

Abstract
Inhibitor of NF-κB kinases β (IKKβ) and α (IKKα) activate distinct NF-κB signaling modules. The IKKβ/canonical NF-κB pathway rapidly responds to stress-like conditions, whereas the IKKα/noncanonical pathway controls adaptive immunity. Moreover, IKKα can attenuate IKKβ-initiated inflammatory responses. High mobility group box 1 (HMGB1), a chromatin protein, is an extracellular signal of tissue damage-attracting cells in inflammation, tissue regeneration, and scar formation. We show that IKKα and IKKβ are each critically important for HMGB1-elicited chemotaxis of fibroblasts, macrophages, and neutrophils in vitro and neutrophils in vivo. By time-lapse microscopy we dissected different parameters of the HMGB1 migration response and found that IKKα and IKKβ are each essential to polarize cells toward HMGB1 and that each kinase also differentially affects cellular velocity in a time-dependent manner. In addition, HMGB1 modestly induces noncanonical IKKα-dependent p52 nuclear translocation and p52/RelB target gene expression. Akin to IKKα and IKKβ, p52 and RelB are also required for HMGB1 chemotaxis, and p52 is essential for cellular orientation toward an HMGB1 gradient. RAGE, a ubiquitously expressed HMGB1 receptor, is required for HMGB1 chemotaxis. Moreover, IKKβ, but not IKKα, is required for HMGB1 to induce RAGE mRNA, suggesting that RAGE is at least one IKKβ target involved in HMGB1 migration responses, and in accord with these results enforced RAGE expression rescues the HMGB1 migration defect of IKKβ, but not IKKα, null cells. Thus, proinflammatory HMGB1 chemotactic responses mechanistically require the differential collaboration of both IKK-dependent NF-κB signaling pathways.
High mobility group box 1 (HMGB1) is a nonhistone nuclear protein expressed by all mammalian cells, passively released by necrotic cells, and actively secreted by immune effector cells (1–4). In necrotic cells, HMGB1 dissociates from chromatin and after the cellular and nuclear membranes break up is released into the extracellular space (1). Moreover, HMGB1 becomes acetylated in activated monocytes, macrophages, and dendritic cells, causing its relocation to specialized cytoplasmic organelles, from where it is secreted upon stimulation (2). Extracellular HMGB1 signals through the receptor for advanced glycation end products (RAGE), TLR2, and TLR4 (3–9), functioning as a major in vivo sensor of tissue damage by eliciting inflammatory reactions as a cytokine and a chemokine (reviewed in Refs. 3, 4, 6, 10, 11). In addition, HMGB1’s chemotactic activity also recruits cells to repair damaged tissues (12).
The signal transduction pathway elicited by HMGB1 is only beginning to unfold. RAGE’s cytoplasmic domain has been found to interact with Diaphanous-1, which is required for activation of Rac-1 and Cdc42 and importantly also for RAGE ligand-induced cell migration (13). We previously reported that, unlike other mediators of cell migration, cellular chemotaxis toward HMGB1 requires canonical NF-κB activation in fibroblasts and mesoangioblasts in vitro and for the emigration of mesoangioblasts to damaged muscle in vivo (14). HMGB1 induction of canonical NF-κB signaling and fibroblast chemotaxis also required ERK activation (14). More recently, we also showed that HMGB1-induced cell migration requires Src family kinases, reorganizes the cellular cytoskeleton, and induces phosphorylation of Src, FAK, and paxillin, a scaffold protein in focal adhesions (15). A dual requirement for Src and canonical NF-κB activation could either indicate that both signaling pathways are needed independent of each other for HMGB1 chemotaxis or that Src is necessary to drive NF-κB activation by an atypical inhibitor of NF-κB kinase (IKK) independent route (16–19). In this study, we have examined the functional contributions of the IKKβ- and IKKα-driven canonical and noncanonical NF-κB signaling pathways in HMGB1-induced cell migration responses.
Members of the NF-κB transcription factor family orchestrate a wide range of stress-like inflammatory responses, participate in cellular differentiation, and regulate the growth and survival of normal and malignant cells (20–23). Selectivity and at times redundancy in NF-κB–mediated transcriptional control arise from the assembly of a variety of homodimers and heterodimers of five different NF-κB proteins (RelA/p65, RelB, c-Rel, NF-κB1/p105, and NF-κB2/p100) that are sequestered in the cytoplasm by one of four inhibitory proteins (IκBα, IκBβ, IκBε, and IκBγ/p100). Proteins p100 and p105 are precursors of the NF-κB p52 and p50 subunits, respectively, and in their unprocessed forms also function as NF-κB inhibitors via their carboxyl-terminal IκB-like domains. In response to extracellular stress-like stimuli, IκBα is phosphorylated by the IKK complex and is targeted for ubiquitination and subsequent proteasomal destruction, resulting in the nuclear translocation of NF-κB heterodimers and the activation of their target genes. The IKK complex consists of two serine–threonine kinases, IKKα and IKKβ, and NEMO/IKKγ, a regulatory or docking protein that facilitates IKK complex assembly and regulates the transmission of upstream activating signals to IKKα and IKKβ (23–25). IKKβ is almost always the IκBα kinase that activates NF-κB–dependent immediate stress-like responses in vivo, although IKKα also occasionally takes on this role (26). In contrast to the positive proinflammatory IKKβ, IKKα instead functions to attenuate or resolve acute inflammatory responses by more than one mechanism (27–29).
Activation of IKKα’s kinase activity occurs in response to a limited set of extracellular signals (including CD40L, lymphotoxin β [LTβ], and BAFF) (reviewed in Ref. 21) and also requires protein synthesis. IKKα is the unique, direct activator of the noncanonical NF-κB pathway, wherein it phosphorylates a pair of serines in NF-κB2/p100, which leads to proteasomal processing into NF-κB p52 and the nuclear translocation of p52–RelB heterodimers, which bind to sequences that diverge considerably from those recognized by other NF-κB heterodimers (30). Interestingly, extracellular stimuli resulting in cellular responses that appear to require sustained or long-lasting NF-κB induction activate both IKKβ-dependent canonical and IKKα-dependent noncanonical signaling pathways (22, 31, 32). In addition to driving RelB/p52 heterodimers into the nucleus, the IKKα-dependent noncanonical pathway has also been reported to activate p65/p52 (32) and recently even a subset of p50/p65 heterodimers (sequestered in the cytoplasm in p100 complexes), suggesting that IKKα might also contribute to the somewhat delayed activation of specific proinflammatory responses (22).
Moreover, unlike IKKβ, IKKα has functions outside of NF-κB activation. In vivo IKKα is essential for keratinocyte differentiation (33–35) where it functions independent of NF-κB and its kinase activity in a Smad2/3-dependent signaling pathway (36–38). IKKα also functions as an NF-κB–independent kinase that affects chromatin activity, specific components of the cell cycle, and effectors of apoptosis (reviewed in Refs. 18, 24).
In this study, we show the surprising findings that IKKβ and IKKα are both essential for HMGB1 chemotactic responses in fibroblasts, macrophages in vitro, and neutrophils in vitro and in vivo. HMGB1 is a modest inducer of the IKKα-dependent noncanonical pathway, and HMGB1 migration requires both noncanonical NF-κB subunits (RelB and p52). Time-lapse microscopy of mouse embryo fibroblasts (MEFs) in an HMGB1 gradient revealed that the two IKKs are both essential for establishing directed cell movement and also have differential effects on cellular velocity. Moreover, fibroblasts migrating toward HMGB1 require IKKβ, but not IKKα, to induce the expression of full-length RAGE mRNA, whose encoded cell surface receptor is necessary for HMGB1 chemotaxis, and enforced RAGE overexpression rescues the HMGB1 migration defect of IKKβ−/− but not IKKα−/− MEFs. Taken together our results suggest that IKKβ and IKKα are essential for HMGB1 chemotactic responses for different reasons.
Materials and Methods
Conditional IKKα and IKKβ knockout mice
Mice with IKKα or IKKβ alleles flanked by LoxP recombination sites (IKKβf/f and IKKαf/f) were generated by Lexicon Genetics (The Woodlands, TX) for Boehringer Ingelheim Pharmaceuticals by strategies previously described for p38a floxed mice (39) (Supplemental Fig. 1). IKKβf/f and IKKαf/f homozygous mice were respectively bred to CreERT1 (40) and CreERT2 (41) mice, which ubiquitously express 4-orthohydroxytamoxifen (4-OHT)-inducible Cre recombinases (42, 43), to produce IKKβf/f:CreERT1 and IKKαf/f:CreERT2 mice. The IKKαf/f and IKKβf/f strains were also interbred with MLysCre mice to produce IKKαf/f:MLysCre and IKKβf/f: MLysCre strains, which conditionally expresses Cre recombinase under the control of the macrophage lysozyme promoter only in mature macrophages (MΦs) and neutrophils (44).
