealed that filament morphology was in a continuous state of flux, with filaments stretching, condensing or merging, and forming new filaments (supplementary material Fig. S1B and Movie 1). Filament migration was continuous and unidirectional, originating from diffuse material at the cell periphery and tracking toward the centre of the cell (supplementary material Fig. S1B). Fluorescence recovery after photobleaching (FRAP) was used to monitor keratin synthesis. GFP-K13 was observed to incorporate into the endogenous keratin IF network at the periphery of the bleached area (supplementary material Fig. S1B and Movie 1) and FRAP followed a similar pattern to that described previously for other cell lines (Windoffer et al., 2004).
Having established that keratin IF network dynamics in SiHa cells are typical of epithelial cells, we sought to assess the effect of 16E1^E4 binding on keratin dynamics. By immunofluorescence, 16E1^E4 was observed to colocalise with GFP-K13 in the IF network, resulting in network reorganisation. To monitor keratin dynamics in the presence of 16E1^E4, visualisation of the 16E1^E4 protein was essential; therefore, we engineered an YFP-16E1^E4 fusion protein. Initially, double-fluorescence studies were carried out using YFP-16E1^E4 in combination with fluorescently labelled K13. The two proteins were observed to co-localise forming filamentous, bundle and aggregate structures, however, the phototoxicity resulting from this double exposure system prevented time-lapse analysis.
Since YFP-16E1^E4, like the wild-type protein, was observed to align with and reorganise both GFP-K13 and the endogenous SiHa keratin IF network (Fig. 3A), time-lapse analysis of YFP-16E1^E4 was used to specifically examine the dynamics of the 16E1^E4-associated keratin IF network. The YFP-16E1^E4-keratin network differed in a number of ways from GFP-K13 incorporated into the endogenous IF network. In contrast to the continual flux, in terms of merging and extension of filaments, which was observed for GFP-K13, the YFP-16E1^E4-keratin filament architecture was static and did not alter significantly over the 2 hour period (Fig. 3B, blue arrows). Indeed, elements of gross 16E1^E4-keratin IF architecture were recognisable from the first to the final frame, in stark contrast to the GFP-K13 time-lapse recording, in which the cell in the last frame was unrecognisable as that in the first frame (supplementary material Fig. S1C).
Fig. 3.

YFP-16E1^E4 associates with keratin and alters normal keratin IF network dynamics in SiHa cells. (A) Fluorescence images of SiHa cells expressing YFP-16E1^E4 (green) with a keratin immunostain (red) and DAPI stain (blue). YFP-16E1^E4 was found to associate with the filamentous keratin IF network (top panels) resulting in reorganisation of the network (bottom panels). Scale bars: 5 μm. (B) FRAP analysis of the YFP-16E1^E4-keratin network recorded over a 2 hour period, 12 hours after transfection, in SiHa cells. The first image was acquired 1 second before bleaching (−1 s); the red arrow indicates the bleached area; subsequent images were acquired after bleaching at the time points indicated. The network remained largely static over this time period (blue arrows). Fluorescence recovery occurs throughout the bleached bar, indicating association of 16E1^E4 with the polymerised keratin IF network. Scale bar: 5 μm. (C) Higher-magnification images of time-lapse frames taken at times indicated. Some peripheral filament assembly is observed (white and yellow arrows); however, on merging with the static network (blue circle) the entire network appears to contract away from the cell edge (dotted line).
Rather than individual filaments migrating towards the centre of the cell through the merging of filaments, the YFP-16E1^E4-keratin network appeared to contract into the centre of the cell with few visible alterations in filament junctions (Fig. 3C, white arrows and supplementary material Movie 2). Although the integration of YFP-16E1^E4 into new filaments at the cell periphery was observed (Fig. 3C, yellow arrows), the static nature of the network limited their migration. It is reasonable to conclude that this process is the initiation of 16E1^E4-mediated reorganisation of the keratin IF network.
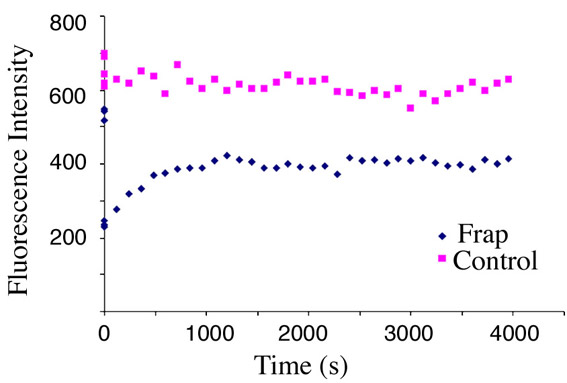
To assess whether the YFP-16E1^E4-keratin filaments, like GFP-K13 filaments, originate solely at the periphery of the cell, fluorescence recovery of the YFP-16E1^E4-keratin network after photobleaching was monitored (Fig. 3B, red arrow). In contrast to the GFP-K13 network described above, which reassembled from the periphery of the cell after photobleaching, YFP-16E1^E4 fluorescence returned uniformly across the photobleached region. Fluorescence recovered rapidly and was complete after 30 minutes. A comparable elevation in fluorescence was not observed in control regions (supplementary material Fig. S2), hence YFP-16E1^E4 fluorescence recovery was not simply due to protein synthesis. The rapid uniform recovery of YFP-16E1^E4 fluorescence suggests that 16E1^E4 is capable of binding to polymerised keratin.
Although the keratin IF network in cells expressing 16E1^E4 looks intact upon immunofluorescence analysis, the 16E1^E4 protein has, by binding to polymerised keratin, orchestrated a dramatic cessation of normal keratin IF network dynamics. These observations are consistent with the model proposed by Wang et al. (Wang et al., 2004) in which 16E1^E4 acts as a keratin crosslinker.
Cells respond to 16E1^E4-induced inhibition of keratin IF network dynamics by activation of SAPKs and phosphorylation of keratin
Studies in both stressed cells and cells expressing keratin mutants, have shown that altered keratin IF network dynamics and keratin reorganisation are accompanied by an elevation in keratin phosphorylation (Omary et al., 2006; Russell et al., 2004; Werner et al., 2004). Since the association of 16E1^E4 with keratin results in altered keratin dynamics, we investigated the phosphorylation status of this malfunctioning 16E1^E4-associated keratin network.
In vivo, 16E1^E4 is expressed in cells having predominantly differentiation-dependent keratins; however, the phosphorylation sites for these keratins have not been characterised, whereas the phosphorylation status of the K8-K18 network, both during normal cellular events and in response to stress stimuli, is well defined. Hence, we chose to use the existing well-developed panel of phospho-specific K8-K18 antibodies to examine the phosphorylation state of keratin in the presence of 16E1^E4.
