

Abstract
Bacterial nicotinate mononucleotide adenylyltransferase encoded by the essential gene nadD plays a central role in the synthesis of the redox cofactor NAD+. The NadD enzyme is conserved in the majority of bacterial species and has been recognized as a novel target for developing new and potentially broad-spectrum antibacterial therapeutics. Here we report the crystal structures of Bacillus anthracis NadD in complex with three NadD inhibitors, including two analogues synthesized in the present study. These structures revealed a common binding site shared by different classes of NadD inhibitors and explored the chemical environment surrounding this site. The structural data obtained here also showed that the subtle changes in ligand structure can lead to significant changes in the binding mode, information that will be useful for future structure-based optimization and design of high affinity inhibitors.
INTRODUCTION
Due to the widespread occurrence of drug resistance in many infectious bacterial pathogens, there is an urgent and continuing need for developing new antibiotics 1–3. In the current post-genomics era, the complete genome sequences of hundreds of bacterial species have become available, allowing for many potentially new antibiotic targets to be identified through comparative genomic studies and experimental gene essentiality analysis 4–8. Such capabilities are of special utility given the significant increase in the number of bacterial strains resistant to common antibiotics 9–11. An approach to combat bacterial drug resistance is to develop new antibiotics against previously unexploited targets that have emerged from genomics studies 4, 12, 13. One such target is the enzyme NaMN adenylyltransferase encoded by gene nadD in the biosynthesis pathways of the ubiquitous cofactor nicotinamide adenine dinucleotide (NAD+)5, 14.
NAD+ is the essential redox cofactor for hundreds of enzymes and has an impact on nearly all aspects of metabolism in the cell. The enzyme NaMN adenylyltransferase, or NadD, occupies a central position in bacterial NAD+ biosynthesis, and is required for both de novo and salvage routes to generate NAD+ 15. NadD has been recognized as a promising new target for developing novel antibiotics due to its crucial role in synthesizing NAD+; its essentiality has been demonstrated experimentally in a number of species 5, 6. Another attractive aspect of targeting NadD is that it is highly conserved in the overwhelming majority of bacterial species including most pathogens. Therefore, drugs developed based on the inhibition of NadD have the potential of possessing wide-spectrum antibacterial activity. Since the first report of the identification of nadD gene in 2000 16, many biochemical and structural studies have been conducted on this enzyme. The crystal structures of NadD from a number of pathogenic bacterial species, such as E. coli, Pseudomonas aeruginosa, Staphylococcus aureus, and Bacillus anthracis have been reported 17–22. More recently, using a structure-based drug design approach, we have identified for the first time several inhibitors of NadD and demonstrated that inhibition of NadD indeed leads to the suppression of bacterial growth 14.
Bacterial NadD, as well as its human counter parts (human Nmnat isoforms −1. −2 and −3), are members of the HxGH-motif containing nucleotidyl transferase superfamily and share the same overall fold 23. However, the sequence identities between the bacterial and human enzymes are low (~22%) and the biochemical properties of the two enzyme subfamilies are also distinct especially with regard to substrate specificity 18, 22, 24, 25. While the bacterial enzyme almost exclusively prefers nicotinic acid mononucleotide (NaMN) as substrate, all three human Nmnat isoforms work equally well on both NaMN and its amidated form, nicotinamide mononucleotide (NMN). Structural analyses have revealed conformational differences in the enzyme’s active sites that may account for their different biochemical properties 26. These differences have allowed development of specific inhibitors against NadD that have no adverse effects on the activity of human Nmnat isoforms 14. Indeed, among the first NadD inhibitors identified, which included two distinct chemical scaffolds (Classes 1 and 3, Table 1) with IC50 values in the low µM range, none of them have detectable inhibitory activity against human Nmnat 14. These results further validated NadD as a tractable target for antibacterial therapeutic development.
Table 1.
Chemical structures of two classes of bacterial NadD inhibitors as represented by compounds 1_02 and 3_02a
| Compound class |
Structure |
|---|---|
| 1 | 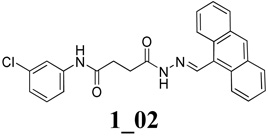 |
| 3 |
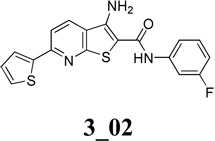 |
The numbering of the compounds follows the scheme: compound class_1st generation analog_2nd generation analog.
Previously we reported the crystal structure of baNadD in complex with product NaAD and with a Class 3 inhibitor (3_02) 14. In the current study, we present crystal structures of NadD in complex with a Class 1 inhibitor (1_02) identified in our previous study and two novel inhibitors (1_02_1 and 1_02_3) designed and synthesized based on the complex structure of 1_02. Comparison of these complex structures revealed a common binding region in NadD shared between the two different classes of inhibitors with distinct chemical scaffolds. Binding of these inhibitors appears to stabilize the enzyme in a catalytically impaired conformation and blocks substrate binding. Interestingly, the overall binding modes of the inhibitors differ, though they all interact via aromatic groups through a common site. The detailed interactions between the inhibitors and enzyme residues as revealed from these structures indicate the potential for the identification of chemically diverse inhibitors that target this region. Such potential is anticipated to facilitate the structure-based design of highly potent and specific NadD inhibitors.
RESULTS
Structure of baNadD in complex with inhibitor 1_02
The complex of baNadD and 1_02 crystallized in the same space group P21212 as the previously reported baNadD-3_02 complex 14 and the protein conformations in the two inhibitor complexes are also very similar with root mean square deviation (RMSD) for all Cα atoms of 0.175Å; they resemble the conformation of the enzyme in its apo state rather than the substrate or product bound state, with RMSD values of 0.494Å and 0.833Å, respectively, compared to the apo and product bound baNadD 14, 21, 22. Inspection of the electron density for the bound compound revealed a symmetrically shaped density much larger than the compound (Fig.1A). This density is located at a symmetrical interface between two baNadD monomers where the inhibitor can bind in one of two different but symmetrically related orientations, with the positions of the central anthracene ring overlapping with each other (Fig. 1A, 1B). These two orientations are in fact equivalent and physically indistinguishable. It can be viewed as such that in the complex crystal, half of the protein molecule population would bind the inhibitor in one orientation, while the other half bind the inhibitor in the second orientation. The resulted electron density is the accumulated average from all the complex molecules in the crystal. Therefore we modeled 1_02 molecule in two orientations each with half occupancy (Fig.1).
Figure 1.