HMGB1 and reagents
Full-length LPS-free rHMGB1 protein was obtained from HMGBiotech (Milano, Italy) and was rigorously tested to be LPS-free by HMGBiotech and independently by the Bianchi laboratory. Other reagents were obtained from the following suppliers: human recombinant platelet-derived growth factor (PDGF) (R&D Systems, Minneapolis, MN); TNF-α (BioSource International, Camarillo, CA); SC-514 (Calbiochem, San Diego, CA); fibronectin (Roche Diagnostics, Monza, Italy); human recombinant complement component 5a (C5a), 4-OHT, and mouse anti–α-tubulin mAb (Sigma-Aldrich, St. Louis, MO); mouse anti-lamin B1 mAb (Abcam, Cambridge, MA); rabbit polyclonal anti-RAGE and goat polyclonal anti–β-actin Abs (Santa Cruz Biotechnology, Santa Cruz, CA); rabbit polyclonal anti-p100/p52, anti-IKKα, and anti-IKKβ Abs (Cell Signaling Technology, Danvers, MA); FITC-conjugated anti-mouse Ly-6G (Gr-1) (eBioscience, San Diego, CA); FITC-conjugated rat IgG2b,κ and purified anti-mouse CD16/32 (Fc block) (BioLegend, San Diego, CA); anti-mouse and anti-rabbit IgG HRP (DakoCytomation, Carpinteria, CA); and agonistic monoclonal anti-mouse LTβR Ab (Biogen, Cambridge, MA) (45).
Cells and tissue culture
Primary MEFs were derived from 14.5 (Rage−/−) or 12–13 d (IKKβf/f: CreERT1 or IKKαf/f:CreERT2) mouse embryos and cultured in DMEM with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cre recombinase excision of LoxP-targeted IKK alleles was induced by exposing cells to 100 nM 4-OHT for ~ 36 h to ensure IKK protein depletion. Bone marrow progenitors from the femurs of IKK wild-type (WT) (IKKαf/f or IKKβf/f), IKKαf/f:MLysCre, and IKKβf/f:MLysCre adult mice were differentiated to MΦs in M-CSF–conditioned DMEM/10% FBS for 7 d.
Retroviruses and retroviral transductions
A human RAGE cDNA was subcloned upstream of an internal ribosome entry site puromycin cassette in the BIP murine Moloney retroviral vector (46, 47). The generation of amphotyped viruses, infection of cells, and selection of stable puromycin-resistant cell populations have been described previously (46, 47).
In vitro chemotaxis assays
Boyden chamber assays with MEFs were performed as described (12) and with MΦs and neutrophils essentially as described (48), except that 48-well chambers were used with 120-μm thickness 8- and 3-μm pore size filters, respectively (Neuroprobe, Gaithersburg, MD) (49). Mature neutrophils were purified to 70–80% by Percoll gradient fractionation of bone marrow cells (50) (see Supplemental Fig. 3 for more details). Migration distances for macrophages and neutrophils were measured by the leading front method as described previously (49).
In time-lapse chemotaxis assays, cells of different genotypes were distinguished from each other by staining with 500 nM CellTracker Green CMFDA or 500 nM CellTracker Orange CMRA (Invitrogen, Carlsbad, CA) for 15 min in a humidified tissue culture incubator. Dye-labeled cells were washed, resuspended at a concentration of 4 × 105 cells per milliliter in DMEM (phenol red-free)/0.1% BSA, mixed, and placed (300 μl) in a μ-Slide (IBIDI/Integrated BioDiagnostics, Martinsried, Germany) precoated with 50 μg/ml fibronectin (μ-Slide setup is depicted in Supplemental Fig. 3). After cells had firmly attached to the substratum, slides were laid on a 37°C humidified stage of an UltraVIEW European Remote Sensing spinning disk confocal microscope (Perkin Elmer, Waltham, MA). A 0–30 μg/ml HMGB1 gradient, mixed with fluorescent beads (Invitrogen, San Diego, CA) for microscope viewing, was allowed to form in the chamber’s channel as described (51) (Supplemental Fig. 4). Pictures of cells at the edge of the gradient were captured every 2 min for up to 3 h (microscope objective, ×5 magnification, numerical aperture 0.15 [Carl Zeiss, Munich, Germany]; C9100 electron multiplying-charge coupled device, camera [Hamamatsu Photonics, Shizuoka, Japan]; UltraVIEW European Remote Sensing acquisition software). Directional tracks are defined as those with ending points closer to the higher HMGB1 concentration in the gradient compared with their starting points, whereas nondirectional tracks are the opposite, and indeterminate tracks starting and ending at the same distance from the HMGB1 gradient (moving laterally) are also considered nondirectional tracks. Cell tracks were analyzed with ImageJ software (Rasband, W.S., ImageJ, National Institutes of Health, Bethesda, MD; http://rsb.info.nih.gov/ij/, 1997–2008).
Immunoblotting and cell fractionation
Immunoblotting and the preparation of nuclear and cytoplasmic fractions were done as described previously (47). HRP-conjugated secondary Ab signals were detected with an enhanced chemiluminescent detection kit (GE Healthcare, Piscataway, NJ)). Nuclear p52 levels were normalized with respect to nuclear lamin B1. Film images were acquired with a Fluor-S MultiImager (Bio-Rad, Hercules, CA). Scanned protein bands were quantified with Quantity One 4.5.0 software (Bio-Rad) and processed with Adobe Creative Suite (Adobe, San Jose, CA).
SYBR Green real-time RT-PCR assays
Total cell RNAs were prepared, and SYBR Green real-time RT-PCR was performed and quantified as described previously (47). Forward (F) and reverse (R) PCR primer sequences were as follows: Gapdh F (5′-GCTCACTGGCATGGCCTTC-3′), Gapdh R (5′-CCTTCTTGATGTCATCATACTTGGC-3′); Rage F (5′-AGTCAGAGGAAGCGGAGATG-3′), Rage R (5′-AAGGAGGAATTGGGATGGAATG-3′); Cxcl12 F (5′-GCACGGCTGAAGAACAACAAC-3′), Cxcl12 R (5′-TTCCTCGGGCGTCTGACTC-3′).
In vivo chemotaxis assays
Mice were anesthetized with isoflurane and injected i.p. with 1 ml of either TNF-α (0.1 μg/ml in 0.9% NaCl) orHMGB1 (9 μg/ml in 0.9%NaCl). After 4–5 h, mice were sacrificed, and the cellular contents of their i.p. cavities were collected by sequential lavages in a total volume of 15 ml PBS. After centrifugation, RBCs were lysed by resuspension with ammonium chloride-potassium carbonate-EDTA buffer (0.15 M NH4Cl, 1 mM KHCO3, and 0.1 mM Na2EDTA in PBS), and the remaining cells were collected by centrifugation. Next, cell pellets were resuspended in PBS and preincubated with purified anti-mouse CD16/32 (Fc Block), stained with either FITC-conjugated anti-mouse Gr-1 Ab or FITC-conjugated IgG2b,κ isotype control Ab, and analyzed with a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Small debris and doublets were excluded by a population gate. Quadrants were added to focus on the Gr-1Hi–staining cells in TNF-α controls (FL1 103), which were then applied to the HMGB1 samples. Statistics were computed from these quadrants to calculate the percentage of Gr-1Hi cells of the parent population using FlowJo analysis software version 8.7.3 (Tree Star, Ashland, OR).
Cells from i.p. lavages were also examined by conventional Diff-Quick (Fisher Scientific, Pittsburgh, PA) staining to score for the typical nuclear morphology of mature neutrophils. Cell suspensions from i.p. lavages were smeared on glass slides, stained with HEMA 3 STAT PACK (Fisher Diagnostics Protocol; Fisher Scientific), and mounted with coverslips. Images were acquired with a Sony DXC-9000 camera (Sony, New York, NY) mounted on a Nikon Eclipse E600 microscope (×100 objective, numerical aperture 1.30; Nikon, Melville, NY) and processed with Adobe Photoshop software.
All of the animal work was approved by Stony Brook University’s Institutional Animal Care and Use Committee in accordance with National Institutes of Health grant guidelines.
Statistical analysis
The p values were determined to four significant figures by unpaired, two-tailed Student t tests (unless otherwise indicated) with Prism version 4.0 software (GraphPad, San Diego, CA).
Results
IKKβ and IKKα are both required for chemotaxis of primary MEFs and macrophages toward HMGB1
In a previous study, we showed that HMGB1 activates the canonical NF-κB pathway in 3T3 fibroblasts and that the translocation of p50/p65 canonical dimers to the nucleus is necessary for the chemotactic response of immortalized MEFs to HMGB1 (14). However, we did not elaborate the individual roles of the NF-κB–activating kinases in the HMGB1 chemotactic response. In this study, we employed primary cells from different strains of conditional IKKα and IKKβ knockout (KO) mice to dissect the individual contributions of each NF-κB–activating IKK for HMGB1 chemotactic responses of fibroblast and immune effector cells.
To begin to assess the contributions of IKKβ and IKKα for HMGB1-induced cell migration, primary MEFs from IKKβf/fCreERT1 and IKKαf/f:CreERT2 mice were used in Boyden chamber cell migration experiments. IKKβf/f:CreERT1 and IKKαf/f:CreERT2 primary WT MEFs migrated in response to HMGB1 and PDGF, whereas 4-OHT–induced IKKβ or IKKα deletion in these respective cell populations blocked their chemotaxis to HMGB1 but not to PDGF (Fig. 1A, 1B). Ablation of IKKβ and IKKα protein expression in primary MEFs derived from IKKβf/f:CreERT1 and IKKαf/f:CreERT2 mice was induced with 4-OHT for 36 h and verified by immunoblotting (Supplemental Fig. 1J, 1E). To rule out chemokinesis (cytokine-activated random cell movement) in our Boyden chamber migration assays, we performed a checkerboard assay by applying cytokines to either side of the filter, but we only observed cytokine-induced cell migration when there was a gradient of chemoattractant below the filter (data not shown).