A number of K8-K18 phosphorylation sites have been identified (S431 and S73 in K8; S33 and S52 in K18), these sites are known to be targeted during mitosis and apoptosis (Omary et al., 2006; Schutte et al., 2004), facilitating reorganisation of the keratin IF network. In addition, these sites are also phosphorylated in response to stress. K8 S73 in particular has been characterised as a ‘phosphate sponge’ for SAPKs in normal tissue (Omary et al., 2006; Ridge et al., 2005). To establish whether the association of 16E1^E4 with keratin alters its phosphorylation status, we used four phospho-epitope-specific keratin antibodies (K8 S431, 2DJ; K8 S73, LJ4; K18 S33, 8250; K18 S52, 3055) (Ku et al., 2004). The previously characterised 16E1^E4 keratin-binding mutant ΔLLKLL (Wang et al., 2004) was included here to determine the importance of 16E1^E4-keratin binding on the keratin phosphorylation status. SiHa cells were infected with either rAd16E1^E4 to express 16E1^E4, rAd16E1^E4ΔLLKLL to express ΔLLKLL or rAdβ-Gal to express β-Gal, which was used as a negative control. Cells were fixed 24 hours after infection and stained for 16E1^E4 and phosphorylated keratin.
Cells infected with both rAd16E1^E4ΔLLKLL and rAdβ-Gal were predominantly negative for all four phospho-keratin antibodies, as expected. However, a small number cells displayed strong distinctive phospho-keratin staining patterns; these cells were identified as either apoptotic or mitotic cells as defined by DAPI staining (data not shown). In cells expressing 16E1^E4, all 16E1^E4 aggregate structures were observed to associated with phosphorylated keratin (Fig. 4B). In addition, some filamentous and bundled 16E1^E4 keratin structures also displayed strong co-localisation of phospho-keratin and 16E1^E4 (Fig. 4A).
Fig. 4.

The association of 16E1^E4 with keratin results in keratin hyperphosphorylation. (A) SiHa cells infected to express 16E1^E4 were fixed and stained for 16E1^E4 (green), nuclei (blue) and phosphorylated keratin as indicated (red). In some, but not all, cells displaying filamentous 16E1^E4 staining, the associated keratin IF network is hyperphosphorylated at each of the four sites. Scale bars: 5 μm. (B) By contrast, all of the 16E1^E4 aggregate structures contain hyperphosphorylated keratin. Scale bars: 5 μm. (C) SiHa cells expressing 16E1^E4, ΔLLKLL (the 16E1^E4 keratin binding mutant) or β-Gal were harvested 12 or 24 hours after infection and analysed by western blotting using antibodies indicated on the right; GAPDH was used as a loading control. 16E1^E4 accumulated as expected over this period (top panel). Expression of 16E1^E4, but not β-Gal, results in an elevation in levels of phosphorylated keratin 8 and keratin 18. (D) SiHa cells expressing either 16E1^E4 or β-Gal were harvested 24 or 48 hours after infection and lysates analysed by western blotting using the antibodies indicated; a GAPDH loading control is also included. The levels of activated p38 MAPK and JNK are elevated in cells expressing 16E1^E4 compared with β-Gal-expressing cells.
16E1^E4 is known to induce G2 arrest, and the nuclei of the 16E1^E4 cells examined looked neither apoptotic nor mitotic. These data therefore indicate that the progression from filamentous and bundled 16E1^E4-keratin structures to aggregated structures is accompanied by the phosphorylation of both K8 and K18. However, given the absence of phospho-keratin in many of the filamentous and bundled 16E1^E4-keratin structures, keratin phosphorylation does not appear to be necessary for 16E1^E4-mediated inhibition of keratin IF network dynamics.
To determine whether phosphorylated keratin does indeed accumulate over time, in response to 16E1^E4-mediated disruption of the network, cell extracts were analysed by western blotting for 16E1^E4 and phosphorylated keratin. SiHa cells infected, as above, with adenovirus to express either 16E1^E4 or β-Gal, were harvested 12 and 24 hours after infection. As shown in Fig. 4C, 16E1^E4, levels dramatically increased over this time period. All of the phospho-keratin species monitored were significantly enriched in the presence of 16E1^E4 at 24 hours in comparison with cells expressing the keratin-binding mutant ΔLLKLL and β-Gal control cells. Levels of phosphorylated keratin were low, as expected, in cells expressing β-Gal. This data supports the hypothesis that phosphorylated keratin accumulates in 16E1^E4-keratin aggregate structures in a time-dependent manner.
A number of SAPKs have been implicated in targeting keratin during cellular stress, resulting in its hyperphosphorylation (Ku et al., 2002a; Ku and Omary, 2006; Ridge et al., 2005). In particular, stress-activated p38 mitogen-activated protein kinase (MAPK), is known to phosphorylate K8 at S73 exclusively (Ku et al., 2002b; Woll et al., 2007). A second SAPK, JNK, is also known to target K8 at S431 (Ku et al., 2002b). Having established that the expression of 16E1^E4 in SiHa cells results in the phosphorylation of both K8-S73 and K8-S431 we looked at the levels of activated p38 and JNK in these cells. SiHa cells infected with either rAd16E1^E4 to express 16E1^E4 or rAdβ-Gal to express β-Gal were harvested 24 or 48 hours after infection. Cell extracts were analysed by western blotting for p38 MAPK, phosphorylated p38 MAPK, JNK, phosphorylated JNK and 16E1^E4 (Fig. 4D). In cells expressing β-Gal the activated forms of these kinases were not detected; however, we observed a rapid and distinctive rise in levels of activated p38 MAPK and JNK in cells expressing 16E1^E4.
Together, these data indicate that in cells expressing 16E1^E4, the association of 16E1^E4 with keratin results in activation of p38 MAPK and JNK, and hyperphosphorylation of the keratin IF network.
Association of 16E1^E4 with keratin culminates in keratin ubiquitylation
Under conditions of stress, keratin hyperphosphorylation and aggregate formation is accompanied by ubiquitin-mediated proteasomal degradation (Jaitovich et al., 2008; Ku and Omary, 2000). Having established that keratin is hyperphosphorylated in the presence of 16E1^E4, we investigated whether it was also ubiquitylated.
In these experiments, we again included the 16E1^E4 keratin-binding mutant ΔLLKLL to establish whether the effects of 16E1^E4 on keratin were directly attributable to binding. Again SiHa cells were infected with either rAd16E1^E4 to express 16E1^E4, rAd16E1^E4ΔLLKLL to express ΔLLKLL or rAdβ-Gal to express β-Gal. Cells were harvested in lysis buffer 12, 24 or 48 hours after infection. Lysates were analysed by SDS-PAGE and western blotting for 16E1^E4, keratin, β-Gal and GAPDH, which was used as a loading control.
ΔLLKLL accumulated more slowly than 16E1^E4 over the first 24 hours, but reached similar levels to 16E1^E4 by 48 hours (Fig. 5A). The keratin western blots, shown at high exposure in Fig. 5A, reveal accumulation of high molecular weight K8 and K18 species in 16E1^E4 compared with cells infected with either ΔLLKLL or β-Gal. These high molecular weight keratin ladders become evident at 24 hours and were more prominent for K18 than for K8. Interestingly, levels of full-length keratin 8 and 18 were also lower in 16E1^E4-expressing cells compared with cells expressing either ΔLLKLL or β-Gal.
Fig. 5.

