

Abstract
The dramatic increase in the prevalence of obesity and its strong association with cardiovascular disease have resulted in unprecedented interest in understanding the effects of obesity on the cardiovascular system. A consistent, but puzzling clinical observation is that obesity confers an increased susceptibility to the development of cardiac disease, while at the same time affording protection against subsequent mortality (termed the obesity paradox). In this review we focus on evidence available from human and animal model studies and summarize the ways in which obesity can influence structure and function of the heart. We also review current hypotheses regarding mechanisms linking obesity and various aspects of cardiac remodeling. There is currently great interest in the role of adipokines, factors secreted from adipose tissue, and their role in the numerous cardiovascular complications of obesity. Here we focus on the role of leptin and the emerging promise of adiponectin as a cardioprotective agent. The challenge of understanding the association between obesity and heart failure is complicated by the multifaceted interplay between various hemodynamic, metabolic, and other physiological factors that ultimately impact the myocardium. Furthermore, the end result of obesity-associated changes in the myocardial structure and function may vary at distinct stages in the progression of remodeling, may depend on the individual pathophysiology of heart failure, and may even remain undetected for decades before clinical manifestation. Here we summarize our current knowledge of this complex yet intriguing topic.
I. INTRODUCTION
A. Changing Prevalence of Obesity and Link to Cardiovascular Disease
Obesity and overweight are most often defined by body mass index (BMI) (25), with underweight being <20 kg/m2, normal 20–25 kg/m2, overweight 25–30 kg/m2, class I obesity 30–35 kg/m2, class II obesity 35–40 kg/m2, and class III obesity >40 kg/m2. Surveys based on self-reported data conservatively estimate that 15 and 33% of adults in Canada were obese and overweight, respectively, yet these values are lower than in the United States where the latest Behavioral Risk Factor Surveillance System (BRFSS) data demonstrate a continued escalation in obesity in each of the states (44). A particularly worrying trend is the striking increase in class III obesity in recent years with 4% of men and 7% of women in the United States meeting this definition (136, 171). Obesity is strongly associated with the development of major risk factors for atherosclerotic disease such as hypertension, hyperlipidemia, and diabetes (79, 89, 358). In addition, some evidence suggests that obesity also has direct effects on the heart that may not be the result of atherosclerosis. It is estimated that in Canada, cardiovascular disease costs the economy over $18.5 million per annum and accounts for over one-third of all deaths (data provided by Heart and Stroke Foundation of Canada). Such statistics have provided great impetus for us to develop our understanding of mechanisms linking obesity with increased cardiovascular morbidity and mortality.
B. Evidence for Causative Role of Obesity in Heart Failure
Abundant evidence shows that obesity is associated with structural and functional changes in the heart in both humans and animal models (see sects. III and IV). Many of these changes, such as left ventricular (LV) hypertrophy, left atrial (LA) enlargement, and subclinical impairment of LV systolic and diastolic function are believed to be precursors to more overt forms of cardiac dysfunction and heart failure. Thus it is generally assumed that longstanding obesity will eventually lead to heart failure. However, in human studies, we mainly have to rely on cross-sectional data at this time, and the conclusions are not firm regarding the direct role of obesity in the development of heart failure (14, 46, 132, 376). One major issue is the methods for diagnosing heart failure. Hospital discharge coding and death certificates are notoriously inaccurate. Furthermore, misdiagnosis of heart failure is common and may be more common in obese subjects because of the frequent occurrences of edema and dyspnea (42). Pulmonary pathologies and right heart failure may easily be mistaken for diastolic heart failure. Perhaps more importantly, obesity is very frequently associated with other risk factors for developing heart failure (e.g., hypertension, diabetes, hyperlipidemia, sleep-disordered breathing, etc.), and office-based blood pressure recordings in obese patients may underestimate the burden of hypertension that would be picked up by 24-h recordings. Only one large, longitudinal, population-based study addressing this issue is available (173). In this study, participants in the Framingham Heart Study were stratified by BMI at the time of enrollment and then followed for incident heart failure (diagnosed by adjudicated clinical criteria). The main findings were that increased BMI was associated with an increased risk of heart failure in both men and women and that this risk was graded across categories of increasing BMI. In the subset of patients who had echocardiography within 30 days of the heart failure diagnosis, the majority had reduced LV ejection fractions. Although the investigators adjusted for the presence of cardiovascular risk factors, we know that traditional risk factors explain only a fraction of the actual myocardial infarctions that occur in the general population. Thus the possibility that coronary artery disease contributed to the development of heart failure has not been excluded. Indeed, central obesity is undeniably a risk marker, and probably a risk factor for the development and progression of coronary artery disease (43, 105, 185–188, 229, 241, 328). A summary of the crude cumulative incidence of heart failure by gender and BMI category is shown in Figure 1.
FIG. 1.

Cumulative incidence of heart failure according to category of body mass index at the baseline examination. The body mass index was 18.5–24.9 in normal subjects, 25.0–29.9 in overweight subjects, and 30.0 or more in obese subjects. The mean age at enrollment ranged between 53 and 60 years in the various categories. Interestingly, the curves for the three groups remain rather close together for almost 15 years in the men. Thereafter, overweight and obese men have significantly more heart failure. Given that the age when the curves begin to diverge is in the late 60s, this again raises the question of whether coronary disease might be a contributing factor. [From Kenchaiah et al. (174), copyright 2004 Massachusetts Medical Society. All rights reserved.]
C. The Obesity Paradox
While it is widely accepted that obesity increases the risk of developing heart disease, a growing number of recent reports document a statistically significant survival benefit in obese patients once they have been diagnosed with cardiovascular diseases (128, 144, 145, 200, 201, 211, 244). For example, follow-up examination of a stable outpatient cohort showed that obese patients had better survival than normal-weight patients with comparable severity of heart failure (201). In another cohort of outpatients with established heart failure, higher BMIs were associated with lower mortality risk (65). Another study tracking 3-yr survival rates in patients post-myocardial infarction (MI) clearly showed improvements as BMI increased after adjusting for severity of illness, age, and gender (128). One group has suggested that the positive effect of obesity on survival of heart failure patients is most significant in those with preserved systolic function (accounting for ~50% of those with heart failure, Ref. 360) (124), although a very recent study demonstrated that higher BMI was also associated with lower in-hospital mortality risk in patients with acute decompensated heart failure (103). In 9,633 consecutive patients undergoing percutaneous coronary interventions, those who were very lean or with BMI in the normal range were found to be at the highest risk for in-hospital complications, cardiac death, and 1-yr mortality. Importantly, there was a graded response manifest as a progressive decrease in mortality as BMI increased (121, 123). In 1,676 consecutive patients with unstable angina or non-ST-elevation MI, all of whom were treated with early revascularization, obese and very obese patients had less than half the long-term mortality of normal BMI patients (35). Thus it appears that these patients might have some degree of resistance to ischemia-reperfusion injury. Lastly, a protective effect of obesity has been seen in hemodialysis patients evidenced by improved survival in those with higher BMIs (164, 307, 311). Of note, these end-stage renal failure patients have an extremely high risk of dying from cardiovascular disease. Thus, although obesity is widely accepted as a risk factor for coronary heart disease and heart failure, accumulating evidence strongly supports a protective role of obesity once patients have developed cardiovascular disease.
Although a great deal of data support the existence of a protective effect of obesity, some authors have questioned the existence of the obesity paradox (85, 233). These authors have suggested that there is a “U-shaped” outcome curve according to BMI for patients with heart failure, in which mortality is greatest in underweight patients; lower in normal, overweight, and mildly obese patients; but higher again in more severely obese patients (85, 126). It has also been posited that more intense treatment regimens are applied in obese patients and that this might explain their increased survival post-MI (85). However, this seems doubtful given the widely accepted and well-publicized guidelines for the treatment of cardiovascular disease. One recent study has suggested that overweight and obese individuals were in fact protected from short-term death yet have a long-term mortality risk that is similar to normal-weight individuals (254).
The conclusion that obesity may both elicit cardiac disease and protect from cardiovascular death clearly now requires further mechanistic analyses at cellular, molecular, and systematic levels. If we can identify the beneficial component, then it might be possible to harness the effect for therapeutic purposes.
II. OVERVIEW OF ANIMAL MODELS THAT HAVE BEEN USED TO STUDY CARDIAC CHANGES ASSOCIATED WITH OBESITY
Many animal models have been studied in attempts to clarify the mechanisms that contribute to cardiac injury and dysfunction in obesity. In evaluating results from animal studies, it is important to bear in mind that similar to the situation in humans, obesity in certain animal models is associated with coincident morbidities such as impaired glucose tolerance, diabetes, and hypertension. In discussing various models, attempts will be made where possible to highlight potential confounding variables. The majority of studies have been performed in rodents (with a smaller number in other species such as rabbits or dogs). Most of the animal data discussed in this review will be based on rodent models with generalized obesity, or transgenic mice with targeted mutations in cardiomyocytes. Models with generalized obesity are of two types: genetic mutations that result in obesity, and diet-induced obesity achieved by feeding animals a high-fat diet.
Commonly used mouse models develop obesity on the basis of mutations in the leptin gene or the leptin receptor. Thus ob/ob mice are null for the leptin gene. Ob/ob mice on the C57BL6/J background develop obesity, hyperinsulinemia, and impaired glucose tolerance shortly after weaning and develop overt diabetes between 10 and 15 wk of age (227). Mice with mutations in the leptin receptor (db/db) developed this mutation on the C57BLK/SJ background and develop diabetes as early as 5 wk of age (34). Thus, in evaluating cardiac phenotypes in these models, it will be important to take into consideration the age at which the studies are performed, given the potentially confounding effects of hyperglycemia on phenotypes that are observed. Moreover, it will be important to determine which changes in these models are a consequence of obesity versus effects that are secondary to loss of leptin-mediated signaling. Another mouse model (KKAy) in which central melanocortin signaling is disrupted by the mutant agouti peptide has been used to elucidate the relationship between obesity and activation of sympathetic nervous system (10).
There are numerous transgenic mouse models that develop obesity, yet only a few have been evaluated in terms of the consequences of the obesity on cardiac function. The few models in which there are data are now discussed. The UCP-DTA mouse in which a diphtheria-toxin transgene is expressed in brown adipose tissue (BAT) resulting in BAT ablation develop a mild degree of obesity and insulin resistance that is less severe than that observed in mice with mutations in leptin or its receptor (215). Expression of 11β-hydroxysteroid dehydrogenase (11β-HSD) in adipose tissue results in a mouse with abdominal obesity that develops hypertension (225). Germ-line knockouts of the adiponectin gene have been particularly informative in terms of elucidating the interaction between adiponectin and cardiovascular injury (192, 208, 218, 226, 320, 321). The relevance of this model stems from the fact that levels of adiponectin are invariably reduced in animals and humans with obesity (161). The Jackson laboratory is currently generating new mouse mutants with obesity and cardiovascular disease by conducting random mutagenesis in mice on various genetic backgrounds. It is expected that these animals will become valuable resources in the quest for elucidating the complex interactions between obesity and cardiovascular disease (334).
Nonobese transgenic mice with mutations or transgenes that mirror critical pathways that are believed to be involved in the pathogenesis of cardiac dysfunction in obesity have been developed and will be discussed. These include mice with targeted mutations in the insulin signaling pathway as well as mice with targeted overexpression of transgenes that precipitate lipotoxic cardiomyopathy (149). Table 1 summarizes recently generated transgenic mouse models that develop lipotoxic cardiomyopathy (47, 50, 51, 101, 102, 127, 202, 203, 280, 363, 381, 382, 386). The majority of these models were generated by introducing mutations that increased fatty acid (FA) uptake to levels that exceeded the capacity of mitochondria to oxidize them. Overexpression of peroxisome proliferator-activated receptor (PPAR)-α induced lipotoxic cardiomyopathy that was worsened on diets that were high in long-chain versus medium-chain FAs, implying that lipotoxicity may be mediated primarily by intermediates derived from long-chain FAs. Although PPAR-α increased in the expression of genes that promote mitochondrial fatty acid oxidation (FAO), increased mitochondrial FAO could not accommodate the increase in FA uptake. Indeed, genetic ablation of expression of the FA transporter (CD36) reversed lipotoxicity in PPAR-α overexpressing mice (382). Mice with mutations that primarily increase lipid uptake have also revealed that specific lipid uptake pathways might mediate differential aspects of lipotoxicity-mediated cardiac dysfunction (50, 51). Two models with mutations that limit FA metabolism or triglyceride degradation are also presented. Impaired mitochondrial FAO in cardiomyocyte-restricted PPAR-δ knockout (KO) mice leads to heart failure and lipid accumulation in the absence of an increase in lipid uptake pathways, underscoring the important role of mitochondrial FAO capacity in contributing to the pathogenesis of lipotoxicity (47). Adipose tissue triglyceride lipase (ATGL) KO mice develop massive myocardial lipid accumulation presumably because of a defect in lipolysis of myocardial triglyceride leading to contractile dysfunction. The mechanism for heart failure in ATGL KO mice remains to be completely characterized, but might be due in part to disruption of contractile units by increased lipid and increased fibrosis (127). It remains to be established if reduced lipolysis of myocardial triglycerides contributes to the pathogenesis of lipotoxic cardiomyopathy in obesity.
TABLE 1.
Transgenic models of lipotoxicity
| Model | Comments |
|---|---|
| Cardiomyocyte overexpression of PPAR-α | Mice develop a lipotoxic cardiomyopathy that is associated with increased FAO, decreased glucose oxidation, and increased peroxisomal production of ROS (102). Phenotype is worsened by diets high in saturated fat, improved by diets enriched in medium-chain FAs (101), and reversed when CD36 is knocked out (382). |
| Cardiomyocyte expression of membrane-anchored LPL | Mice develop lipotoxic cardiomyopathy (381). Overexpression of apolipoprotein B in hearts reverses lipid overload and restores heart function (386). PPAR-γ but not PPAR-α agonists also reduce lipotoxicity and restore heart function (363). Most of the lipid that contributes to lipotoxicity in this model is derived from triglyceride-containing lipoprotein particles (280). |
| Cardiomyocyte overexpression of acyl CoA synthase | These mice develop lipotoxic cardiomyopathy, lipoapoptosis, and systolic dysfunction (51). Increasing leptin levels via adenoviral overexpression in livers reverses the lipotoxicity and restores cardiac function (202). α-Lipoic acid also reverses lipotoxic cardiomyopathy (203). |
| Cardiomyocyte overexpression of FATP | These mice develop lipotoxicity, increased FAO, decreased glucose oxidation but develop diastolic dysfunction with relatively preserved systolic function. Free FAs but not triglycerides are increased in these hearts (50). |
| Cardiomyocyte-restricted KO of PPAR-δ | Animals develop cardiac lipotoxicity and heart failure on the basis of a reduction in mitochondrial FAO (47). |
| Adipose tissue triglyceride lipase KO mice | Develop massive cardiac lipid accumulation because of inability to break down triglycerides (127). |
PPAR, peroxisome proliferator-activated receptor; KO, knockout; FAO, fatty acid oxidation; ROS, reactive oxygen species; FA, fatty acid.
The most commonly studied genetic rat model of obesity is the Zucker or fa/fa rat, which has a mutation in the leptin receptor. Zucker fatty rats are models of obesity, insulin resistance, and glucose intolerance that develop glucose intolerance and relatively mild hyperglycemia as they age. Zucker diabetic fatty rats (ZDF) were originally derived from Zucker fatty rats by selective inbreeding of rats that developed severe hyperglycemia. These animals have an additional defect in beta-cells that accelerates beta-cell dysfunction leading to the early onset of severe type 2 diabetes (305).
High fat feeding has been used by many groups to study the consequences of obesity in the heart, and some of these data are discussed in more detail in subsequent sections of this review. Interpretation of the impact of dietary manipulation on cardiac structure and function in animal models is confounded by differences in dietary lipid composition such as saturated versus unsaturated fats, and whether or not the high-fat diets are isocaloric or promote obesity. For example, isocaloric high-fat diets that are associated with reduced levels of insulin and leptin appear to attenuate LV hypertrophy in pressure overload hypertrophy and minimize LV remodeling in a rat infarct model (239, 261, 262). In contrast, high-fat feeding that is associated with the development of insulin resistance and obesity is associated with development of LV dysfunction in many (269, 273, 296) but not all studies (40). Studies examining the effects of altering dietary FA composition have suggested that diets with increased saturated fat content, or exposure of cultured cells to saturated FA promote apoptosis, which can be reversed by the addition of mono- or polyunsaturated FA (PUFA) (74, 212, 236, 260). Although there is broad interest in the cardiovascular benefits of dietary supplementation with n-3 and n-6 PUFA (32), most studies have focused on potential antiarrthymic effects of n-3 and n-6 PUFAs particularly in the context of myocardial ischemia, and few have specifically examined the effects on cardiac structure and function in obesity. A small number of studies have suggested that increased dietary n-3 PUFA may increase cardiac efficiency and enhance recovery following ischemia/reperfusion (230, 277) and may attenuate LV hypertrophy (LVH) and reverse LV dysfunction in a mouse model of cardiac lipid accumulation induced by systemic carnitine deficiency (juvenile visceral steatosis) by remodeling the cardiac lipid pool and reducing the activation of protein kinase C (337).
III. STRUCTURAL CHANGES IN THE HEART IN OBESITY
A. Human
1. Quantification of cardiac hypertrophy in obesity
Cardiac hypertrophy is usually defined as an increase in the size of the entire heart or more commonly of a specific cardiac chamber relative to body size. In the past, body surface area (BSA) was often chosen as the index of body size against which to judge cardiac size or mass. However, in the setting of significant obesity, LV weight/BSA is often normal or lower than normal because BSA increases more than LV weight (72, 73, 197). Because of this, many investigators now choose to index heart size to lean body mass, height, or height raised to the power of 2.7 (72, 73). The latter method is proposed to be an optimal allometric correction factor that minimizes gender differences in cardiac size and geometry (70). Commonly accepted cut-off values for increased LV mass are >50 g/m2in men and 47 g/m2.7in women (72, 197).
2. Patterns of LVH in obesity
Many published studies have concluded that obesity is independently associated with LV hypertrophy (22, 28, 63, 72, 116, 235, 242, 266, 279, 284, 377). A few studies suggest that LV mass may be increased in obesity, but that the increase is appropriate for body size if obesity is truly “uncomplicated” (i.e., lack of comorbid conditions such as hypertension, diabetes, coronary artery disease, etc.) (151, 155, 156). There are somewhat divergent views about the degree of hypertrophy and the particular LV geometric patterns that are seen in obesity. Early studies in relatively small numbers of patients suggested that obese subjects predominantly had dilated hearts (11, 13, 14, 92). These reports may have been affected by a referral bias in which patients with heart failure symptoms were selectively referred for evaluation. Thus the dilated phenotype may conceivably have been a result of comorbid conditions rather than a direct result of obesity (151, 156). More recently, several studies have prospectively evaluated larger cohorts of younger and older subjects with obesity who did not have evidence of organic cardiovascular disease (22, 151, 156, 242, 279, 377). These studies have generally relied on noninvasive imaging such as echocardiography, radionucleotide studies, or magnetic resonance imaging (MRI) to assess cardiac size, geometry, and function. The more recent data confirm the increased prevalence of LV hypertrophy in obese subjects. In addition, the results indicate that both LV cavity size and wall thickness are increased in obese subjects compared with age-matched controls (22, 134, 151, 156, 242, 279, 377). However, wall thickness is commonly increased to a greater extent than cavity size. Thus there appears to be a slight predominance of concentric cardiac hypertrophy (increased wall thickness relative to chamber size) compared with an eccentric pattern of hypertrophy (chamber enlargement is more prominent than the increase in wall thickness). The pathophysiological mechanisms proposed to account for the presence of LV hypertrophy and the different patterns of LV geometry are discussed subsequently. The pattern of hypertrophy that is present may be clinically meaningful, since accurate phenotypic characterization could help us to better understand underlying mechanisms, which might allow more specific and targeted therapeutic approaches.
3. Cardiac tissue composition in obesity
Relatively few studies have compared the biochemical and structural composition of the heart in obese and normal subjects. This is not too surprising given the large obstacles inherent in obtaining human cardiac tissue, particularly from control subjects without organic heart disease. Thus we have largely been forced to rely on data from animal models that may not accurately reflect the human condition (see below). However, autopsy series present the opportunity to study heart tissue from obese and nonobese subjects, and in general, these studies have shown cardiac hypertrophy plus a variable extent of coronary artery disease (9, 64, 91, 147, 185–188, 300). However, studies relying on autopsy may be biased towards the presence of coexisting conditions and/or unexpected causes of death.
4. Cardiac adiposity
Increased cardiac mass has been postulated to result from increased epicardial fat and fatty infiltration of the myocardium (11). It is doubtful whether excess epicardial fat should be considered as a true form of cardiac hypertrophy. However, in autopsy studies, it might be difficult to separate adherent and infiltrating fat from underlying cardiac muscle. Most imaging methodologies such as echocardiography and MRI can now allow a relatively clear separation of myocardium from fat and allow calculation of a “fat free” cardiac mass. Not surprisingly, increased epicardial fat is a common finding in severe obesity. Iacobellis and colleagues (152, 155) have argued that the amount of epicardial fat parallels the amount of visceral adipose tissue and that the amount of epicardial fat is correlated with the severity of LVH. Some authors have reported that epicardial fat may penetrate into the right ventricular (RV) free wall and cause replacement of RV myocardium (327). However, widespread use of cardiac MRI and computed tomography, particularly when evaluating for possible arrhythmogenic right ventricular dysplasia, have revealed that epicardial fat anterior to the right ventricle is extremely common (178, 342, 344). Moreover, epicardial fat commonly interpolates into the RV free wall. An example of a cardiac MRI showing a moderate accumulation of epicardial fat is seen in Figure 2. One study used MRI spectroscopy to quantify triglyceride content in human myocardium and found it to be significantly increased in obese compared with normal-weight subjects (152). This observation is supported by the findings of Peterson et al. (278) who showed that obese subjects, particularly those with insulin resistance, have increased myocardial fatty acid uptake and utilization (278). Using a different approach, Kasper et al. (170) performed endomyocardial biopsy to assess myocardial histology, and obese subjects were found to have mild myocyte hypertrophy but no evidence of abnormal collagen accumulation. In contrast, Quilliot et al. (288) reported that serum markers thought to reflect cardiac collagen turnover were increased in obese subjects. Taken together, there does not appear to be a specific pathological change in the human heart which is clearly associated with obesity, other than mild myocyte hypertrophy and perhaps intra- and extracellular fat accumulation.