FIGURE 1.

HMGB1 chemotaxis of MEFs requires IKKβ, IKKα, p52, and RelB. Cell migrations with IKKβf/f:CreERT1 (A) or IKKαf/f:CreERT2 MEFs (B) or the same cells pre-exposed to 4-OHT for 36 h (to delete IKKβ or IKKα, respectively) were done in Boyden chambers in SF media or in the same media containing HMGB1 (30 ng/ml) or PDGF (10 ng/ml) (fibroblast migration positive control). Three experiments were done in duplicate. C, Migration responses of WT, p100/p52−/−, and RelB−/− MEFs in SF media, HMGB1 (30 ng/ml), or PDGF (10 ng/ml) for 3 h in Boyden chambers. Two experiments were done in duplicate. In all cases, bars are the mean cell numbers of five high-power fields. All error bars are SEM. *p = 0.02; ***p = 0.002; ****p = 0.0006. HPF, high-power fields.
Because IKKα has multiple functions that are dependent on and independent of its critical role in the activation of noncanonical NF-κB signaling, we investigated whether the two noncanonical NF-κB subunits, p52 and RelB, were also required for MEFs to migrate toward HMGB1. Boyden chamber assays were performed with WT, p52−/−, and RelB−/− immortalized MEFs. As shown in Fig. 1C, akin to IKKα, p52 and RelB are also each essential for HMGB1-mediated chemotaxis but migrate equally well as WT MEFs in response to a PDGF positive control. Thus, HMGB1-induced cell migration requires IKKα to drive NF-κB noncanonical signaling.
Next, we investigated the requirements for each IKK in the HMGB1-mediated migration of immune effector cells by employing MΦs derived from IKKαf/f:MLysCre and IKKβf/f: MLysCre mice. Bone marrow-derived hematopoietic progenitor cells were prepared and differentiated into MΦs in M-CSF-1–conditioned media. The selective ablation of IKKα or IKKβ expression in MΦs, derived from IKKαf/f:MLysCre and IKKβf/f: MLysCre mice, respectively, was verified by immunoblotting (Fig. 2). Chemotaxis assays were carried out in 48-well micro-chambers using 120-μm thickness cellulose nitrate filters with the depth of filter penetration as a measure of migration (49). These thick, tortuous pore filters better mimic the cell–matrix interactions through which cells migrate in vivo than thin, straight pore (10 μm) polycarbonate filters. Fig. 2 shows that, although WTM Φs respond well to HMGB1, MΦs from IKKαf/f:MLysCre or IKKβf/f:MLysCre mice fail to migrate toward HMGB1. As a positive control, WT and IKK mutant MΦs all migrated in response to C5a, a potent chemotactic factor that does not signal through NF-κB. Thus, HMGB1 migration responses of primary fibroblasts and immune effector cells simultaneously require both IKKα and IKKβ.
FIGURE 2.
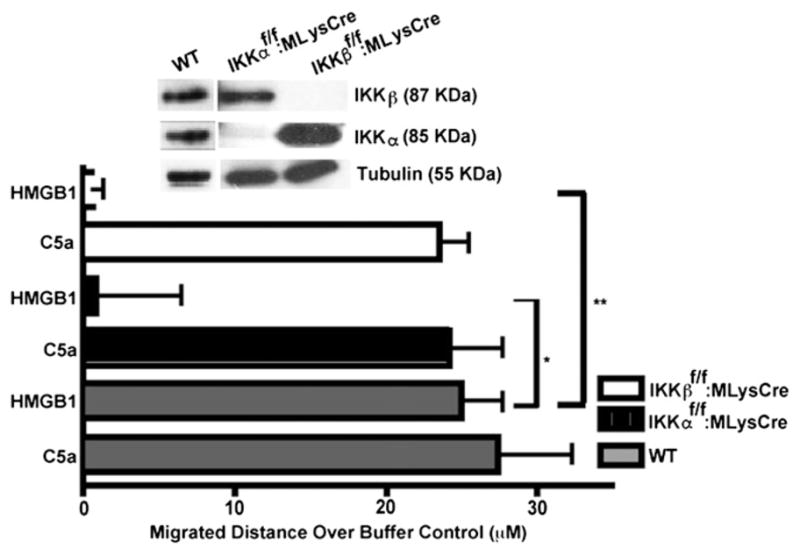
IKKα and IKKβ are both essential for the migration of primary macrophages toward HMGB1. Migration assays of primary WT and IKK mutant MΦs were performed in 48-well Boyden chambers with 120-μm thickness 8-μm pore size filters in response to HMGB1 (30 ng/ml) or C5a (2 nM) (MΦ migration positive control) for 3 h. Bars are distances migrated through filter pores after subtracting basal migration in SF media. WT MΦs migrated in response to HMGB1 or C5a, and IKKα or IKKβ mutant MΦs migrated to C5a at least two to three times more than their matched SF controls (data not shown). Three independent experiments were done in duplicate. Immunobloting shows the absence of IKKα or IKKβ protein in MΦs derived from IKKαf/f:MLysCre and IKKβf/f:MLysCre mice, respectively. All error bars are SEM. *p = 0.02, **p = 0.007.
IKKβ and IKKα are both required for the HMGB1-mediated recruitment of neutrophils in vivo
To assess the physiological relevance of our in vitro results with primary cells, we investigated the dual importance of IKKβ and IKKα for chemotaxis to HMGB1 in an in vivo context. HMGB1 was previously shown to rapidly elicit neutrophil-dependent peritonitis, after injection into the peritoneal cavity of WT mice (52). We employed this peritonitis model to assess the individual contributions of IKKβ and IKKα for HMGB1-induced neutrophil migration within 5 h, because the recruitment of immune effector cells (including neutrophils, monocytes, and macrophages) at later times would be confounded by other endogenous cytokines subsequently released in response to HMGB1, which would invoke chemotactic responses independent of the NF-κB signaling capacity of the migrating cells. Approximately 4 h after i.p. instillation of HMGB1 or a TNF-α positive control into WT (IKKβf/f or IKKαf/f), IKKβf/f:MLysCre, or IKKαf/f:MLysCre mice, cells were recovered by i.p. lavage. As previously described for assessing neutrophil emigration in HMGB1-induced peritonitis (52), neutrophils were quantified by flow cytometry for cell surface expression of Gr-1, a marker of the myeloid-specific Ly-6G locus (53), which identifies mature neutrophils as Gr-1Hi cells (54). The i.p. instillation of either HMGB1 or TNF-α recruited comparable numbers of neutrophils in WT (IKKβf/f or IKKαf/f) mice (Fig. 3A). However, in either IKKβf/f:MLysCre or IKKαf/f:MLysCre mutant mice, neutrophil recruitment in response to HMGB1 was greatly reduced compared with that in WT (IKKαf/f or IKKβf/f) mice, although the migration responses of IKKβ- or IKKα-deficient neutrophils in vivo were comparable to those of neutrophils in IKK WT mice (Fig. 3A). The primary FACS profiles identifying the Gr-1Hi cell subsets in these i.p. lavages are shown in Supplemental Fig. 2. In addition, these quantitative results were also corroborated in other experiments in which mature neutrophils were instead identified by their characteristic nuclear morphology in conventional Diff-Quick–stained cell smears of these i.p. lavages (Fig. 3B). Finally, primary neutrophils purified from mouse bone marrow also showed a similar pronounced defect in HMGB1 chemotaxis in the absence of either IKKβ or IKKα (Supplemental Fig. 3). We conclude from these experiments that mice with conditional IKKα and IKKβ deficits specifically in mature neutrophils and macrophages are refractory to peritonitis induced by HMGB1, but not by TNF-α, most likely due to intrinsic cell defects in these immune effector cells that specifically preclude their HMGB1-mediated chemotaxis.
FIGURE 3.

IKKα and IKKβ are both necessary to elicitHMGB1-induced neutrophil peritonitis in mice. A, HMGB1 (~ 9 μg) or TNF-α (100 ng) were injected in the peritoneal cavities of WT, IKKαf/f:MLysCre, and IKKβf/f: MLysCre mice. The i.p. lavages were done after ~4 h. Cells were stained with a FITC-conjugated anti–Gr-1 Ab and submitted to flow cytometry to quantify neutrophils (Gr-1Hi cell subset). Bars are the mean numbers of Gr-1Hi cells in three to four animals per group. Error bars are SEM. **p = 0.01. B, Diff-Quick–stained slides of the same i.p. lavages in A were prepared to distinguish polymorphonuclear neutrophils from other immune effector cells on the basis of their unique nuclear morphology. Three to five mice were analyzed in each group. Error bars are SEM. *p = 0.03
IKKβ and IKKα are differentially required to initiate cellular chemotaxis in response to an HMGB1 gradient
Because the canonical and noncanonical NF-κB pathways control distinct target genes and cellular responses, we suspected that they might control different aspects of HMGB1-induced cell migration. Thus, to more precisely delineate the mechanistic contributions of IKKβ and IKKα, specific migration parameters including cell polarization, velocity, and Euclidean distance (i.e., the straight line distance between starting and arriving points) were tracked in an HMGB1 gradient inside a μ-Slide by time-lapse microscopy (see μ-Slide setup and how the directionality of cell movement was assessed in Materials and Methods and in Supplemental Fig. 4). Importantly, WT versus IKKβ−/− or WT versus IKKα−/− MEFs (generated from WT cells by 4-OHT–induced IKK deletions) were directly compared in the same HMGB1 gradient by distinguishing them with different fluorescent tags. Analogous versions of this technique have been extensively used to tease out fine details of cellular movement in response to cytokine and chemokine gradients in physiologically well-controlled environments (51, 55–57).