16E1^E4-keratin association results in the observation of high molecular weight K8-K18 ladders, which accumulate upon proteasomal inhibition. (A) SiHa cells were infected to express 16E1^E4, ΔLLKLL, or β-Gal and harvested 12, 24 or 48 hours after infection. Western blots were probed to detect proteins as indicated on the right, both high and low exposures of K8 and K18 blots are shown (bold); GAPDH was used as a loading control. In the high-exposure blots, accumulation of high molecular weight K8 and K18 ladders is evident only in cells expressing 16E1^E4. (B) SiHa cells, infected to express 16E1^E4 or β-Gal, were treated with the proteasomal inhibitor ALLN 18 hours before harvest. Accumulation of high molecular weight K18 species on proteasomal inhibition indicates that they are ubiquitylated.
Keratin laddering is generally attributed to its conjugation to ubiquitin (Jaitovich et al., 2008; Ku and Omary, 2000). Typically, ubiquitylated species are enriched on inhibition of the proteasome, prompting us to look at the effect of proteasomal inhibition on the K18 ladders observed in cells expressing 16E1^E4. SiHa cells infected with either rAd16E1^E4 or rAdβ-Gal, were treated overnight with the proteasomal inhibitor acetyl-leucyl-leucyl-norleucinal (ALLN) and harvested 24 hours after infection. Upon ALLN treatment of rAdβ-Gal-infected cells, a small number of high molecular weight K18 species, typical of keratin turnover by ubiquitylation, were observed at very high exposure. By contrast, the keratin laddering observed in rAd16E1^E4-infected cells was dramatically enhanced upon proteasomal inhibition and was clearly visible at high exposure (Fig. 5B). Additionally treatment of these lysates with isopeptidase, which cleaves ubiquitin chains, significantly reduced the number of high molecular weight K18 species (data not shown).
Having shown that the expression of 16E1^E4 results in keratin laddering, which is enhanced upon proteasomal inhibition, we sought to establish whether these keratin species were indeed ubiquitylated. Keratin was immunoprecipitated from lysates of SiHa cells infected with either rAd16E1^E4 or rAdβ-Gal using either a PanKeratin antibody (C2562) or a K8-K18-specific antibody (L1A2). Immunoprecipitates (IPs) were separated by SDS-PAGE and probed for ubiquitin (Fig. 6Ai,Bi). In both keratin IPs, ladders of ubiquitylated keratin were greatly enhanced in extracts from cells expressing 16E1^E4 compared with β-Gal. Cell extracts probed for K8 and K18 are shown to represent keratin input (Fig. 6Ai,Bi). As observed in Fig. 5, high molecular weight K18 ladders were much more prominent than K8 ladders in 16E1^E4-expressing cells.
Fig. 6.

The high molecular weight keratin species observed in the presence of 16E1^E4 are ubiquitylated. (A,B), SiHa cells expressing 16E1^E4 or β-Gal were harvested and lysed 24 hours after infection; keratin was immunoprecipitated using either C2562, a pan-keratin antibody (A) or L2A1, a K8-K18 antibody (B). A ubiquitin probe of the immunoprecipitates reveals accumulation of high molecular weight ubiquitylated keratin, Ub-K (panels i). High molecular weight K18 laddering was also apparent in K18 probed cell extracts. IPs reprobed for K18 revealed that these ladders were immunoprecipitated, indicating that they are ubiquitylated (panels ii). Alignment of the IPs probed with K18 and ubiquitin identified a number of discrete bands, common to both blots, thus confirming the presence of ubiquitylated K18 in 16E1^E4-expressing cells (panels iii).
To identify individual ubiquitylated K18 species, the IPs were re-probed for K18 (Fig. 6Aii,Bii). Extracts were also re-probed for GAPDH to demonstrate that comparable protein levels were used as input for each IP (Fig. 6Aii). The very high molecular weight ubiquitylated species which dominate the keratin IPs (Fig. 6Ai,Bi) are less apparent in samples reprobed for K18 (Fig. 6Aii,Bii). This is probably because their polyubiquitylated nature elevates the ratio of ubiquitin to keratin epitopes. However, several prominent bands observed in both the ubiquitin and K18 re-probes were aligned (Fig. 6Aiii,Biii), thus identifying individual ubiquitylated keratin species. These data confirm that the high molecular weight K18 species observed in the presence of 16E1^E4 are covalently linked to ubiquitin.
The distribution of ubiquitylated protein in SiHa cells infected with rAd16E1^E4 was also examined by immunostaining for 16E1^E4 and ubiquitin. Ubiquitin staining was diffuse in cells displaying filamentous 16E1^E4 staining and in uninfected cells. However, 16E1^E4 aggregates reacted strongly with the ubiquitin antibody (Fig. 7A). The distribution of ubiquitylated keratin was also examined in cells expressing 16E1^E4 by fractionation and western blotting. As shown in Fig. 7A, the high molecular weight keratin species fractionated exclusively in the insoluble fraction. The ubiquitylated keratin species, which accumulated as a result of 16E1^E4-keratin association, was localised to insoluble 16E1^E4-keratin aggregate structures.
Fig. 7.

Ubiquitylated keratin associates with 16E1^E4 aggregates and its accumulation results in proteasomal inhibition (A) SiHa cells infected to express 16E1^E4 were immunostained for 16E1^E4 (green) and ubiquitin (red); DAPI stain is blue. The merged images clearly show that 16E1^E4-keratin aggregate structures co-localise with ubiquitin (arrows). Scale bars: 5 μm. (B) Fractionation of 16E1^E4 and keratin from SiHa cells expressing 16E1^E4 using 1% NP40 (N), 1% empigen (E) and 9M urea (U). The high molecular weight keratin species, which accumulate in the presence of E1^E4, fractionate exclusively in the insoluble fraction. (C) SiHa cells were infected to express 16E1^E4 or the keratin-binding mutant ΔLLKLL and harvested at the times shown. Cell lysates were probed to detect proteins as indicated. Accumulation of 16E1^E4 coincides with accumulation of K18 ladders and ubiquitylated protein. Accumulation of ubiquitylated protein is not observed in cells expressing the keratin binding mutant, indicating that 16E1^E4-keratin association results in an impairment of proteasomal function.
The accumulation of hyperphosphorylated, ubiquitylated keratin is often associated with impairment of proteasome function. Using western blotting, we compared the levels of ubiquitylated protein in cells expressing 16E1^E4, with cells expressing the keratin-binding mutant (ΔLLKLL) (Fig. 7C). In ΔLLKLL-expressing cells, the level of ubiquitylated protein was low and remained constant over the time period shown, indicating a functioning proteasome. However, ubiquitylated protein was observed to accumulate at much higher levels in the 16E1^E4-expressing cells, indicating an impairment in proteasome function.
Association of 16E1^E4 with the keratin IF network results not only in keratin re-organisation, but also in keratin depletion in HPV16-infected cervical tissue
Keratin IF network reorganisation has been documented in HPV16-infected epithelium. In addition, keratin depletion has also been observed in 16E1^E4-positive cells in organotypic raft cultures containing the HPV16 genome (Nakahara et al., 2005). Having here established that the association of 16E1^E4 with keratin results in keratin ubiquitylation, we carried out a detailed comparison of keratin and 16E1^E4 immunofluorescence staining in normal cervical epithelium and cervical epithelium harbouring a productive HPV16 lesion.