FIG. 2.
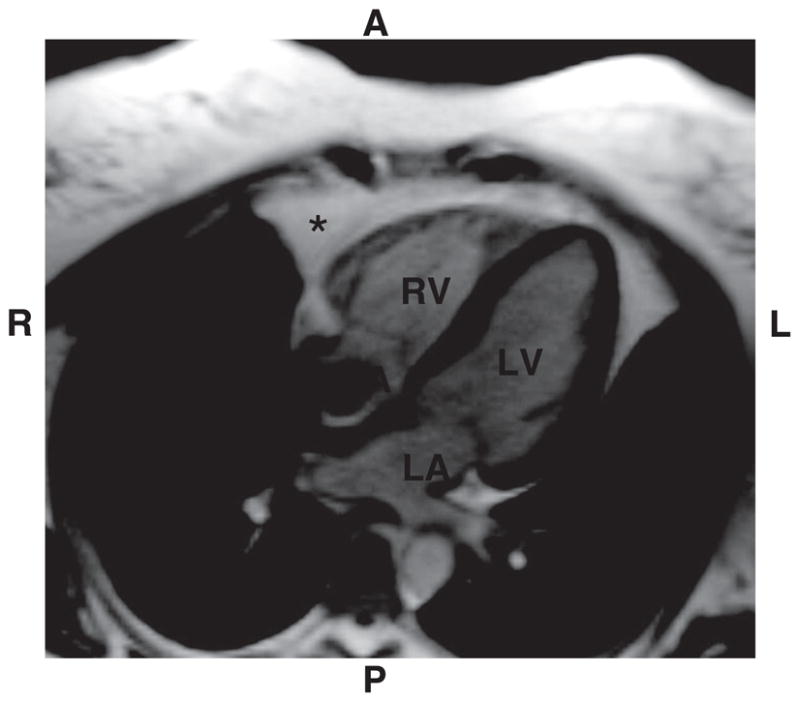
Cardiac magnetic resonance image showing epicardial fat (*) with the largest accumulation being seen anterior (A) to the right ventricle (RV). Fat appears white on this steady-state free precision imaging sequence, while myocardium is gray. Smaller amounts of fat are seen around the left ventricular (LV) free wall and in the atrioventricular groove. RA, right atrium; LA, left atrium; R, right; L, left; P, posterior. This example shows a “typical” amount of fat in an average-weight indivdual; however, the extent of epicardial fat varies widely from person to person and correlates better with the amount of visceral abdominal fat than with body mass index.
5. Right ventricular size in obesity
A few studies have focused on the right ventricle in uncomplicated obesity. Wong et al. (378) reported that RV cavity size and wall thickness were mildly increased in obese subjects compared with a normal-weight reference group. Alpert and co-workers (16, 17) reached similar conclusions. Although it seems likely that RV enlargement could result from obstructive sleep apnea and chronic pulmonary hypertension (see below), the available published data surprisingly do not consistently support this hypothesis (266, 378). In fact, in one study the majority of obese subjects did not have enough tricuspid valve regurgitation to even estimate pulmonary artery pressures, and those that did have tricuspid regurgitation generally had normal pressures (378).
6. Left atrial size in obesity
Many studies have shown increased LA dimensions in obese subjects compared with normal-weight control subjects (16, 17, 22, 140, 274, 371, 375). Unlike LV mass, LA size is usually not indexed to body size, so this finding could be misleading. The most commonly reported measure of LA size is the uniaxial anterior-posterior dimension, which is generally measured from the parasternal long axis echocardiographic view. This is a well-accepted, reproducible measure of LA size that has known clinical relevance with respect to long-term event rate, survival, and the risk of developing atrial fibrillation (114, 180). The mechanisms of increased LA size appear to be similar to those causing LVH: increasing BMI, hypertension, volume overload, and possibly LV diastolic filling abnormalities. Interestingly, obese subjects in the Framingham heart study were found to have an increased risk of developing atrial fibrillation, and this risk was entirely explained by the increase in LA size (371).
7. Valvular heart disease in obesity
Only limited data exist regarding the direct effects of obesity on the heart valves. Nevertheless, the topic of valvular disease in obesity has received increased attention in recent years because of the finding that anorexigenic drugs used to facilitate weight loss are associated with mitral and aortic valve regurgitation (57). Somewhat counterintuitively, a widely cited paper that evaluated the frequency of valvular abnormalities in a relatively large cohort of subjects undergoing echocardiography showed a lower prevalence of valvular regurgitation in obese than in normal-weight subjects (323). The problem of difficult echocardiographic imaging windows in obese subjects complicates the quantitative assessment of valvular disease in this population.
B. Animal
1. LVH
LVH has often been observed in most models reported to date, such as mice and rats (to be discussed in more detail below) as well as rabbits (38) and dogs (364). Within a given species there are strain differences and age-dependent effects. For example, while some groups have reported cardiac hypertrophy in hearts of young ob/ob mice (30, 34, 227), others have only observed this phenotype in older animals (23, 24). Similarly, cardiac hypertrophy has been reported in old but not in young db/db mice (23, 24). Likewise, high-fat feeding of Sprague-Dawley rats for up to 14 wk has not been associated with LVH despite the development of obesity (40, 299), whereas 7 wk of high-fat feeding in Wistar rats results in LVH (269). Also, isocaloric high-fat diets that do not lead to hyperinsulinemia or weight gain seem to attenuate pressure overload LVH (261, 262). The pathogenesis of cardiac hypertrophy is complex and may involve the presence of coexisting hypertension, plasma volume expansion, and activation of the sympathetic nervous system. In models with leptin deficiency, there are data emerging that leptin may have independent effects on cardiac hypertrophy, and this is discussed further in section VIC. Other cytokines that play a role in energy expenditure or appetite regulation may also modulate cardiac hypertrophy. For example, activation of ciliary neurotrophic factor receptors induces regression of cardiac hypertrophy in ob/ob and db/db mice, independently of its effects on weight loss (294), and adiponectin mediates antihypertrophic effects in the heart in part through activation of AMPK signaling (320).
2. Lipid accumulation
A commonly described feature of most animal models of obesity is increased accumulation of intramyocellular triglycerides in the heart. This has been reported in the hearts of genetic models such as ob/ob and db/db mice (29, 31, 34, 53), Zucker or fa/fa rats (394), and following high-fat feeding (346). Accumulation of myocardial triglyceride and lipid metabolites such as ceramides has been associated with cardiomyocyte apoptosis in Zucker rats, and reduced levels correlate with reduced apoptosis following thiazolidinedione (TZD) treatment (394). Moreover, a diet that was high in saturated fat was associated with increased triglyceride and ceramide concentrations that correlated with an increase in apoptosis (260). There is no evidence that triglyceride accumulation per se is damaging and may represent an adaptive mechanism to sequester increased lipids, thereby representing a marker but not necessarily a mediator of lipotoxicity (214). Although there is much evidence that increased concentrations of ceramide may mediate apoptosis in the heart in response to ischemia or drugs or cytokines that promote apoptosis, will promote cytochrome c release and apoptosis when directly applied to cardiomyocytes, and that inhibition of de novo ceramide synthesis prevents palmitate-induced apoptosis in L6 myotubes (75, 82, 276, 355, 368), it remains to be established if ceramides induce apoptosis in lipotoxic heart disease. Evidence exists that increased palmitate may mediate apoptosis via ceramide-independent mechanisms despite the associated increase in ceramide content, suggesting the existence of other lipid mediators of lipoapoptosis (184, 213). The existence of mouse mutants with impaired ceramide biosynthesis and the availability of pharmacological inhibitors of ceramide biosynthesis that can be safely administered to animals (142) now provide the opportunity to clarify the role of ceramide accumulation in mediating lipoapoptosis that characterizes lipotoxic cardiomyopathies.
3. Fibrosis
Increased interstitial fibrosis has been described in the hearts of Zucker (fa/fa) rats (350, 394) and in UCP-DTA mice (54). In ob/ob mice, fibrosis occurs mainly in the coronary perivascular region (391, 392). The fibrosis is correlated with increased expression of plasminogen activator inhibitor 1 (PAI-1), increased levels of transforming growth factor (TGF)-β, and increased activation of stress signaling pathways such as Jun NH2-terminal kinase (JNK). It is likely that the fibrotic response is due in part to local activation of the renin-angiotensin system as treatment of animals with angiotensin converting enzyme inhibitors (ACEI) or angiotensin II receptor blockers (ARBs) result in regression of fibrosis and normalization of PAI-1 and TGF-β levels. Fibrotic responses may also ensue as a result of replacement fibrosis following apoptotic cardiomyocyte death.
IV. FUNCTIONAL CHANGES IN THE HEART IN OBESITY
A. Human
1. LV systolic function
Many studies have evaluated LV systolic function in obesity. The findings of these studies are quite variable. Various authors have reported depressed LV ejection fraction (EF) (15, 68, 100), normal EF (22, 111, 116, 191, 242, 266, 279, 377), and supernormal EF (28, 155, 274, 375) in obese subjects. The findings of the various studies likely depend to some extent on the population being evaluated. The technique used for measuring EF seems to matter less since similar findings have been obtained with echocardiography, nuclear imaging, and cardiac MRI. As mentioned in the preceding discussions, it is always a challenge to separate out the effects of obesity from the effects of comorbidities such as hypertension, diabetes, and vascular disease. All of the latter are important causes of LV dysfunction.
The early reports of cardiac enlargement and systolic dysfunction led to the development of a notion that there was a specific “cardiomyopathy of obesity” (11). More recently, however, several studies have found that LV ejection fraction is normal or even supranormal in the majority of obese subjects, even those with severe obesity (22, 28, 155, 191, 242, 274, 279, 375, 377). These results need to be interpreted with caution since increased endocardial shortening is a common finding in concentric LVH due to the relatively enhanced excursion of the endocardium in a thick-walled ventricle (21, 71). Thus, even if the EF is normal, myocardial function is often reduced when it is measured with more sensitive methods such as midwall LV fractional shortening, systolic velocity measured with tissue Doppler, or systolic strain rate. Indeed, obese subjects have generally been found to have subclinical contractile abnormalities when assessed with the aforementioned techniques (21, 22, 71, 279, 377). Invasive studies have also led to the conclusion that myocardial contractility is reduced (111). Interestingly, LVH and mild abnormalities of myocardial systolic function have been observed in obese children and adolescents (48, 367), and the severity of the dysfunction is comparable to that seen in obese subjects in their 30s, 40s, and 50s. Thus it is unclear whether the relatively mild alterations of myocardial systolic function progress with longer durations of obesity. Alpert et al. (14) have argued that duration of obesity is the factor that determines the likelihood of developing systolic dysfunction and heart failure. Although this is an attractive hypothesis, not all studies have found such a relationship (377). Moreover, no longitudinal studies in obese subjects are available to delineate the natural history of the contractile abnormalities in obesity. Lastly, obesity in older individuals appears to pose less of a mortality risk than it does in younger subjects (367). In summary, the jury is still out on whether long-term obesity leads to heart failure independent of coronary disease or other morbidities.
2. LV diastolic function
Several studies have also assessed LV diastolic filling and diastolic function in obesity. As with the findings in systolic function, results are variable and somewhat conflicting. Mitral inflow velocities measured with pulsed-wave Doppler give information about LV relaxation rate and atrial contractile strength, but the effects of altered loading conditions (i.e., LA pressure) tend to dominate over the effects of LV relaxation abnormalities. Some studies have found reduced early diastolic (E-wave) velocities (155) while others have found them to be unchanged (242, 266, 274, 279, 314). Similarly, E-wave deceleration times have been reported to be increased (274) or unchanged (279) in obese subjects. Lastly, late diastolic (A-wave) velocities have been reported to be increased (28, 155, 242) or unchanged (266, 274, 279, 314). Prolongation of the isovolumic relaxation time is probably the most consistent diastolic abnormality seen in obesity (28, 242, 375). Because increased age predictably is associated with reduced E-velocity, prolonged E-deceleration time, and increased A-velocity, controlling for age is mandatory in the assessment of mitral flow patterns.
Over the last decade, echocardiographic techniques have been developed that allow us to quantify relatively load-independent indexes of myocardial diastolic function (141, 245). These are based mainly on tissue Doppler imaging, a robust method of recording the velocity and amplitude of myocardial movements with high temporal resolution. From the parent tissue Doppler velocities, strain and strain rate at different locations in the heart can be derived. Using these approaches, at least two groups have reported evidence of reduced early diastolic tissue velocities and diastolic strain rate in obese compared with normal-weight subjects (279, 377). It is believed that decreases in these parameters predominantly reflect slowing in the rate of LV relaxation. Recently, speckle tracking techniques have been added to the ultrasound-based methods for assessing myocardial function, but they have not yet been studied in obesity.
Limited studies are available that compare intracardiac pressures in obese and normal-weight subjects. Resting pulmonary capillary wedge pressures were found to be normal in obese subjects (165). However, compared with normal-weight control subjects, the obese subjects had an exaggerated rise in wedge pressure during passive leg raising or during exercise. These data were interpreted as showing reduced distensibility of the central circulation. Noninvasive methods have also been used to estimate resting LV filling pressures in obese subjects. The ratio of the mitral E-wave velocity to the early diastolic mitral annular tissue velocity (E′) is a well validated index of pulmonary capillary wedge pressure (87, 264). E/E′ measured at rest has been reported to be normal (<10) in obese subjects (377). Serum levels of brain natriuretic peptide (BNP) are widely used in the clinical evaluation of patients with known or suspected heart failure. Elevated levels of this hormone are indicative of volume overload and higher LV filling pressures. Interestingly, obese subjects have lower levels of serum BNP than normal-weight controls with similar pulmonary capillary wedge pressure (232). Moreover, BNP levels are consistently reported to be in the normal range or below normal in the majority of obese subjects (66). Thus the bulk of evidence points to the conclusion that even though obesity is associated with diastolic dysfunction at the myocardial level, LV filling pressures remain normal at rest. The vast majority of obese patients, even those with severe obesity, do not have clinical heart failure.
3. RV function
One recent study found that RV ejection fraction was not altered in obese subjects (378). However, those with BMI >35 kg/m2 had reduced RV function compared with a reference population as evidenced by reduced systolic tissue Doppler velocities, and reduced measures of systolic strain and strain rate. Surprisingly, these changes occurred irrespective of the presence of sleep apnea. Similar but lesser degrees of reduced systolic function were present in overweight and mildly obese groups. Differences in RV diastolic velocities were also seen in obese versus a reference population. BMI remained independently related to RV changes after adjusting for age, log insulin, and mean arterial pressures. In obese patients, these changes were associated with reduced exercise capacity but not the duration of obesity or the severity of sleep apnea. Two other studies showed preserved RV systolic function in obesity (137). However, one of these did find altered RV diastolic filling characteristics (266).
4. Vascular function
The topic of vascular dysfunction in obesity is large, and we will not attempt to review it in any detail in this paper but will refer the reader to a comprehensive recent review in this area (247). Suffice it to say that obesity, especially abdominal obesity, is a well-defined risk factor for the development of atherosclerotic coronary artery disease (12, 188, 241). However, since abdominal obesity is part of the diagnostic criteria for the metabolic syndrome, it is nearly impossible to dissect out the independent contributions of the different components of this syndrome. In addition, endothelial dysfunction is widely present in obesity and the metabolic syndrome (84, 325, 358). Both macro- and microvascular abnormalities likely contribute to the many structural and functional problems of the heart in obesity.
B. Animal
Obesity is associated with alterations in cardiac function, and in animal models, studies have been performed 1) in vivo, 2) in isolated perfused hearts or in papillary muscles, or 3) in vitro using isolated cardiomyocyte preparations. Furthermore, studies have been performed under normoxic conditions or following ischemia. This section will focus primarily on studies performed in obese animal models. Transgenic models designed to isolate abnormalities of various pathways that are hypothesized to be involved in obesity-related pathologies are discussed in sections II and VC.
1. In vivo studies
Echocardiography has been the most widely used modality to investigate cardiac function in murine models of obesity-related cardiac dysfunction. A smaller number of studies have also performed invasive hemodynamic assessment of LV function or used cardiac MRI. In most studies in vivo cardiac function has been evaluated in anesthetized animals. Anesthetic agents vary from study to study and may have variable effects on heart rate, which could influence echocardiographic findings. Generally, most studies reveal evidence of mildly impaired systolic function in vivo that is absent in younger animals (<12 wk of age) but become evident between 12 and 20 wk of age (34, 53, 313, 357, 361, 388). Fewer studies reveal changes in diastolic function (53, 313). Ob/ob mice on the LDLR null background have reduced functional shortening (FS) by echocardiography at 24 wk of age. These animals are hypercholesterolemic and develop atherosclerosis. In addition, there is a loss of the normal diurnal variation in heart rate and blood pressure. Caloric restriction leads to a 45% reduction in body weight, reduced atherosclerosis, improved glucose homeostasis, reduced markers of inflammation, and normalization of cardiac function (361). These studies suggest that many of the associated changes in cardiac function are dependent on the effects that may be secondary to obesity and glucose intolerance as opposed to leptin deficiency per se. Similar beneficial effects were also reported following treatment with a dual PPAR-α/-γ agonist (362). In 10- to 11-wk-old female ob/ob mice, Christoffersen et al. (53) reported minimal changes in LV systolic function under baseline conditions but a reduction in the inotropic response to dobutamine. They reported evidence of diastolic dysfunction manifested by reduced E/A ratios (53). Similar findings have been reported in db/db mice with evidence of systolic and diastolic function by echocardiography that was absent in 6-wk-old (shortly after onset of hyperglycemia) but present in 12-wk-old mice (313). The study of ob/ob mice was performed under anesthesia, which reduced heart rates to between 300 and 400/min, whereas the db/db mouse studies were performed in conscious mice at heart rates of 600–700/min.
In a study of ob/ob and db/db mice in which cardiac function was evaluated using invasive LV catheterization, Buchanan et al. (34) reported that LV function differed by strain and varied with age. In younger mice of both strains, rates of LV contraction and relaxation (dP/dt) were actually increased in young animals (4–5 wk of age) despite the presence of significant obesity. Moreover, LV systolic pressures were increased, without any significant differences in LV end-diastolic pressures. In 15-wk-old ob/ob mice, the increase in dP/dt remained and LV end-diastolic pressure did not increase. Thus these studies do not support the existence of significant in vivo diastolic dysfunction in ob/ob animals. In contrast, db/db mice that develop a much earlier onset of hyperglycemia exhibited a progressive decline in indices of LV contractility so that by 15 wk of age +dP/dt were equivalent to controls and −dP/dt were reduced (34). The mechanisms for the hypercontractile phenotype in younger ob/ob and db/db mice are most likely the result of load-dependent variables such as preload and afterload, as was recently shown by Van den Bergh et al. (357), who also demonstrated load-independent cardiac dysfunction in db/db mice that developed in 24-wk-old but not in 12-wk-old animals. These data are consistent with a progressive reduction in cardiac function, which may represent the cumulative effects of persistent hyperglycemia, insulin resistance, and increased myocardial lipid supply.
In vivo changes in cardiac function in mouse models with lesser degrees of obesity are more subtle. Thus C57BL6 mice when placed on a high-fat diet develop LV dysfunction as evidenced by reduced fractional shortening only after 20 wk of high-fat feeding (273). UCP-DTA mice are a model of the insulin resistance syndrome with lesser degrees of obesity and insulin resistance than leptin-deficient mouse models. These mice have a modest increase in blood pressure, eccentric LVH (with increased LV chamber size), and increased cardiac output (54).
Studies in rats have shown a similar pattern to mouse studies. In genetically obese Zucker fatty rats, LVH is present in young animals shortly after the development of diabetes. This is associated with increased LV function by echocardiography (104); however, they eventually develop significant LV dysfunction in vivo as evidenced by decreased fractional shortening. This can be reversed with TZD treatment (394). The beneficial effect of TZD treatment was ascribed to the reversal of lipid accumulation and prevention of lipotoxicity (lipid accumulation and subsequent apoptosis). Studies in high-fat fed rats have not revealed such striking findings. Thus 12 wk of high-fat feeding of Sprague-Dawley rats revealed no changes in invasive LV hemodynamics, e.g., dP/dt, echocardiographic parameters, LV hypertrophy, or LV fibrosis despite significant obesity and dyslipidemia (40). A similar lack of LVH was observed independently by another group following a 14-wk high-fat diet in Sprague-Dawley rats (299).
2. Studies in isolated hearts
These studies have revealed subtle changes in cardiac function at time points when significant changes are not apparent when evaluated in vivo. Examination of cardiac function in isolated working db/db mouse hearts revealed significant reduction in cardiac power despite preserved in vivo cardiac function (34). Similarly, when perfused with 5 mM glucose and 0.4 or 1.0 mM palmitate, significant reductions in cardiac performance were also observed in ob/ob mouse hearts (227). In isovolumic ob/ob hearts, although cardiac performance was similar at low work loads, there was a significant reduction at increased work loads (30). In Langendorff-perfused hearts from fa/fa (Zucker) rats prior to the onset of diabetes, cardiac function was also depressed (369), and in rats studied after the onset of diabetes, increased FA utilization and decreased glucose and lactate utilization were observed (370). Treatment of Zucker rats with a TZD improved systemic glucose homeostasis and intracardiac triglyceride accumulation, increased glucose oxidation rates, and improved cardiac function (115). Similar studies in db/db mice revealed that treatment of these mice with PPAR-γ or PPAR-α agonists can improve cardiac metabolism, but do not necessarily restore cardiac function (1, 2, 36).