IKKβ and IKKα are both essential in primary MEFs to establish the direction and efficiency of cell motion in response to HMGB1. Within the initial 60 min of their exposure to an HMGB1 gradient, primary MEFs lacking either IKK move randomly, whereas in contrast the majority of their WT counterparts move toward the gradient (Fig. 4A–D). Moreover, the noncanonical NF-κB p100/p52 subunit is also required for the directionality of cell movement (Fig. 4E versus Fig. 4F). In addition, the Euclidean distance (which is shorter in a random walk compared with a directional walk) was significantly different between WT and IKKβ null cells, as it was between WT and IKKα−/− and WT and p52−/− cells (Supplemental Fig. 5A–C). WT and mutant cells were directly compared with each other in each μ-Slide to control for any variations in the absolute degrees of cell migration between different experiments (see Materials and Methods). Interestingly, the velocity of cell movement within the initial 30–60 min was also significantly reduced by the loss of IKKβ (Fig. 5A) and modestly blunted by the loss of p100/p52 (Fig. 5B). In contrast, IKKα ablation did not have an immediate effect on cell velocity (Fig. 5C), because IKKα-compromised cells began to move as vigorously as WT cells even though they lack directionality. By 180 min of the migration response, IKKα also eventually contributed to the speed of cell movement like IKKβ (Supplemental Fig. 5D, 5E). However, although these observations indicate that both IKKs made statistically significant, differential time-dependent contributions to the velocity of cell movement, their effects on the cellular velocity parameter were modest in comparison with the dual critical need of both IKKα and IKKβ to determine cellular orientation to the HMGB1 gradient.
FIGURE 4.

IKKα, IKKβ, and p100/p52 are essential for the directionality of cell movement toward HMGB1. Cells were exposed to 0–30 ng/ml HMGB1 gradients in μ-Slides for 1 h, and their movement was recorded by time-lapse microscopy. Red and black lines are cell tracks moving toward the HMGB1 gradient or moving in other directions, respectively. Directional tracks are defined as those with ending points closer to the higher HMGB1 concentration in the gradient compared with their starting points, whereas nondirectional tracks are the opposite, and indeterminate tracks starting and ending at the same distance from the HMGB1 gradient (i.e., moving laterally) are also considered nondirectional tracks. The effects of IKKα, IKKβ, or p100/p52 ablation on the percentage of tracks moving toward the HMGB1 gradient or in other irrelevant directions are shown in bar graphs adjacent to cell tracks plots. The differences in the numbers of directional versus nondirectional tracks between WT and each mutant MEF cell population were always statistically significant (χ2 test with p < 0.01 in all comparisons). A and B, IKKβf/f:CreERT1 MEFs versus the same cell population pre-exposed to 4-OHT for 36 h to ablate IKKβ expression. C and D, IKKαf/f:CreERT2 MEFs versus their 4-OHT–treated IKKα-ablated counterparts. E and F, Immortalized WT versus p100/p52−/− MEFs. Experiments were repeated two to four times with comparable results. A descriptive view of μ-Slides and how cell movement was followed by time-lapse microscopy is also provided in Supplemental Fig. 4.
FIGURE 5.
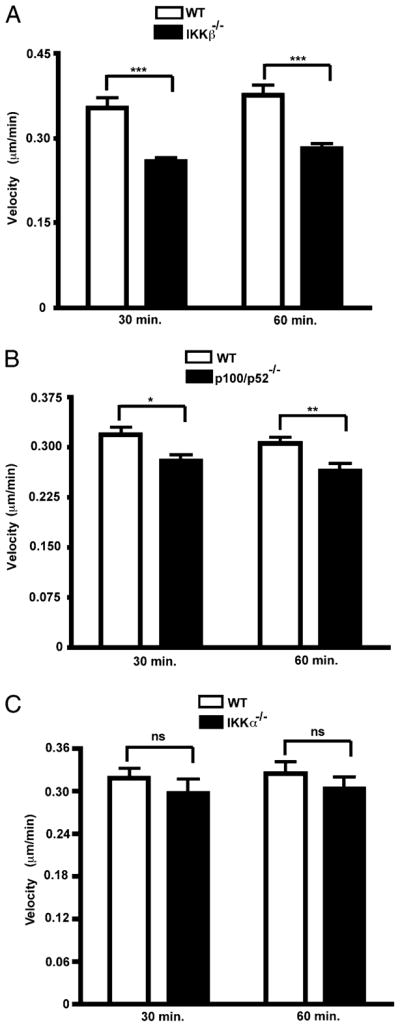
Requirements for IKKβ, IKKα, and p52 for cellular velocity in the initial phase of HMGB1-induced cell migration. A–C, All cell tracks of WT MEFs versus their IKKβ, IKKα, and p52 null counterparts in Fig. 4 were analyzed for velocities 30 and 60 min after exposure to their respective HMGB1 gradients. Velocity is the total path traveled divided by the time of observation. WT refers to either primary IKKαf/f:CreERT2 or IKKβf/f:CreERT1 MEFs. IKKα−/− and IKKβ−/− MEFs were generated from the latter respective WT cells by exposure to 4-OHT for 36 h. Bars are mean velocities, and error bars are SEs of the mean.*p = 0.009; **p = 0.003; ***p < 0.0001. Experiments were repeated two to four times with comparable results.
IKKβ, but not IKKα, is necessary for HMGB1 to induce RAGE, and RAGE is required for cell migration toward HMGB1
Next, we sought to further elaborate the mechanistic basis for a dual IKKα and IKKβ requirement for HMGB1 chemotactic responses. NF-κB signaling is generally not required for the mechanic aspects of cellular chemotactic responses in vitro and in vivo, being unnecessary for cell migration responses elicited by a variety of other chemoattractants, including TNF-α, PDGF, C5a, or fMLP (14) (Figs. 1–3). Because our results revealed that both IKKs are needed very early in the migration response to HMGB1 (Figs. 4, 5), we reasoned that specific aspects of how cells sense HMGB1 could be one of the causes for NF-κB signaling dependency. Thus, we turned our attention to RAGE, a ubiquitously expressed HMGB1 receptor (3, 5, 6, 10), that is upregulated upon engaging its ligands (6) and whose transcriptional promoter contains functional NF-κB binding sites (58). Real-time RT-PCR experiments were performed with RAGE PCR primer pairs that would detect all RAGE mRNA isoforms, among which full-length RAGE is the most predominant (59). Total RAGE mRNA expression in HMGB1-stimulated and unstimulated WT, IKKβ−/−, and IKKα−/− MEFs indicate that RAGE induction by HMGB1 requires IKKβ but is independent of IKKα (Fig. 6A, 6B), whereas basal RAGE expression was not influenced by either IKK (data not shown). Moreover, we also observed a similar degree of IKKβ dependency for HMGB1 induction of RAGE mRNA by MΦs (Supplemental Fig. 6). Cell migration to HMGB1 (but not PDGF) was compromised in the absence of RAGE (Fig. 7A). Although RAGE null cells may perhaps retain some ability to migrate toward HMGB1, this residual migration response did not appear to reach statistical significance (p value of 0.06 in Fig. 7A). Taken together, the above results suggest that RAGE is at least one of the targets of IKKβ required for chemotaxis to HMGB1, but IKKα is required for a different reason. An early IKKβ requirement to maintain sufficient RAGE expression is also supported by the lack of HMGB1 migration of cells briefly pre-exposed to a specific IKKβ kinase inhibitor (Fig. 7B).
FIGURE 6.

IKKβ, but not IKKα, is required to induce RAGE mRNA expression in MEFs in response to HMGB1. A and B, Real-time RT-PCR analysis of RAGE mRNA expression in WT versus IKKβ−/− and IKKα−/− MEFs in response to HMGB1 (50 ng/ml) for 3 h. Primary WT IKKβf/f CreERT1 MEFs were pre-exposed to 4-OHT to delete IKKβ in A, and immortalized WT and IKKα−/− MEFs were used in B. Experiments were performed three times each in duplicate. *p = 0.04.
FIGURE 7.

RAGE and IKKβ kinase activity are essential for MEF chemotaxis to HMGB1 A, Boyden chamber migration assays of primary WT and RAGE−/− MEFs in response to HMGB1 or PDGF versus matched SF controls. B, WT IKKβf/f primary MEFs were briefly exposed for 30 min to 100 nM SC-514, a specific IKKβ kinase inhibitor (66). SC-514–pretreated and untreated matched controls were subjected to Boyden chamber migration assays in response to HMGB1 (30 ng/ml) or PDGF (10 ng/ml) in comparison with SF media. Bars are mean numbers of cells in five high-power fields from three experiments each run in duplicate with error bars representing SEM. **p = 0.02; ***p = 0.003; ****p = 0.0001.