Paraffin-embedded formalin-fixed tissue sections were stained for keratin (C2562) and 16E1^E4 (TVG405). At high magnification, we observed that the keratin was reorganised to the edge of the cell in some 16E1^E4-positive cells (supplementary material Fig. S3A), as has been shown previously (Wang et al., 2004). These images also clearly showed the co-localisation of 16E1^E4 with keratin filaments (supplementary material Fig. S3B), illustrating that the association of 16E1^E4 with keratin is preserved in these sections and that the epitopes for both antibodies are adequately exposed.
The multiple keratin types present in normal cervical epithelium are detected by the pan-keratin antibody, resulting in solid staining throughout the section (Fig. 8A). By contrast, in HPV16-infected tissue, we noted that the keratin staining was broken near the surface of the section (yellow arrows). Upon overlaying the keratin and 16E1^E4 stains, it was clear that the break in keratin staining coincided with strong 16E1^E4 staining (Fig. 8A). This apparent depletion of keratin became evident just after the onset of 16E1^E4 expression and gradually culminates in the observation of cells, which despite rigorous epitope exposure conditions, showed no keratin staining. The accumulation of 16E1^E4 towards the surface of the epithelium did not however correlate with continued loss of keratin immunofluorescence; on the contrary, cells at the epithelial surface had strong staining for both keratin and 16E1^E4.
Fig. 8.

Quantitative analysis of 16E1^E4 and keratin levels in HPV16-infected tissue sections. (A) Tissue sections of both normal (top) and HPV16-infected (bottom) cervical epithelium were immunostained as indicated. All keratin networks were detected using the pan-keratin antibody. The solid keratin staining throughout the normal tissue contrasts with the broken staining observed in HPV16-infected tissue. Scale bars: 10 μm. (B) Plots of both 16E1^E4 and keratin fluorescence intensity (y-axis) from the surface of the epithelium (0 μm) toward the basal layer (x-axis) clearly show depletion and reaccumulation of keratin in HPV16-infected tissue (yellow arrow).
To quantify immunofluorescence levels for 16E1^E4 and keratin, we selected a number of cross-sectional vectors, starting at the epithelial surface (0 μM) and recorded both red and green channel fluorescence intensity (Fig. 8B). Comparison of the keratin levels highlighted the very different keratin immunofluorescence patterns observed in infected and uninfected tissue, differences were particularly evident in the 0-70 μm region, spanning the epithelial surface and the uppermost layers of the epithelium. In this region, the keratin signal remained high in normal cervical tissue, whereas in HPV16-infected tissue, the keratin signal was observed to decline to only ~10% of its original value. This dramatic decrease was followed by an equally dramatic recovery in keratin levels. In this region also, 16E1^E4 was observed to steadily accumulate, with levels peaking at the surface of the epithelium.
We propose that keratin depletion is not only coincident with, but also dependent on 16E1^E4 expression and that this process is initiated by 16E1^E4-mediated keratin phosphorylation, leading to keratin ubiquitylation and degradation. The area of keratin depletion highlighted in Fig. 8 spanned the length of the tissue and was clearly detrimental to the integrity of the tissue.
Discussion
16E1^E4-mediated keratin IF reorganisation has been attributed to key amino acid sequences (Wang et al., 2004). Conservation of these sequences (Fig. 1A) suggests that other HPV alpha-group E1^E4 proteins also facilitate keratin IF network reorganisation. Here, we have shown that a broad panel of HPV alpha-group E1^E4 proteins [α9 (HPV31, HPV33 and HPV11), α7 (HPV45 and HPV18) and α4 (HPV2)] co-localise with the keratin IF network (Fig. 1B). In addition, the observation of E1^E4-keratin bundle and aggregate structures suggests that all of these E1^E4 proteins effect keratin IF network reorganisation. Conservation of both the N-terminal keratin-binding motif and C-terminal multimerisation sequences indicates that the HPV alpha-group E1^E4 proteins share a common mode of keratin association and most probably mediate network re-organisation in a similar manner.
HPV16 infection occurs in the cervix and the vulva, hence 16E1^E4 is expressed and accumulates in cells in which the differentiation-dependent keratins K4-K13 and K1-K10 are found (Esquius et al., 1991; Kuppers et al., 1998; Smedts et al., 1990). Previous work demonstrated co-localisation of 16E1^E4 with K13 in HPV16-infected cervical tissue (Wang et al., 2004) and here we have shown co-localisation of 16E1^E4 with K10 in organotypic rafts containing the HPV16 genome (Fig. 2A). In addition, our study shows that 16E1^E4-mediated IF network reorganisation in HaCaT cells, which contain both differentiation-dependent (K1-K10, K4-K13) and simple epithelial keratins (K8-K18), follows a similar pattern to its reorganisation of the K8-K18 network (Fig. 2B,C).
As 16E1^E4 has been shown to associate, via its ‘LLKLL’ motif, preferentially with K18 (Wang et al., 2004), it is reasonable to assume that this motif is also used to bind the type I keratin partners of the differentiation-dependent networks. Similarly, 16E1^E4-mediated reorganisation of these keratin networks is most probably facilitated by its multimerisation. These findings establish that 16E1^E4 can associate with and disrupt the differentiation-dependent keratin IF networks present in the upper layers of mucosal epithelium, where 16E1^E4 is expressed and accumulates, in the course of a productive HPV infection.
Although the stabilising properties of the IF network provide integrity to both cells and tissues, the network is also very dynamic (Magin et al., 2007). To gain an insight into the events leading to 16E1^E4 mediated keratin IF network reorganisation we looked at keratin network dynamics in SiHa cells, using time-lapse imaging. In contrast to the continuous flux of GFP-K13 filaments observed in the absence of 16E1^E4 (supplementary material Fig. S1B), time-lapse imaging of YFP-16E1^E4 revealed a largely static 16E1^E4-keratin IF network, where filaments and junctions were locked in place (Fig. 3B,C; supplementary material Movie 2). 16E1^E4-mediated keratin reorganisation has been shown to be dependent on C-terminal amino acids (Wang et al., 2004); these residues facilitate multimerisation into amyloid-like fibrils when not restrained by the N-terminal residues (McIntosh et al., 2008). The formation of 16E1^E4 fibrils on keratin might facilitate keratin IF crosslinking, accounting for the static nature of the network. We propose that the contraction of this static network away from the cell periphery observed here (Fig. 3B,C) constitutes the initial stages of 16E1^E4-mediated keratin IF network reorganisation. The observation of peripheral 16E1^E4-keratin filament assembly revealed that 16E1^E4 can associate with soluble keratin and that these 16E1^E4-associated subunits can assemble into filaments (Fig. 3B). This process might provide an efficient way of mopping up soluble keratin subunits to expedite network reorganisation. Although this system is limited to monitoring 16E1^E4-associated keratin and so does not reflect total cellular keratin, immunofluorescence studies have shown that 16E1^E4 decorates the entire visible keratin network. In addition, FRAP analysis revealed the incorporation of 16E1^E4 into the assembled keratin IF network (Fig. 3B; supplementary material Movie 2). Association of 16E1^E4 with polymerised keratin means that 16E1^E4 can target terminally differentiated keratin IF networks, thus expediting the process of network disruption. This is likely to be necessary given the variable and sometimes late onset of 16E1^E4 expression in the life cycle.