Inhibitor 1_02 binds between two monomers of baNadD. A). The Fo-Fc omit map for 1_02. Two 1_02 molecules, colored green and yellow, respectively, each with half occupancy are modeled in the density. B). 1_02 binds at a baNadD monomer-monomer interface formed in the crystal of the complex. The two baNadD monomers are colored cyan and green respectively.
baNadD structures have been reported recently in its apo form, in complex with substrate NaMN, with product NaAD, as well as with inhibitor 3_02 14, 21, 22. The overall baNadD structure contains a Rossman-fold core with a central six-stranded parallel β-sheet and two or three α helices on each side of the sheet (Fig. 2A). Following the sixth and the last β strand, two α helices (α6 and α7) form a small C-terminal subdomain that is characteristic of the nucleotidyltransferase superfamily. The signature HxGH motif (15HYGH18) is located in the loop connecting the first β-strand (β1) and succeeding α helix (α1). This motif is involved in the interaction with the phosphate groups of the substrates (ATP and NaMN) and participates in the catalysis.
Figure 2.

Interactions of 1_02 with baNadD. A). Ribbon representation of baNadD-1_02 complex. Inhibitor 1_02 is shown as sticks. B). Detailed interactions between 1_02 and baNadD residues. The Cα trace of the protein is shown; relevant side chains are shown as sticks. Hydrogen bonds are shown as dotted lines. Water molecules are shown as small red spheres. C). Surface representation of the inhibitor binding site on baNadD, colored by the electrostatic potentials. Three water molecules adjacent to 1_02 are shown as green spheres.
In the baNadD-1_02 complex structure determined in the present work, 1_02 sits at a central cleft between strands β1 and β4 of the β sheet, which is the catalytic and substrate binding sites of the enzyme (Fig. 2A). The compound is bent at the acylhydrazone linkage and follows the contour of the crevice of the substrate binding site (Fig. 2B and 2C). The anthryl rings together with the acylhydrazone of the compound stack against the side chains of Trp 116, Tyr112 and Met109 (Fig. 2B). A single direct hydrogen bond is formed between the amide group of the carboxyamide moiety of 1_02 to the main chain carbonyl of Gly8. The chloride of the terminal chlorophenyl group appears to interact favorably with the side chain of His18 of the HxGH motif. There are two indirect hydrogen bonds between the compound and protein atoms. One is formed between the hydrazone amide and the side chain of Thr85 via a water molecule (wat2), and the other between the acyl oxygen group and Asn39 side chain through wat1. The chlorophenyl ring is also in contact with the side chains of Ile 7 and Ile 21, which may provide additional stabilizing van der Waals interactions with the compound (Fig. 2B).
Comparison of the binding modes of 1_02 and 3_02
Comparison of the binding mode of 1_02 and that of 3_02 reported previously 14 shows that the nearly coplanar anthracene rings and the hydrazone portion of 1_02 overlaps with the largely planar 3_02 (Fig.3A). They form similar stacking and van der Waals interactions with multiple protein residues including Trp116, Tyr112, Met109 and Lys115. This shared binding site corresponds to the region that binds nicotinic acid riboside portion of NaMN substrate in the absence of the inhibitors (Fig. 3B). In particular, Trp116 would stack against the pyridine ring of NaMN and is critical for the proper positioning of the substrate. Therefore binding of the inhibitors would prevent NaMN binding all together. Notably, the two classes of compounds do not overlap completely and each has additional interactions with the enzyme that are not present in the other compound (Fig. 3A). While 3_02 largely overlaps with the NaMN substrate binding site, 1_02 also intrudes into the ATP binding pocket and its chlorophenyl group would overlap with the ribose of ATP (Fig. 3B). The inhibitory efficiencies of the two compounds against baNadD have been determined previously, with 1_02 having Ki of 9 µM and 10 µM, respectively, with regard to NaMN and ATP substrates; while 3_02 has Ki of 18 µM and 32 µM against NaMN and ATP, respectively 14. These values are consistent with the structural observation that 1_02 interferes with binding of both NaMN and ATP whereas 3_02 mostly interferes with NaMN binding.
Figure 3.

Comparison of the binding modes of 1_02 (magenta), 3_02 (yellow), and the product deamido-NAD (blue). A). Superposition of baNadD bound 1_02 with 3_02 showing the overlapping binding mode. The protein conformations of the two structures are essentially identical and a single ribbon diagram is shown. B). Superposition of the baNadD-1_02 complex (orange) with the baNadD-product complex (cyan). 1_02 is in magenta; the product deamido-NAD is in blue.
Binding of both 1_02 and 3_02 appears to stabilize the enzyme in a conformation that is significantly different from its substrate or product bound form, and is apparently catalytic incompetent (Fig. 3B). The different conformation associated with inhibitor binding as compared to the substrate or product bound conformations has been suggested to lead to mixed inhibition kinetics that contains both competitive and non-competitive characters 14.
Structure of baNadD in complex with inhibitor 1_02_1
Because 1_02 must adopt either of the two symmetrically related orientations with half occupancy in the crystal due to the overlapping position of the anthracene rings, we hypothesized that a symmetrical compound that fit the observed density of 1_02 would bind to the enzyme with full occupancy and higher affinity. A compound was designed to retain the central planer ring system with an acylhydrazone arm and terminal chlorophenyl ring on each side. The resulting compound, designated 1_02_1 (Scheme 1), was synthesized and subjected to biochemical and crystallographic analysis. Compound 1_02_1 replaced the anthracene ring with a benzene and includes a Cl atom at the ortho position of the terminal phenyl rings; this selection was based on availability of chemical precursors. 1_02_1 was then tested as a NadD inhibitor. According to the structure activity relationship (SAR) data from a limited set of analogs of Class 1 compounds (Fig. 4A), it was expected that 1_02_1 should have an improved activity compared to the different asymmetric “monomeric” compounds. Inhibition assay on 1_02_1 yielded an IC50 of 13±2 µM and 16±4 µM against ecNadD and baNadD, respectively (Fig. 4B). Compared to the Class 1 analogs shown in Fig. 4A, 1_02_1 is significantly better than those compounds with either a benzene or naphthalene rings, while its activity is similar to those compounds containing an antharcene ring, including 1_02. As 1_13 and 1_15 in Fig. 4A contain only benzene rings and linkers identical to 1_02_1, they may be considered as “precursors” of 1_02_1. Therefore the design strategy to create a symmetrical compound may be considered successful, as a more than 10 fold improvement in activity was achieved. 1_02_1 is also slightly more active than compound 1_02, which has an IC50 of 25 µM.