3. Studies in isolated cardiomyocytes or papillary muscles
Cardiomyocyte or papillary muscle function has been examined in Zucker rats, ob/ob mice, and models of diet-induced obesity. In ob/ob mice, cardiomyocyte contractile function is reduced and is associated with reduced calcium sensitivity and altered calcium transients (88, 207). In addition, others have shown that there is reduced β-adrenergic responsiveness in cardiomyocytes isolated from ob/ob mice (238). Ob/ob cardiomyocytes are also more susceptible to nitric oxide (NO)-mediated depression in contractility relative to nonobese controls (330). Treatment of Wistar rats with a high-fat diet for 7 wk was associated with LV hypertrophy triglyceride accumulation and insulin resistance. Papillary muscles revealed an increase in baseline and maximal force generation, but impaired recovery from high work loads, impaired insulin-mediated augmentation of calcium-mediated contractility, and reduced insulin-mediated phosphorylation of phospholamban (269).
4. Normoxia versus ischemia
Given the possible association between obesity, insulin resistance, and increased mortality following cardiac ischemia, many groups have examined the recovery of hearts from rodent models of obesity following ischemia/reperfusion in vitro and in vivo. Findings have been somewhat variable depending on the model examined. Thus isolated working hearts from db/db mice exhibit reduced recovery following ischemia and reperfusion after diabetes develops, but recovery is normal in the pre-diabetic stage (3). Treatment with PPAR-γ or PPAR-α agonists while improving myocardial substrate utilization did not increase postischemic recovery in db/db hearts (3, 36), but acute treatment with high concentrations of glucose and insulin, which substantially increased glucose oxidation rates, increased postischemic recovery and cardiac efficiency (90). In UCP-DTA mice, functional recovery following low-flow ischemia was impaired (54). In fa/fa rats studied very shortly after the onset of hyperglycemia, ischemia was associated with increased ischemic contracture, but functional recovery following reperfusion was enhanced in the obese mice despite the fact that at baseline, cardiac function was reduced (369). The mechanism for cardioprotection is unclear. In contrast, studies in older Zucker rats demonstrated reduced recovery from ischemia, which could be prevented by treating these animals with a TZD (159, 322, 389). Studies in nondiabetic rats have also shown a positive benefit of TZD treatment upon ischemia/reperfusion (158, 176, 390), although this has not been shown to be true for other models such as pigs (380). Studies performed in vivo using coronary artery ligation show that obesity and insulin resistance are associated with impaired recovery of cardiac function. This is true in genetic models such as Zucker rats, db/db mice, and mice with diet-induced obesity (119, 146, 346). Infarct size is unchanged in high-fat fed models (347) but increased in db/db mice (160). The existence of impaired leptin signaling in the hearts of many models of obesity has prompted some investigators to evaluate the role of leptin in mediating cardioprotection in the context of ischemia and reperfusion. Thus leptin administration has been shown to reduce infarct size and enhance myocardial functional recovery following coronary artery ligation in mice (4, 326).
Taken together, these studies provide strong evidence for an association between obesity and disturbances in cardiac function. Earliest changes are seen in isolated perfused hearts and precede detectable changes in vivo. One reason for this is the presence of hemodynamic compensations such as changes in preload or afterload, which might mask intrinsic contractile defects. The extent of dysfunction in vivo or in vitro becomes more evident as the duration of the obesity increases, or following an ischemic insult.
C. Comparison Between Animal and Human Data
It is a challenge to directly compare the findings of human compared with animal studies. Generally, the abnormalities of cardiac structure and function uncovered in animal studies tend to be significantly more pronounced than those in human studies. This may result from the fact that the specific genetic abnormalities present in many of the animal models are not seen in most humans with obesity. For example, leptin deficiency or absence of functional leptin receptors is the basis of obesity in the widely studied ob/ob and db/db mice, whereas leptin deficiency is an extremely rare cause of obesity in humans. To further emphasize this point, leptin therapy has been associated with marked reversal of the obesity phenotype in ob/ob mice, whereas administration of leptin has unfortunately been largely ineffective in treating human obesity (27). With the use of similar logic, it is not surprising that complete knockout or marked overexpression of a specific target in a mouse may produce a more exaggerated phenotype than is seen in the less dramatic polygenic/environmental causes of obesity that are usually present in humans. The environmental impact on heart failure in humans is another factor that is difficult to mimic in laboratory animals. Lastly, end-organ effects of obesity that require a long duration to become evident are much easier to study and observe in small rodents with a life span of 1–2 years than in humans with a much longer life span. Thus many cardiac findings may be more apparent in the animal models of obesity than in humans. Despite these caveats, the wealth of data that can be derived from animal studies allows for “proof of concept” experiments that are often impossible to perform in humans. Thus the two lines of investigation are felt to be synergistic, even though establishing disease pathogenesis and final proof of the therapeutic benefits of any treatment must ultimately be obtained in clinical studies.
V. MECHANISMS CONTRIBUTING TO STRUCTURAL AND FUNCTIONAL CHANGES IN THE HEART IN OBESITY
A. Changes in Cardiac Metabolism
It has long been known that diabetes is associated with shifts in myocardial substrate utilization characterized by an increase in FA utilization and a decrease in glucose utilization (37, 315). Only recently have similar studies been performed in genetic models of obesity, prior to the onset of significant hyperglycemia. We have evaluated myocardial substrate utilization in ob/ob and db/db mouse hearts prior to the onset of hyperglycemia. In both instances we observed changes in myocardial substrate utilization that mimic the changes that occur in diabetes. Indeed, reduced glucose oxidation rates and increased FA oxidation rates were present in 4-wk-old obese mice (34). The reduction in glucose oxidation did not decline any further as diabetes developed in these models, although FA oxidation rates increased further. Changes in substrate metabolism seem to be the earliest measurable abnormality in the hearts of these mice and precede measurable changes in in vivo cardiac function (see sect. IVB) and in isolated hearts (34), thereby suggesting that altered myocardial substrate utilization might be a critical mediator of subsequent contractile dysfunction. Ob/ob mice also have increased capacity to oxidize FA in response to increasing delivery of FA substrates, which exceeds that of wild-type hearts (227). The mechanisms responsible for the substrate switching that characterizes the diabetic heart likely involve increased delivery of fatty acids, decreased insulin signaling, and activation of transcriptional pathways such as the PPAR-α/peroxisome proliferator-activated receptor-γ coactivator (PGC-1) signaling network that regulate myocardial substrate utilization. In studies of young ob/ob and db/db mice, changes in substrate utilization were not associated with any increase in the expression of PPAR-α, PGC-1, or its transcriptional targets (34). In a study of high-fat feeding, a reduction in myocardial glucose utilization was noted as early as 10 days after the initiation of high-fat feeding, and the high-fat fed animals had evidence of reduced insulin signaling (273). Thus early in the course of obesity, it is likely that changes in myocardial substrate utilization reflect changes in substrate availability, such as increased myocardial delivery of fatty acids and triglycerides. The reciprocal reduction in glucose utilization likely reflects allosteric inhibition of glucose utilization in the face of an increase in FA utilization (Randle phenomena). It is also possible that impaired insulin signaling may have independent effects to reduce myocardial glucose utilization as was observed in the hearts of mice with cardiomyocyte-restricted deletion of insulin receptors (26). As caloric excess and/or obesity becomes more long-standing, activation of transcriptional pathways, namely, activation of PPAR-α/PGC-1 mediated signaling increases the expression of genes involved in FAO and FA import such as CPT1, LCAD, MCAD, and FA transporters such as FATP1 and CD36, which contributes further to the metabolic changes in these hearts. A second mechanism that may contribute to increased myocardial FA uptake in obesity is redistribution of CD36 to the plasma membrane (59).
An important recent contribution to the field is the observation that alterations in myocardial substrate utilization in mouse models of obesity are associated with increased myocardial oxygen consumption and decreased cardiac efficiency (30, 31, 34, 148, 227). Potential mechanisms for reduced cardiac efficiency will be discussed in the section on mitochondria (sect. VB). The implication for reduced cardiac efficiency in these hearts is the potential consequence of a limitation in myocardial reserve, which may increase the likelihood of worsening of cardiac function in the face of hemodynamic stressors such as cardiac hypertrophy. In Zucker rats, a reduction in glucose (and lactate) metabolism has also been described prior to the onset of significant hyperglycemia (115, 370). Changes in FA utilization in Zucker rats relative to observations in mouse models are distinct. Zucker rats appear to have a defect in their ability to upregulate FA oxidative capacity in response to increasing FA delivery that has been postulated to contribute to accumulation of myocardial triglycerides and lipotoxicity (316, 387). Recent studies in obese humans have yielded results that mirror the changes described in mice. In a study of severely obese females, Peterson et al. (278) reported that obesity was associated with increased rates of FA oxidation, increased myocardial oxygen consumption, and reduced cardiac efficiency, and the extent of these changes was proportionate to the degree of insulin resistance or obesity. Despite the increase in myocardial FAO in mice and humans with obesity, there is an increase in the triglyceride accumulation within the heart. Many animal studies have demonstrated this, and recent studies in humans using either NMR spectroscopy or lipid staining of postmortem tissue have provided strong evidence for the existence of lipid accumulation in human hearts (228, 316, 336). It is postulated that an imbalance between FA oxidation and lipid uptake exists in the heart in obesity, with uptake exceeding oxidation. This has also been supported by studies in transgenic mouse models of lipotoxicity (see Table 1). Examination of substrate utilization has been performed in some but not all of these models and has demonstrated the existence of increased rates of FA oxidation that occur within the heart, despite increased intramyocardial triglyceride accumulation. Impaired mitochondrial function may contribute in part to the inability of the heart to oxidize the increased lipid burden and will be discussed further below.
B. Mitochondrial Dysfunction and Oxidative Stress
Obesity is associated with changes in mitochondrial morphology. Increased mitochondrial number has been described in the hearts of ob/ob mice (29, 88), db/db mice (31), and UCP-DTA mice (93). In UCP-DTA mice, the increase in mitochondrial number is due to upregulation of PGC-1α and PPAR-α signaling and is blocked by the introduction of the PPAR-α null allele. Despite the increase in number, there is accumulating evidence that mitochondrial dysfunction exists. Studies in ob/ob and db/db mice revealed two distinct defects (30, 31). In hearts that were perfused with glucose prior to analysis, mitochondria were characterized by reduced rates of oxygen consumption and ATP generation. Analysis of protein content of OXPHOS subunits in ob/ob mice revealed significant reduction in protein levels of mitochondrial complexes I, III, and V, and in db/db mice, although there was an increase in the expression of PGC1-α and genes that regulate FAO, there was not a coordinate increase in OXPHOS gene expression. Thus mitochondria isolated from ob/ob and db/db mice exhibit striking defects in mitochondrial oxidative capacity. When mitochondria were studied following perfusion of hearts with fatty acids, mitochondrial oxygen consumption increased, but ATP production declined further. Reduced ATP/O ratios are consistent with mitochondrial uncoupling, which was confirmed by measuring proton leak kinetics (31). In these studies there was no increase in the protein content of uncoupling protein 3 (UCP3), but evidence was obtained that mitochondrial uncoupling proteins were activated in these hearts. Mechanisms that could activate uncoupling include increased exposure of mitochondria to fatty acids and increased generation of superoxides (29, 31). Increased levels of UCP3 have been reported in db/db mice (249), suggesting that activation as well as increased expression of uncoupling proteins both contribute to mitochondrial uncoupling in db/db mouse hearts. A recent report in UCP-DTA mice, which is a model of insulin resistance and mild obesity, revealed reduced ATP/O ratios, which are also consistent with mitochondrial uncoupling (93). Uncoupled mitochondria exhibit increased oxygen consumption rates. Moreover, FA utilization is increased in uncoupled mitochondria. Thus uncoupling of mitochondria represents a novel mechanism contributing to changes in substrate utilization and reduction in cardiac efficiency that exists in the heart in obesity. Mitochondrial proliferation and dysfunction have also been described in the heart in animal models with type I diabetes (318, 319, 353, 354), indicating that obesity and diabetes (both of which are associated with increased delivery of FA to the myocardium) ultimately lead to mitochondrial alterations that ultimately become mal-adaptive. Given the high rates of energy utilization within the heart, it is reasonable to hypothesize that mitochondrial dysfunction and impaired myocardial energetics may contribute to contractile dysfunction in the heart of obese individuals and increase their susceptibility to heart failure. Studies in humans with type 2 diabetes who do not have underlying coronary artery disease demonstrating reductions on PCr/ATP ratios support this hypothesis (83, 310).
Mitochondria also represent an important source of superoxide in cells. There are a number of reports indicating increased indices of oxidative stress markers in the myocardium in obesity (110, 207, 308, 361). We recently demonstrated that mitochondrial ROS production is increased in the hearts of db/db mice and is generated in complex I and possibly in complex III (31). Nonmitochondrial mechanisms may also contribute to increased oxidative stress in the hearts of models of obesity. Suggested mechanisms include reduced NOS1 activity and increased xanthine oxidoreductase activity in ob/ob mice that are normalized with leptin treatment and increased oxidative modifications of SERCA which is reversed following exposure of ob/ob cardiomyocytes to inhibitors of NADPH oxidase (207, 308).
C. Impaired Insulin Signaling
It is increasingly accepted that insulin signaling is impaired in the heart in various rodent models of obesity and insulin resistance (5, 6). Impaired insulin-mediated activation of intracellular signaling molecules or glucose uptake have been described in ob/ob mouse hearts and cardiomyocytes (227), db/db mouse cardiomyocytes (41), hearts of Zucker fatty rats (52, 133, 322, 389), Goto-Kakizaki (GK) rats (81), and mice and rats with diet-induced obesity (273). Impaired insulin signaling occurs within 2 wk of high-fat feeding and therefore represents an early adaptation of the heart to caloric excess. The consequences of acquired insulin resistance are not completely understood. However, there is evidence that reversal of myocardial insulin resistance with pharmacological insulin sensitizers will increase myocardial glucose utilization, decrease FA utilization, and reduce injury and enhance recovery following myocardial ischemia (322, 389).
Using knockout techniques in mice, insulin receptors have been selectively deleted from cardiomyocytes (26). These animals represent a genetic model of myocardial insulin resistance. Although complete loss of insulin signaling might not necessarily recapitulate the insulin resistance that develops in the heart in obesity, the model has been informative in highlighting important roles of insulin signaling in the heart. Specifically, deletion of insulin receptors in the heart is associated with reduced rates of FA and glucose oxidation and reduced expression of FAO genes, consistent with mitochondrial dysfunction (26). Insulin receptor-deficient hearts also have increased susceptibility to injury when exposed to hypertrophic stimuli such as pressure overload and chronic adrenergic stimulation with isoproterenol (150, 231). Studies in isoproterenol-treated CIRKO hearts also highlighted the important role of insulin signaling in promoting angiogenesis in the heart (231). Impaired angiogenic signaling was also independently confirmed in the hearts of Zucker rats and in muscle-restricted insulin receptor KO mice that also lack insulin receptor expression in the myocardium (133, 349).
D. Diabetes (Hyperglycemia)
The development of insulin resistance and type 2 diabetes in obese individuals (163, 219), the significant contribution of diabetes to cardiovascular disease (234, 255, 352), and potential mechanisms by which diabetes may lead to cardiac dysfunction (31) have been extensively reviewed. Nevertheless, the precise role of diabetes in producing structural changes in the adult heart is quite controversial. As with many aspects of cardiovascular disease in obesity, it is a challenge to separate the direct effects of obesity from its frequent traveling companions. The presence of diabetes, or measures of insulin resistance, was not predictive of LV mass in the mainly Caucasian, middle-aged women studies by Avelar et al. (22). In other populations, such as those in the STRONG heart study (native Americans of the western United States) and the Framingham heart study, diabetes or the metabolic syndrome do appear to be significant risk factors for LV hypertrophy (272, 306, 358). In the population involved in the STRONG heart study, diabetes is almost universal and may have a genetic underpinning. Thus it is likely that multiple predisposing factors for LV hypertrophy and genetic modifiers may summate in a complex and nonlinear fashion that produces different end effects, depending on the population under study. Advanced glycation end products (AGE) mediate the detrimental influence of hyperglycemia on many diabetic complications, and Laakso’s group (177) conducted a comprehensive 18-yr follow-up study in Finnish subjects which was the first to demonstrate that AGE can predict total, cardiovascular disease and coronary heart disease mortality in nondiabetic women. Furthermore, it has been suggested that in cardiomyocytes AGEs regulate the response to ischemia-reperfusion injury via modulation of cardiac energy metabolism (33). Ren’s group recently demonstrated that a 12-wk high-fat diet in rats induced obesity and elevated serum AGE levels which correlated with increased O-Glc-NAc modifications and apoptosis in cardiomyocytes (205) in a similar manner to the apoptosis induced by hyperglycemia (206). Many of the changes in cardiac metabolism, discussed in section VA, that have recently been described in obesity, were long recognized to occur in animals and humans with type1 and type 2 diabetes. Thus the development of diabetes in individuals with obesity is likely to have synergistic and deleterious effects on cardiac metabolism and function.
E. Inflammation
There is ever-increasing interest in the link between obesity and inflammation (78, 80, 204, 221), and an inflammatory state is both a causative factor in inducing MI and a primary constituent of the response to MI (210, 341, 385). Several studies have established clear correlations between markers of inflammation and mortality in patients with heart failure (157, 246). C-reactive protein, a heavily studied serum marker of inflammation, may serve as a useful prognostic tool and as a marker of long-term mortality in patients with acute MI (329, 332). It appears that a disparity in the inflammatory cytokine system occurs in obese individuals with heart failure since, while pro-inflammatory cytokines such as interleukin (IL)-6, IL-1β, atrial natriuretic peptide (ANP), and tumor necrosis factor (TNF)-α increase, there is not a corresponding increase in anti-inflammatory cytokines such as IL-10 and TGF-β (122, 366). The degree and nature of inflammation may influence further progression of disease via both direct myocardial effects and on other components of the cardiovascular system. There are now many reports indicating that the various proinflammatory cytokines can play a role in the myocardial remodeling process by directly influencing aspects such as hypertrophy, apoptosis, fibrosis, and ultimately contractility (20, 138). Despite the potential promise of targeting inflammation, studies to date employing anti-TNF-α therapeutic approaches have thus far proven ineffective in treating heart failure. This has somewhat tempered enthusiasm regarding the possibility of targeting the imbalance in inflammatory cytokines, although other aspects of the cytokine imbalance besides TNF-α may still prove to be effective targets for therapeutic interventions (19, 122).
F. Pressure/Volume Overload
Hypertension is highly prevalent in obese subjects (>60%), and it is probably the most common cause of LV hypertrophy in the general population. Typically, we think of pressure overload as causing a concentric pattern of hypertrophy (120). Given the relatively high frequency of concentric LV geometry in obese patients, there are compelling reasons to believe that hypertension plays a major role. Even if daytime or office blood pressures are normal, 24-h blood pressure recordings show that obese patients often have a “nondipping” pattern in which they lose the normal nocturnal decline in blood pressure (190). Sleep-disordered breathing may explain this phenomenon (see below). Thus the true prevalence of hypertension in obesity may be even higher than what has been estimated in the literature. Higher systolic blood pressures, even if they are not in the hypertensive range, are associated with a greater extent of LV hypertrophy in obesity (22). Several studies have shown synergistic effects between increasing BMI and increasing systolic blood pressure (22, 72).
Obese individuals have expanded central blood volume (165). In addition, stroke volume and cardiac output are both increased (13, 49, 165, 170, 284). These changes in blood volume and cardiac output are most likely due to the increased metabolic demand that result from increases in both lean and fat mass. The large fat depots in significant obesity produce a low-resistance vascular circuit that may further increase cardiac output. There may also be changes in renal absorption of salt and water. The combination of these factors is proposed to produce a form of volume overload similar to that which occurs with regurgitant valvular heart disease, beriberi, or arterial-venous fistulas. Chronic volume overload or high output failure is classically felt to produce an eccentric form of cardiac hypertrophy with enlarged cardiac chambers, but normal wall thickness (120). However, dilated cardiomyopathy of any etiology will produce a similar cardiac geometry. Even in high output failure, the cardiac output may decline in the end stage of the disease. Several authors have argued that volume overload is a primary mechanism contributing to the hypertrophy in obesity (13, 58, 199). Unfortunately, this relatively straightforward and logical hypothesis is complicated by the fact that multiple recent studies have shown a predominance of concentric geometry in obese patients (22, 63, 72, 279, 377).
G. Sleep Apnea
Sleep-disordered breathing is increasingly recognized as an important cause of cardiovascular abnormalities in obesity (287). Obstructive sleep apnea is very common in obesity and is nearly universal in severe obesity. There are multiple routes by which sleep apnea could lead to LV hypertrophy, including exacerbation of nighttime and daytime hypertension, increased sympathetic tone, chronic hypoxemia, and exaggerated swings in intrathoracic pressure during obstructive episodes (287). Although some studies have failed to show a relationship between sleep apnea and LV hypertrophy, the majority do support such an association (18, 22, 69, 250, 256–258, 284, 287, 331, 373). One of the confounding factors in these analyses results from difficulties in the quantification of the severity of sleep apnea. Most studies have relied on the apnea-hypopnea index (the number of apneic and hypopneic episodes per hour of sleep). However, the number of such episodes may not be the actual cause of hypertrophy. Rather, growing data suggest that nocturnal oxygen de-saturation may be the true culprit (22, 69). Cloward et al. (55) found that the use of continuous positive airway pressure at night for 6 mo was associated with a reduction in LV wall thickness (55). Avelar et al. (22) reported that in a predominantly Caucasian female population with severe obesity, the degree of LV hypertrophy was related to BMI, systolic blood pressure, and the severity of nocturnal hypoxemia (22). The majority of these patients had concentric LV hypertrophy. In this study, a diagnosis of hypertension or an average nocturnal O2 saturation <85% each had synergistic interactions with increasing BMI.