Normal keratin dynamics are altered in response to a number of stimuli, including mitosis, apoptosis and cell stress (Herrmann et al., 2007). Reorganisation of the IF network in response to these stimuli is orchestrated largely by keratin phosphorylation, which facilitates filament disassembly by solubilisation (Omary et al., 2006). The association of 16E1^E4 with keratin has previously been shown to result in phosphorylation of K18 at S33; however, this event did not result in keratin solubilisation, despite the recruitment of 14-3-3 (Wang et al., 2004). Here, we have found elevated phosphorylation of, not only S33, but also S52 in K18, and S431 and S73 in K8, in cells expressing 16E1^E4. We show that phosphorylation of these sites accompanied 16E1^E4-mediated disruption of the IF network; phosphorylated species accumulated over time and target sites were occupied in all 16E1^E4-keratin aggregate structures (Fig. 4B,C) and in many 16E1^E4-keratin bundle structures. Since some filamentous 16E1^E4-keratin structures also displayed strong phospho-keratin staining (Fig. 4A), 16E1^E4-mediated disruption of normal keratin dynamics might act as the initial impetus for keratin phosphorylation.
Keratin hyperphosphorylation, as observed here in cells expressing 16E1^E4, is commonly associated with cell stress. Stress-activated p38 MAPK is known to specifically target S73 in K8 (Ku et al., 2002a; Liao et al., 1997; Ridge et al., 2005). K8 S73 is a unique marker of stress because it is unphosphorylated under basal conditions (Liao et al., 1997). Here, we found that 16E1^E4-mediated keratin IF network reorganisation was accompanied by activation of not only p38 MAPK, but also JNK, which is known to target K8 S431 (Fig. 4D). These data indicate that the association of 16E1^E4 with the keratin IF network elicits a similar response to stress stimuli, resulting in SAPK activation, keratin hyperphosphorylation and reorganisation of the keratin IF network. Notably also, the K8 S73 motif is conserved in other type II epidermal keratins (Toivola et al., 2002), including K4, the predominant type II keratin in cervical epithelium. As we have shown here, 16E1^E4 associates with and reorganises a number of keratin IF networks, including K4-K13 (Fig. 2). It is probable that this process is in part facilitated by activation of p38 MAPK, resulting in phosphorylation of the highly conserved phospho-epitope corresponding to K8-S73.
The expression of 16E1^E4 also results in accumulation of high molecular weight K18, and to a lesser extent K8 species (Fig. 5A). These high molecular weight species were enhanced upon proteasomal inhibition (Fig. 5B) and K18 immunoprecipitation revealed that they were covalently linked to ubiquitin (Fig. 6A,B). The relatively low level of high molecular weight K8 species prevented their detection post immunoprecipitation; therefore, it is still unclear if K8 is ubiquitylated.
Proteasomal turnover of the K8-K18 network is modulated by phosphorylation, with phosphorylation of K8-S73 again playing a key role (Jaitovich et al., 2008; Ku and Omary, 2006). In cells expressing 16E1^E4, we observed the accumulation of both phosphorylated and ubiquitylated keratin; however, ubiquitylated keratin was restricted to insoluble 16E1^E4-keratin aggregates (Fig. 7A,C), whereas both filamentous and aggregated E1^E4-keratin structures were hyperphosphorylated (Fig. 4A,B). It is likely that 16E1^E4-mediated keratin hyperphosphorylation precedes its ubiquitylation.
Elevated protein ubiquitylation is typically accompanied by proteasomal degradation, indeed in mechanically stimulated cells, keratin phosphorylation and ubiquitylation culminates in its depletion (Jaitovich et al., 2008). Although the keratin species profile is significantly altered in the presence of 16E1^E4, depletion of total keratin is not immediately obvious, this would appear to be inconsistent with the observation of phosphorylated and ubiquitylated keratin. A number of factors peculiar to this system might contribute to these observations. First, sustained 16E1^E4 expression is not feasible using these transient expression systems, as the keratin network was reinstated at later time points (Fig. 2C). This is not the case in vivo where 16E1^E4 is observed to accumulate in the upper layers of the epithelium (Fig. 8A). Second, 16E1^E4 expression resulted in the aggregation of 16E1^E4-keratin; aggregates and inclusion bodies in general, are resistant to proteasomal degradation. Ubiquitylation might, however, contribute to the depletion of soluble keratin observed in 16E1^E4-expressing cells; this is likely to restrict reinstatement of the keratin IF network.
A number of studies have indicated that the differentiation program in epithelial tissue harbouring HPV infection is disrupted (Nakahara et al., 2005; Roberts et al., 1993; Bryan and Brown, 2000; Doorbar et al., 1991; Wang et al., 2004). Interestingly, we also observed extensive keratin depletion in 16E1^E4-positive cells in HPV16-infected cervical tissue and in HPV16 organotypic raft tissue (Fig. 2A, Fig. 8). We propose that 16E1^E4-mediated keratin IF network disruption, phosphorylation and ubiquitylation leads to the depletion of keratin in those cells where accumulation of 16E1^E4 is observed. Surprisingly however, in these lesions, the continued accumulation of 16E1^E4 did not correlate with further loss of keratin; in fact keratin levels recovered towards the surface of the epithelium.
In monolayer cells, we found that at later time points the presence of hyperphosphorylated and ubiquitylated keratin also corresponded to abnormally high levels of protein destined for proteasomal degradation. Similarly, in liver tissue, the formation of Mallory bodies, which are composed of hyperphosphorylated and ubiquitylated keratin, is also associated with impairment of the proteasome (Bardag-Gorce et al., 2002; Riley et al., 2002). It is possible that levels of ubiquitylated keratin eventually exceed the capacity of the proteasome, resulting in a general impairment of the ubiquitin-proteasome system, allowing reaccumulation of keratin.
It is well established that the association of the keratin IF network with desmosomal junctions contributes to epidermal integrity. This is particularly evident in disorders associated with keratin mutations, such as EBS (epidermolysis bullosa simplex), where keratin hyperphosphorylation, ubiquitylation and depletion is observed and cells have an increased susceptibility to mechanical stress resulting in increased epidermal fragility (Pekny and Lane, 2007; Russell et al., 2004). These data clearly show that 16E1^E4 expression results in similar modifications of the keratin IF network. Given that the keratinocytes of the upper layers of HPV-infected epithelium harbour not only 16E1^E4, but also assembled virus, the impact of E1^E4-mediated keratin IF reorganisation on intracellular junctions remains an important question.
Taken together, these results provide a molecular basis for 16E1^E4-mediated keratin IF reorganisation and reveal that keratin is modified by phosphorylation and ubiquitylation in much the same manner as observed in cells subjected to stress stimuli.