Scheme 1.

a) benzene-1,4-dicarbaldehyde, ethanol, reflux
Figure 4.

A). Structure and activities of representative Class 1 compounds. The compounds cocrystallized with baNadD are boxed. B). Dose dependent inhibition by compound 1_02_1 against baNadD (left panel) and ecNadD (right panel).
To understand the binding mode of 1_02_1, we determined the crystal structure of baNadD in complex with the compound. The baNadD-1_02_1 complex has the same crystal form as the 1_02 complex and retains the crystal lattice packing involving the same monomer-monomer interface to which the inhibitor binds. 1_02_1 has well defined electron density and is modeled with full occupancy (Fig.5A). As predicted, 1_02_1 binds at the same site as 1_02 and overlaps with the two orientations of that molecule (Fig. 5B). The conformations of the acylhydrazone arms of the two compounds are very similar despite the presence of several rotatable bonds (Fig. 5B). 1_02_1 also interacts with the protein similarly as 1_02. Most van der Waals interactions, especially the stacking interactions with Trp116 and Tyr112, are preserved (Fig. 5C). However, due to the difference in the central ring systems and the restriction of the covalent linkage to the central benzene ring, the acylhydrazone arms of 1_02_1 displays a slight rigid body rotation (~15°) compared to 1_02. As a result, there are differences in the hydrogen bond patterns and in the orientation of the end chlorophenyl group. The hydrogen bond between the carboxyamide nitrogen of 1_02 and Gly8 main chain (shown in Fig. 2B) is lost in the 1_02_1 complex structure, whereas a new hydrogen bond is formed between the carboxyamide oxygen and Gly106 main chain amide (Fig. 5C). The smaller single six-membered central ring of 1_02_1 may lead to a decrease in the van der Waals interactions with the protein as compared to the anthracene ring in 1_02.
Figure 5.

Structure of baNadD-1_02_1 complex. A). The 2Fo-Fc map of 1_02_1, the two formate molecules (For) and the surrounding regions. B). Superposition of the enzyme bound 1_02_1 (blue) with 1_02 in its two orientations (represented in two different shades of gray). C). Detailed interactions between 1_02_1 and baNadD residues.
Interestingly, in the 1_02_1 complex structure, two well-ordered formate molecules are observed mediating specific interactions between the acylhydrazone amide group and the main chain amide and side chain hydroxyl of residue Thr85 (Fig. 5C). In the NaMN or NaAD complex structure, the carboxylate group of the nicotinic acid binds in this region and interacts with the main chain amide of Thr85. Thus, the formate molecule observed in the 1_02_1 complex structure mimics the interaction between the nicotinic acid carboxylate group of the substrate NaMN and the enzyme.
Carboxylate containing analogs of 1_02 and 1_02_1
Motivated by the presence of the formates in the 1_02_1 complex structure, additional analogs were designed. These analogs (1_02_2 and 1_02_3 in Scheme 3) were designed to include a carboxylate moiety to approximate the location of the formates in the baNadD-1_02_1 complex structure. In that structure, one formate oxygen is 2.76 Å from the side chain hydroxyl group of Thr85 and the other oxygen is 2.91 Å from the backbone amide nitrogen of Thr85, forming a well-defined ion-dipole interactions. Accordingly, it was hypothesized that the carboxylate moieties would mimic these interactions, thereby further improving binding. In addition, the inclusion of the carboxylate moieties would enhance the solubility of the compounds, making them more suitable for biochemical experiments and potentially enhance their bioavailability. This led to the design and synthesis of 1_02_2 and 1_02_3 shown in Scheme 3. 1_02_2 was a direct mimic of 1_02_1 while 1_02_3 was designed as an analog of 1_02, to test if the presence of the carboxylate could improve the affinity of the monomeric species.
Scheme 3.

a) napthalene-1-carbaldehyde, ethanol, reflux. b) benzene-1,4-dicarbaldehyde, ethanol, reflux
Experiments were then undertaken on the two new 1_02 analogs to measure the inhibitory activity against baNadD. Surprisingly, 1_02_2 did not inhibit baNadD at concentrations up to 100 µM, while 1_02_3 only weakly inhibits baNadD activity (IC50>200 µM). Thus, the inclusion of the carboxylates did not lead to improved binding with the symmetric, dimeric analog 1_02_02, although some binding affinity of the monomer analog, 1_02_3 is present.
To understand the unexpected results, both 1_02_2 and 1_02_3 were subjected to crystallographic analysis. All attempts to cocrystallize 1_02_2 with baNadD were unsuccessful; however, cocrystals of 1_02_3 bound to baNadD were obtained and the complex structure was determined to 2.55Å resolution. The 1_02_3 complex crystal is in a different space group (C2) from all other baNadD-inhibitor complexes, and contained eight baNadD monomers in the asymmetric unit. Notably, the baNadD monomer-monomer interface to which 1_02_3 binds is different from that observed in all other inhibitor complex structures (Fig. 6A), indicating that packing of the enzyme molecules in the crystal can be influenced by inhibitor binding. The electron density for 1_02_3 was well-defined allowing unambiguous modeling of the compound in the complex (Fig. 6B). Interestingly, the binding mode of 1_02_3 differs significantly from that of 1_02 and 1_02_1, although some overlap is present (Fig. 6C). In particular, the naphthalene ring of 1_02_3 binds to the same site as the aromatic rings of the other Class 1 compounds, and form similar van der Waals contacts with Trp116, Tyr112, Met 109, as well as with Lys115. However, the acylhydrazone arm and the end chlorobenzene ring of 1_02_3 adopt completely different conformations from that of compounds 1_02 and 1_02_1, and interact with different functional groups on the protein. In this binding mode, the carboxylate group of the compound, though occupying a similar position as the formate molecule in the 1_02_1 complex, interacts with the enzyme somewhat differently. One oxygen of the carboxylate still interacts with the side chain hydroxyl of Thr85 (3.13 Å). In addition, there are ion-dipole interactions of the carboxylate with the main chain amides of Thr85 (3.30 Å) and of Tyr117 (2.81 Å) (Fig 6D). These interactions are reminiscent of those observed in the NaMN substrate complex where the carboxylate of the substrate also forms two hydrogen bonds with the main chain amides of both Thr85 and Tyr117 14, 22. An additional hydrogen bond between the amide group of 1_02_3 and the main chain carbonyl of Lys115 is also observed (2.8 Å). Overall, these interactions lead to a different binding mode for 1_02_3 even though the aromatic and acidic groups bind to the sites as predicted based on the 1_02_1 complex. In this mode the carboxy amide moiety and adjacent chlorophenyl ring of the compound are largely exposed to the solvent while their counterpart in the 1_02 and 1_02_1 complexes binds to the adenosine binding site of the enzyme and is much less solvent accessible.