Enforced RAGE expression rescues the HMGB1 migration defect of IKKβ, but not IKKα, null cells
In light of the above results, we next investigated whether enforced RAGE overexpression in IKKβ−/− or IKKα−/− MEFs affected their HMGB1 migration activity. We stably transduced MEFs with a murine Moloney retroviral vector coexpressing a human full-length RAGE cDNA and a puromycin resistance gene (RAGE-BIP). To detect RAGE expression, we used a rabbit polyclonal anti-RAGE Ab raised against RAGE’s N-proximal 300 aas. RAGE immunoblotting clearly indicated that each RAGE-BIP retrovirus-transduced, puromycin-resistant cell population overexpressed comparable levels of the human full-length RAGE protein, which comigrated with the major species of full-length, membrane-bound RAGE in mouse lung extracts (60) (Fig. 8A). Because the RAGE expressed by the retroviral vector harboring the full-length cDNA can only specify the full-length protein, the presence of two forms of the full-length RAGE protein, which are also apparent in WT mouse lung extract, is most likely due to differential degrees of posttranslational modification (Fig. 8A).
FIGURE 8.

Enforced RAGE expression rescues the defective HMGB1 migration response of IKKβ−/− but not of IKKα−/− MEFs. A, Immunoblot of RAGE protein expression in RAGE-BIP retrovirus stably transduced MEFs compared with RAGE KO MEFs and WT mouse lung extract, which expresses full-length membrane forms of RAGE (60). B–D, Migration assays in response to HMGB1 and PDGF of RAGE-BIP and empty BIP vector control populations of IKKβ−/−, IKKα−/−, and WT MEFs, respectively, each relative to their matched SF controls. All experiments were done three times in duplicate in Boyden chambers for 3 h. Error bars are SEM. *p = 0.006; **p = 0.0006; ***p < 0.0001.
RAGE overexpression rescued the HMGB1 chemotactic response of IKKβ null MEFs (in comparison with IKKβ−/− cells with an empty BIP retrovector) (Fig. 8B). RAGE-BIP and BIP IKKβ−/− cells responded equally well to a PDGF positive control with respect to their matched serum-free (SF) controls. The SF background migration of RAGE-BIP versus BIP IKKβ−/− cells appeared to be modestly higher for the former (Fig. 8B) but was not statistically significant. In contrast to these results with IKKβ−/− cells, RAGE-BIP IKKα−/− cells still failed to respond to HMGB1 (Fig. 8C), even though the latter cells and their BIP control responded equally well to a PDGF positive control. Moreover and importantly, RAGE-BIP and BIP WT MEFs migrated toward HMGB1 to similar degrees with respect to their individual SF negative controls (which was somewhat higher for the WT RAGE-BIP cell population) (Fig. 8D). Thus, enforced RAGE overexpression fully rescued the HMGB1 migration defect of IKKβ null MEFs to WT levels but did not enhance the HMGB1 chemotactic response of WT MEFs.
Taken together, our data in fibroblasts suggest that IKKβ/canonical NF-κB signaling is necessary to induce and subsequently maintain RAGE expression for these cells to migrate in response to HMGB1, but IKKα is required for a different reason.
HMGB1 modestly induces the NF-κB noncanonical pathway
Because the IKKα-dependent NF-κB noncanonical pathway was required for the chemotactic response of MEFs to HMGB1, we next assessed whether HMGB1 induces IKKα-dependent p52 nuclear translocation. MEFs were stimulated for 8 h with either HMGB1 or an agonistic anti-murine LTβR Ab as a positive control (45, 61). Representative examples of immunoblots of nuclear and cytoplasmic extracts of WT versus IKKα−/− cells indicate that HMGB1 (Fig. 9A, 9B, Supplemental Fig. 7) is a modest inducer of p52 nuclear translocation in WT but not in IKKα−/− MEFs. In contrast (and as expected), LTβR stimulation induced robust p100 > p52 processing in WT MEFs, although no effect was seen in IKKα null cells (Supplemental Fig. 7). We also observed a reduced level of p52 in IKKα null MEFs (Fig. 9A, 9B, Supplemental Fig. 7), which is in agreement with prior reports (61, 62) that IKKα is required to maintain a constitutive, basal level of cytoplasmic and nuclear p52. Immunoblots were performed multiple times with independent batches of cells and quantified by densitometric analysis confirming that HMGB1 is a statistically significant (albeit modest) inducer of p52 nuclear translocation (Fig. 9C). In accord with these results, CXCL12/SDF-1, a direct target of p52/RelB (21, 61), was strongly induced by LTβR engagement and modestly upregulated by HMGB1 with a dependency on IKKα (Fig. 10).
FIGURE 9.

HMGB1 modestly induces IKKα-dependent p52 nuclear translocation. A and B, Immortalized WT and IKKα−/− MEFs, respectively, were stimulated with HMGB1 (50 ng/ml). An asterisk denotes a cross-reactive band in the anti-p52 nuclear blots. Immunoblots were stripped and reprobed for lamin B1 or α-tubulin as protein reference controls for nuclear and cytoplasmic cell fractions, respectively. Efficacies of nuclear (lamin B1 positive/tubulin negative) and cytoplasmic (tubulin positive/lamin B1 negative) cell fractionation are shown in A and B. C, Densitometry quantification of p52 nuclear import by WT and IKKα−/− MEFs in response to HMGB1 or LTβR activation (immunoblots of LTβR’s effects are shown in Supplemental Fig. 6). p52 signals were normalized to lamin B1 as a nuclear protein reference control. Experiments were done three to four times and include 2–8 h time points for HMGB1 and 8 h for LTβ. All error bars are SEM. #p = 0.02; ##p = 0.004; ###p = 0.0006.
FIGURE 10.
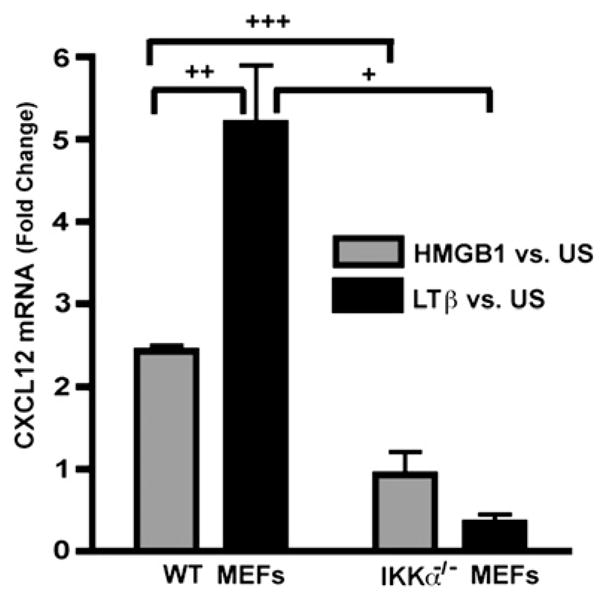
HMGB1 induces IKKα-dependent CXCL12/SDF-1 expression. Real-time RT-PCR analysis of CXCL12/SDF-1 mRNA expression in WT and IKKα−/− MEFs in response to HMGB1 or LTβR stimulation. +p = 0.02; ++p = 0.01; +++p = 0.006.
Discussion
Cell migration is a complex, multistep process that is essential for the orchestration of embryogenesis, innate and adaptive immune responses, tissue repair, and regeneration. Abnormal cell migration is associated with the progression of a variety of diseases, including cancer (reviewed in Refs. 63, 64). Cells migrate directionally in response to growth factors, chemotactic factors, and extracellular matrix molecules. The mechanics of cellular migration have been well documented: the process begins with cells establishing polarity toward the chemotactic gradient by rearranging the actin cytoskeleton and then extending broad lamellipodia and spike-like filopodia toward the direction of migration. Moreover, chemotaxis is a very rapid process and as such typically does not require the synthesis of new proteins for cells to initiate and maintain directed movement.
HMGB1 is a nonhistone chromosomal architectural protein inside the cell, whereas outside the cell it has other potent functions akin to cytokines and chemokines. Extracellular HMGB1 functions as a danger signal that plays a central role in the orchestration of damaged tissue repair by recruiting inflammatory cells and other cells involved in tissue regeneration and scar formation (3, 4, 12, 65). HMGB1 engagement of its known receptors can activate Erk and NF-κB signaling pathways (14), but the molecular requirements for HMGB1’s chemotactic activity remain unclear. We recently reported that, unlike a number of other well-known chemotactic stimuli, HMGB1 elicits cell migration after the nuclear translocation and functional activation of canonical NF-κB p50/RelA heterodimers; HMGB1-elicited cell migration requires transcription and new protein synthesis (14).
The pivotal functions of NF-κB transcription factors in immunity have been extensively documented (reviewed in Refs. 18, 21, 25), but their roles in cell migration and tissue regeneration are less well-known. Interestingly, extracellular stimuli leading to cellular responses that require sustained or more long-lasting NF-κB induction appear to activate both NF-κB signaling pathways (22, 31, 32). In this study, we show that both IKKβ- and IKKα-driven NF-κB signaling pathways are required for different reasons for cells to rapidly initiate and maintain their migration in response toHMGB1.