H. Neurohumoral Activation
Numerous studies show evidence that obese subjects have activation of the sympathetic nervous system. This appears to result at least in part from the effects of sleep-disordered breathing (117, 118, 199, 252). Increased sympathetic tone may contribute to the high incidence of concentric LV geometry because of hemodynamic factors such as elevated blood pressure and increased cardiac contractility. In addition, catecholamines may have direct hypertrophic effects that are independent of hemodynamic factors. There also appears to be activation of the renin-angiotensin system in obesity (304). These two pathways are interrelated and are both prohypertrophic via direct signaling effects and hemodynamic effects (i.e., vasoconstriction and elevation of blood pressure). As mentioned above, sympathetic activation likely results from sleep apnea and other indirect factors. In contrast, activation of the renin-angiotensin system may occur directly via signals from adipose tissue. Engeli et al. have suggested that adipose tissue contains the major components of a local renin-angiotensin system (96). Furthermore, increased activity of this system has been implicated in human obesity hypertension (67, 96). One proposed mechanism for such an effect is increased secretion of angiotensinogen from adipocytes, especially those in visceral fat (359). Angiotensin is thought to cause sympathoexcitation, so there may be additive effects of renin-angiotensin and sympathetic activation with respect to blood pressure elevation and cardiac remodeling in obesity (67).
I. Changes in Extracellular Matrix and Fibrosis
Alterations in the composition and structure of the extracellular matrix (ECM) can make an important contribution to changes in the cardiac size, structure, and function in heart failure (98, 237). Since changes in ECM composition clearly appear to be an important component of cardiac remodeling in heart failure, it is important to delineate the potential mechanisms contributing to these changes. With respect to obesity, it is important to consider the concept that regulation of myocardial ECM by adipokines is of great importance (77, 99). Although relatively little is known to date, Tyagi’s group (243) has suggested that elevated matrix metalloproteinase (MMP)-9 activity in the left ventricle of the human heart appears to be an early trigger of pathological remodeling leading to end-stage human heart failure. Serum markers thought to reflect cardiac collagen turnover have been shown to be elevated in obese subjects (288). Furthermore, in a pressure-overload model of heart failure in mice, an increased collagen degradation as a result of elevated expression and activity of MMP-2/9 correlated with development of heart failure (135). Increased fibrosis in coronary vessels as well as accumulation of collagen in the cardiac interstitium was detected in a high-fat diet-induced rabbit model of obesity (39). Leptin signaling may contribute to regulation of cardiac ECM in obesity, since increased fibrosis has been described in the hearts of ob/ob mice (391, 392) and in Zucker (fa/fa) rats (350, 394). We have recently demonstrated using primary human pediatric ventricular myocytes that leptin increased pro-collagen type III and type IV mRNA and decreased pro-collagen type I levels but did not change total collagen synthesis (217). Leptin also increased MMP2 isoform expression and activity (217). Further studies to examine direct effects of adipokines on myocardial ECM and alterations in animal models lacking or overexpressing individual adipokines are likely to be of great interest.
J. Apoptosis
The mechanisms leading to cardiomyocyte death in heart failure have recently become more clearly characterized (301, 302, 333). Myocyte apoptosis has been suggested to be an important etiological component of heart failure, particularly in the transition from compensatory remodeling to heart failure (112), although this concept has been somewhat contentious (95, 189, 295, 301). Endomyocardial biopsies from patients with dilated or ischemic cardiomyopathy and end-stage heart failure demonstrated apoptotic cardiomyocyte death (253, 263), and there was increased susceptibility to hypoxia-induced apoptosis in cardiomyocytes isolated from failing human hearts (351). Besides the obvious role of apoptotic cell death in myocyte loss, activation of several proteases in the apoptotic cascade can cleave various contractile proteins (α-actin, α-actinin, α, β-myosin heavy chain, tropomyosin and troponins, Ref. 56), leading to deterioration in contractile function of the existing myocytes. Accordingly, inhibiting caspases can both reduce the occurrence of heart failure or slow its progression after MI (45, 131, 374, 383, 384) as well as improve contractile function (45). Thus there is great therapeutic potential to preserve cardiac function by reducing apoptosis and furthering our current understanding of mechanisms regulating myocyte apoptosis is essential (240).
Our understanding of obesity-induced alterations in cardiomyocyte apoptosis is, surprisingly, still in its infancy. Studies in Zucker fatty rats have shown increased apoptosis that was associated with increased cardiomyocyte levels of ceramide and triglycerides. Treatment of these mice with a TZD reduced triglyceride and ceramide levels and reduced apoptosis (394). Studies using TUNEL and caspase-3 activity assays demonstrated increased levels of apoptosis in young ob/ob and db/db mice compared with control. In older obese mice, more DNA damage and less DNA repair were noted, and the degree of apoptosis became more enhanced and correlated with decreased survival, which suggests a potential role for apoptosis in myocardial dysfunction and early mortality in these models of obesity (24). Another very recent study used the high-fat diet-induced hyperinsulinemic and insulin-resistant rat model of obesity and did not detect significant differences in apoptosis despite impaired cardiomyocyte function (296). Future studies are required to define changes in apoptosis, or necrosis, occurring in obesity and their pathophysiological significance.
VI. ROLE OF ADIPOKINES IN REGULATION OF CARDIAC REMODELING
A. Adipose Tissue as an Endocrine Organ
Adipose tissue, once considered simply a lipid storage depot, is now known to produce and secrete an ever increasing range of factors, collectively termed “adipokines” (175). There is currently great research and clinical interest in the endocrine effects of these factors on cardiac tissue and their role in heart failure (86, 129, 267). Importantly, there is also a growing appreciation for the potential importance of autocrine and paracrine effects mediated by adipokines derived from myocardium itself or the larger pericardial fat mass in obese subjects (153, 220, 281, 285, 293). It is important to consider the temporal nature of changes in cardiac structure and function occurring in obese individuals, since the obesity paradox might well be in part explained by potentially protective effects of adipokines on the myocardium (343). This was addressed by Kim’s group via a high-fat feeding regimen in mice (273). They found that obesity-associated defects in cardiac function did not develop in concert with insulin resistance and concluded that changes in circulating adipokines could be an important contributing factor in cardiac dysfunction. Although evidence is accumulating on mechanisms via which various adipokines may influence cardiac remodeling in obesity (Fig. 3), we will focus here on adiponectin and leptin (129, 298, 317).
FIG. 3.

Potential direct and indirect effects of adipokines on cardiac remodeling. Various circulating adipokines, the profile of which alters in obesity, may directly (solid lines) influence remodeling events known to occur in heart failure: hypertrophy, apoptosis, fibrosis, and metabolic alterations. Another potential mechanism whereby adipokines may influence cardiac structure and function is via indirect effects (broken lines) on parameters known to influence cardiac remodeling, such as hypertension, insulin resistance, and renal effects.
B. Effects of Adiponectin on the Heart
Interest in the direct myocardial effects of adiponectin has escalated greatly in recent years (345), and the promise of many initial studies is such that it is likely adiponectin may represent a fruitful therapeutic target in heart failure (267, 268). First, an important pathophysiological contribution was suggested by the recent explosion in the number of clinical studies that have established correlations between plasma adiponectin levels and various aspects and severity of heart failure (107, 113, 143, 166, 172, 183, 198, 209, 251, 275, 282, 286, 303, 312, 366). Genetic studies have also pointed to an important role of adiponectin in heart failure with increased left ventricular mass observed in uncomplicated obese subjects carrying the G/G genotype at position 276 of the human adiponectin gene (154). Indeed, adiponectin has been proposed as a biomarker that might serve as a suitable screening test facilitating early intervention and prevention of heart failure (130, 275, 283). In particular, given our appreciation of the significant roles played by the various multimeric forms of adiponectin (97), it will be most interesting to observe if any aspects of coronary heart disease (CHD) correlate most strongly with oligomeric, hexameric, or trimeric forms. Nevertheless, some studies reported little correlation with adiponectin and risk factors for CHD, whereas adiponectin was clearly inversely associated with diabetic risk factors in all studies (309, 372), leading some to suggest that adiponectin’s influence on CHD may principally occur secondary to diabetes. In relation to the role of adiponectin in the obesity paradox, it was found that a high circulating adiponectin level was a predictor of mortality in patients with congestive heart failure (179). In another case, low plasma adiponectin was related to an increased risk of 5-yr mortality after first-ever ischemic stroke (94).
Several impressive in vivo studies have demonstrated potent direct effects of adiponectin on the heart. With the use of adiponectin KO mice, it was shown that adiponectin deficiency enhanced progressive cardiac remodeling induced by surgical aortic constriction (208). Similarly, pressure overload-induced concentric hypertrophy was enhanced in adiponectin-deficient mice and led to increased mortality that was corrected by adiponectin replacement (320). Administration of adiponectin diminished the infarct size, apoptosis, and TNF-α production induced by ischemia-reperfusion in both wild-type and adiponectin KO mice (321). Studies in cultured cells suggest that the modulating effects of adiponectin on LV remodeling might play a role only when additional hypertrophic signaling pathways are activated. Thus, whereas adiponectin may mediate a direct in vitro hypertrophic effect on primary cardiomyocytes (320), another study showed that both full-length and globular adiponectin attenuated endothelin (ET)-1-induced hypertrophy in cultured cardiomyocytes, suggesting a potential role in the pathogenesis of ET-1-related cardiomyocyte hypertrophy after MI (108).
Adiponectin is known to mediate potent metabolic effects in skeletal muscle and liver (97, 161), and an understanding of direct metabolic effects on cardiomyocytes is now emerging. Adiponectin caused a small but significant increase in glucose and fatty acid uptake and induced AMPK phosphorylation in cultured neonatal rat cardiomyocytes and mouse HL-1 cells (281). Another study demonstrated a role for the COOH-terminal globular fragment of adiponectin in regulating cardiac fatty acid oxidation in rabbit hearts in the immediate newborn period (265). We have recently examined the effect of specific forms of adiponectin on metabolism in neonatal rat cardiomyocytes and found that both full-length and globular adiponectin elicited an acute increase in glucose uptake and oxidation (271). After prolonged treatment, increased fatty acid uptake and oxidation predominated and correlated with decreased PDH activity and glucose oxidation. Collectively, these effects were mediated via both AdipoR1 and AdipoR2 and occurred via signaling mechanisms involving AMPK, ACC, and Akt. Hence, from studies to date, we may speculate that acute administration of adiponectin post-MI may confer beneficial metabolic effects.
Adiponectin may be synthesized and secreted by cardiomyocytes (281), and myocardial adiponectin expression was recently analyzed in a small number of autopsy specimens. Adiponectin was not found in individuals without cardiac lesions, but it was detected in patients who suffered MI or dilated cardiomyopathy (339). Furthermore, myocardial necrosis in obese mice with viral myocarditis was attenuated upon endogenous cardiac induction of adiponectin expression, suggesting this may represent a compensatory protective response (167, 338, 340). It is clear that further analysis of effects of adiponectin on the heart will be of interest and ultimately perhaps of significant therapeutic value.
Finally, indirect effects of adiponectin may contribute significantly to the development of heart failure and progression of myocardial remodeling. Adiponectin mediates a variety of cardiovascular effects including anti-atherogenic effects via a combination of effects on endothelial cells, macrophages, and vascular smooth muscle cells (195). Antidiabetic (162), anti-inflammatory (348), and antihypertensive (259) effects have all been established to occur in response to adiponectin. Ferguson’s group (106) has shown that adiponectin can directly act on the area postrema, which lies out with the blood-brain barrier, to ultimately regulate cardiovascular function (106), and the possibility that adiponectin also controls peripheral effects via the central nervous system was alluded to recently with the demonstration of adiponectin, albeit at lower levels and with a different multimer profile, in cerebrospinal fluid (193).
C. Effects of Leptin on the Heart
Elevated circulating levels of leptin, which act via a family of receptor (ob.R) isoforms (335) to mediate an ever-growing wide range of physiological effects (8, 312), are found in obese individuals. Importantly, leptin may impact on myocardial function via direct peripheral effects or via secondary central nervous system-mediated responses, and there is an extensive literature documenting that leptin can influence many parameters associated with obesity and heart failure (216, 289). Lack of leptin or leptin resistance leads to accumulation of lipids in non-adipose peripheral tissues due to loss of leptin’s antisteatotic effects and results in a variety of lipotoxic effects (356). For example, leptin-deficient ob/ob mice exhibit a higher degree of cardiomyocyte apoptosis that can be restored to normal levels by leptin administration (24). The metabolic consequences observed in obese models lacking leptin function are discussed above. Leptin can also induce generation of reactive oxygen species in the heart (379). Another characteristic marker of heart failure, depressed cardiac response to β-adrenergic agonists, is also observed in ob/ob mice and can be corrected by leptin replacement therapy (238). Several reports have suggested that leptin directly induced hypertrophy in both human and rodent cardiomyocytes (217, 292, 293, 379). One report concluded that leptin mediates antihypertrophic effects since infusion of leptin to ob/ob mice, which exhibit LVH, decreased LV wall thickness (23). The original hypertrophy in these mice may not have resulted from lack of leptin per se, but instead from other systemic alterations. Similarly, recovery upon leptin administration does not necessarily imply a direct effect on myocytes. In general, the ob/ob mouse model is rather unique (plasma leptin levels are increased in almost all other obese rodent models and in humans, Ref. 297) and may not precisely reflect the alterations observed in human obesity. Another caveat of this model for the study of hypertrophy is the presence of low arterial pressure, while the opposite is found in most forms of obesity (224). In summary, it would appear that leptin mediates a myriad of effects that can impinge on development or progression of heart failure, and it is likely that too much or too little is detrimental.
An intriguing aspect to consider with respect to the influence of leptin on the myocardium is the concept that selective leptin resistance occurs in obesity (60, 61, 222, 223). Initial studies purporting this concept suggested that while the effects of leptin on satiety and energy metabolism were resistant, the sympathoexcitatory effects were maintained (60, 61, 222, 223, 290, 291). Indeed, using a high-fat induced model of obesity in mice, leptin resistance measured by STAT-3 activation was shown to be restricted to the arcuate nucleus of the hypothalamus (60, 194), and this correlated with expression of the suppressor of leptin signaling SOCS-3 (248). It was also recently suggested that leptin resistance in obesity is restricted to the metabolic actions of leptin (62), and impaired leptin signaling was observed in one study using ventricular cardiomyocytes isolated from rats subjected to 10-wk dietary sucrose feeding to generate hyperleptinemia and insulin resistance (139). Notably, leptin administration in mice has been shown to reduce infarct size and enhance functional recovery following coronary artery ligation (4, 326). Nevertheless, whether the various actions of leptin on the heart are enhanced or suppressed in hyperleptinemic obese individuals is uncertain and most certainly deserving of further investigation.
VII. CLINICAL ISSUES
Many studies have shown that weight loss improves blood pressure, diabetes, sleep apnea, hyperlipidemia, and sympathetic/parasympathetic tone (196, 284, 324, 365). If they are maintained, these improvements are expected to translate into benefits for cardiac structure and function (181, 284). Indeed, several studies have reported beneficial effects of weight loss on cardiac structure and LV function. Weight loss achieved via life-style modifications or through bariatric surgery have both been consistently associated with reductions in LV dimensions, LV wall thickness, LV mass, and LA dimension (14, 16, 17, 169). Perhaps more importantly, patients can exercise longer and they report less dyspnea and chest pain after significant weight loss (168). It is widely believed that cardiovascular and other health benefits accrue when weight loss is achieved by reduced caloric intake and increased exercise. In fact, even modest amounts of weight loss or increased exercise may produce salutary effects. The removal of subcutaneous fat by liposuction does not appear to produce beneficial metabolic changes (182). Pharmacotherapy with the two currently approved drugs for obesity treatment, orlistat (a gastrointestinal lipase inhibitor) and sibutramine (a monoamine reuptake inhibitor), have both been associated with weight loss, improvements in serum lipids, and insulin sensitivity (76, 109, 270). Orlistat treatment usually reduces blood pressure, while sibutramine is commonly associated with increased heart rate and blood pressure (109, 393). Neither drug has significant effects on cardiac dimensions, valvular function, or pulmonary artery pressures when given for up to 6 mo (125, 393). This topic has been reviewed elsewhere recently (270). The full extent and duration of benefits that can be achieved when weight loss is induced by pharmacotherapy or bariatric surgery is not clear at this point in time. Nonetheless, bariatric surgery in general produces much greater amounts of weight loss than life-style modification or pharmacotherapy, and these losses are usually sustained for much longer (324). Given the rapid increase in the prevalence of severe obesity worldwide (136, 171) and the growing popularity of surgical treatments, further studies of the cardiovascular outcomes after bariatric surgery are clearly warranted (7).
VIII. PERSPECTIVES
The extraordinarily rapid increase in the worldwide prevalence of obesity has profound public health implications. The full scope of the problem is hard to realize because many obesity-related complications require decades to become manifest. Therefore, it is critical that we further our understanding of the pathophysiological bases for the multiorgan diseases that are associated with or caused by obesity. In this review, we have discussed the complex constellation of physical, metabolic, neuronal, and hormonal alterations in obesity that may lead to structural and functional alterations in the heart. In particular, we have focused on processes that might link obesity to various aspects of cardiac remodeling and the eventual development of heart failure (summarized in Fig. 4). It seems likely that obesity influences cardiac remodeling both directly and via its closely associated comorbidities such as hypertension, diabetes, sleep-disordered breathing, kidney dysfunction, atherosclerosis, and endothelial dysfunction. This multifarious web of interconnected processes begins with the onset of obesity, which increasingly occurs in childhood or adolescence. However, the cumulative effects on the heart may not become clinically apparent for as much as 50 years or more. Thus, in terms of the actual impact of obesity on the cardiovascular system, we are only seeing the tip of the proverbial iceberg today. Further complicating our ability to unravel the impact of obesity on human health is the growing body of data showing that obesity may have protective as well as deleterious effects on the cardiovascular system. Multiple studies showing improved survival in obese patients with established cardiovascular disease has led to the concept of “the obesity paradox.” Understanding the nature of the beneficial and the detrimental components of obesity is arguably one of the most pressing agendas for our current biomedical and scientific communities. We hope this review will serve as a catalyst for further research into this fascinating, perplexing, and troubling condition.
FIG. 4.

Summary of potential mechanisms via which obesity can influence structure and function of the heart.
Acknowledgments
We greatly appreciate the contributions of Hanna Park and Min Park in producing Figures 3 and 4 and Ying Liu in formatting this manuscript.
We regret not being able to cite many relevant studies due to space restriction.
GRANTS
G. Sweeney thanks the Heart and Stroke Foundation of Canada, Canadian Diabetes Association, and Canadian Institutes of Health Research for supporting related work in his laboratory. E. D. Abel is an Established Investigator of the American Heart Association. Research studies in the Abel laboratory that have contributed to this review were supported by National Heart, Lung, and Blood Institute Grants UO1-HL-70525, UO1-HL-087947 (Animal Models of Diabetes Complications Consortium), RO1-HL-70070, and RO1-HL-73167. This work was also supported by a grant from the Department of Veterans Affairs (to S. E. Litwin).