Figure 6.

Structure of baNadD-1_02_3 complex. A). Compound 1_02_3 (blue sticks) binds between two baNadD monomers (colored cyan and light cyan), which have a difference interface from that in the 1_02_1 complex. The two monomers of baNadD in the 1_02_1 complex are colored light pink with one monomer superimposed onto the cyan monomer of the 1_02_3 complex. B). The Fo-Fc omit map for 1_02_3. C). Superposition of the three enzyme-bound Class 1 inhibitors showing the common aromatic binding site as well as differences in the binding mode of each compound. The protein molecules in the 1_02_1 and 1_02_3 complexes are shown in light pink and cyan, respectively. D). Detailed interactions between 1_02_3 and baNadD residues.
The 1_02_3 complex structure provides a possible explanation as to why 1_02_2 does not bind as anticipated. While the naphthalene ring and carboxylate moieties bind to the anticipated sites, the geometrical restraints to achieve these interactions leads to a reorientation of the compound and a significant change in the overall binding mode of 1_02_3 (Fig. 6C and 6D). Potential binding of 1_02_2 in the same orientation as 1_02_3 would disallow the second arm of the hydrazine linker to access the binding pocket occupied by 1_02 and 1_02_1, thereby abolishing binding.
DISCUSSION
In an effort to develop inhibitors targeting the essential bacterial NadD enzymes, we have identified three classes of bacterial NadD inhibitors with distinct scaffolds in a structure-based in sillico screen 14. We have now also obtained the crystals structures of B. anthracis NadD in complex with inhibitors from two different chemical classes: 3_02 from Class 3 (reported in Ref. 14), and three different Class 1 compounds (1_02, 1_02_1 and 1_02_3) presented in the current work. The complex structures of baNadD with different inhibitors revealed a common binding site near residues Trp117, Try112, and Met109, as shown in Fig. 7, which appears to have an affinity for aromatic groups from different small molecules. This site overlaps but is distinct from the substrate NaMN binding pocket, and may serve as a primary site to be targeted in future inhibitor design efforts. Such design efforts would target compounds whose aromatic moieties interact with the identified “aromatic” site, with the remainder of those putative molecules sampling various binding modes in the vicinity of this site.
Figure 7.

Superposition of the baNadD bound 3_02 (yellow), 1_02 (magenta), 1_02_1 (blue) and 1_02_3 (green). The surface presentation of the enzyme (colored according to electrostatic potentials) in the 1_02 complex structure is shown. The image also includes three nearby water molecules observed in the 1_02 complex structure (cyan spheres) and the formate molecule observed in the 1_02_1 complex structure. The orientation of deamino-NAD+ (thin, atom-colored licorice representation) from the product-complex structure (pdb code 3e27) is also shown.
The complex structures of three Class 1 compounds provide useful information about the chemical characters of the inhibitor-binding site of NadD. Compounds 1_02 and 1_02_1 bind to the aromatic site with their central anthracene or benzene rings and hydrazone groups; while the linker and the terminal chlorobenzene ring intrude into a deep groove on the enzyme and interact directly with the conserved active site HxGH motif residues. In addition to this groove, the binding potential of a small pocket adjacent to the primary binding site is highlighted in the 1_02_1 and 1_02_3 complex structures, where it is revealed that this pocket favors binding of a carboxylate group. In the 1_02_3 complex structure, binding of the carboxylate group at this site comes at the expense of completely reorienting the acylhydrazone arms of the compound, which results in an overall decreased affinity. This reorientation is also proposed to disallow binding of the dimeric 1_02_2 to the protein.
Although the activities of the current NadD inhibitors are only in the low micromolar IC50 range at best, there are several attractive features in their binding modes. Binding of both classes of inhibitors appears to stabilize the enzyme in a catalytically incompetent conformation, significantly different from its substrate or product bound conformation, resulting in a mixed inhibition kinetics behavior that contains both competitive and non-competitive characters. As such the binding pocket can accommodate small molecules with structures very different from the natural ligands of the enzymes. Therefore, such small molecule binders are anticipated to have minimal adverse effects on the numerous other NAD+ or ATP utilizing enzymes. The non-competitive character of inhibition by these inhibitors also indicates that once higher affinity compounds are found, they may not be strongly influenced by cellular ATP or NAD+ concentrations, which are on the order of ~10–103 µM 27–29. Such inhibitors could have better in vivo efficacy than purely competitive inhibitors.
Although a non-native dimer interface is observed in several baNadD-inhibitor (e.g., 3_02, 1_02 and 1_02_1) complex crystal structures, it has become clear that this dimerization mode is due to crystal lattice packing interactions under specific crystallization conditions since such dimerization is not observed in solution in an analytical ultracentrifugation study14. Crystal structures of the 1_02_3 complex and apo-baNadD obtained in different space groups also do not have the same dimerization mode21, 22. This observation partially explains the moderate improvement of the activity of the much larger dimeric 1_02_1 as compared to its monomeric precursor. Therefore future inhibitor design and optimization effort should be focused on engineering specific direct interactions between the inhibitors and enzyme monomer. Toward this goal, the complex structures of NadD with different inhibitors provided useful information on a common primary target site and the chemical environment of the vicinity of this site, which can be exploited to improve on the existing inhibitor scaffolds or design high affinity inhibitors with novel scaffolds for maximum interaction with the enzyme.
EXPERIMENTAL SECTION
Protein Crystallography
The expression and purification of Bacillus anthracis NadD (baNadD) has been reported elsewhere14. For co-crystallization of baNadD with compounds 1_02, 1_02_1 and 1_02_3, appropriate amount of the stock compound solutions (20 mM in DMSO) was mixed with the protein to the final concentration of 1 mM, while the final protein concentration is 19 mg/ml. The PEG/Ion Crystallization Screening kit (Hampton Research) was used for the initial screens of the complex crystals. Hanging drop vapor diffusion methods were used for the crystallization where equal volume (1.5 µl) of the complex and reservoir solution was mixed and equilibrated against the reservoir at 20°C. The baNadD-1_02 cocrystals were obtained in conditions that contain 0.2–0.25 M magnesium formate and 20% −24% PEG 3350. The baNadD-1_02_1 complex crystals were obtained from 0.2 M potassium formate and 20% PEG 3350. Both crystals were cryoprotected in solutions that contained an increased concentration of PEG 3350 (40%) and original components of the reservoir and frozen in liquid propane. The baNadD-1_02_3 complex formed crystals in 0.2 M potassium citrate and 20% PEG 3350, and the crystal was frozen in the cryoprotectant containing original components of the reservoir supplemented with 10% DMSO and flash frozen in liquid nitrogen.