IKKβ is essential for the HMGB1-dependent migration responses in vitro and in vivo
IKKβ was essential for the in vitro migration toward HMGB1 of primary MEFs (Fig. 1A), MΦs (Fig. 2A), and neutrophils (Supplemental Fig. 3) and for the migration of neutrophils in vivo (Fig. 3, Supplemental Fig. 1). Blocking IKKβ kinase activity with the specific inhibitor SC-514 (66) also prevented the chemotactic response of primary MEFs to HMGB1 (Fig. 7B). Parsing out individual cell migration parameters by time-lapse microscopy revealed that IKKβ is essential very early (i.e., within the initial 30–60 min) for sensing an HMGB1 gradient (Fig. 4A, 4B) and also contributes to the speed of cell movement (Fig. 5A) and Euclidean migration distance (Supplemental Fig. 5A). Importantly, the rapid recruitment of mature neutrophils in an in vivo murine model of HMGB1- but not TNF-α–induced peritonitis was also blocked in mice lacking IKKβ in mature neutrophils and macrophages (Fig. 3A, 3B, Supplemental Fig. 2).
Evidence that IKKβ is necessary to drive RAGE expression to maintain HMGB1 chemotaxis
An IKKβ requirement for HMGB1 to upregulate RAGE, a ubiquitously expressed HMGB1 receptor (3, 5, 6, 9, 10), helps to explain the IKKβ/canonical NF-κB requirement for HMGB1 chemotaxis. IKKβ was necessary for HMGB1 to induce RAGE mRNA expression in MEFs (Fig. 6A) and MΦs (Supplemental Fig. 6), and RAGE was also required for the migration of fibroblasts to HMGB1 (Fig. 7A). Moreover, enforced RAGE overexpression functionally rescued the HMGB1 chemotactic response of IKKβ−/− but not IKKα−/− MEFs to WT levels (Fig. 8).
Except for mouse lung tissue, a number of reports indicate that RAGE RNA and protein are, respectively, expressed at low or barely detectable levels in a variety of different cell types (6, 60, 67–69), and ligand engagement is known to cause RAGE internalization (70). However, despite its very low expression level, the ubiquitously expressed RAGE receptor has been reported to mediate HMGB1 responses by a variety of cells, including fibroblasts, macrophages, myoblasts, and neutrophils (9, 71–73). In multiple experiments performed with a variety of polyclonal or monoclonal anti-RAGE Abs, we could only detect endogenous RAGE protein barely above background noise in MEFs or MΦs by either immunoblotting or FACS analysis in agreement with other published work (60, 69) (data not shown). Although our real-time PCR experiments clearly show that IKKβ is necessary for HMGB1 induction of RAGE mRNA (Fig. 6A, Supplemental Fig. 6), RAGE protein expression essentially remained at the limit of detection in either HMGB1 prestimulated MEFs or macrophages. Nevertheless, our observations are in full agreement with a recent report revealing that cell surface RAGE is not upregulated by homogenous rHMGB1 alone but only in conjunction with limiting amounts of another proinflammatory stimulus, such as LPS (69). We interpret our results in conjunction with this other published evidence to suggest that IKKβ/canonical NF-κB signaling in the context of rHMGB1 alone helps to maintain the balance of cell surface RAGE levels that are simultaneously subject to rapid depletion by HMGB1 engagement resulting in RAGE shedding (60) and internalization (70). Consistent with this hypothesis, we previously demonstrated that in comparison with other archetypical, strong inducers of IKKβ-dependent canonical NF-κB signaling, rHMGB1 alone is a modest activator of this pathway (12). Moreover, we also previously reported that protein synthesis and transcription were required in MEFs for their HMGB1 migration (but not to a PDGF control) (14), which would also fit with RAGE activation and resynthesis occurring downstream of HMGB1-mediated IKKβ activation. In addition, we have also observed that the directionality, velocity, and distance of cell movement in response to an HMGB1 gradient are each inhibited within 60 min of blocking protein synthesis (data not shown). Thus, from all of this collective evidence, we suggest that HMGB1-induced RAGE expression provides a means to counteract receptor depletion from the cell surface as cells migrate in response to the ligand, and we posit that IKKβ-dependent canonical NF-κB activation could be necessary for maintaining RAGE expression in the early phase of HMGB1 chemotactic responses to sustain them.
In addition to IKKβ, IKKα, p52, and RelB are required for HMGB1-elicited cell migration
Multiple cell types require IKKα for their migration response toward HMGB1. Primary MEFs and MΦ deficient for IKKα failed to migrate in response to HMGB1 in vitro (Figs. 1B, 2, respectively). In addition, in an in vivo model of HMGB1-induced peritonitis, mice lacking IKKα in MΦs and neutrophils were severely compromised for the rapid recruitment of IKKα null neutrophils in response to HMGB1 but not to TNF-α (Fig. 3A, 3B, Supplemental Fig. 2), and primary IKKα-deficient neutrophils also failed to migrate to HMGB1 but not C5a (Supplemental Fig. 3). These results indicate that, in addition to IKKβ, IKKα is also required for the initial chemotactic response to HMGB1 in vitro and in vivo. We also demonstrated that p100/p52−/− and RelB−/− immortalized MEFs are not attracted by HMGB1 in vitro (Fig. 1C), indicating that the IKKα-dependent NF-κB noncanonical pathway is required for the chemotactic movement of MEFs toward HMGB1.
The in vivo migration response of neutrophils to HMGB1 in mice lacking either IKKα or IKKβ in mature neutrophils or macrophages could potentially be more complex than an intrinsic requirement for either IKK in neutrophils. For instance, HMGB1 may induce peritoneal cavity macrophages to produce chemokines that attract neutrophils, and the latter could conceivably be compromised in both the IKKαf/f:MLysCre and IKKβf/f:MLysCre strain backgrounds, in which mature neutrophils and macrophages are deficient in either IKKα or IKKβ, respectively. In addition, it is also conceivable that the defective HMGB1 migration response of either IKKα- or IKKβ-deficient neutrophils in vivo could involve alterations in their margination to or diapedesis through the endothelium, in addition to directly affecting their chemotaxis. Although it could be argued that these latter possibilities are not formally ruled out by our experiments, we believe that our findings showing rapid (within 4 h), compromised peritoneal neutrophil recruitment in response to HMGB1, but not to TNF-α, in both conditional IKKα and IKKβ deleter strains are best explained by, and more consistent with, an intrinsic cellular role for each IKK in neutrophils for their HMGB1 chemotaxis. Moreover, this latter interpretation is also in agreement with our other in vitro primary cell data showing intrinsic needs for each IKK in mature neutrophils, macrophages, and fibroblasts for their HMGB1-induced migration.
The contributions of IKKα for HMGB1 chemotaxis were further defined by time-lapse microscopy. Akin to IKKβ, IKKα was also essential for determining the directionality of cell movement (Fig. 4A–D) within 60 min after exposure to an HMGB1 gradient. Unlike IKKβ (Fig. 5A), the loss of IKKα in MEFs had no significant effect on cell velocity at early times (Fig. 5C). However, as the migration response progressed, IKKα (like IKKβ) also eventually affected the velocity of cell movement (Supplemental Fig. 4E). Akin to IKKα, NF-κB p100/p52 was also essential within the initial 60 min to establish the directionality of cell movement (Fig. 4E, 4F). Because HMGB1 induction of RAGE expression required IKKβ, but not IKKα (Fig. 6A, 6B), and each IKK also differentially contributed to cellular velocity in the early phase of the HMGB1 migration response, our data suggest that the two NF-κB–activating kinases regulate different downstream targets, with somewhat overlapping but different essential mechanistic roles for HMGB1 migration responses. IKKα was necessary for basal and HMGB1-upregulated expression of CXCL12/SDF-1 in MEFs (Fig. 9). Interestingly, HMGB1 release by macrophages and dendritic cells was recently shown to be required for their chemotaxis toward rSDF-1 (74), supporting the notion that HMGB1 and SDF-1 signaling may collaborate to elicit some cell migration responses, and in light of those findings we hypothesize that IKKα-dependent SDF-1 expression by some cells may also be required for their chemotaxis toward HMGB1. Interestingly, cell migration toward HMGB1 was previously found to be pertussis toxin-sensitive, suggesting the functional contribution of a G protein-coupled receptor in addition to RAGE in HMGB1 chemotactic responses (12), and CXCL12/SDF-1 chemotactic responses are well known to require the CXCR4 G protein-coupled receptor. It is also important to point out in this context that a number of reports from multiple groups have revealed that, in addition to interacting with CXCL12/SDF-1 (74), HMGB1 also forms specific, functional complexes with a selected number of endogenous and exogenous mediators (including CpG-oligodeoxynucleotides, LPS, IL-1β, and nucleosomes) to dramatically enhance the induction of other proinflammatory cytokines by responding cells, thereby leading to more pronounced inflammatory responses (69, 75–81). Moreover, the barely detectable levels of cell surface RAGE on peritoneal macrophages were reported to be modestly upregulated by HMGB1, but only in combination with another limiting proinflammatory mediator, such as LPS (69). How such functional complexes with HMGB1 are recognized by specific cellular receptors to enhance proinflammatory signaling remains to be elaborated, but the need for direct physical interactions with HMGB1 suggests that dual receptor recognition may be involved (81).