References
- 1.Aasum E, Belke DD, Severson DL, Riemersma RA, Cooper M, Andreassen M, Larsen TS. Cardiac function and metabolism in Type 2 diabetic mice after treatment with BM 17.0744, a novel PPAR-alpha activator. Am J Physiol Heart Circ Physiol. 2002;283:H949–H957. doi: 10.1152/ajpheart.00226.2001. [DOI] [PubMed] [Google Scholar]
- 2.Aasum E, Cooper M, Severson DL, Larsen TS. Effect of BM 17.0744, a PPARalpha ligand, on the metabolism of perfused hearts from control and diabetic mice. Can J Physiol Pharmacol. 2005;83:183–190. doi: 10.1139/y04-139. [DOI] [PubMed] [Google Scholar]
- 3.Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes. 2003;52:434–441. doi: 10.2337/diabetes.52.2.434. [DOI] [PubMed] [Google Scholar]
- 4.Abe Y, Ono K, Kawamura T, Wada H, Kita T, Shimatsu A, Hasegawa K. Leptin induces elongation of cardiac myocyte and causes eccentric left ventricular dilatation with compensation. Am J Physiol Heart Circ Physiol. 2007;292:H2387–H2396. doi: 10.1152/ajpheart.00579.2006. [DOI] [PubMed] [Google Scholar]
- 5.Abel ED. Insulin signaling in heart muscle: lessons from genetically engineered mouse models. Curr Hypertens Rep. 2004;6:416–423. doi: 10.1007/s11906-004-0034-4. [DOI] [PubMed] [Google Scholar]
- 6.Abel ED. Myocardial insulin resistance and cardiac complications of diabetes. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:219–226. doi: 10.2174/1568008054064869. [DOI] [PubMed] [Google Scholar]
- 7.Adams TD, Avelar E, Cloward T, Crosby RD, Farney RJ, Gress R, Halverson RC, Hopkins PN, Kolotkin RL, Lamonte MJ, Litwin S, Nuttall RT, Pendleton R, Rosamond W, Simper SC, Smith SC, Strong M, Walker JM, Wiebke G, Yanowitz FG, Hunt SC. Design and rationale of the Utah obesity study. A study to assess morbidity following gastric bypass surgery. Contemp Clin Trials. 2005;26:534–551. doi: 10.1016/j.cct.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413–437. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- 9.Ahmed Q, Chung-Park M, Tomashefski JF., Jr Cardiopulmonary pathology in patients with sleep apnea/obesity hypoventilation syndrome. Hum Pathol. 1997;28:264–269. doi: 10.1016/s0046-8177(97)90122-2. [DOI] [PubMed] [Google Scholar]
- 10.Aizawa-Abe M, Ogawa Y, Masuzaki H, Ebihara K, Satoh N, Iwai H, Matsuoka N, Hayashi T, Hosoda K, Inoue G, Yoshimasa Y, Nakao K. Pathophysiological role of leptin in obesity-related hypertension. J Clin Invest. 2000;105:1243–1252. doi: 10.1172/JCI8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexander JK. The cardiomyopathy of obesity. Prog Cardiovasc Dis. 1985;27:325–334. doi: 10.1016/s0033-0620(85)80002-5. [DOI] [PubMed] [Google Scholar]
- 12.Allison MA, Michael Wright C. Body morphology differentially predicts coronary calcium. Int J Obes Relat Metab Disord. 2004;28:396–401. doi: 10.1038/sj.ijo.0802571. [DOI] [PubMed] [Google Scholar]
- 13.Alpert MA, Hashimi MW. Obesity and the heart. Am J Med Sci. 1993;306:117–123. doi: 10.1097/00000441-199308000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Alpert MA, Lambert CR, Panayiotou H, Terry BE, Cohen MV, Massey CV, Hashimi MW, Mukerji V. Relation of duration of morbid obesity to left ventricular mass, systolic function, and diastolic filling, and effect of weight loss. Am J Cardiol. 1995;76:1194–1197. doi: 10.1016/s0002-9149(99)80338-5. [DOI] [PubMed] [Google Scholar]
- 15.Alpert MA, Lambert CR, Terry BE, Cohen MV, Mukerji V, Massey CV, Hashimi MW, Panayiotou H. Interrelationship of left ventricular mass, systolic function and diastolic filling in normotensive morbidly obese patients. Int J Obes Relat Metab Disord. 1995;19:550–557. [PubMed] [Google Scholar]
- 16.Alpert MA, Terry BE, Kelly DL. Effect of weight loss on cardiac chamber size, wall thickness and left ventricular function in morbid obesity. Am J Cardiol. 1985;55:783–786. doi: 10.1016/0002-9149(85)90156-0. [DOI] [PubMed] [Google Scholar]
- 17.Alpert MA, Terry BE, Mulekar M, Cohen MV, Massey CV, Fan TM, Panayiotou H, Mukerji V. Cardiac morphology and left ventricular function in normotensive morbidly obese patients with and without congestive heart failure, and effect of weight loss. Am J Cardiol. 1997;80:736–740. doi: 10.1016/s0002-9149(97)00505-5. [DOI] [PubMed] [Google Scholar]
- 18.Amin RS, Kimball TR, Bean JA, Jeffries JL, Willging JP, Cotton RT, Witt SA, Glascock BJ, Daniels SR. Left ventricular hypertrophy and abnormal ventricular geometry in children and adolescents with obstructive sleep apnea. Am J Respir Crit Care Med. 2002;165:1395–1399. doi: 10.1164/rccm.2105118. [DOI] [PubMed] [Google Scholar]
- 19.Aukrust P, Gullestad L, Ueland T, Damas JK, Yndestad A. Inflammatory and anti-inflammatory cytokines in chronic heart failure: potential therapeutic implications. Ann Med. 2005;37:74–85. doi: 10.1080/07853890510007232. [DOI] [PubMed] [Google Scholar]
- 20.Aukrust P, Yndestad A, Damas JK, Gullestad L. Inflammation and chronic heart failure: potential therapeutic role of intravenous immunoglobulin. Autoimmun Rev. 2004;3:221–227. doi: 10.1016/S1568-9972(03)00103-4. [DOI] [PubMed] [Google Scholar]
- 21.Aurigemma GP, Silver KH, Priest MA, Gaasch WH. Geometric changes allow normal ejection fraction despite depressed myocardial shortening in hypertensive left ventricular hypertrophy. J Am Coll Cardiol. 1995;26:195–202. doi: 10.1016/0735-1097(95)00153-q. [DOI] [PubMed] [Google Scholar]
- 22.Avelar E, Cloward TV, Walker JM, Farney RJ, Strong M, Pendleton RC, Segerson N, Adams TD, Gress RE, Hunt SC, Litwin SE. Left ventricular hypertrophy in severe obesity: interactions among blood pressure, nocturnal hypoxemia, body mass. Hypertension. 2007;49:34–39. doi: 10.1161/01.HYP.0000251711.92482.14. [DOI] [PubMed] [Google Scholar]
- 23.Barouch LA, Berkowitz DE, Harrison RW, O’Donnell CP, Hare JM. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation. 2003;108:754–759. doi: 10.1161/01.CIR.0000083716.82622.FD. [DOI] [PubMed] [Google Scholar]
- 24.Barouch LA, Gao D, Chen L, Miller KL, Xu W, Phan AC, Kittleson MM, Minhas KM, Berkowitz DE, Wei C, Hare JM. Cardiac myocyte apoptosis is associated with increased DNA damage and decreased survival in murine models of obesity. Circ Res. 2006;98:119–124. doi: 10.1161/01.RES.0000199348.10580.1d. [DOI] [PubMed] [Google Scholar]
- 25.Belanger-Ducharme F, Tremblay A. Prevalence of obesity in Canada. Obes Rev. 2005;6:183–186. doi: 10.1111/j.1467-789X.2005.00179.x. [DOI] [PubMed] [Google Scholar]
- 26.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, Zhang D, Cooksey RC, McClain DA, Litwin SE, Taegtmeyer H, Severson D, Kahn CR, Abel ED. Insulin signaling coordinately regulates cardiac size, metabolism, contractile protein isoform expression. J Clin Invest. 2002;109:629–639. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bell-Anderson KS, Bryson JM. Leptin as a potential treatment for obesity: progress to date. Treat Endocrinol. 2004;3:11–18. doi: 10.2165/00024677-200403010-00002. [DOI] [PubMed] [Google Scholar]
- 28.Berkalp B, Cesur V, Corapcioglu D, Erol C, Baskal N. Obesity and left ventricular diastolic dysfunction. Int J Cardiol. 1995;52:23–26. doi: 10.1016/0167-5273(95)02431-u. [DOI] [PubMed] [Google Scholar]
- 29.Boudina S, Abel ED. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology. 2006;21:250–258. doi: 10.1152/physiol.00008.2006. [DOI] [PubMed] [Google Scholar]
- 30.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 31.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 32.Breslow JL. n-3 fatty acids and cardiovascular disease. Am J Clin Nutr. 2006;83:1477S–1482S. doi: 10.1093/ajcn/83.6.1477S. [DOI] [PubMed] [Google Scholar]
- 33.Bucciarelli LG, Kaneko M, Ananthakrishnan R, Harja E, Lee LK, Hwang YC, Lerner S, Bakr S, Li Q, Lu Y, Song F, Qu W, Gomez T, Zou YS, Yan SF, Schmidt AM, Ramasamy R. Receptor for advanced-glycation end products: key modulator of myocardial ischemic injury. Circulation. 2006;113:1226–1234. doi: 10.1161/CIRCULATIONAHA.105.575993. [DOI] [PubMed] [Google Scholar]
- 34.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 35.Buettner HJ, Mueller C, Gick M, Ferenc M, Allgeier J, Comberg T, Werner KD, Schindler C, Neumann FJ. The impact of obesity on mortality in UA/non-ST-segment elevation myocardial infarction. Eur Heart J. 2007;28:1694–1701. doi: 10.1093/eurheartj/ehm220. [DOI] [PubMed] [Google Scholar]
- 36.Carley AN, Semeniuk LM, Shimoni Y, Aasum E, Larsen TS, Berger JP, Severson DL. Treatment of type 2 diabetic db/db mice with a novel PPARgamma agonist improves cardiac metabolism but not contractile function. Am J Physiol Endocrinol Metab. 2004;286:E449–E455. doi: 10.1152/ajpendo.00329.2003. [DOI] [PubMed] [Google Scholar]
- 37.Carley AN, Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys Acta. 2005;1734:112–126. doi: 10.1016/j.bbalip.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 38.Carroll JF, Summers RL, Dzielak DJ, Cockrell K, Montani JP, Mizelle HL. Diastolic compliance is reduced in obese rabbits. Hypertension. 1999;33:811–815. doi: 10.1161/01.hyp.33.3.811. [DOI] [PubMed] [Google Scholar]
- 39.Carroll JF, Tyagi SC. Extracellular matrix remodeling in the heart of the homocysteinemic obese rabbit. Am J Hypertens. 2005;18:692–698. doi: 10.1016/j.amjhyper.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 40.Carroll JF, Zenebe WJ, Strange TB. Cardiovascular function in a rat model of diet-induced obesity. Hypertension. 2006;48:65–72. doi: 10.1161/01.HYP.0000224147.01024.77. [DOI] [PubMed] [Google Scholar]
- 41.Carroll R, Carley AN, Dyck JR, Severson DL. Metabolic effects of insulin on cardiomyocytes from control and diabetic db/db mouse hearts. Am J Physiol Endocrinol Metab. 2005;288:E900–E906. doi: 10.1152/ajpendo.00491.2004. [DOI] [PubMed] [Google Scholar]
- 42.Caruana L, Petrie MC, Davie AP, McMurray JJ. Do patients with suspected heart failure and preserved left ventricular systolic function suffer from “diastolic heart failure” or from misdiagnosis? A prospective descriptive study. Bone Miner J. 2000;321:215–218. doi: 10.1136/bmj.321.7255.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cassidy AE, Bielak LF, Zhou Y, Sheedy PF, 2nd, Turner ST, Breen JF, Araoz PA, Kullo IJ, Lin X, Peyser PA. Progression of subclinical coronary atherosclerosis: does obesity make a difference? Circulation. 2005;111:1877–1882. doi: 10.1161/01.CIR.0000161820.40494.5D. [DOI] [PubMed] [Google Scholar]
- 44.CDC. CfDCaP. State-specific prevalence of obesity among adults–United States, 2005. MMWR Morb Mortal Wkly Rep. 2006;55:985–988. [PubMed] [Google Scholar]
- 45.Chandrashekhar Y, Sen S, Anway R, Shuros A, Anand I. Long-term caspase inhibition ameliorates apoptosis, reduces myocardial troponin-I cleavage, protects left ventricular function, attenuates remodeling in rats with myocardial infarction. J Am Coll Cardiol. 2004;43:295–301. doi: 10.1016/j.jacc.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 46.Chen YT, Vaccarino V, Williams CS, Butler J, Berkman LF, Krumholz HM. Risk factors for heart failure in the elderly: a prospective community-based study. Am J Med. 1999;106:605–612. doi: 10.1016/s0002-9343(99)00126-6. [DOI] [PubMed] [Google Scholar]
- 47.Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, Chen YE, Yang Q. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–1250. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- 48.Chinali M, de Simone G, Roman MJ, Lee ET, Best LG, Howard BV, Devereux RB. Impact of obesity on cardiac geometry and function in a population of adolescents: the Strong Heart Study. J Am Coll Cardiol. 2006;47:2267–2273. doi: 10.1016/j.jacc.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 49.Chinali M, Devereux RB, Howard BV, Roman MJ, Bella JN, Liu JE, Resnick HE, Lee ET, Best LG, de Simone G. Comparison of cardiac structure and function in American Indians with and without the metabolic syndrome (the Strong Heart Study) Am J Cardiol. 2004;93:40–44. doi: 10.1016/j.amjcard.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 50.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, Schaffer JE. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005;96:225–233. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- 51.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chou E, Suzuma I, Way KJ, Opland D, Clermont AC, Naruse K, Suzuma K, Bowling NL, Vlahos CJ, Aiello LP, King GL. Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulin-resistant and diabetic States: a possible explanation for impaired collateral formation in cardiac tissue. Circulation. 2002;105:373–379. doi: 10.1161/hc0302.102143. [DOI] [PubMed] [Google Scholar]
- 53.Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003;144:3483–3490. doi: 10.1210/en.2003-0242. [DOI] [PubMed] [Google Scholar]
- 54.Cittadini A, Mantzoros CS, Hampton TG, Travers KE, Katz SE, Morgan JP, Flier JS, Douglas PS. Cardiovascular abnormalities in transgenic mice with reduced brown fat: an animal model of human obesity. Circulation. 1999;100:2177–2183. doi: 10.1161/01.cir.100.21.2177. [DOI] [PubMed] [Google Scholar]
- 55.Cloward TV, Walker JM, Farney RJ, Anderson JL. Left ventricular hypertrophy is a common echocardiographic abnormality in severe obstructive sleep apnea and reverses with nasal continuous positive airway pressure. Chest. 2003;124:594–601. doi: 10.1378/chest.124.2.594. [DOI] [PubMed] [Google Scholar]
- 56.Communal C, Sumandea M, de Tombe P, Narula J, Solaro RJ, Hajjar RJ. Functional consequences of caspase activation in cardiac myocytes. Proc Natl Acad Sci USA. 2002;99:6252–6256. doi: 10.1073/pnas.092022999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, Schaff HV. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med. 1997;337:581–588. doi: 10.1056/NEJM199708283370901. [DOI] [PubMed] [Google Scholar]
- 58.Contaldo F, Pasanisi F, Finelli C, de Simone G. Obesity, heart failure and sudden death. Nutr Metab Cardiovasc Dis. 2002;12:190–197. [PubMed] [Google Scholar]
- 59.Coort SL, Hasselbaink DM, Koonen DP, Willems J, Coumans WA, Chabowski A, van der Vusse GJ, Bonen A, Glatz JF, Luiken JJ. Enhanced sarcolemmal FAT/CD36 content and triacylglycerol storage in cardiac myocytes from obese Zucker rats. Diabetes. 2004;53:1655–1663. doi: 10.2337/diabetes.53.7.1655. [DOI] [PubMed] [Google Scholar]
- 60.Correia ML, Haynes WG. Obesity-related hypertension: is there a role for selective leptin resistance? Curr Hypertens Rep. 2004;6:230–235. doi: 10.1007/s11906-004-0074-9. [DOI] [PubMed] [Google Scholar]
- 61.Correia ML, Haynes WG, Rahmouni K, Morgan DA, Sivitz WI, Mark AL. The concept of selective leptin resistance: evidence from agouti yellow obese mice. Diabetes. 2002;51:439–442. doi: 10.2337/diabetes.51.2.439. [DOI] [PubMed] [Google Scholar]
- 62.Correia ML, Rahmouni K. Role of leptin in the cardiovascular and endocrine complications of metabolic syndrome. Diabetes Obes Metab. 2006;8:603–610. doi: 10.1111/j.1463-1326.2005.00562.x. [DOI] [PubMed] [Google Scholar]
- 63.Crisostomo LL, Araujo LM, Camara E, Carvalho C, Silva FA, Vieira M, Rabelo A., Jr Comparison of left ventricular mass and function in obese versus nonobese women <40 years of age. Am J Cardiol. 1999;84:1127–1129. doi: 10.1016/s0002-9149(99)00519-6. [DOI] [PubMed] [Google Scholar]
- 64.Cummings PM, Le BH, Lopes MB. Postmortem findings in morbidly obese individuals dying after gastric bypass procedures. Hum Pathol. 2007;38:593–597. doi: 10.1016/j.humpath.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 65.Curtis JP, Selter JG, Wang Y, Rathore SS, Jovin IS, Jadbabaie F, Kosiborod M, Portnay EL, Sokol SI, Bader F, Krumholz HM. The obesity paradox: body mass index and outcomes in patients with heart failure. Arch Intern Med. 2005;165:55–61. doi: 10.1001/archinte.165.1.55. [DOI] [PubMed] [Google Scholar]
- 66.Das SR, Drazner MH, Dries DL, Vega GL, Stanek HG, Abdullah SM, Canham RM, Chung AK, Leonard D, Wians FH, Jr, de Lemos JA. Impact of body mass and body composition on circulating levels of natriuretic peptides: results from the Dallas Heart Study. Circulation. 2005;112:2163–2168. doi: 10.1161/CIRCULATIONAHA.105.555573. [DOI] [PubMed] [Google Scholar]
- 67.Davy KP, Hall JE. Obesity and hypertension: two epidemics or one? Am J Physiol Regul Integr Comp Physiol. 2004;286:R803–R813. doi: 10.1152/ajpregu.00707.2003. [DOI] [PubMed] [Google Scholar]
- 68.De la Maza MP, Estevez A, Bunout D, Klenner C, Oyonarte M, Hirsch S. Ventricular mass in hypertensive and normotensive obese subjects. Int J Obes Relat Metab Disord. 1994;18:193–197. [PubMed] [Google Scholar]
- 69.De Simone G. Morbid obesity and left ventricular geometry. Hypertension. 2007;49:7–9. doi: 10.1161/01.HYP.0000251714.60547.06. [DOI] [PubMed] [Google Scholar]
- 70.De Simone G, Daniels SR, Devereux RB, Meyer RA, Roman MJ, de Divitiis O, Alderman MH. Left ventricular mass and body size in normotensive children and adults: assessment of allometric relations and impact of overweight. J Am Coll Cardiol. 1992;20:1251–1260. doi: 10.1016/0735-1097(92)90385-z. [DOI] [PubMed] [Google Scholar]
- 71.De Simone G, Devereux RB, Koren MJ, Mensah GA, Casale PN, Laragh JH. Midwall left ventricular mechanics. An independent predictor of cardiovascular risk in arterial hypertension. Circulation. 1996;93:259–265. doi: 10.1161/01.cir.93.2.259. [DOI] [PubMed] [Google Scholar]
- 72.De Simone G, Devereux RB, Roman MJ, Alderman MH, Laragh JH. Relation of obesity and gender to left ventricular hypertrophy in normotensive and hypertensive adults. Hypertension. 1994;23:600–606. doi: 10.1161/01.hyp.23.5.600. [DOI] [PubMed] [Google Scholar]
- 73.De Simone G, Kizer JR, Chinali M, Roman MJ, Bella JN, Best LG, Lee ET, Devereux RB. Normalization for body size and population-attributable risk of left ventricular hypertrophy: the Strong Heart Study. Am J Hypertens. 2005;18:191–196. doi: 10.1016/j.amjhyper.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 74.De Vries JE, Vork MM, Roemen TH, de Jong YF, Cleutjens JP, van der Vusse GJ, van Bilsen M. Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J Lipid Res. 1997;38:1384–1394. [PubMed] [Google Scholar]
- 75.Delpy E, Hatem SN, Andrieu N, de Vaumas C, Henaff M, Rucker-Martin C, Jaffrezou JP, Laurent G, Levade T, Mercadier JJ. Doxorubicin induces slow ceramide accumulation and late apoptosis in cultured adult rat ventricular myocytes. Cardiovasc Res. 1999;43:398–407. doi: 10.1016/s0008-6363(99)00142-x. [DOI] [PubMed] [Google Scholar]
- 76.Derosa G, Cicero AF, Murdolo G, Piccinni MN, Fogari E, Bertone G, Ciccarelli L, Fogari R. Efficacy and safety comparative evaluation of orlistat and sibutramine treatment in hypertensive obese patients. Diabetes Obes Metab. 2005;7:47–55. doi: 10.1111/j.1463-1326.2004.00372.x. [DOI] [PubMed] [Google Scholar]
- 77.Deschamps AM, Spinale FG. Pathways of matrix metalloproteinase induction in heart failure: bioactive molecules and transcriptional regulation. Cardiovasc Res. 2006;69:666–676. doi: 10.1016/j.cardiores.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 78.Despres JP. Inflammation and cardiovascular disease: is abdominal obesity the missing link? Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S22–S24. doi: 10.1038/sj.ijo.0802495. [DOI] [PubMed] [Google Scholar]
- 79.Despres JP. Intra-abdominal obesity: an untreated risk factor for Type 2 diabetes and cardiovascular disease. J Endocrinol Invest. 2006;29:77–82. [PubMed] [Google Scholar]