The X-ray diffraction data of the baNadD-1_02 complex crystal was collected at beamline 19BM, Advanced Photon Source, Argonne National Laboratory, whereas the data for baNadD-1_02_1 and baNadD–1_02_3 crystals were collected in-house on a Rigaku FRE rotating anode X-ray generator equipped with Osmic focusing device and RAXIS IV++ image plate detector. The data were further processed using HKL3000 software 30.
Both the baNadD-1_02 and baNadD-1_02_1 complexes were crystallized in the P21212 space group, isomorphous to the crystals of baNadD-3_02 complex reported recently 14. Therefore, the model of the baNadD-3_02 (pdb code 3hfj), excluding ligand and solvent molecules was used as the initial model for the refinement of both new complexes using the program Refmac of the CCP4 package 31–33. The solution of baNadD -1_02_3 complex was found by the molecular replacement method of Phaser 34 using apo baNadD as the starting model. Model inspection and adjustment was performed with Coot 35. The electron densities for compound 1_02, 1_02_1 and 1_02_03 were clearly visible in the early stage of the refinement. The PRODRG server 36 was used to generate the models for the compounds to be included in the complex structure. Final rounds of refinements were performed using PHENIX 37, 38 and the model geometry was monitored by Molprobity 39. The crystal data and refinement statistics of these complexes are list in Table 2. The coordinates have been deposited in the Protein Data Bank 40 with accession codes 3MLA, 3MLB, and 3MMX.
Table 2.
Crystal Data and refinement statistics
| Datasets | baNadD-1_02 | baNadD·1_02_1 | baNadD 1_02_03 |
|---|---|---|---|
| Data Statistics | |||
| Space group | P21212 | P21212 | C2 |
| Unit cell Dimensions |
a=88.6 Å b=97.53 Å c=44.30 Å |
a=88.34 Å,b=96.64 Å,c=44.13 Å |
a=295.15 Å b=46.45 Å c=114.95 Å |
| β=91.42° | |||
| Resolution (Å) | 50–1.70 | 50–1.80 | 50–2.55 |
| Total observations | 230680 | 73589 | 191091 |
| Unique Reflections | 74116 | 47493 | 97476 |
| Completeness | 99.0 (97.0) | 88.8 (79.3) | 99.6 (100) |
| (outershell) (%) | |||
| Rsym (outer shell) | 0.040 (0.574) | 0.040 (0.271) | 0.075/0.571 |
| I/δ (outer shell) | 39.2 (2.6) | 23.8 (2.7) | 20.53/2.06 |
| Refinement | |||
| Rworkb | 0.190 | 0.183 | 0.205 |
| Rfreec | 0.230 | 0.232 | 0.270 |
| r.m.s.d bond length (Å) | 0.006 | 0.007 | 0.008 |
| r.m.s.d bond angle (°) | 1.049 | 1.121 | 1.073 |
| Protein atoms | 3051 | 3067 | 11783 |
| Water molecules | 308 | 337 | 232 |
| Ligand atoms | 85 | 48 | 191 |
| Average B-factors (Å2) | |||
| Protein | 32.5 | 26.3 | 57.31 |
| Ligands | 39.7 | 24.6 | 67.81 |
| Water | 32.6 | 33.2 | 44.19 |
| Ramachandran Plot | |||
| Favored region (%) | 98.6 | 98.6 | 97.4 |
| Allowed region (%) | 100.0 | 100.0 | 99.8 |
Rsym = ΣhklΣj|Ij – <I>| /ΣhklΣj|Ij|.
Rwork = Σhkl|Fo – Fc| /Σhkl|Fo |, where Fo and Fc are the observed and calculated structure factors, respectively.
Five percent randomly selected reflections were excluded from refinement and used in the calculation of Rfree.
Enzyme inhibition assay
A general phosphate detection assay method using Malachite Green reagent was adapted to measure the activity of NaMN adenylyltransferase 14. Briefly, the byproduct of NadD catalyzed reaction, inorganic pyrophosphate (PPi), was hydrolysed by inorganic pyrophosphatase and the resulting orthophosphate was detected by the Malachite Green dye. The reaction mixture contained 2.3 nM ecNadD (or 1.2 nM baNadD) in 100 mM Hepes, pH 7.5 buffer, 0.2 mM ATP, 0.07 or 0.2 mM NaMN, 10 mM MgCl2, 0.1 mg/ml Bovine Serum Albumin, 0.2 U inorganic pyrophosphatase. Appropriate amount of inhibitors were added to the reaction mixture to assess their effect on enzyme activity. After preincubation of the enzyme with the compounds for 5 min at room temperature, the reaction was started by adding NaMN substrate. The reaction was quenched with two volumes of Malachite Green Reagent in 1.2 M sulfuric acid prepared as described by Cogan et al.41. After 20–30 min incubation to allow for complex/color formation, the absorbance was measured at 620 nm. To account for possible contribution of free phosphate and/or pyrophosphate (present in the sample or released due to non-specific hydrolysis of ATP) as well as of background absorbance (color) of the tested compounds, parallel reactions were run for each experimental point without addition of NadD enzymes, and their OD620 values were subtracted from the measurements of enzyme activity in respective samples. Reaction in the presence of 2% DMSO but without inhibitor served as the positive control.
For IC50 determination, the initial rate of the enzymatic reaction was measured at fixed NaMN and ATP concentrations (equal to two-fold Kmvalues) in the absence and presence of various concentrations of inhibitors.The IC50 value was determined by plotting the rates versus inhibitor concentration and fitting to the equation (1) using GraphPad Prism®.
| (1) |
v0 and vi represent initial rates in the absence and presence of inhibitors at concentration [I].