IKKα-dependent p52 nuclear translocation is associated with chemotaxis to HMGB1
Because the ability of cells to migrate toward HMGB1 requires IKKα and the p52/RelB noncanonical NF-κB subunits, HMGB1 signaling might be expected to induce IKKα-dependent p100 processing to drive more p52 into cell nuclei. Quantitative analysis of multiple cell fractionation experiments performed over an 8 h time course showed that HMGB1 is a statistically significant enhancer of p52 nuclear translocation. HMGB1’s activity as an inducer of the noncanonical pathway is modest in comparison with a strong, archetypical stimulus of IKKα-dependent p100 > p52 processing like an LTβR agonist (Fig. 9C, Supplemental Fig. 7). In accord with these results, HMGB1 also induced the expression of CXCL12/SDF-1, a direct target gene of the IKKα–p52/RelB axis (21, 61) (Fig. 10). Even though these results help to explain how the p52/RelB pathway becomes invoked in an inducible manner, this is only part of the reason why IKKα is so critical in this cell migration response. In fact, cells require IKKα at the earliest measurable phase of their chemotactic responses to HMGB1: orientation/polarization toward the cytokine gradient. Thus, we believe that constitutive yet IKKα-dependent p100 > p52 processing is very important here, and in keeping with prior reports (61, 62), we also found reduced levels of nuclear and cytoplasmic p52 in IKKα null cells. Thus, IKKα null cells begin their migration response with an inherently reduced level of p52. This also helps to explain why p52 genetic ablation (unlike IKKα loss) modestly blunted cell velocity within the initial 30–60 min of HMGB1 chemotaxis (Fig. 5B).
Collaboration between different signaling pathways in HMGB1-induced cell migration
Several signaling pathways are activated by extracellular HMGB1, including MAPKs, canonical NF-κB, and Src, and each of these appears to be essential for HMGB1-induced cell migration (14, 15). We hypothesize that the requirement for Src is more likely connected to the mechanical aspects of cell migration, and not to specific requirements for gene expression, and we posit that Src signaling would be parallel to canonical NF-κB signaling to elicit HMGB1 chemotaxis. Herein, we show that the noncanonical NF-κB pathway is also required in HMGB1 chemotactic responses and also provide mechanistic evidence in MEFs that the functional dependencies on both NF-κB signaling pathways are for different reasons.
The collaboration between both NF-κB signaling pathways is particularly interesting in this context. Unlike other proinflammatory responses wherein IKKα acts in an attenuating capacity (27–29), in this cell migration response IKKα plays an activating role in concert with IKKβ signaling to allow for chemotaxis toward HMGB1. We posit that this type of collaboration could be particularly well suited to sustain chemotactic responses that may initially be used by inflammatory cells and subsequently by stem cells engaged in tissue repair.
Supplementary Material
Acknowledgments
We thank Francesco De Marchis and Dr. Patricio Mena for technical advice. We also thank Drs. Amer Beg, Michael Karin, Alexander Hoffmann, Falk Weih, and Jeffery Browning for providing WT, IKKα−/−, p100/p52−/−, and RelB−/− immortalized MEFs and agonistic anti-murine LTβR Ab, respectively, and Mary McFarland and Lynn Pantages (Boehringer Ingelheim Pharmaceuticals) for early help with animal care.
Abbreviations used in this paper
- 4-OHT
4-orthohydroxytamoxifen
- C5a
complement component 5a
- F
forward
- HMGB1
high mobility group box 1
- HPF
high-power fields
- IKK
inhibitor of NF-κB kinase
- KO
knockout
- LTβ
lymphotoxin β
- MEF
mouse embryo fibroblast
- MΦ
mature macrophage
- PDGF
platelet-derived growth factor
- R
reverse
- RAGE
receptor for advanced glycation end products
- SF
serum-free
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures
M.E.B. is founder and part owner of HMGBiotech, a company that provides goods and services related to HMGB proteins.
References
- 1.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 2.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 4.Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009;86:573–576. doi: 10.1189/jlb.1008585. [DOI] [PubMed] [Google Scholar]
- 5.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of RAGE and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 6.Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83:876–886. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 7.Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–C924. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 8.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 9.Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, Nawroth P, Andersson U, Harris RA, Harris HE. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 10.Andersson U, Erlandsson-Harris H, Yang H, Tracey KJ. HMGB1 as a DNA-binding cytokine. J Leukoc Biol. 2002;72:1084–1091. [PubMed] [Google Scholar]
- 11.Herold K, Moser B, Chen Y, Zeng S, Yan SF, Ramasamy R, Emond J, Clynes R, Schmidt AM. Receptor for advanced glycation end products (RAGE) in a dash to the rescue: inflammatory signals gone awry in the primal response to stress. J Leukoc Biol. 2007;82:204–212. doi: 10.1189/jlb.1206751. [DOI] [PubMed] [Google Scholar]
- 12.Palumbo R, Sampaolesi M, De Marchis F, Tonlorenzi R, Colombetti S, Mondino A, Cossu G, Bianchi ME. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol. 2004;164:441–449. doi: 10.1083/jcb.200304135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, Schmidt AM. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem. 2008;283:34457–34468. doi: 10.1074/jbc.M801465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palumbo R, Galvez BG, Pusterla T, De Marchis F, Cossu G, Marcu KB, Bianchi ME. Cells migrating to sites of tissue damage in response to the danger signal HMGB1 require NF-kappaB activation. J Cell Biol. 2007;179:33–40. doi: 10.1083/jcb.200704015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palumbo R, De Marchis F, Pusterla T, Conti A, Alessio M, Bianchi ME. Src family kinases are necessary for cell migration induced by extracellular HMGB1. J Leukoc Biol. 2009;86:617–623. doi: 10.1189/jlb.0908581. [DOI] [PubMed] [Google Scholar]
- 16.Lluis JM, Buricchi F, Chiarugi P, Morales A, Fernandez-Checa JC. Dual role of mitochondrial reactive oxygen species in hypoxia signaling: activation of nuclear factor-kappaB via c-SRC and oxidant-dependent cell death. Cancer Res. 2007;67:7368–7377. doi: 10.1158/0008-5472.CAN-07-0515. [DOI] [PubMed] [Google Scholar]
- 17.Lee HS, Moon C, Lee HW, Park EM, Cho MS, Kang JL. Src tyrosine kinases mediate activations of NF-kappaB and integrin signal during lipopolysaccharide-induced acute lung injury. J Immunol. 2007;179:7001–7011. doi: 10.4049/jimmunol.179.10.7001. [DOI] [PubMed] [Google Scholar]
- 18.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 19.Chiu YC, Fong YC, Lai CH, Hung CH, Hsu HC, Lee TS, Yang RS, Fu WM, Tang CH. Thrombin-induced IL-6 production in human synovial fibroblasts is mediated by PAR1, phospholipase C, protein kinase C alpha, c-Src, NF-kappa B and p300 pathway. Mol Immunol. 2008;45:1587–1599. doi: 10.1016/j.molimm.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 21.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Basak S, Kim H, Kearns JD, Tergaonkar V, O’Dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. 2007;128:369–381. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto Y, Gaynor RB. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29:72–79. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25:6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- 26.Hansberger MW, Campbell JA, Danthi P, Arrate P, Pennington KN, Marcu KB, Ballard DW, Dermody TS. IkappaB kinase subunits alpha and gamma are required for activation of NF-kappaB and induction of apoptosis by mammalian reovirus. J Virol. 2007;81:1360–1371. doi: 10.1128/JVI.01860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 28.Li Q, Lu Q, Bottero V, Estepa G, Morrison L, Mercurio F, Verma IM. Enhanced NF-kappaB activation and cellular function in macrophages lacking IkappaB kinase 1 (IKK1) Proc Natl Acad Sci USA. 2005;102:12425–12430. doi: 10.1073/pnas.0505997102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu B, Yang Y, Chernishof V, Loo RR, Jang H, Tahk S, Yang R, Mink S, Shultz D, Bellone CJ, et al. Proinflammatory stimuli induce IKKalpha-mediated phosphorylation of PIAS1 to restrict inflammation and immunity. Cell. 2007;129:903–914. doi: 10.1016/j.cell.2007.03.056. [DOI] [PubMed] [Google Scholar]
- 30.Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. EMBO J. 2004;23:4202–4210. doi: 10.1038/sj.emboj.7600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saitoh T, Nakayama M, Nakano H, Yagita H, Yamamoto N, Yamaoka S. TWEAK induces NF-kappaB2 p100 processing and long lasting NF-kappaB activation. J Biol Chem. 2003;278:36005–36012. doi: 10.1074/jbc.M304266200. [DOI] [PubMed] [Google Scholar]
- 32.Yang CH, Murti A, Pfeffer LM. Interferon induces NF-kappa B-inducing kinase/tumor necrosis factor receptor-associated factor-dependent NF-kappa B activation to promote cell survival. J Biol Chem. 2005;280:31530–31536. doi: 10.1074/jbc.M503120200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 34.Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S. Limb and skin abnormalities in mice lacking IKKalpha. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- 35.Li Q, Lu Q, Hwang JY, Büscher D, Lee KF, Izpisua-Belmonte JC, Verma IM. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu Y, Baud V, Oga T, Kim KI, Yoshida K, Karin M. IKKalpha controls formation of the epidermis independently of NF-kappaB. Nature. 2001;410:710–714. doi: 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- 37.Sil AK, Maeda S, Sano Y, Roop DR, Karin M. IkappaB kinase-alpha acts in the epidermis to control skeletal and craniofacial morphogenesis. Nature. 2004;428:660–664. doi: 10.1038/nature02421. [DOI] [PubMed] [Google Scholar]