Materials and Methods
Cell culture and immunofluorescence
SiHa cells are a transformed cervical epithelial cell line that constitutively express HPV16 E6 and HPV16 E7 (Friedl et al., 1970). HaCaT are a human keratinocyte cell line (Boukamp et al., 1988). All cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 1% penicillin and streptomycin; cells were incubated at 37°C in a 5% CO2 environment. SiHa cells were infected with adenovirus plasmids at a multiplicity of infection of 100 and harvested at the times indicated after infection. Transfections were performed using the GeneJuice transfection reagent according to the manufacturer's protocol (Novagen, Nottingham, UK). The Proteasome inhibitor ALLN (Calbiochem) was used at a concentration of 10 μM, 18 hours before harvest. For indirect immunofluorescence, cells were grown on coverslips, fixed with methanol for 2 minutes at −20°C, rehydrated in PBS and blocked in 2% (w/v) BSA in PBS for 30 minutes. Cells were stained at room temperature for 1 hour with primary antibody followed by secondary antibody before mounting. HPV16 genome-containing organotypic raft cultures were generated as previously described (Wang et al., 2009). Tissue sectioning, dewaxing, rehydration and epitope exposure and blocking were carried out as described previously (Wang et al., 2004). Sections were stained in a humidified chamber at 4°C overnight, with primary antibody followed by a 2 hour incubation with secondary antibody.
Protein expression
16E1^E4 was expressed either by transfection using the pMV11.16E1^E4 plasmid (Davy et al., 2002) or infection using the rAd16E1^E4 plasmid (Doorbar et al., 2000). ΔLLKLL was expressed by infection using the rAd16E1^E4ΔLLKLL plasmid (Wang et al., 2004). The HPV type 2, 18, 31, 35, 45 and 11 E1^E4 were expressed by transfection of corresponding pcDNA vectors. Vectors were constructed using the pcDNA 3.1 Directional TOPO Expression kit (Invitrogen). YFP-16E1^E4 was expressed by transfection of the pYFP-16E1^E4 plasmid. pYFP-16E1^E4 was constructed by subcloning a HindIII-BamH1 fragment containing full-length 16E1^E4 into the HindIII-BamH1 sites of pEYFP-C1 vector (Clontech). β-gal was expressed by infection with rAdβ-Gal (Davy et al., 2002). GFP-K13 was expressed by transfection of the pHK13DP.EGFP plasmid (Windoffer and Leube, 1999) kindly provided by Rudolf Leube (Institute of Molecular and Cellular Anatomy, RWTH Aachen University, Aachen, Germany).
Antibodies
The keratin antibodies used for immunofluorescence microscopy are as follows: keratin 4 (Novacastra, NCL-CK4), keratin 5 (Novacastra, NCL-CK5), keratin 10 (Novacastra, NCL-CK10), keratin 13 (Novacastra, NCL-CK13), keratin 14 (Novacastra, NCL-11002), pan-keratin (Sigma, C2562). Keratin 8 (MRC, Cam 5.2), keratin 18 (Sigma, Cy 90) and the phospho-epitope-specific keratin 8 and 18 antibodies [LJ4 (K8-S73), 2D6 (K8-S431), 3305 (K18-S52), 8250 (K18-S33)] (kindly provided by Bishar M. Omary, Palo Alto VA Medical Centre, Palo Alto, CA) (Ku et al., 2004) were used for both immunofluorescence microscopy and immunoblotting. For immunofluorescence microscopy, the high-risk HPV16 E1^E4 proteins were detected using the TVG405 (Doorbar et al., 1992) antibody conjugated to Alexa Fluor 488. Specificity of TVG405 for these proteins was demonstrated by ELISA on a panel of MBP-E1^E4 fusion proteins. HPV2 E1^E4 was detected using a rabbit polyclonal antibody developed in-house and HPV11 E1^E4 was detected using a rabbit polyclonal antibody (Peh et al., 2002). Nuclei were stained using DAPI (4-6-diamidino-2-phenylindole) (Sigma). For immunoblotting, 16E1^E4 was detected using TVG402 (Doorbar et al., 2000) and GAPDH using MAB374 (Chemicon). The ubiquitin antibodies Sc-8017 (Santa Cruz) and Z0458 (DAKO) were used for immunoblotting and immunofluorescence microscopy, respectively.
Fractionation, immunoprecipitation and immunoblotting
For protein immunoprecipitation, cells were lysed in 1% (w/v) SDS in PBS, treated with benzonase and centrifuged at 1300 g for 10 minutes. The soluble fraction was incubated (3 hours, 4°C) with protein-G-Sepharose beads covalently conjugated (1 hours, 4°C) to either the PanKeratin (Sigma, C2562) or the K8-K18 [L2A1 (Chou and Omary, 1991)] antibody. Mock immunoprecipitations, without antibody, were also carried out to demonstrate specificity. To assess protein solubility, cell extracts were sequentially fractionated in 1% NP40, 1% empigen and 9 M urea. For analysis of active kinases, cell extracts were prepared in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% NP40, 1 mM Vanadate and 1 tablet of miniprotease inhibitor cocktail (Roche) per 10 ml. Cell extracts and immunoprecipitated proteins were analysed using immunoblotting, as described previously (Wang et al., 2004).
Immunofluorescence microscopy
Low-power images of tissue sections were acquired using a Labophot II microscope (Nikon, Kingston-upon-Thames, UK). Cells were imaged using a Leica DMRXE upright microscope (Leica Microsystems, Milton Keynes, UK). Specimens were scanned at 0.5 μm intervals between Z-sections with four accumulations per frame. Images are presented as overlays, resulting in a composite image.
Time-lapse microscopy
Time-lapse observations were made with a DeltaVision microscope (Olympus Ltd., London, UK) equipped with a 100×, 1.4 NA oil-immersion objective. Confluent 60 mm glass dishes (MatTek, Ashland, MA) of transfected live cells were transferred into the chamber of the microscope at 37°C. Cells were left to equilibrate for 1 hour before imaging. GFP and YFP images were acquired by excitation and emission using a FITC filter at 515-545 nm. Images were collected every 2 minutes over a period of 2 hours in the same focal plane. Each frame was composed of five Z-sections, giving a focal depth of 2 μm.
Fluorescence recovery after photobleaching
For cells expressing both YFP-16E1^E4 and GFP-K13, bar-shaped regions were bleached at 488 nm for 0.05 seconds, recovery was monitored using time-lapse imaging. Images were captured using softWoRx QLM software (Applied Precision, LLC, Marlborough, UK). Bleached targets were tracked using a custom-built patch (provided by Dan Zue, NIMR, London, UK) in ImageJ and data analysed using Microsoft Office Excel software.
Sequence alignment
Alignments were carried out using MultalAlin (Multiple sequence alignment with hierarchical clustering) software (Corpet, 1988).