Chemistry
Proton NMR spectra were recorded on Varian 500 MHz FT NMR spectrometers. Mass spectra were recorded on a LCQ mass spectrometer (Finnigan MAT, San Jose, CA). Element analyses were performed by Atlantic Mircolab, Inc. (Norcross, GA). Flash column chromatography was performed using Silica Gel 60 (230–400 mesh) from Thomas Scientific (Swedesboro, NJ). Analytical thin layer chromatography (TLC) was performed on precoated glass backed plates from Analtech Inc. (Newark, DE) (TLC uniplates, Silica gel GHLF, 250 µ). Plates were visualized using ultraviolet, iodine vapors, phosphomolybdic acid or ninhydrin. Compound 1 was available from commercial supplier. The purity of the final compounds tested for biological activity was determined by elemental analysis (Atlantic Microlab) and confirmed by HPLC. The purity was found to be ≥ 95%.
Synthesis of N-(2-Chloro-phenyl)-3-(4-{[3-(2-chloro-phenylcarbamoyl)-propionyl]-hydrazonomethyl}-benzylidene-hydrazinocarbonyl)-propionamide (1_02_1, Scheme 1)
Benzene-1,4-dicarbaldehyde (0.01 g, 0.07 mmol) and N-(2-Chloro-phenyl)-3-hydrazinocarbonyl-propionamide 1 (0.036 g, 0.14 mmol) in ethanol (5mL) were heated to reflux for 2 h. After cooling to room temperature, the precipitate was filtered off and washed with ethanol to give 1_02_1 as a pale white solid (0.03 g, 69%). 1H NMR (500MHz, DMSO-d6) 12.31 (2H, s), 8.21 (2H, s), 7.94 (4H, s), 7.60–8.20 (8H, br); MS Anal. Mol. Wt. 580.14 (604.2 M+Na). Elemental Analysis Calculated for C28H26Cl2N6O4 0.4H2O: C, 57.13; H, 4.58; N, 14.27. Found: C, 57.36; H, 4.49; N, 14.00.
Synthesis of (N'-tert-Butoxycarbonyl-hydrazino)-acetic acid ethyl ester (4, Scheme 2)
Scheme 2.

a) water. b) succinic anhydride, DMF, 70°C. c) HBTU, DIPEA, DMF. d) TFA/CH2Cl2. e) Ethanol, 1N NaOH.
Ethyl bromoacetate 3 (6.97 mL, 62.8 mmol) was added to a stirred solution of tert-butylcarbazate 2 (24.9 g, 188.6 mmol) in water (25 mL) at room temperature. The mixture was stirred for 30 min. Water layer was then extracted with ethyl acetate (3x). Ethyl acetate extracts were pooled together and washed with brine. Ethyl acetate was evaporated under vacuum to get crude product which was purified by flash column chromatography using hexane:ethyl acetate (70:30) as an eluent (yield 70 %). 1H NMR (500 MHz, CDCl3) 1.28 (3H, CH2-CH3, t), 1.45 (9H, C-CH3, s), 3.64 (2H, NH-CH2-CO, s), 4.20 (2H, CH2CH3, q); MS Anal. Mol. Wt. 218.25 (M+1).
Synthesis of 4-(N'-tert-Butoxycarbonyl-N-ethoxycarbonylmethyl-hydrazino)-4-oxo-butyric acid (5, Scheme 2)
Into a solution of (N'-tert-Butoxycarbonyl-hydrazino)-acetic acid ethyl ester 4 (1.85 g, 18.5 mmol) in DMF (30 mL) was added succinic anhydride (4.84 g, 22.2 mmol) and the mixture was stirred at 75 °C for 18 h. DMF was evaporated and the crude mixture was purified by flash column chromatography using hexane:ethylacetate (1 % acetic acid) as an eluent (yield 50 %). 1H NMR (500 MHz, CDCl3) 1.28 (3H, CH2-CH3, t), 1.48 (9H, C-CH3, s), 2.55–3.00 (6H, NH-CH2-CO, N-CH2-CH2-CO, m), 4.20 (2H, CH2CH3, q); MS Anal. Mol. Wt. 318.25 (M-1).
Synthesis of {N'-tert-Butoxycarbonyl-N-[3-(2-chloro-phenylcarbamoyl)-propionyl]-hydrazino}-acetic acid ethyl ester (6, Scheme 2)
Into a mixture of compound 5, HBTU and DIPEA in DMF was added 2-chloroaniline and the solution was stirred for 48 h. DMF was evaporated under vacuum and the mixture was dissolved in ethyl acetate and washed with water (2x), 1M KHSO4 (2x) and water (2x). Ethyl acetate was then evaporated to get crude compound which was purified by flash column chromatography using hexane:ethyl acetate (50:50) as an eluent (yield 38 %). 1H NMR (500 MHz, CDCl3) 1.27 (3H, CH2-CH3, t), 1.48 (9H, C-CH3, s), 2.58–3.06 (6H, NH-CH2-CO, N-CH2-CH2-CO, m), 4.20 (2H, CH2CH3, q), 7.01(1H, ArH, t), 7.22–7.27(1H, ArH, m) 7.34(1H, ArH, d), 8.06 (1H, ArNH, s), 8.33(1H, ArH, d); MS Anal. Mol. Wt. 427.88 (M+23).
Synthesis of {N-[3-(2-Chloro-phenylcarbamoyl)-propionyl]-hydrazino}-acetic acid ethyl ester (7, Scheme 2)
Compound 6 (0.3 g, 0.7 mmol) was dissolved in 5 mL of 20 % TFA in dichloromethane and the solution was stirred for 1 h. TFA was then evaporated under vacuum and the crude mixture was purified by flash column chromatography using ethyl acetate as an eluent (yield 87 %). 1H NMR (500 MHz, CDCl3) 1.27 (3H, CH2-CH3, t), 2.75 (2H, N-CH2-CH2-CO, t), 3.11(2H, N-CH2-CH2-CO, t), 4.20 (2H, CH2CH3, q), 4.36 (2H, NH-CH2-CO, s), 7.00 (1H, ArH, t), 7.23 (1H, ArH, t) 7.33(1H, ArH, d) 8.24–8.40 (2H, ArNH, ArH, m); MS Anal. Mol. Wt. 327.76 (M+1).