- 38.Descargues P, Sil AK, Sano Y, Korchynskyi O, Han G, Owens P, Wang XJ, Karin M. IKKalpha is a critical coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc Natl Acad Sci USA. 2008;105:2487–2492. doi: 10.1073/pnas.0712044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–1187. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Badea TC, Wang Y, Nathans J. A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J Neurosci. 2003;23:2314–2322. doi: 10.1523/JNEUROSCI.23-06-02314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seibler J, Zevnik B, Kuter-Luks B, Andreas S, Kern H, Hennek T, Rode A, Heimann C, Faust N, Kauselmann G, et al. Rapid generation of inducible mouse mutants. Nucleic Acids Res. 2003;31:e12. doi: 10.1093/nar/gng012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci USA. 1996;93:10887–10890. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, Metzger D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ERT and Cre-ERT2 recombinases. Nucleic Acids Res. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 45.Browning JL, Miatkowski K, Sizing I, Griffiths D, Zafari M, Benjamin CD, Meier W, Mackay F. Signaling through the lymphotoxin beta receptor induces the death of some adenocarcinoma tumor lines. J Exp Med. 1996;183:867–878. doi: 10.1084/jem.183.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Massa PE, Li X, Hanidu A, Siamas J, Pariali M, Pareja J, Savitt AG, Catron KM, Li J, Marcu KB. Gene expression profiling in conjunction with physiological rescues of IKKalpha-null cells with wild type or mutant IKKalpha reveals distinct classes of IKKalpha/NF-kappaB-dependent genes. J Biol Chem. 2005;280:14057–14069. doi: 10.1074/jbc.M414401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Penzo M, Massa PE, Olivotto E, Bianchi F, Borzi RM, Hanidu A, Li X, Li J, Marcu KB. Sustained NF-kappaB activation produces a short-term cell proliferation block in conjunction with repressing effectors of cell cycle progression controlled by E2F or FoxM1. J Cell Physiol. 2009;218:215–227. doi: 10.1002/jcp.21596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shah AB, DiMartino SJ, Trujillo G, Kew RR. Selective inhibition of the C5a chemotactic cofactor function of the vitamin D binding protein by 1,25(OH)2 vitamin D3. Mol Immunol. 2006;43:1109–1115. doi: 10.1016/j.molimm.2005.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zigmond SH, Hirsch JG. Leukocyte locomotion and chemotaxis. New methods for evaluation, and demonstration of a cell-derived chemotactic factor. J Exp Med. 1973;137:387–410. doi: 10.1084/jem.137.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boxio R, Bossenmeyer-Pourié C, Steinckwich N, Dournon C, Nüsse O. Mouse bone marrow contains large numbers of functionally competent neutrophils. J Leukoc Biol. 2004;75:604–611. doi: 10.1189/jlb.0703340. [DOI] [PubMed] [Google Scholar]
- 51.Zantl R, Rädler U, Horn E. Chemotaxis in mu channels. Imaging & Microscopy. 2006;8:30–32. [Google Scholar]
- 52.Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, Chavakis T. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007;26:1129–1139. doi: 10.1038/sj.emboj.7601552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol. 1993;151:2399–2408. [PubMed] [Google Scholar]
- 54.May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT. Urokinase receptor (CD87) regulates leukocyte recruitment via beta 2 integrins in vivo. J Exp Med. 1998;188:1029–1037. doi: 10.1084/jem.188.6.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woolf E, Grigorova I, Sagiv A, Grabovsky V, Feigelson SW, Shulman Z, Hartmann T, Sixt M, Cyster JG, Alon R. Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat Immunol. 2007;8:1076–1085. doi: 10.1038/ni1499. [DOI] [PubMed] [Google Scholar]
- 56.Heit B, Robbins SM, Downey CM, Guan Z, Colarusso P, Miller BJ, Jirik FR, Kubes P. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat Immunol. 2008;9:743–752. doi: 10.1038/ni.1623. [DOI] [PubMed] [Google Scholar]
- 57.Müller N, van den Brandt J, Odoardi F, Tischner D, Herath J, Flügel A, Reichardt HM. A CD28 superagonistic antibody elicits 2 functionally distinct waves of T cell activation in rats. J Clin Invest. 2008;118:1405–1416. doi: 10.1172/JCI32698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem. 1997;272:16498–16506. doi: 10.1074/jbc.272.26.16498. [DOI] [PubMed] [Google Scholar]
- 59.Kalea AZ, Reiniger N, Yang H, Arriero M, Schmidt AM, Hudson BI. Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J. 2009;23:1766–1774. doi: 10.1096/fj.08-117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, Reiss K, Saftig P, Bianchi ME. A soluble form of the receptor for advanced glycation end products (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10) FASEB J. 2008;22:3716–3727. doi: 10.1096/fj.08-109033. [DOI] [PubMed] [Google Scholar]
- 61.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 62.Qing G, Xiao G. Essential role of IkappaB kinase alpha in the constitutive processing of NF-kappaB2 p100. J Biol Chem. 2005;280:9765–9768. doi: 10.1074/jbc.C400502200. [DOI] [PubMed] [Google Scholar]
- 63.Horwitz R, Webb D. Cell migration. Curr Biol. 2003;13:R756–R759. doi: 10.1016/j.cub.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 64.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 65.Dumitriu IE, Bianchi ME, Bacci M, Manfredi AA, Rovere-Querini P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol. 2007;81:84–91. doi: 10.1189/jlb.0306171. [DOI] [PubMed] [Google Scholar]
- 66.Kishore N, Sommers C, Mathialagan S, Guzova J, Yao M, Hauser S, Huynh K, Bonar S, Mielke C, Albee L, et al. A selective IKK-2 inhibitor blocks NF-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J Biol Chem. 2003;278:32861–32871. doi: 10.1074/jbc.M211439200. [DOI] [PubMed] [Google Scholar]
- 67.Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- 68.Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, Rieber EP. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2006;323:475–488. doi: 10.1007/s00441-005-0069-0. [DOI] [PubMed] [Google Scholar]
- 69.Qin YH, Dai SM, Tang GS, Zhang J, Ren D, Wang ZW, Shen Q. HMGB1 enhances the proinflammatory activity of lipopolysaccharide by promoting the phosphorylation of MAPK p38 through receptor for advanced glycation end products. J Immunol. 2009;183:6244–6250. doi: 10.4049/jimmunol.0900390. [DOI] [PubMed] [Google Scholar]
- 70.Sevillano N, Girón MD, Salido M, Vargas AM, Vilches J, Salto R. Internalization of the receptor for advanced glycation end products (RAGE) is required to mediate intracellular responses. J Biochem. 2009;145:21–30. doi: 10.1093/jb/mvn137. [DOI] [PubMed] [Google Scholar]
- 71.Riuzzi F, Sorci G, Donato R. The amphoterin (HMGB1)/receptor for advanced glycation end products (RAGE) pair modulates myoblast proliferation, apoptosis, adhesiveness, migration, and invasiveness. Functional inactivation of RAGE in L6 myoblasts results in tumor formation in vivo. J Biol Chem. 2006;281:8242–8253. doi: 10.1074/jbc.M509436200. [DOI] [PubMed] [Google Scholar]
- 72.Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G, Nawroth PP, Bierhaus A, Schwaninger M. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008;28:12023–12031. doi: 10.1523/JNEUROSCI.2435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Zoelen MAD, Yang H, Florquin S, Meijers JCM, Akira S, Arnold B, Nawroth PP, Bierhaus A, Tracey KJ, van der Poll T. Role of Toll-like receptors 2 and 4, and the receptor for advanced glycation end products in high-mobility group box 1-induced inflammation in vivo. Shock. 2009;31:280–284. doi: 10.1097/SHK.0b013e318186262d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Campana L, Bosurgi L, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 for stromal cell-derived factor-1/CXCL12-dependent migration of macrophages and dendritic cells. J Leukoc Biol. 2009;86:609–615. doi: 10.1189/jlb.0908576. [DOI] [PubMed] [Google Scholar]
- 75.Rouhiainen A, Tumova S, Valmu L, Kalkkinen N, Rauvala H. Pivotal advance: analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (amphoterin) J Leukoc Biol. 2007;81:49–58. doi: 10.1189/jlb.0306200. [DOI] [PubMed] [Google Scholar]
- 76.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 77.Ivanov S, Dragoi AM, Wang X, Dallacosta C, Louten J, Musco G, Sitia G, Yap GS, Wan Y, Biron CA, et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 2007;110:1970–1981. doi: 10.1182/blood-2006-09-044776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Youn JH, Oh YJ, Kim ES, Choi JE, Shin JS. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-alpha production in human monocytes. J Immunol. 2008;180:5067–5074. doi: 10.4049/jimmunol.180.7.5067. [DOI] [PubMed] [Google Scholar]
- 79.Sha Y, Zmijewski J, Xu Z, Abraham E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol. 2008;180:2531–2537. doi: 10.4049/jimmunol.180.4.2531. [DOI] [PubMed] [Google Scholar]
- 80.Urbonaviciute V, Fürnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–3018. doi: 10.1084/jem.20081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hreggvidsdottir HS, Ostberg T, Wähämaa H, Schierbeck H, Aveberger AC, Klevenvall L, Palmblad K, Ottosson L, Andersson U, Harris HE. The alarminHMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J Leukoc Biol. 2009;86:655–662. doi: 10.1189/jlb.0908548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.