Supplementary Material
Acknowledgments
The authors would like to thank Bishar Omary for providing the phospho-epitope-specific antibodies and Rudolph Leube for providing the GFP-keratin 13 plasmid. We would also like to thank Jonathan Stoye in the Division of Virology for supporting this work at NIMR. This work was funded by the UK Medical Research Council. Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/16/2810/DC1
References
- Bardag-Gorce F., van Leeuwen F. W., Nguyen V., French B. A., Li J., Riley N., McPhaul L. W., Lue Y. H., French S. W. (2002). The role of the ubiquitin-proteasome pathway in the formation of mallory bodies. Exp. Mol. Pathol. 73, 75-83 [DOI] [PubMed] [Google Scholar]
- Boukamp P., Petrussevska R. T., Breitkreutz D., Hornung J., Markham A., Fusenig N. E. (1988). Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106, 761-771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden P. E., Woodworth C. D., Doniger J., DiPaolo J. A. (1992). Down-regulation of keratin 14 gene expression after v-Ha-ras transfection of human papillomavirus-immortalized human cervical epithelial cells. Cancer Res. 52, 5865-5871 [PubMed] [Google Scholar]
- Breitkreutz D., Boukamp P., Ryle C. M., Stark H. J., Roop D. R., Fusenig N. E. (1991). Epidermal morphogenesis and keratin expression in c-Ha-ras-transfected tumorigenic clones of the human HaCaT cell line. Cancer Res. 51, 4402-4409 [PubMed] [Google Scholar]
- Breitkreutz D., Stark H. J., Plein P., Baur M., Fusenig N. E. (1993). Differential modulation of epidermal keratinization in immortalized (HaCaT) and tumorigenic human skin keratinocytes (HaCaT-ras) by retinoic acid and extracellular Ca2+. Differentiation 54, 201-217 [DOI] [PubMed] [Google Scholar]
- Bryan J. T., Brown D. R. (2000). Association of the human papillomavirus type 11 E1()E4 protein with cornified cell envelopes derived from infected genital epithelium. Virology 277, 262-269 [DOI] [PubMed] [Google Scholar]
- Chou C. F., Omary M. B. (1991). Phorbol acetate enhances the phosphorylation of cytokeratins 8 and 18 in human colonic epithelial cells. FEBS Lett. 282, 200-204 [DOI] [PubMed] [Google Scholar]
- Corpet F. (1988). Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881-10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy C. E., Jackson D. J., Wang Q., Raj K., Masterson P. J., Fenner N. F., Southern S., Cuthill S., Millar J. B., Doorbar J. (2002). Identification of a G(2) arrest domain in the E1 wedge E4 protein of human papillomavirus type 16. J. Virol. 76, 9806-9818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorbar J. (2005). The papillomavirus life cycle. J. Clin. Virol. 32, S7-S15 [DOI] [PubMed] [Google Scholar]
- Doorbar J. (2006). Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 110, 525-541 [DOI] [PubMed] [Google Scholar]
- Doorbar J., Ely S., Sterling J., McLean C., Crawford L. (1991). Specific interaction between HPV-16 E1-E4 and cytokeratins results in collapse of the epithelial cell intermediate filament network. Nature 352, 824-827 [DOI] [PubMed] [Google Scholar]
- Doorbar J., Ely S., Coleman N., Hibma M., Davies D. H., Crawford L. (1992). Epitope-mapped monoclonal antibodies against the HPV16E1-E4 protein. Virology 187, 353-359 [DOI] [PubMed] [Google Scholar]
- Doorbar J., Foo C., Coleman N., Medcalf L., Hartley O., Prospero T., Napthine S., Sterling J., Winter G., Griffin H. (1997). Characterization of events during the late stages of HPV16 infection in vivo using high-affinity synthetic Fabs to E4. Virology 238, 40-52 [DOI] [PubMed] [Google Scholar]
- Doorbar J., Elston R. C., Napthine S., Raj K., Medcalf E., Jackson D., Coleman N., Griffin H. M., Masterson P., Stacey S., et al. (2000). The E1E4 protein of human papillomavirus type 16 associates with a putative RNA helicase through sequences in its C terminus. J. Virol. 74, 10081-10095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquius J., Brisigotti M., Matias-Guiu X., Prat J. (1991). Keratin expression in normal vulva, non-neoplastic epithelial disorders, vulvar intraepithelial neoplasia, and invasive squamous cell carcinoma. Int. J. Gynecol. Pathol. 10, 341-355 [DOI] [PubMed] [Google Scholar]
- Fausther M., Villeneuve L., Cadrin M. (2004). Heat shock protein 70 expression, keratin phosphorylation and Mallory body formation in hepatocytes from griseofulvin-intoxicated mice. Comp. Hepatol. 3, 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl F., Kimura I., Osato T., Ito Y. (1970). Studies on a new human cell line (SiHa) derived from carcinoma of uterus. I. Its establishment and morphology. Proc. Soc. Exp. Biol. Med. 135, 543-545 [DOI] [PubMed] [Google Scholar]
- Fuchs E., Weber K. (1994). Intermediate filaments: structure, dynamics, function, and disease. Annu. Rev. Biochem. 63, 345-382 [DOI] [PubMed] [Google Scholar]
- Getsios S., Huen A. C., Green K. J. (2004). Working out the strength and flexibility of desmosomes. Nat. Rev. Mol. Cell Biol. 5, 271-281 [DOI] [PubMed] [Google Scholar]
- Green K. J., Gaudry C. A. (2000). Are desmosomes more than tethers for intermediate filaments? Nat. Rev. Mol. Cell Biol. 1, 208-216 [DOI] [PubMed] [Google Scholar]
- Herrmann H., Bar H., Kreplak L., Strelkov S. V., Aebi U. (2007). Intermediate filaments: from cell architecture to nanomechanics. Nat. Rev. Mol. Cell Biol. 8, 562-573 [DOI] [PubMed] [Google Scholar]
- Izawa I., Inagaki M. (2006). Regulatory mechanisms and functions of intermediate filaments: a study using site- and phosphorylation state-specific antibodies. Cancer Sci. 97, 167-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitovich A., Mehta S., Na N., Ciechanover A., Goldman R. D., Ridge K. M. (2008). Ubiquitin-proteasome-mediated degradation of keratin intermediate filaments in mechanically stimulated A549 cells. J. Biol. Chem. 283, 25348-25355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N. O., Omary M. B. (2000). Keratins turn over by ubiquitination in a phosphorylation-modulated fashion. J. Cell Biol. 149, 547-552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N. O., Omary M. B. (2006). A disease- and phosphorylation-related nonmechanical function for keratin 8. J. Cell Biol. 174, 115-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N. O., Azhar S., Omary M. B. (2002a). Keratin 8 phosphorylation by p38 kinase regulates cellular keratin filament reorganization: modulation by a keratin 1-like disease causing mutation. J. Biol. Chem. 277, 10775-10782 [DOI] [PubMed] [Google Scholar]
- Ku N. O., Michie S., Resurreccion E. Z., Broome R. L., Omary M. B. (2002b). Keratin binding to 14-3-3 proteins modulates keratin filaments and hepatocyte mitotic progression. Proc. Natl. Acad. Sci. USA 99, 4373-4378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N. O., Toivola D. M., Zhou Q., Tao G. Z., Zhong B., Omary M. B. (2004). Studying simple epithelial keratins in cells and tissues. Methods Cell Biol. 78, 489-517 [DOI] [PubMed] [Google Scholar]
- Kuppers V., Koldovsky U., Somville T., Bender H. G. (1998). Pattern of various cytokeratins of normal vulva, vulvar intraepithelial neoplasia (VIN) and vulvar carcinoma. Pathologe 19, 279-285 [DOI] [PubMed] [Google Scholar]