Synthesis of {N-[3-(2-Chloro-phenylcarbamoyl)-propionyl]-hydrazino}-acetic acid (8)
Compound 7 was dissolved in 10 mL ethanol followed by addition of 1.2 mL of 1N NaOH. The mixture was stirred for 1 h. Ethanol was then evaporated under vacuum to obtain crude compound which was dissolved in water and the solution was neutralized using 1N HCl. Evaporation of the water followed by separation of salt by precipitation in ethanol yielded compound 8 (yield 82 %) which was used without any purification for next step. 1H NMR (500 MHz, CD3OD) 2.50–3.21 (4H, N-CH2-CH2-CO, m), 4.18–4.44 (2H, NH-CH2-CO, m), 7.14 (1H, ArH, t), 7.27 (1H, ArH, t) 7.43(1H, ArH, d) 7.78 (1H, ArH, s); MS Anal. Mol. Wt. 299.07 (M+1).
Synthesis of {N'-(4-{Carboxymethyl-[3-(2-chloro-phenylcarbamoyl)-propionyl]-hydrazonomethyl}-benzylidene)-N-[3-(2-chloro-phenylcarbamoyl)-propionyl]-hydrazino}-acetic acid (1_02_2, Scheme 3)
Benzene-1,4-dicarbaldehyde (0.006 g, 0.05 mmol) and compound 8 (0.032 g, 0.10 mmol) in ethanol (5 mL) were heated to reflux for 12 h. After cooling to room temperature, the precipitate was filtered off and washed with ethanol to give 1_02_2 as a pale white solid (0.015 g, yield 44%). 1H NMR (500 MHz, DMSO-d6) 2.72 (4H, N-CH2-CH2-CO, t), 3.15 (4H, N-CH2-CH2-CO, t), 4.52 (4H, NH-CH2-CO, s), 7.15 (2H, ArH, t), 7.29 (2H, ArH, t), 7.47(2H, ArH, d) 7.65–7.77(6H, ArH, m); 9.56(2H, Ar-CH=N); MS Anal. Mol. Wt. 696.15 (M-2) Elemental Analysis Calculated for C32H28Cl2N6O8Na2 1.5H2O: C, 50.01; H, 4.06; N, 10.93. Found: C, 50.07; H, 4.09; N, 11.10.
Synthesis of {N-[3-(2-Chloro-phenylcarbamoyl)-propionyl]-N'-naphthalen-1-ylmethylene-hydrazino}-acetic acid (1_02_3, Scheme 3)
naphthalene-1-carbaldehyde (0.015 g, 0.10 mmol) and compound 8(0.031 g, 0.10 mmol) in ethanol (5 mL) were heated to reflux for 12 h. After cooling to room temperature, the precipitate was filtered off and washed with ethanol to give 1_02_3 as a pale white solid (0.02 g, yield 47%). 1H NMR (500 MHz, DMSO-d6) 2.64 (1H, N-CH2-CH2-CO, t), 2.79 (1H, N-CH2-CH2-CO, t), 3.17(1H, N-CH2-CH2-CO, t), 3.23(1H, N-CH2-CH2-CO, t), 4.89–4.97 (2H, NH-CH2-CO,m), 7.17 (1H, ArH, t), 7.31 (1H, ArH, t), 7.48(1H, ArH, d) 7.56–7.68 (3H, ArH, m), 7.75(1H, ArH, d), 7.96–8.04 (3H, ArH, m), 8.50 (1H, ArNH, s), 8.70 (1H, ArH, t), 9.56(2H, Ar-CH=N); MS Anal. Mol. Wt. 437.11 (M-1). Elemental Analysis Calculated for C23H20ClN3O4 0.8H2O: C, 61.07; H, 4.81; N, 9.29. Found: C, 61.06; H, 4.81; N, 9.27.
ACKNOWLEDGEMENT
We thank Dominika Borek for help with X-ray data collection at APS synchrotron, Chuo Chen for helpful discussion. This work is supported by grants from the NIH (AI059146), the Welch Foundation (I-5105), and the University of Maryland Computer-Aided Drug Design Center. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by University of Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Abbreviations
- NAD
nicotinamide adenine dinucleotide
- NaAD
nicotinic acid adenine dinucleotide or deamido-NAD
- NaMN
nicotinic acid mononucleotide
- NMN
nicotinamide mononucleotide
- DMSO
dimethyl sulfoxide
- PEG
polyethylene glycol
- PPi
inorganic pyrophosphate
- RMSD
root mean square deviation
Footnotes
The atomic coordinates and structure factors (accession codes 3MLA, 3MLB and 3MMX) have been deposited in the RCSB Protein Data Bank (www.rcsb.org).
REFERENCES
- 1.Vicente M, Hodgson J, Massidda O, Tonjum T, Henriques-Normark B, Ron EZ. The fallacies of hope: will we discover new antibiotics to combat pathogenic bacteria in time? FEMS Microbiol Rev. 2006;30:841–852. doi: 10.1111/j.1574-6976.2006.00038.x. [DOI] [PubMed] [Google Scholar]
- 2.Wright GD, Sutherland AD. New strategies for combating multidrug-resistant bacteria. Trends Mol Med. 2007;13:260–267. doi: 10.1016/j.molmed.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDevitt D, Rosenberg M. Exploiting genomics to discover new antibiotics. Trends Microbiol. 2001;9:611–617. doi: 10.1016/s0966-842x(01)02235-1. [DOI] [PubMed] [Google Scholar]
- 5.Gerdes SY, Scholle MD, D'Souza M, Bernal A, Baev MV, Farrell M, Kurnasov OV, Daugherty MD, Mseeh F, Polanuyer BM, Campbell JW, Anantha S, Shatalin KY, Chowdhury SA, Fonstein MY, Osterman AL. From genetic footprinting to antimicrobial drug targets: examples in cofactor biosynthetic pathways. J Bacteriol. 2002;184:4555–4572. doi: 10.1128/JB.184.16.4555-4572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerdes S, Edwards R, Kubal M, Fonstein M, Stevens R, Osterman A. Essential genes on metabolic maps. Curr Opin Biotechnol. 2006;17:448–456. doi: 10.1016/j.copbio.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Osterman AL, Begley TP. A subsystems-based approach to the identification of drug targets in bacterial pathogens. Prog Drug Res. 2007;64:133–170. doi: 10.1007/978-3-7643-7567-6_6. 131. [DOI] [PubMed] [Google Scholar]
- 8.Osterman AL, Gerdes SY. Comparative approach to analysis of gene essentiality. Methods Mol Biol. 2008;416:459–466. doi: 10.1007/978-1-59745-321-9_31. [DOI] [PubMed] [Google Scholar]
- 9.Falagas ME, Bliziotis IA, Kasiakou SK, Samonis G, Athanassopoulou P, Michalopoulos A. Outcome of infections due to pandrug-resistant (PDR) Gram-negative bacteria. BMC Infect Dis. 2005;5(1):24. doi: 10.1186/1471-2334-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 11.Dorman SE, Chaisson RE. From magic bullets back to the magic mountain: the rise of extensively drug-resistant tuberculosis. Nat Med. 2007;13:295–298. doi: 10.1038/nm0307-295. [DOI] [PubMed] [Google Scholar]