- 80.Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881–887. doi: 10.1038/nature05488. [DOI] [PubMed] [Google Scholar]
- 81.Desrois M, Sidell RJ, Gauguier D, Davey CL, Radda GK, Clarke K. Gender differences in hypertrophy, insulin resistance and ischemic injury in the aging type 2 diabetic rat heart. J Mol Cell Cardiol. 2004;37:547–555. doi: 10.1016/j.yjmcc.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 82.Di Paola M, Zaccagnino P, Montedoro G, Cocco T, Lorusso M. Ceramide induces release of pro-apoptotic proteins from mitochondria by either a Ca2+-dependent or a Ca2+-independent mechanism. J Bioenerg Biomembr. 2004;36:165–170. doi: 10.1023/b:jobb.0000023619.97392.0c. [DOI] [PubMed] [Google Scholar]
- 83.Diamant M, Lamb HJ, Groeneveld Y, Endert EL, Smit JW, Bax JJ, Romijn JA, de Roos A, Radder JK. Diastolic dysfunction is associated with altered myocardial metabolism in asymptomatic normotensive patients with well-controlled type 2 diabetes mellitus. J Am Coll Cardiol. 2003;42:328–335. doi: 10.1016/s0735-1097(03)00625-9. [DOI] [PubMed] [Google Scholar]
- 84.Diamant M, Tushuizen ME. The metabolic syndrome and endothelial dysfunction: common highway to type 2 diabetes and CVD. Curr Diab Rep. 2006;6:279–286. doi: 10.1007/s11892-006-0061-4. [DOI] [PubMed] [Google Scholar]
- 85.Diercks DB, Roe MT, Mulgund J, Pollack CV, Jr, Kirk JD, Gibler WB, Ohman EM, Smith SC, Jr, Boden WE, Peterson ED. The obesity paradox in non-ST-segment elevation acute coronary syndromes: results from the Can Rapid risk stratification of unstable angina patients suppress adverse outcomes with early implementation of the American College of Cardiology/American Heart Association Guidelines Quality Improvement Initiative. Am Heart J. 2006;152:140–148. doi: 10.1016/j.ahj.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 86.Do D, Alvarez J, Chiquette E, Chilton R. The good fat hormone: adiponectin and cardiovascular disease. Curr Atheroscler Rep. 2006;8:94–99. doi: 10.1007/s11883-006-0045-5. [DOI] [PubMed] [Google Scholar]
- 87.Dokainish H, Zoghbi WA, Lakkis NM, Al-Bakshy F, Dhir M, Quinones MA, Nagueh SF. Optimal noninvasive assessment of left ventricular filling pressures: a comparison of tissue Doppler echocardiography and B-type natriuretic peptide in patients with pulmonary artery catheters. Circulation. 2004;109:2432–2439. doi: 10.1161/01.CIR.0000127882.58426.7A. [DOI] [PubMed] [Google Scholar]
- 88.Dong F, Zhang X, Yang X, Esberg LB, Yang H, Zhang Z, Culver B, Ren J. Impaired cardiac contractile function in ventricular myocytes from leptin-deficient ob/ob obese mice. J Endocrinol. 2006;188:25–36. doi: 10.1677/joe.1.06241. [DOI] [PubMed] [Google Scholar]
- 89.Douketis JD, Sharma AM. Obesity and cardiovascular disease: pathogenic mechanisms and potential benefits of weight reduction. Semin Vasc Med. 2005;5:25–33. doi: 10.1055/s-2005-871739. [DOI] [PubMed] [Google Scholar]
- 90.Dragoy Hafstad A, Khalid AM, How OJ, Larsen TS, Aasum E. Glucose and insulin improve cardiac efficiency and post-ischemic functional recovery in perfused hearts from type 2 diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2007;292:E1288–E1294. doi: 10.1152/ajpendo.00504.2006. [DOI] [PubMed] [Google Scholar]
- 91.Drenick EJ, Fisler JS. Sudden cardiac arrest in morbidly obese surgical patients unexplained after autopsy. Am J Surg. 1988;155:720–726. doi: 10.1016/s0002-9610(88)80029-1. [DOI] [PubMed] [Google Scholar]
- 92.Duflou J, Virmani R, Rabin I, Burke A, Farb A, Smialek J. Sudden death as a result of heart disease in morbid obesity. Am Heart J. 1995;130:306–313. doi: 10.1016/0002-8703(95)90445-x. [DOI] [PubMed] [Google Scholar]
- 93.Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation. 2007;115:909–917. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Efstathiou SP, Tsioulos DI, Tsiakou AG, Gratsias YE, Pefanis AV, Mountokalakis TD. Plasma adiponectin levels and five-year survival after first-ever ischemic stroke. Stroke. 2005;36:1915–1919. doi: 10.1161/01.STR.0000177874.29849.f0. [DOI] [PubMed] [Google Scholar]
- 95.Elsasser A, Suzuki K, Schaper J. Unresolved issues regarding the role of apoptosis in the pathogenesis of ischemic injury and heart failure. J Mol Cell Cardiol. 2000;32:711–724. doi: 10.1006/jmcc.2000.1125. [DOI] [PubMed] [Google Scholar]
- 96.Engeli S, Negrel R, Sharma AM. Physiology and pathophysiology of the adipose tissue renin-angiotensin system. Hypertension. 2000;35:1270–1277. doi: 10.1161/01.hyp.35.6.1270. [DOI] [PubMed] [Google Scholar]
- 97.Fang X, Sweeney G. Mechanisms regulating energy metabolism by adiponectin in obesity and diabetes. Biochem Soc Trans. 2006;34:798–801. doi: 10.1042/BST0340798. [DOI] [PubMed] [Google Scholar]
- 98.Fedak PW, Verma S, Weisel RD, Li RK. Cardiac remodeling and failure From molecules to man (Part II) Cardiovasc Pathol. 2005;14:49–60. doi: 10.1016/j.carpath.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 99.Felkin LE, Birks EJ, George R, Wong S, Khaghani A, Yacoub MH, Barton PJ. A quantitative gene expression profile of matrix metalloproteinases (MMPS) and their inhibitors (TIMPS) in the myocardium of patients with deteriorating heart failure requiring left ventricular assist device support. J Heart Lung Transplant. 2006;25:1413–1419. doi: 10.1016/j.healun.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 100.Ferraro S, Perrone-Filardi P, Desiderio A, Betocchi S, D’Alto M, Liguori L, Trimigliozzi P, Turco S, Chiariello M. Left ventricular systolic and diastolic function in severe obesity: a radio-nuclide study. Cardiology. 1996;87:347–353. doi: 10.1159/000177118. [DOI] [PubMed] [Google Scholar]
- 101.Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci USA. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fonarow GC, Srikanthan P, Costanzo MR, Cintron GB, Lopatin M. An obesity paradox in acute heart failure: analysis of body mass index and inhospital mortality for 108,927 patients in the Acute Decompensated Heart Failure National Registry. Am Heart J. 2007;153:74–81. doi: 10.1016/j.ahj.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 104.Fredersdorf S, Thumann C, Ulucan C, Griese DP, Luchner A, Riegger GA, Kromer EP, Weil J. Myocardial hypertrophy and enhanced left ventricular contractility in Zucker diabetic fatty rats. Cardiovasc Pathol. 2004;13:11–19. doi: 10.1016/S1054-8807(03)00109-1. [DOI] [PubMed] [Google Scholar]
- 105.Freedman DS, Dietz WH, Tang R, Mensah GA, Bond MG, Urbina EM, Srinivasan S, Berenson GS. The relation of obesity throughout life to carotid intima-media thickness in adulthood: the Bogalusa Heart Study. Int J Obes Relat Metab Disord. 2004;28:159–166. doi: 10.1038/sj.ijo.0802515. [DOI] [PubMed] [Google Scholar]
- 106.Fry M, Smith PM, Hoyda TD, Duncan M, Ahima RS, Sharkey KA, Ferguson AV. Area postrema neurons are modulated by the adipocyte hormone adiponectin. J Neurosci. 2006;26:9695–9702. doi: 10.1523/JNEUROSCI.2014-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Frystyk J, Berne C, Berglund L, Jensevik K, Flyvbjerg A, Zethelius B. Serum adiponectin is a predictor of coronary heart disease: a population-based 10-year follow-up study in elderly men. J Clin Endocrinol Metab. 2007;92:571–576. doi: 10.1210/jc.2006-1067. [DOI] [PubMed] [Google Scholar]
- 108.Fujioka D, Kawabata K, Saito Y, Kobayashi T, Nakamura T, Kodama Y, Takano H, Obata JE, Kitta Y, Umetani K, Kugiyama K. Role of adiponectin receptors in endothelin-induced cellular hypertrophy in cultured cardiomyocytes and their expression in infarcted heart. Am J Physiol Heart Circ Physiol. 2006;290:H2409–H2416. doi: 10.1152/ajpheart.00987.2005. [DOI] [PubMed] [Google Scholar]
- 109.Fujioka K, Seaton TB, Rowe E, Jelinek CA, Raskin P, Lebovitz HE, Weinstein SP. Weight loss with sibutramine improves glycaemic control and other metabolic parameters in obese patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2000;2:175–187. doi: 10.1046/j.1463-1326.2000.00081.x. [DOI] [PubMed] [Google Scholar]
- 110.Fujita A, Sasaki H, Ogawa K, Okamoto K, Matsuno S, Matsumoto E, Furuta H, Nishi M, Nakao T, Tsuno T, Taniguchi H, Nanjo K. Increased gene expression of antioxidant enzymes in KKAy diabetic mice but not in STZ diabetic mice. Diabetes Res Clin Pract. 2005;69:113–119. doi: 10.1016/j.diabres.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 111.Garavaglia GE, Messerli FH, Nunez BD, Schmieder RE, Grossman E. Myocardial contractility and left ventricular function in obese patients with essential hypertension. Am J Cardiol. 1988;62:594–597. doi: 10.1016/0002-9149(88)90662-5. [DOI] [PubMed] [Google Scholar]
- 112.Garg S, Narula J, Chandrashekhar Y. Apoptosis and heart failure: clinical relevance and therapeutic target. J Mol Cell Cardiol. 2005;38:73–79. doi: 10.1016/j.yjmcc.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 113.George J, Patal S, Wexler D, Sharabi Y, Peleg E, Kamari Y, Grossman E, Sheps D, Keren G, Roth A. Circulating adiponectin concentrations in patients with congestive heart failure. Heart. 2006;92:1420–1424. doi: 10.1136/hrt.2005.083345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gerdts E, Wachtell K, Omvik P, Otterstad JE, Oikarinen L, Boman K, Dahlof B, Devereux RB. Left atrial size and risk of major cardiovascular events during antihypertensive treatment: losartan intervention for endpoint reduction in hypertension trial. Hypertension. 2007;49:311–316. doi: 10.1161/01.HYP.0000254322.96189.85. [DOI] [PubMed] [Google Scholar]
- 115.Golfman LS, Wilson CR, Sharma S, Burgmaier M, Young ME, Guthrie PH, Van Arsdall M, Adrogue JV, Brown KK, Taegtmeyer H. Activation of PPARgamma enhances myocardial glucose oxidation and improves contractile function in isolated working hearts of ZDF rats. Am J Physiol Endocrinol Metab. 2005;289:E328–E336. doi: 10.1152/ajpendo.00055.2005. [DOI] [PubMed] [Google Scholar]
- 116.Grandi AM, Zanzi P, Piantanida E, Gaudio G, Bertolini A, Guasti L, Venco A. Obesity and left ventricular diastolic function: noninvasive study in normotensives and newly diagnosed never-treated hypertensives. Int J Obes Relat Metab Disord. 2000;24:954–958. doi: 10.1038/sj.ijo.0801261. [DOI] [PubMed] [Google Scholar]
- 117.Grassi G, Facchini A, Trevano FQ, Dell’Oro R, Arenare F, Tana F, Bolla G, Monzani A, Robuschi M, Mancia G. Obstructive sleep apnea-dependent and -independent adrenergic activation in obesity. Hypertension. 2005;46:321–325. doi: 10.1161/01.HYP.0000174243.39897.6c. [DOI] [PubMed] [Google Scholar]
- 118.Grassi G, Seravalle G, Quarti-Trevano F, Dell’Oro R, Bolla G, Mancia G. Effects of hypertension and obesity on the sympathetic activation of heart failure patients. Hypertension. 2003;42:873–877. doi: 10.1161/01.HYP.0000098660.26184.63. [DOI] [PubMed] [Google Scholar]
- 119.Greer JJ, Ware DP, Lefer DJ. Myocardial infarction and heart failure in the db/db diabetic mouse. Am J Physiol Heart Circ Physiol. 2006;290:H146–H153. doi: 10.1152/ajpheart.00583.2005. [DOI] [PubMed] [Google Scholar]
- 120.Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975;56:56–64. doi: 10.1172/JCI108079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gruberg L, Weissman NJ, Waksman R, Fuchs S, Deible R, Pinnow EE, Ahmed LM, Kent KM, Pichard AD, Suddath WO, Satler LF, Lindsay J., Jr The impact of obesity on the short-term and long-term outcomes after percutaneous coronary intervention: the obesity paradox? J Am Coll Cardiol. 2002;39:578–584. doi: 10.1016/s0735-1097(01)01802-2. [DOI] [PubMed] [Google Scholar]
- 122.Gullestad L, Kjekshus J, Damas JK, Ueland T, Yndestad A, Aukrust P. Agents targeting inflammation in heart failure. Expert Opin Invest Drugs. 2005;14:557–566. doi: 10.1517/13543784.14.5.557. [DOI] [PubMed] [Google Scholar]
- 123.Gurm HS, Brennan DM, Booth J, Tcheng JE, Lincoff AM, Topol EJ. Impact of body mass index on outcome after percutaneous coronary intervention (the obesity paradox) Am J Cardiol. 2002;90:42–45. doi: 10.1016/s0002-9149(02)02384-6. [DOI] [PubMed] [Google Scholar]
- 124.Gustafsson F, Kragelund CB, Torp-Pedersen C, Seibaek M, Burchardt H, Akkan D, Thune JJ, Kober L. Effect of obesity and being overweight on long-term mortality in congestive heart failure: influence of left ventricular systolic function. Eur Heart J. 2005;26:58–64. doi: 10.1093/eurheartj/ehi022. [DOI] [PubMed] [Google Scholar]
- 125.Guven A, Koksal N, Cetinkaya A, Sokmen G, Ozdemir R. Effects of the sibutramine therapy on pulmonary artery pressure in obese patients. Diabetes Obes Metab. 2004;6:50–55. doi: 10.1111/j.1463-1326.2004.00314.x. [DOI] [PubMed] [Google Scholar]
- 126.Habbu A, Lakkis NM, Dokainish H. The obesity paradox: fact or fiction? Am J Cardiol. 2006;98:944–948. doi: 10.1016/j.amjcard.2006.04.039. [DOI] [PubMed] [Google Scholar]
- 127.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 128.Hall JA, French TK, Rasmusson KD, Vesty JC, Roberts CA, Rimmasch HL, Kfoury AG, Renlund DG. The paradox of obesity in patients with heart failure. J Am Acad Nurse Pract. 2005;17:542–546. doi: 10.1111/j.1745-7599.2005.00084.x. [DOI] [PubMed] [Google Scholar]
- 129.Han SH, Quon MJ, Kim JA, Koh KK. Adiponectin and cardiovascular disease: response to therapeutic interventions. J Am Coll Cardiol. 2007;49:531–538. doi: 10.1016/j.jacc.2006.08.061. [DOI] [PubMed] [Google Scholar]
- 130.Hashimoto N, Kanda J, Nakamura T, Horie A, Kurosawa H, Hashimoto T, Sato K, Kushida S, Suzuki M, Yano S, Iwai R, Takahashi H, Yoshida S. Association of hypoadiponectinemia in men with early onset of coronary heart disease and multiple coronary artery stenoses. Metabolism. 2006;55:1653–1657. doi: 10.1016/j.metabol.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 131.Hayakawa K, Takemura G, Kanoh M, Li Y, Koda M, Kawase Y, Maruyama R, Okada H, Minatoguchi S, Fujiwara T, Fujiwara H. Inhibition of granulation tissue cell apoptosis during the subacute stage of myocardial infarction improves cardiac remodeling and dysfunction at the chronic stage. Circulation. 2003;108:104–109. doi: 10.1161/01.CIR.0000074225.62168.68. [DOI] [PubMed] [Google Scholar]
- 132.He J, Ogden LG, Bazzano LA, Vupputuri S, Loria C, Whelton PK. Risk factors for congestive heart failure in US men and women: NHANES I epidemiologic follow-up study. Arch Intern Med. 2001;161:996–1002. doi: 10.1001/archinte.161.7.996. [DOI] [PubMed] [Google Scholar]
- 133.He Z, Opland DM, Way KJ, Ueki K, Bodyak N, Kang PM, Izumo S, Kulkarni RN, Wang B, Liao R, Kahn CR, King GL. Regulation of vascular endothelial growth factor expression and vascularization in the myocardium by insulin receptor and PI3K/Akt pathways in insulin resistance and ischemia. Arterioscler Thromb Vasc Biol. 2006;26:787–793. doi: 10.1161/01.ATV.0000209500.15801.4e. [DOI] [PubMed] [Google Scholar]
- 134.Heckbert SR, Post W, Pearson GD, Arnett DK, Gomes AS, Jerosch-Herold M, Hundley WG, Lima JA, Bluemke DA. Traditional cardiovascular risk factors in relation to left ventricular mass, volume, systolic function by cardiac magnetic resonance imaging: the Multiethnic Study of Atherosclerosis. J Am Coll Cardiol. 2006;48:2285–2292. doi: 10.1016/j.jacc.2006.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Henderson BC, Tyagi N, Ovechkin A, Kartha GK, Moshal KS, Tyagi SC. Oxidative remodeling in pressure overload induced chronic heart failure. Eur J Heart Failure. 2007;9:450–457. doi: 10.1016/j.ejheart.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 136.Hensrud DD, Klein S. Extreme obesity: a new medical crisis in the United States. Mayo Clin Proc. 2006;81:S5–S10. doi: 10.1016/s0025-6196(11)61175-0. [DOI] [PubMed] [Google Scholar]
- 137.Her C, Cerabona T, Bairamian M, McGoldrick KE. Right ventricular systolic function is not depressed in morbid obesity. Obes Surg. 2006;16:1287–1293. doi: 10.1381/096089206778663887. [DOI] [PubMed] [Google Scholar]
- 138.Hilfiker-Kleiner D, Landmesser U, Drexler H. Molecular mechanisms in heart failure focus on cardiac hypertrophy, inflammation, angiogenesis, apoptosis. J Am Coll Cardiol. 2006;48:A56–66. [Google Scholar]
- 139.Hintz KK, Aberle NS, Ren J. Insulin resistance induces hyperleptinemia, cardiac contractile dysfunction but not cardiac leptin resistance in ventricular myocytes. Int J Obes Relat Metab Disord. 2003;27:1196–1203. doi: 10.1038/sj.ijo.0802389. [DOI] [PubMed] [Google Scholar]
- 140.Hirschler V, Acebo HL, Fernandez GB, de Lujan Calcagno M, Gonzalez C, Jadzinsky M. Influence of obesity and insulin resistance on left atrial size in children. Pediatr Diabetes. 2006;7:39–44. doi: 10.1111/j.1399-543X.2006.00139.x. [DOI] [PubMed] [Google Scholar]
- 141.Ho CY, Solomon SD. A clinician’s guide to tissue Doppler imaging. Circulation. 2006;113:e396–398. doi: 10.1161/CIRCULATIONAHA.105.579268. [DOI] [PubMed] [Google Scholar]
- 142.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 143.Hong SJ, Park CG, Seo HS, Oh DJ, Ro YM. Associations among plasma adiponectin, hypertension, left ventricular diastolic function and left ventricular mass index. Blood Pressure. 2004;13:236–242. doi: 10.1080/08037050410021397. [DOI] [PubMed] [Google Scholar]
- 144.Horwich TB, Fonarow GC. The impact of obesity on survival in patients with heart failure. Heart Fail Monit. 2002;3:8–14. [PubMed] [Google Scholar]
- 145.Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Woo MA, Tillisch JH. The relationship between obesity and mortality in patients with heart failure. J Am Coll Cardiol. 2001;38:789–795. doi: 10.1016/s0735-1097(01)01448-6. [DOI] [PubMed] [Google Scholar]
- 146.Hoshida S, Yamashita N, Otsu K, Kuzuya T, Hori M. Cholesterol feeding exacerbates myocardial injury in Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol. 2000;278:H256–H262. doi: 10.1152/ajpheart.2000.278.1.H256. [DOI] [PubMed] [Google Scholar]
- 147.House AA, Walley VM. Right heart failure due to ventricular adiposity: “adipositas cordis”—an old diagnosis revisited. Can J Cardiol. 1996;12:485–489. [PubMed] [Google Scholar]
- 148.How OJ, Aasum E, Severson DL, Chan WY, Essop MF, Larsen TS. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes. 2006;55:466–473. doi: 10.2337/diabetes.55.02.06.db05-1164. [DOI] [PubMed] [Google Scholar]
- 149.Hsueh W, Abel ED, Breslow JL, Maeda N, Davis RC, Fisher EA, Dansky H, McClain DA, McIndoe R, Wassef MK, Rabadan-Diehl C, Goldberg IJ. Recipes for creating animal models of diabetic cardiovascular disease. Circ Res. 2007;100:1415–1427. doi: 10.1161/01.RES.0000266449.37396.1f. [DOI] [PubMed] [Google Scholar]
- 150.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol. 2003;285:H1261–H1269. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 151.Iacobellis G. True uncomplicated obesity is not related to increased left ventricular mass and systolic dysfunction. J Am Coll Cardiol. 2004;44:2257–2258. doi: 10.1016/j.jacc.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 152.Iacobellis G, Leonetti F. Epicardial adipose tissue and insulin resistance in obese subjects. J Clin Endocrinol Metab. 2005;90:6300–6302. doi: 10.1210/jc.2005-1087. [DOI] [PubMed] [Google Scholar]
- 153.Iacobellis G, Leonetti F, Singh N. Relationship of epicardial adipose tissue with atrial dimensions and diastolic function in morbidly obese subjects. Int J Cardiol. 2007;115:272–273. doi: 10.1016/j.ijcard.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 154.Iacobellis G, Petrone A, Leonetti F, Buzzetti R. Left ventricular mass and +276 G/G single nucleotide polymorphism of the adiponectin gene in uncomplicated obesity. Obesity. 2006;14:368–372. doi: 10.1038/oby.2006.48. [DOI] [PubMed] [Google Scholar]