- Lane E. B., McLean W. H. (2004). Keratins and skin disorders. J. Pathol. 204, 355-366 [DOI] [PubMed] [Google Scholar]
- Liao J., Ku N. O., Omary M. B. (1997). Stress, apoptosis, and mitosis induce phosphorylation of human keratin 8 at Ser-73 in tissues and cultured cells. J. Biol. Chem. 272, 17565-17573 [DOI] [PubMed] [Google Scholar]
- Magin T. M., Vijayaraj P., Leube R. E. (2007). Structural and regulatory functions of keratins. Exp. Cell Res. 313, 2021-2032 [DOI] [PubMed] [Google Scholar]
- McIntosh P. B., Martin S. R., Jackson D. J., Khan J., Isaacson E. R., Calder L., Raj K., Griffin H. M., Wang Q., Laskey P., et al. (2008). Structural analysis reveals an amyloid form of the human papillomavirus type 16 E1-E4 protein and provides a molecular basis for its accumulation. J. Virol. 82, 8196-8203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton K., Peh W., Southern S., Griffin H., Sotlar K., Nakahara T., El-Sherif A., Morris L., Seth R., Hibma M., et al. (2003). Organization of human papillomavirus productive cycle during neoplastic progression provides a basis for selection of diagnostic markers. J. Virol. 77, 10186-10201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll R., Franke W. W., Volc-Platzer B., Krepler R. (1982). Different keratin polypeptides in epidermis and other epithelia of human skin: a specific cytokeratin of molecular weight 46,000 in epithelia of the pilosebaceous tract and basal cell epitheliomas. J. Cell Biol. 95, 285-295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara T., Peh W. L., Doorbar J., Lee D., Lambert P. F. (2005). Human papillomavirus type 16 E1circumflexE4 contributes to multiple facets of the papillomavirus life cycle. J. Virol. 79, 13150-13165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omary M. B., Ku N. O., Tao G. Z., Toivola D. M., Liao J. (2006). “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem. Sci. 31, 383-394 [DOI] [PubMed] [Google Scholar]
- Peh W. L., Middleton K., Christensen N., Nicholls P., Egawa K., Sotlar K., Brandsma J., Percival A., Lewis J., Liu W. J., et al. (2002). Life cycle heterogeneity in animal models of human papillomavirus-associated disease. J. Virol. 76, 10401-10416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peh W. L., Brandsma J. L., Christensen N. D., Cladel N. M., Wu X., Doorbar J. (2004). The viral E4 protein is required for the completion of the cottontail rabbit papillomavirus productive cycle in vivo. J. Virol. 78, 2142-2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M., Lane E. B. (2007). Intermediate filaments and stress. Exp. Cell Res. 313, 2244-2254 [DOI] [PubMed] [Google Scholar]
- Ridge K. M., Linz L., Flitney F. W., Kuczmarski E. R., Chou Y. H., Omary M. B., Sznajder J. I., Goldman R. D. (2005). Keratin 8 phosphorylation by protein kinase C delta regulates shear stress-mediated disassembly of keratin intermediate filaments in alveolar epithelial cells. J. Biol. Chem. 280, 30400-30405 [DOI] [PubMed] [Google Scholar]
- Riley N. E., Li J., Worrall S., Rothnagel J. A., Swagell C., van Leeuwen F. W., French S. W. (2002). The Mallory body as an aggresome: in vitro studies. Exp. Mol. Pathol. 72, 17-23 [DOI] [PubMed] [Google Scholar]
- Roberts S., Ashmole I., Johnson G. D., Kreider J. W., Gallimore P. H. (1993). Cutaneous and mucosal human papillomavirus E4 proteins form intermediate filament-like structures in epithelial cells. Virology 197, 176-187 [DOI] [PubMed] [Google Scholar]
- Roberts S., Ashmole I., Gibson L. J., Rookes S. M., Barton G. J., Gallimore P. H. (1994). Mutational analysis of human papillomavirus E4 proteins: identification of structural features important in the formation of cytoplasmic E4/cytokeratin networks in epithelial cells. J. Virol. 68, 6432-6445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S., Ashmole I., Rookes S. M., Gallimore P. H. (1997). Mutational analysis of the human papillomavirus type 16 E1-E4 protein shows that the C terminus is dispensable for keratin cytoskeleton association but is involved in inducing disruption of the keratin filaments. J. Virol. 71, 3554-3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugg E. L., Leigh I. M. (2004). The keratins and their disorders. Am. J. Med. Genet. C Semin. Med. Genet. 131, 4-11 [DOI] [PubMed] [Google Scholar]
- Russell D., Andrews P. D., James J., Lane E. B. (2004). Mechanical stress induces profound remodelling of keratin filaments and cell junctions in epidermolysis bullosa simplex keratinocytes. J. Cell Sci. 117, 5233-5243 [DOI] [PubMed] [Google Scholar]
- Schutte B., Henfling M., Kolgen W., Bouman M., Meex S., Leers M. P., Nap M., Bjorklund V., Bjorklund P., Bjorklund B., et al. (2004). Keratin 8/18 breakdown and reorganization during apoptosis. Exp. Cell Res. 297, 11-26 [DOI] [PubMed] [Google Scholar]
- Smedts F., Ramaekers F., Robben H., Pruszczynski M., van Muijen G., Lane B., Leigh I., Vooijs P. (1990). Changing patterns of keratin expression during progression of cervical intraepithelial neoplasia. Am. J. Pathol. 136, 657-668 [PMC free article] [PubMed] [Google Scholar]
- Toivola D. M., Zhou Q., English L. S., Omary M. B. (2002). Type II keratins are phosphorylated on a unique motif during stress and mitosis in tissues and cultured cells. Mol. Biol. Cell 13, 1857-1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walboomers J. M., Jacobs M. V., Manos M. M., Bosch F. X., Kummer J. A., Shah K. V., Snijders P. J., Peto J., Meijer C. J., Munoz N. (1999). Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 189, 12-19 [DOI] [PubMed] [Google Scholar]
- Wang Q., Griffin H., Southern S., Jackson D., Martin A., McIntosh P., Davy C., Masterson P. J., Walker P. A., Laskey P., et al. (2004). Functional analysis of the human papillomavirus type 16 E1=E4 protein provides a mechanism for in vivo and in vitro keratin filament reorganization. J. Virol. 78, 821-833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Kennedy A., Das P., McIntosh P. B., Howell S. A., Isaacson E. R., Hinz S. A., Davy C., Doorbar J. (2009). Phosphorylation of the human papillomavirus type 16 E1-E4 protein at T57 by ERK triggers a structural change that enhances keratin binding and protein stability. J. Virol. 83, 3668-3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner N. S., Windoffer R., Strnad P., Grund C., Leube R. E., Magin T. M. (2004). Epidermolysis bullosa simplex-type mutations alter the dynamics of the keratin cytoskeleton and reveal a contribution of actin to the transport of keratin subunits. Mol. Biol. Cell 15, 990-1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windoffer R., Leube R. E. (1999). Detection of cytokeratin dynamics by time-lapse fluorescence microscopy in living cells. J. Cell Sci. 112, 4521-4534 [DOI] [PubMed] [Google Scholar]
- Windoffer R., Woll S., Strnad P., Leube R. E. (2004). Identification of novel principles of keratin filament network turnover in living cells. Mol. Biol. Cell 15, 2436-2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woll S., Windoffer R., Leube R. E. (2007). p38 MAPK-dependent shaping of the keratin cytoskeleton in cultured cells. J. Cell Biol. 177, 795-807 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}