- 12.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 13.Mills SD. When will the genomics investment pay off for antibacterial discovery? Biochem Pharmacol. 2006;71:1096–1102. doi: 10.1016/j.bcp.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 14.Sorci L, Pan Y, Eyobo Y, Rodionova I, Huang N, Kurnasov O, Zhong S, MacKerell AD, Jr, Zhang H, Osterman AL. Targeting NAD biosynthesis in bacterial pathogens: Structure-based development of inhibitors of nicotinate mononucleotide adenylyltransferase NadD. Chem Biol. 2009;16:849–861. doi: 10.1016/j.chembiol.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Begley TP, Kinsland C, Mehl RA, Osterman A, Dorrestein P. The biosynthesis of nicotinamide adenine dinucleotides in bacteria. Vitam. Horm. 2001;61:103–119. doi: 10.1016/s0083-6729(01)61003-3. [DOI] [PubMed] [Google Scholar]
- 16.Mehl RA, Kinsland C, Begley TP. Identification of the Escherichia coli nicotinic acid mononucleotide adenylyltransferase gene. J Bacteriol. 2000;182:4372–4374. doi: 10.1128/jb.182.15.4372-4374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H, Zhou T, Kurnasov O, Cheek S, Grishin NV, Osterman A. Crystal structures of E. coli nicotinate mononucleotide adenylyltransferase and its complex with deamido-NAD. Structure. 2002;10:69–79. doi: 10.1016/s0969-2126(01)00693-1. [DOI] [PubMed] [Google Scholar]
- 18.Olland AM, Underwood KW, Czerwinski RM, Lo MC, Aulabaugh A, Bard J, Stahl ML, Somers WS, Sullivan FX, Chopra R. Identification characterization and crystal structure of Bacillus subtilis nicotinic acid mononucleotide adenylyltransferase. J Biol Chem. 2002;277:3698–3707. doi: 10.1074/jbc.M109670200. [DOI] [PubMed] [Google Scholar]
- 19.Yoon HJ, Kim HL, Mikami B, Suh SW. Crystal structure of nicotinic acid mononucleotide adenylyltransferase from Pseudomonas aeruginosa in its Apo and substrate-complexed forms reveals a fully open conformation. J Mol Biol. 2005;351:258–265. doi: 10.1016/j.jmb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Han S, Forman MD, Loulakis P, Rosner MH, Xie Z, Wang H, Danley DE, Yuan W, Schafer J, Xu Z. Crystal structure of nicotinic acid mononucleotide adenylyltransferase from Staphyloccocus aureus: structural basis for NaAD interaction in functional dimer. J Mol Biol. 2006;360:814–825. doi: 10.1016/j.jmb.2006.05.055. [DOI] [PubMed] [Google Scholar]
- 21.Lu S, Smith CD, Yang Z, Pruett PS, Nagy L, McCombs D, Delucas LJ, Brouillette WJ, Brouillette CG. Structure of nicotinic acid mononucleotide adenylyltransferase from Bacillus anthracis. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:893–898. doi: 10.1107/S1744309108029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sershon VC, Santarsiero BD, Mesecar AD. Kinetic and X-ray structural evidence for negative cooperativity in substrate binding to nicotinate mononucleotide adenylyltransferase (NMAT) from Bacillus anthracis. J Mol Biol. 2009;385:867–888. doi: 10.1016/j.jmb.2008.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Structure and function of nicotinamide mononucleotide adenylyltransferase. Curr. Med. Chem. 2004;11:873–885. doi: 10.2174/0929867043455666. [DOI] [PubMed] [Google Scholar]
- 24.Sorci L, Cimadamore F, Scotti S, Petrelli R, Cappellacci L, Franchetti P, Orsomando G, Magni G. Initial-rate kinetics of human NMN-adenylyltransferases: substrate and metal ion specificity, inhibition by products and multisubstrate analogues, and isozyme contributions to NAD+ biosynthesis. Biochemistry. 2007;46:4912–4922. doi: 10.1021/bi6023379. [DOI] [PubMed] [Google Scholar]
- 25.Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 26.Zhou T, Kurnasov O, Tomchick DR, Binns DD, Grishin NV, Marquez VE, Osterman AL, Zhang H. Structure of human nicotinamide/nicotinic acid mononucleotide adenylyltransferase. Basis for the dual substrate specificity and activation of the oncolytic agent tiazofurin. J Biol Chem. 2002;277:13148–13154. doi: 10.1074/jbc.M111469200. [DOI] [PubMed] [Google Scholar]
- 27.Ryll T, Wagner R. Improved ion-pair high-performance liquid chromatographic method for the quantification of a wide variety of nucleotides and sugar-nucleotides in animal cells. J Chromatogr. 1991;570:77–88. doi: 10.1016/0378-4347(91)80202-n. [DOI] [PubMed] [Google Scholar]
- 28.Tong L, Lee S, Denu JM. Hydrolase regulates NAD+ metabolites and modulates cellular redox. J Biol Chem. 2009;284:11256–11266. doi: 10.1074/jbc.M809790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang N, De Ingeniis J, Galeazzi L, Mancini C, Korostelev YD, Rakhmaninova AB, Gelfand MS, Rodionov DA, Raffaelli N, Zhang H. Structure and function of an ADP-ribose-dependent transcriptional regulator of NAD metabolism. Structure. 2009;17:939–951. doi: 10.1016/j.str.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 31.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Collaborative Computational Project Number 4. The CCP4 Suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 34.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, Gopal K, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Pai RK, Read RJ, Romo TD, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC. Recent developments in the PHENIX software for automated crystallographic structure determination. J. Synchrotron. Radiat. 2004;11:53–55. doi: 10.1107/s0909049503024130. [DOI] [PubMed] [Google Scholar]
- 38.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 39.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cogan EB, Birrell GB, Griffith OH. A Robotics-Based Automated Assay for Inorganic and Organic Phosphates. Analytical Biochemistry. 1999;271:29–35. doi: 10.1006/abio.1999.4100. [DOI] [PubMed] [Google Scholar]