- 155.Iacobellis G, Ribaudo MC, Leto G, Zappaterreno A, Vecci E, Di Mario U, Leonetti F. Influence of excess fat on cardiac morphology and function: study in uncomplicated obesity. Obes Res. 2002;10:767–773. doi: 10.1038/oby.2002.104. [DOI] [PubMed] [Google Scholar]
- 156.Iacobellis G, Ribaudo MC, Zappaterreno A, Iannucci CV, Di Mario U, Leonetti F. Adapted changes in left ventricular structure and function in severe uncomplicated obesity. Obes Res. 2004;12:1616–1621. doi: 10.1038/oby.2004.201. [DOI] [PubMed] [Google Scholar]
- 157.Ingelsson E, Arnlov J, Sundstrom J, Lind L. Inflammation, as measured by the erythrocyte sedimentation rate, is an independent predictor for the development of heart failure. J Am Coll Cardiol. 2005;45:1802–1806. doi: 10.1016/j.jacc.2005.02.066. [DOI] [PubMed] [Google Scholar]
- 158.Ito H, Nakano A, Kinoshita M, Matsumori A. Pioglitazone, a peroxisome proliferator-activated receptor-gamma agonist, attenuates myocardial ischemia/reperfusion injury in a rat model. Lab Invest. 2003;83:1715–1721. doi: 10.1097/01.lab.0000106724.29121.da. [DOI] [PubMed] [Google Scholar]
- 159.Johns DG, Ao Z, Eybye M, Olzinski A, Costell M, Gruver S, Smith SA, Douglas SA, Macphee CH. Rosiglitazone protects against ischemia/reperfusion-induced leukocyte adhesion in the Zucker diabetic fatty rat. J Pharmacol Exp Ther. 2005;315:1020–1027. doi: 10.1124/jpet.105.090993. [DOI] [PubMed] [Google Scholar]
- 160.Jones SP, Girod WG, Granger DN, Palazzo AJ, Lefer DJ. Reperfusion injury is not affected by blockade of P-selectin in the diabetic mouse heart. Am J Physiol Heart Circ Physiol. 1999;277:H763–H769. doi: 10.1152/ajpheart.1999.277.2.H763. [DOI] [PubMed] [Google Scholar]
- 161.Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–451. doi: 10.1210/er.2005-0005. [DOI] [PubMed] [Google Scholar]
- 162.Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, the metabolic syndrome. J Clin Invest. 2006;116:1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. 2000;106:473–481. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kalantar-Zadeh K, Abbott KC, Salahudeen AK, Kilpatrick RD, Horwich TB. Survival advantages of obesity in dialysis patients. Am J Clin Nutr. 2005;81:543–554. doi: 10.1093/ajcn/81.3.543. [DOI] [PubMed] [Google Scholar]
- 165.Kaltman AJ, Goldring RM. Role of circulatory congestion in the cardiorespiratory failure of obesity. Am J Med. 1976;60:645–653. doi: 10.1016/0002-9343(76)90499-x. [DOI] [PubMed] [Google Scholar]
- 166.Kanaya AM, Wassel Fyr C, Vittinghoff E, Havel PJ, Cesari M, Nicklas B, Harris T, Newman AB, Satterfield S, Cummings SR. Serum adiponectin and coronary heart disease risk in older Black and White Americans. J Clin Endocrinol Metab. 2006;91:5044–5050. doi: 10.1210/jc.2006-0107. [DOI] [PubMed] [Google Scholar]
- 167.Kanda T, Saegusa S, Takahashi T, Sumino H, Morimoto S, Nakahashi T, Iwai K, Matsumoto M. Reduced-energy diet improves survival of obese KKAy mice with viral myocarditis: Induction of cardiac adiponectin expression. Int J Cardiol. 2007;119:310–318. doi: 10.1016/j.ijcard.2006.07.181. [DOI] [PubMed] [Google Scholar]
- 168.Karason K, Lindroos AK, Stenlof K, Sjostrom L. Relief of cardiorespiratory symptoms and increased physical activity after surgically induced weight loss: results from the Swedish Obese Subjects study. Arch Intern Med. 2000;160:1797–1802. doi: 10.1001/archinte.160.12.1797. [DOI] [PubMed] [Google Scholar]
- 169.Karason K, Wallentin I, Larsson B, Sjostrom L. Effects of obesity and weight loss on left ventricular mass and relative wall thickness: survey and intervention study. BMJ. 1997;315:912–916. doi: 10.1136/bmj.315.7113.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kasper EK, Hruban RH, Baughman KL. Cardiomyopathy of obesity: a clinicopathologic evaluation of 43 obese patients with heart failure. Am J Cardiol. 1992;70:921–924. doi: 10.1016/0002-9149(92)90739-l. [DOI] [PubMed] [Google Scholar]
- 171.Katzmarzyk PT, Mason C. Prevalence of class I, II and III obesity in Canada. CMAJ. 2006;174:156–157. doi: 10.1503/cmaj.050806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kawano T, Saito T, Yasu T, Nakamura T, Namai K, Tamemoto H, Kawakami M, Saito M, Ishikawa SE. Close association of hypoadiponectinemia with arteriosclerosis obliterans and ischemic heart disease. Metabolism. 2005;54:653–656. doi: 10.1016/j.metabol.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 173.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS. Obesity and the risk of heart failure. N Engl J Med. 2002;347:305–313. doi: 10.1056/NEJMoa020245. [DOI] [PubMed] [Google Scholar]
- 174.Kenchaiah S, Gaziano JM, Vasan RS. Impact of obesity on the risk of heart failure and survival after the onset of heart failure. Med Clin North Am. 2004;88:1273–1294. doi: 10.1016/j.mcna.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 175.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 176.Khandoudi N, Delerive P, Berrebi-Bertrand I, Buckingham RE, Staels B, Bril A. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma, inhibits the Jun NH(2)-terminal kinase/activating protein 1 pathway and protects the heart from ischemia/reperfusion injury. Diabetes. 2002;51:1507–1514. doi: 10.2337/diabetes.51.5.1507. [DOI] [PubMed] [Google Scholar]
- 177.Kilhovd BK, Juutilainen A, Lehto S, Ronnemaa T, Torjesen PA, Birkeland KI, Berg TJ, Hanssen KF, Laakso M. High serum levels of advanced glycation end products predict increased coronary heart disease mortality in nondiabetic women but not in nondiabetic men: a population-based 18-year follow-up study. Arterioscler Thromb Vasc Biol. 2005;25:815–820. doi: 10.1161/01.ATV.0000158380.44231.fe. [DOI] [PubMed] [Google Scholar]
- 178.Kim E, Choe YH, Han BK, Kim SM, Kim JS, Park SW, Sung J. Right ventricular fat infiltration in asymptomatic subjects: observations from ECG-gated 16-slice multidetector CT. J Comput Assist Tomogr. 2007;31:22–28. doi: 10.1097/01.rct.0000236416.05267.6c. [DOI] [PubMed] [Google Scholar]
- 179.Kistorp C, Faber J, Galatius S, Gustafsson F, Frystyk J, Flyvbjerg A, Hildebrandt P. Plasma adiponectin, body mass index, mortality in patients with chronic heart failure. Circulation. 2005;112:1756–1762. doi: 10.1161/CIRCULATIONAHA.104.530972. [DOI] [PubMed] [Google Scholar]
- 180.Kizer JR, Bella JN, Palmieri V, Liu JE, Best LG, Lee ET, Roman MJ, Devereux RB. Left atrial diameter as an independent predictor of first clinical cardiovascular events in middle-aged and elderly adults: the Strong Heart Study (SHS) Am Heart J. 2006;151:412–418. doi: 10.1016/j.ahj.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 181.Klein S, Burke LE, Bray GA, Blair S, Allison DB, Pi-Sunyer X, Hong Y, Eckel RH. Clinical implications of obesity with specific focus on cardiovascular disease: a statement for professionals from the American Heart Association Council on Nutrition, Physical Activity, Metabolism: endorsed by the American College of Cardiology Foundation. Circulation. 2004;110:2952–2967. doi: 10.1161/01.CIR.0000145546.97738.1E. [DOI] [PubMed] [Google Scholar]
- 182.Klein S, Fontana L, Young VL, Coggan AR, Kilo C, Patterson BW, Mohammed BS. Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N Engl J Med. 2004;350:2549–2557. doi: 10.1056/NEJMoa033179. [DOI] [PubMed] [Google Scholar]
- 183.Koenig W, Khuseyinova N, Baumert J, Meisinger C, Lowel H. Serum concentrations of adiponectin and risk of type 2 diabetes mellitus and coronary heart disease in apparently healthy middle-aged men: results from the 18-year follow-up of a large cohort from southern Germany. J Am Coll Cardiol. 2006;48:1369–1377. doi: 10.1016/j.jacc.2006.06.053. [DOI] [PubMed] [Google Scholar]
- 184.Kong JY, Rabkin SW. Mitochondrial effects with ceramide-induced cardiac apoptosis are different from those of palmitate. Arch Biochem Biophys. 2003;412:196–206. doi: 10.1016/s0003-9861(03)00008-0. [DOI] [PubMed] [Google Scholar]
- 185.Kortelainen ML. Adiposity, cardiac size and precursors of coronary atherosclerosis in 5- to 15-year-old children: a retrospective study of 210 violent deaths. Int J Obes Relat Metab Disord. 1997;21:691–697. doi: 10.1038/sj.ijo.0800463. [DOI] [PubMed] [Google Scholar]
- 186.Kortelainen ML. Association between cardiac pathology and fat tissue distribution in an autopsy series of men without premortem evidence of cardiovascular disease. Int J Obes Relat Metab Disord. 1996;20:245–252. [PubMed] [Google Scholar]
- 187.Kortelainen ML. Myocardial infarction and coronary pathology in severely obese people examined at autopsy. Int J Obes Relat Metab Disord. 2002;26:73–79. doi: 10.1038/sj.ijo.0801852. [DOI] [PubMed] [Google Scholar]
- 188.Kortelainen ML, Sarkioja T. Visceral fat and coronary pathology in male adolescents. Int J Obes Relat Metab Disord. 2001;25:228–232. doi: 10.1038/sj.ijo.0801466. [DOI] [PubMed] [Google Scholar]
- 189.Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klovekorn WP, Schaper J. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–724. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 190.Kotsis V, Stabouli S, Bouldin M, Low A, Toumanidis S, Zakopoulos N. Impact of obesity on 24-hour ambulatory blood pressure and hypertension. Hypertension. 2005;45:602–607. doi: 10.1161/01.HYP.0000158261.86674.8e. [DOI] [PubMed] [Google Scholar]
- 191.Krishnan R, Becker RJ, Beighley LM, Lopez-Candales A. Impact of body mass index on markers of left ventricular thickness and mass calculation: results of a pilot analysis. Echocardiography. 2005;22:203–210. doi: 10.1111/j.0742-2822.2005.03138.x. [DOI] [PubMed] [Google Scholar]
- 192.Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, Eto K, Yamashita T, Kamon J, Satoh H, Yano W, Froguel P, Nagai R, Kimura S, Kadowaki T, Noda T. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002;277:25863–25866. doi: 10.1074/jbc.C200251200. [DOI] [PubMed] [Google Scholar]
- 193.Kusminski CM, McTernan PG, Schraw T, Kos K, O’Hare JP, Ahima R, Kumar S, Scherer PE. Adiponectin complexes in human cerebrospinal fluid: distinct complex distribution from serum. Diabetologia. 2007;50:634–642. doi: 10.1007/s00125-006-0577-9. [DOI] [PubMed] [Google Scholar]
- 194.Ladyman SR, Grattan DR. Region-specific reduction in leptin-induced phosphorylation of signal transducer and activator of transcription-3 (STAT3) in the rat hypothalamus is associated with leptin resistance during pregnancy. Endocrinology. 2004;145:3704–3711. doi: 10.1210/en.2004-0338. [DOI] [PubMed] [Google Scholar]
- 195.Lam KS, Xu A. Adiponectin: protection of the endothelium. Curr Diab Rep. 2005;5:254–259. doi: 10.1007/s11892-005-0019-y. [DOI] [PubMed] [Google Scholar]
- 196.Lara MD, Kothari SN, Sugerman HJ. Surgical management of obesity: a review of the evidence relating to the health benefits and risks. Treat Endocrinol. 2005;4:55–64. doi: 10.2165/00024677-200504010-00006. [DOI] [PubMed] [Google Scholar]
- 197.Lauer MS, Anderson KM, Larson MG, Levy D. A new method for indexing left ventricular mass for differences in body size. Am J Cardiol. 1994;74:487–491. doi: 10.1016/0002-9149(94)90909-1. [DOI] [PubMed] [Google Scholar]
- 198.Laughlin GA, Barrett-Connor E, May S, Langenberg C. Association of adiponectin with coronary heart disease and mortality: the Rancho Bernardo study. Am J Epidemiol. 2007;165:164–174. doi: 10.1093/aje/kwk001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lavie CJ, Messerli FH. Cardiovascular adaptation to obesity and hypertension. Chest. 1986;90:275–279. doi: 10.1378/chest.90.2.275. [DOI] [PubMed] [Google Scholar]
- 200.Lavie CJ, Milani RV. Obesity and cardiovascular disease: the Hippocrates paradox? J Am Coll Cardiol. 2003;42:677–679. doi: 10.1016/s0735-1097(03)00784-8. [DOI] [PubMed] [Google Scholar]
- 201.Lavie CJ, Osman AF, Milani RV, Mehra MR. Body composition and prognosis in chronic systolic heart failure: the obesity paradox. Am J Cardiol. 2003;91:891–894. doi: 10.1016/s0002-9149(03)00031-6. [DOI] [PubMed] [Google Scholar]
- 202.Lee Y, Naseem RH, Duplomb L, Park BH, Garry DJ, Richardson JA, Schaffer JE, Unger RH. Hyperleptinemia prevents lipotoxic cardiomyopathy in acyl CoA synthase transgenic mice. Proc Natl Acad Sci USA. 2004;101:13624–13629. doi: 10.1073/pnas.0405499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lee Y, Naseem RH, Park BH, Garry DJ, Richardson JA, Schaffer JE, Unger RH. Alpha-lipoic acid prevents lipotoxic cardiomyopathy in acyl CoA-synthase transgenic mice. Biochem Biophys Res Commun. 2006;344:446–452. doi: 10.1016/j.bbrc.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 204.Lee YH, Pratley RE. The evolving role of inflammation in obesity and the metabolic syndrome. Curr Diab Rep. 2005;5:70–75. doi: 10.1007/s11892-005-0071-7. [DOI] [PubMed] [Google Scholar]
- 205.Li SY, Liu Y, Sigmon VK, McCort A, Ren J. High-fat diet enhances visceral advanced glycation end products, nuclear O-Glc-Nac modification, p38 mitogen-activated protein kinase activation and apoptosis. Diabetes Obes Metab. 2005;7:448–454. doi: 10.1111/j.1463-1326.2004.00387.x. [DOI] [PubMed] [Google Scholar]
- 206.Li SY, Sigmon VK, Babcock SA, Ren J. Advanced glycation endproduct induces ROS accumulation, apoptosis, MAP kinase activation and nuclear O-GlcNAcylation in human cardiac myocytes. Life Sci. 2007;80:1051–1056. doi: 10.1016/j.lfs.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 207.Li SY, Yang X, Ceylan-Isik AF, Du M, Sreejayan N, Ren J. Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco (endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch. Diabetologia. 2006;49:1434–1446. doi: 10.1007/s00125-006-0229-0. [DOI] [PubMed] [Google Scholar]
- 208.Liao Y, Takashima S, Maeda N, Ouchi N, Komamura K, Shimomura I, Hori M, Matsuzawa Y, Funahashi T, Kitakaze M. Exacerbation of heart failure in adiponectin-deficient mice due to impaired regulation of AMPK and glucose metabolism. Cardiovasc Res. 2005;67:705–713. doi: 10.1016/j.cardiores.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 209.Lindsay RS, Resnick HE, Zhu J, Tun ML, Howard BV, Zhang Y, Yeh J, Best LG. Adiponectin and coronary heart disease: the Strong Heart Study. Arterioscler Thromb Vasc Biol. 2005;25:e15–16. doi: 10.1161/01.ATV.0000153090.21990.8c. [DOI] [PubMed] [Google Scholar]
- 210.Lisman KA, Stetson SJ, Koerner MM, Farmer JA, Torre-Amione G. The role of inflammation in the pathogenesis of heart failure. Curr Cardiol Rep. 2002;4:200–205. doi: 10.1007/s11886-002-0051-3. [DOI] [PubMed] [Google Scholar]
- 211.Lissin LW, Gauri AJ, Froelicher VF, Ghayoumi A, Myers J, Giacommini J. The prognostic value of body mass index and standard exercise testing in male veterans with congestive heart failure. J Card Fail. 2002;8:206–215. doi: 10.1054/jcaf.2002.126812. [DOI] [PubMed] [Google Scholar]
- 212.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. 2001;276:14890–14895. doi: 10.1074/jbc.M010286200. [DOI] [PubMed] [Google Scholar]
- 214.Listenberger LL, Schaffer JE. Mechanisms of lipoapoptosis: implications for human heart disease. Trends Cardiovasc Med. 2002;12:134–138. doi: 10.1016/s1050-1738(02)00152-4. [DOI] [PubMed] [Google Scholar]
- 215.Lowell BB, S-Susulic V, Hamann A, Lawitts JA, Himms-Hagen J, Boyer BB, Kozak LP, Flier JS. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature. 1993;366:740–742. doi: 10.1038/366740a0. [DOI] [PubMed] [Google Scholar]
- 216.Luo JD, Zhang GS, Chen MS. Leptin and cardiovascular diseases. Timely Top Med Cardiovasc Dis. 2005;9:E34. [PubMed] [Google Scholar]
- 217.Madani S, De Girolamo S, Munoz DM, Li RK, Sweeney G. Direct effects of leptin on size and extracellular matrix components of human pediatric ventricular myocytes. Cardiovasc Res. 2006;69:716–725. doi: 10.1016/j.cardiores.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 218.Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, Komuro R, Ouchi N, Kihara S, Tochino Y, Okutomi K, Horie M, Takeda S, Aoyama T, Funahashi T, Matsuzawa Y. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–737. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- 219.Maggio CA, Pi-Sunyer FX. Obesity and type 2 diabetes. Endocrinol Metab Clin North Am. 2003;32:805–822. doi: 10.1016/s0889-8529(03)00071-9. [DOI] [PubMed] [Google Scholar]
- 220.Malavazos AE, Ermetici F, Coman C, Corsi MM, Morricone L, Ambrosi B. Influence of epicardial adipose tissue and adipocytokine levels on cardiac abnormalities in visceral obesity. Int J Cardiol. 2007;121:132–134. doi: 10.1016/j.ijcard.2006.08.061. [DOI] [PubMed] [Google Scholar]
- 221.Marette A. Molecular mechanisms of inflammation in obesity-linked insulin resistance. Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S46–S48. doi: 10.1038/sj.ijo.0802500. [DOI] [PubMed] [Google Scholar]
- 222.Mark AL, Correia ML, Rahmouni K, Haynes WG. Loss of leptin actions in obesity: two concepts with cardiovascular implications. Clin Exp Hypertens. 2004;26:629–636. doi: 10.1081/ceh-200031948. [DOI] [PubMed] [Google Scholar]
- 223.Mark AL, Correia ML, Rahmouni K, Haynes WG. Selective leptin resistance: a new concept in leptin physiology with cardiovascular implications. J Hypertens. 2002;20:1245–1250. doi: 10.1097/00004872-200207000-00001. [DOI] [PubMed] [Google Scholar]
- 224.Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG. Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens. 1999;17:1949–1953. doi: 10.1097/00004872-199917121-00026. [DOI] [PubMed] [Google Scholar]
- 225.Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, Shinyama H, Sharp MG, Fleming S, Mullins JJ, Seckl JR, Flier JS. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest. 2003;112:83–90. doi: 10.1172/JCI17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Matsuda M, Shimomura I, Sata M, Arita Y, Nishida M, Maeda N, Kumada M, Okamoto Y, Nagaretani H, Nishizawa H, Kishida K, Komuro R, Ouchi N, Kihara S, Nagai R, Funahashi T, Matsuzawa Y. Role of adiponectin in preventing vascular stenosis. The missing link of adipo-vascular axis. J Biol Chem. 2002;277:37487–37491. doi: 10.1074/jbc.M206083200. [DOI] [PubMed] [Google Scholar]
- 227.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 228.McGavock JM, Victor RG, Unger RH, Szczepaniak LS. Adiposity of the heart, revisited. Ann Intern Med. 2006;144:517–524. doi: 10.7326/0003-4819-144-7-200604040-00011. [DOI] [PubMed] [Google Scholar]
- 229.McGill HC, Jr, McMahan CA, Herderick EE, Zieske AW, Malcom GT, Tracy RE, Strong JP. Obesity accelerates the progression of coronary atherosclerosis in young men. Circulation. 2002;105:2712–2718. doi: 10.1161/01.cir.0000018121.67607.ce. [DOI] [PubMed] [Google Scholar]
- 230.McLennan PL, Barnden LR, Bridle TM, Abeywardena MY, Charnock JS. Dietary fat modulation of left ventricular ejection fraction in the marmoset due to enhanced filling. Cardiovasc Res. 1992;26:871–877. doi: 10.1093/cvr/26.9.871. [DOI] [PubMed] [Google Scholar]
- 231.McQueen AP, Zhang D, Hu P, Swenson L, Yang Y, Zaha VG, Hoffman JL, Yun UJ, Chakrabarti G, Wang Z, Albertine KH, Abel ED, Litwin SE. Contractile dysfunction in hypertrophied hearts with deficient insulin receptor signaling: possible role of reduced capillary density. J Mol Cell Cardiol. 2005;39:882–892. doi: 10.1016/j.yjmcc.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 232.Mehra MR, Uber PA, Park MH, Scott RL, Ventura HO, Harris BC, Frohlich ED. Obesity and suppressed B-type natriuretic peptide levels in heart failure. J Am Coll Cardiol. 2004;43:1590–1595. doi: 10.1016/j.jacc.2003.10.066. [DOI] [PubMed] [Google Scholar]
- 233.Mehta RH, Califf RM, Garg J, White HD, Van de Werf F, Armstrong PW, Pieper KS, Topol EJ, Granger CB. The impact of anthropomorphic indices on clinical outcomes in patients with acute ST-elevation myocardial infarction. Eur Heart J. 2007;28:415–424. doi: 10.1093/eurheartj/ehl329. [DOI] [PubMed] [Google Scholar]
- 234.Mensah GA, Mokdad AH, Ford E, Narayan KM, Giles WH, Vinicor F, Deedwania PC. Obesity, metabolic syndrome, type 2 diabetes: emerging epidemics and their cardiovascular implications. Cardiol Clin. 2004;22:485–504. doi: 10.1016/j.ccl.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 235.Mensah GA, Treiber FA, Kapuku GK, Davis H, Barnes VA, Strong WB. Patterns of body fat deposition in youth and their relation to left ventricular markers of adverse cardiovascular prognosis. Am J Cardiol. 1999;84:583–588. doi: 10.1016/s0002-9149(99)00383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Miller TA, LeBrasseur NK, Cote GM, Trucillo MP, Pimentel DR, Ido Y, Ruderman NB, Sawyer DB. Oleate prevents palmitate-induced cytotoxic stress in cardiac myocytes. Biochem Biophys Res Commun. 2005;336:309–315. doi: 10.1016/j.bbrc.2005.08.088. [DOI] [PubMed] [Google Scholar]
- 237.Miner EC, Miller WL. A look between the cardiomyocytes: the extracellular matrix in heart failure. Mayo Clin Proc. 2006;81:71–76. doi: 10.4065/81.1.71. [DOI] [PubMed] [Google Scholar]
- 238.Minhas KM, Khan SA, Raju SV, Phan AC, Gonzalez DR, Skaf MW, Lee K, Tejani AD, Saliaris AP, Barouch LA, O’Donnell CP, Emala CW, Berkowitz DE, Hare JM. Leptin repletion restores depressed β-adrenergic contractility in ob/ob mice independently of cardiac hypertrophy. J Physiol. 2005;565:463–474. doi: 10.1113/jphysiol.2005.084566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Morgan EE, Rennison JH, Young ME, McElfresh TA, Kung TA, Tserng KY, Hoit BD, Stanley WC, Chandler MP. Effects of chronic activation of peroxisome proliferator-activated receptor-alpha or high-fat feeding in a rat infarct model of heart failure. Am J Physiol Heart Circ Physiol. 2006;290:H1899–H1904. doi: 10.1152/ajpheart.01014.2005. [DOI] [PubMed] [Google Scholar]
- 240.Morissette MR, Rosenzweig A. Targeting survival signaling in heart failure. Curr Opin Pharmacol. 2005;5:165–170. doi: 10.1016/j.coph.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 241.Morricone L, Donati C, Hassan T, Cioffi P, Caviezel F. Relationship of visceral fat distribution to angiographically assessed coronary artery disease: results in subjects with or without diabetes or impaired glucose tolerance. Nutr Metab Cardiovasc Dis. 2002;12:275–283. [PubMed] [Google Scholar]
- 242.Morricone L, Malavazos AE, Coman C, Donati C, Hassan T, Caviezel F. Echocardiographic abnormalities in normotensive obese patients: relationship with visceral fat. Obes Res. 2002;10:489–498. doi: 10.1038/oby.2002.67. [DOI] [PubMed] [Google Scholar]
- 243.Moshal KS, Tyagi N, Moss V, Henderson B, Steed M, Ovechkin A, Aru GM, Tyagi SC. Early induction of matrix metalloproteinase-9 transduces signaling in human heart end stage failure. J Cell Mol Med. 2005;9:704–713. doi: 10.1111/j.1582-4934.2005.tb00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Mosterd A, Cost B, Hoes AW, de Bruijne MC, Deckers JW, Hofman A, Grobbee DE. The prognosis of heart failure in the general population: The Rotterdam Study. Eur Heart J. 2001;22:1318–1327. doi: 10.1053/euhj.2000.2533. [DOI] [PubMed] [Google Scholar]
- 245.Mottram PM, Marwick TH. Assessment of diastolic function: what the general cardiologist needs to know. Heart. 2005;91:681–695. doi: 10.1136/hrt.2003.029413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Mueller C, Laule-Kilian K, Christ A, Brunner-La Rocca HP, Perruchoud AP. Inflammation and long-term mortality in acute congestive heart failure. Am Heart J. 2006;151:845–850. doi: 10.1016/j.ahj.2005.06.046. [DOI] [PubMed] [Google Scholar]
- 247.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28:463–491. doi: 10.1210/er.2007-0006. [DOI] [PubMed] [Google Scholar]
- 248.Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- 249.Murray AJ, Panagia M, Hauton D, Gibbons GF, Clarke K. Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes. 2005;54:3496–3502. doi: 10.2337/diabetes.54.12.3496. [DOI] [PubMed] [Google Scholar]
- 250.Myslinski W, Duchna HW, Rasche K, Dichmann M, Mosiewicz J, Schultze-Werninghaus G. Left ventricular geometry in patients with obstructive sleep apnea coexisting with treated systemic hypertension. Respiration. 2007;74:176–183. doi: 10.1159/000091187. [DOI] [PubMed] [Google Scholar]
- 251.Nakamura T, Funayama H, Kubo N, Yasu T, Kawakami M, Saito M, Momomura S, Ishikawa SE. Association of hyperadi-ponectinemia with severity of ventricular dysfunction in congestive heart failure. Circ J. 2006;70:1557–1562. doi: 10.1253/circj.70.1557. [DOI] [PubMed] [Google Scholar]
- 252.Narkiewicz K, van de Borne PJ, Cooley RL, Dyken ME, Somers VK. Sympathetic activity in obese subjects with and without obstructive sleep apnea. Circulation. 1998;98:772–776. doi: 10.1161/01.cir.98.8.772. [DOI] [PubMed] [Google Scholar]
- 253.Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW, Khaw BA. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182–1189. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- 254.Nigam A, Wright RS, Allison TG, Williams BA, Kopecky SL, Reeder GS, Murphy JG, Jaffe AS. Excess weight at time of presentation of myocardial infarction is associated with lower initial mortality risks but higher long-term risks including recurrent re-infarction and cardiac death. Int J Cardiol. 2006;110:153–159. doi: 10.1016/j.ijcard.2005.06.040. [DOI] [PubMed] [Google Scholar]
- 255.Nilsson PM. Diabetes and obesity: new data on mechanisms and intervention trials. Expert Rev Cardiovasc Ther. 2005;3:243–247. doi: 10.1586/14779072.3.2.243. [DOI] [PubMed] [Google Scholar]
- 256.Niroumand M, Kuperstein R, Sasson Z, Hanly PJ. Impact of obstructive sleep apnea on left ventricular mass and diastolic function. Am J Respir Crit Care Med. 2001;163:1632–1636. doi: 10.1164/ajrccm.163.7.2007014. [DOI] [PubMed] [Google Scholar]
- 257.Noda A, Okada T, Yasuma F, Nakashima N, Yokota M. Cardiac hypertrophy in obstructive sleep apnea syndrome. Chest. 1995;107:1538–1544. doi: 10.1378/chest.107.6.1538. [DOI] [PubMed] [Google Scholar]
- 258.Nunes JP, Oliveira NP. Cardiac structure and apnea/hypopnea index in patients with arterial hypertension and excessive weight. Kidney Blood Press Res. 2006;29:159–164. doi: 10.1159/000095349. [DOI] [PubMed] [Google Scholar]
- 259.Ohashi K, Kihara S, Ouchi N, Kumada M, Fujita K, Hiuge A, Hibuse T, Ryo M, Nishizawa H, Maeda N, Maeda K, Shibata R, Walsh K, Funahashi T, Shimomura I. Adiponectin replenishment ameliorates obesity-related hypertension. Hypertension. 2006;47:1108–1116. doi: 10.1161/01.HYP.0000222368.43759.a1. [DOI] [PubMed] [Google Scholar]
- 260.Okere IC, Chandler MP, McElfresh TA, Rennison JH, Sharov V, Sabbah HN, Tserng KY, Hoit BD, Ernsberger P, Young ME, Stanley WC. Differential effects of saturated and unsaturated fatty acid diets on cardiomyocyte apoptosis, adipose distribution, serum leptin. Am J Physiol Heart Circ Physiol. 2006;291:H38–H44. doi: 10.1152/ajpheart.01295.2005. [DOI] [PubMed] [Google Scholar]
- 261.Okere IC, Chess DJ, McElfresh TA, Johnson J, Rennison J, Ernsberger P, Hoit BD, Chandler MP, Stanley WC. High-fat diet prevents cardiac hypertrophy and improves contractile function in the hypertensive Dahl salt-sensitive rat. Clin Exp Pharmacol Physiol. 2005;32:825–831. doi: 10.1111/j.1440-1681.2005.04272.x. [DOI] [PubMed] [Google Scholar]
- 262.Okere IC, Young ME, McElfresh TA, Chess DJ, Sharov VG, Sabbah HN, Hoit BD, Ernsberger P, Chandler MP, Stanley WC. Low carbohydrate/high-fat diet attenuates cardiac hypertrophy, remodeling, altered gene expression in hypertension. Hypertension. 2006;48:1116–1123. doi: 10.1161/01.HYP.0000248430.26229.0f. [DOI] [PubMed] [Google Scholar]
- 263.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 264.Ommen SR, Nishimura RA, Appleton CP, Miller FA, Oh JK, Redfield MM, Tajik AJ. Clinical utility of Doppler echocardiography and tissue Doppler imaging in the estimation of left ventricular filling pressures: a comparative simultaneous Doppler-catheterization study. Circulation. 2000;102:1788–1794. doi: 10.1161/01.cir.102.15.1788. [DOI] [PubMed] [Google Scholar]
- 265.Onay-Besikci A, Altarejos JY, Lopaschuk GD. gAd-globular head domain of adiponectin increases fatty acid oxidation in newborn rabbit hearts. J Biol Chem. 2004;279:44320–44326. doi: 10.1074/jbc.M400347200. [DOI] [PubMed] [Google Scholar]
- 266.Otto ME, Belohlavek M, Khandheria B, Gilman G, Svatikova A, Somers V. Comparison of right and left ventricular function in obese and nonobese men. Am J Cardiol. 2004;93:1569–1572. doi: 10.1016/j.amjcard.2004.02.073. [DOI] [PubMed] [Google Scholar]
- 267.Ouchi N, Shibata R, Walsh K. Cardioprotection by adiponectin. Trends Cardiovasc Med. 2006;16:141–146. doi: 10.1016/j.tcm.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Ouchi N, Shibata R, Walsh K. Targeting adiponectin for cardio-protection. Expert Opin Ther Targets. 2006;10:573–581. doi: 10.1517/14728222.10.4.573. [DOI] [PubMed] [Google Scholar]
- 269.Ouwens DM, Boer C, Fodor M, de Galan P, Heine RJ, Maassen JA, Diamant M. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia. 2005;48:1229–1237. doi: 10.1007/s00125-005-1755-x. [DOI] [PubMed] [Google Scholar]
- 270.Padwal RS, Majumdar SR. Drug treatments for obesity: orlistat, sibutramine, rimonabant. Lancet. 2007;369:71–77. doi: 10.1016/S0140-6736(07)60033-6. [DOI] [PubMed] [Google Scholar]
- 271.Palanivel R, Fang X, Park M, Eguchi M, Pallan S, De Girolamo S, Liu Y, Wang Y, Xu A, Sweeney G. Globular and full-length adiponectin mediate specific changes in glucose and fatty acid uptake and metabolism in cardiomyocytes. Cardiovasc Res. 2007;75:148–157. doi: 10.1016/j.cardiores.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 272.Palmieri V, Bella JN, Arnett DK, Liu JE, Oberman A, Schuck MY, Kitzman DW, Hopkins PN, Morgan D, Rao DC, Devereux RB. Effect of type 2 diabetes mellitus on left ventricular geometry and systolic function in hypertensive subjects: Hypertension Genetic Epidemiology Network (HyperGEN) study. Circulation. 2001;103:102–107. doi: 10.1161/01.cir.103.1.102. [DOI] [PubMed] [Google Scholar]
- 273.Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, Dey A, Rothermel B, Kim YB, Kalinowski A, Russell KS, Kim JK. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. 2005;54:3530–3540. doi: 10.2337/diabetes.54.12.3530. [DOI] [PubMed] [Google Scholar]
- 274.Pascual M, Pascual DA, Soria F, Vicente T, Hernandez AM, Tebar FJ, Valdes M. Effects of isolated obesity on systolic and diastolic left ventricular function. Heart. 2003;89:1152–1156. doi: 10.1136/heart.89.10.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Patel DA, Srinivasan SR, Xu JH, Chen W, Berenson GS. Adiponectin and its correlates of cardiovascular risk in young adults: the Bogalusa Heart Study. Metabolism. 2006;55:1551–1557. doi: 10.1016/j.metabol.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 276.Pchejetski D, Kunduzova O, Dayon A, Calise D, Seguelas MH, Leducq N, Seif I, Parini A, Cuvillier O. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ Res. 2007;100:41–49. doi: 10.1161/01.RES.0000253900.66640.34. [DOI] [PubMed] [Google Scholar]
- 277.Pepe S, McLennan PL. Cardiac membrane fatty acid composition modulates myocardial oxygen consumption and postischemic recovery of contractile function. Circulation. 2002;105:2303–2308. doi: 10.1161/01.cir.0000015604.88808.74. [DOI] [PubMed] [Google Scholar]
- 278.Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, Dence C, Klein S, Marsala J, Meyer T, Gropler RJ. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation. 2004;109:2191–2196. doi: 10.1161/01.CIR.0000127959.28627.F8. [DOI] [PubMed] [Google Scholar]
- 279.Peterson LR, Waggoner AD, Schechtman KB, Meyer T, Gropler RJ, Barzilai B, Davila-Roman VG. Alterations in left ventricular structure and function in young healthy obese women: assessment by echocardiography and tissue Doppler imaging. J Am Coll Cardiol. 2004;43:1399–1404. doi: 10.1016/j.jacc.2003.10.062. [DOI] [PubMed] [Google Scholar]
- 280.Pillutla P, Hwang YC, Augustus A, Yokoyama M, Yagyu H, Johnston TP, Kaneko M, Ramasamy R, Goldberg IJ. Perfusion of hearts with triglyceride-rich particles reproduces the metabolic abnormalities in lipotoxic cardiomyopathy. Am J Physiol Endocrinol Metab. 2005;288:E1229–E1235. doi: 10.1152/ajpendo.00273.2004. [DOI] [PubMed] [Google Scholar]
- 281.Pineiro R, Iglesias MJ, Gallego R, Raghay K, Eiras S, Rubio J, Dieguez C, Gualillo O, Gonzalez-Juanatey JR, Lago F. Adiponectin is synthesized and secreted by human and murine cardiomyocytes. FEBS Lett. 2005;579:5163–5169. doi: 10.1016/j.febslet.2005.07.098. [DOI] [PubMed] [Google Scholar]
- 282.Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. J Am Med Assoc. 2004;291:1730–1737. doi: 10.1001/jama.291.14.1730. [DOI] [PubMed] [Google Scholar]
- 283.Pischon T, Rimm EB. Adiponectin: a promising marker for cardiovascular disease. Clin Chem. 2006;52:797–799. doi: 10.1373/clinchem.2006.067819. [DOI] [PubMed] [Google Scholar]
- 284.Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, Eckel RH. Obesity and cardiovascular disease: pathophysiology, evaluation, effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, Metabolism. Circulation. 2006;113:898–918. doi: 10.1161/CIRCULATIONAHA.106.171016. [DOI] [PubMed] [Google Scholar]
- 285.Purdham DM, Zou MX, Rajapurohitam V, Karmazyn M. Rat heart is a site of leptin production and action. Am J Physiol Heart Circ Physiol. 2004;287:H2877–H2884. doi: 10.1152/ajpheart.00499.2004. [DOI] [PubMed] [Google Scholar]
- 286.Qi L, Li T, Rimm E, Zhang C, Rifai N, Hunter D, Doria A, Hu FB. The +276 polymorphism of the APM1 gene, plasma adiponectin concentration, cardiovascular risk in diabetic men. Diabetes. 2005;54:1607–1610. doi: 10.2337/diabetes.54.5.1607. [DOI] [PubMed] [Google Scholar]
- 287.Quan SF, Gersh BJ. Cardiovascular consequences of sleep-disordered breathing: past, present and future: report of a workshop from the National Center on Sleep Disorders Research and the National Heart, Lung, Blood Institute. Circulation. 2004;109:951–957. doi: 10.1161/01.CIR.0000118216.84358.22. [DOI] [PubMed] [Google Scholar]
- 288.Quilliot D, Alla F, Bohme P, Bruntz JF, Hammadi M, Dousset B, Ziegler O, Zannad F. Myocardial collagen turnover in normotensive obese patients: relation to insulin resistance. Int J Obes. 2005;29:1321–1328. doi: 10.1038/sj.ijo.0803022. [DOI] [PubMed] [Google Scholar]
- 289.Rahmouni K, Haynes WG. Leptin and the cardiovascular system. Recent Prog Horm Res. 2004;59:225–244. doi: 10.1210/rp.59.1.225. [DOI] [PubMed] [Google Scholar]
- 290.Rahmouni K, Haynes WG, Morgan DA, Mark AL. Selective resistance to central neural administration of leptin in agouti obese mice. Hypertension. 2002;39:486–490. doi: 10.1161/hy0202.102836. [DOI] [PubMed] [Google Scholar]
- 291.Rahmouni K, Morgan DA, Morgan GM, Mark AL, Haynes WG. Role of selective leptin resistance in diet-induced obesity hypertension. Diabetes. 2005;54:2012–2018. doi: 10.2337/diabetes.54.7.2012. [DOI] [PubMed] [Google Scholar]
- 292.Rajapurohitam V, Gan XT, Kirshenbaum LA, Karmazyn M. The obesity-associated peptide leptin induces hypertrophy in neonatal rat ventricular myocytes. Circ Res. 2003;93:277–279. doi: 10.1161/01.RES.0000089255.37804.72. [DOI] [PubMed] [Google Scholar]
- 293.Rajapurohitam V, Javadov S, Purdham DM, Kirshenbaum LA, Karmazyn M. An autocrine role for leptin in mediating the cardiomyocyte hypertrophic effects of angiotensin II and endothelin-1. J Mol Cell Cardiol. 2006;41:265–274. doi: 10.1016/j.yjmcc.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 294.Raju SV, Zheng M, Schuleri KH, Phan AC, Bedja D, Saraiva RM, Yiginer O, Vandegaer K, Gabrielson KL, O’Donnell CP, Berkowitz DE, Barouch LA, Hare JM. Activation of the cardiac ciliary neurotrophic factor receptor reverses left ventricular hypertrophy in leptin-deficient and leptin-resistant obesity. Proc Natl Acad Sci USA. 2006;103:4222–4227. doi: 10.1073/pnas.0510460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Reeve JL, Duffy AM, O’Brien T, Samali A. Don’t lose heart—therapeutic value of apoptosis prevention in the treatment of cardiovascular disease. J Cell Mol Med. 2005;9:609–622. doi: 10.1111/j.1582-4934.2005.tb00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Relling DP, Esberg LB, Fang CX, Johnson WT, Murphy EJ, Carlson EC, Saari JT, Ren J. High-fat diet-induced juvenile obesity leads to cardiomyocyte dysfunction and upregulation of Foxo3a transcription factor independent of lipotoxicity and apoptosis. J Hypertens. 2006;24:549–561. doi: 10.1097/01.hjh.0000203846.34314.94. [DOI] [PubMed] [Google Scholar]
- 297.Ren J. Leptin and hyperleptinemia—from friend to foe for cardiovascular function. J Endocrinol. 2004;181:1–10. doi: 10.1677/joe.0.1810001. [DOI] [PubMed] [Google Scholar]
- 298.Ren J. Lessons from the leptin paradox in cardiac regulation—too much versus too little. J Physiol. 2005;565:347. doi: 10.1113/jphysiol.2005.086678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Ricci E, Smallwood S, Chouabe C, Mertani HC, Raccurt M, Morel G, Bonvallet R. Electrophysiological characterization of left ventricular myocytes from obese Sprague-Dawley rat. Obesity. 2006;14:778–786. doi: 10.1038/oby.2006.90. [DOI] [PubMed] [Google Scholar]
- 300.Roberts WC. The heart at necropsy in massive obesity (>300 pounds or >136 kilograms) Am J Cardiol. 2002;89:1451. doi: 10.1016/s0002-9149(02)02416-5. [DOI] [PubMed] [Google Scholar]
- 301.Rodriguez M, Lucchesi BR, Schaper J. Apoptosis in myocardial infarction. Ann Med. 2002;34:470–479. doi: 10.1080/078538902321012414. [DOI] [PubMed] [Google Scholar]
- 302.Rodriguez M, Schaper J. Apoptosis: measurement and technical issues. J Mol Cell Cardiol. 2005;38:15–20. doi: 10.1016/j.yjmcc.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 303.Rothenbacher D, Brenner H, Marz W, Koenig W. Adiponectin, risk of coronary heart disease and correlations with cardiovascular risk markers. Eur Heart J. 2005;26:1640–1646. doi: 10.1093/eurheartj/ehi340. [DOI] [PubMed] [Google Scholar]
- 304.Ruano M, Silvestre V, Castro R, Garcia-Lescun MC, Rodriguez A, Marco A, Garcia-Blanch G. Morbid obesity, hypertensive disease and the renin-angiotensin-aldosterone axis. Obes Surg. 2005;15:670–676. doi: 10.1381/0960892053923734. [DOI] [PubMed] [Google Scholar]
- 305.Russell JC, Proctor SD. Small animal models of cardiovascular disease: tools for the study of the roles of metabolic syndrome, dyslipidemia, atherosclerosis. Cardiovasc Pathol. 2006;15:318–330. doi: 10.1016/j.carpath.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 306.Rutter MK, Parise H, Benjamin EJ, Levy D, Larson MG, Meigs JB, Nesto RW, Wilson PW, Vasan RS. Impact of glucose intolerance and insulin resistance on cardiac structure and function: sex-related differences in the Framingham Heart Study. Circulation. 2003;107:448–454. doi: 10.1161/01.cir.0000045671.62860.98. [DOI] [PubMed] [Google Scholar]
- 307.Salahudeen AK. The obesity paradox as it relates to survival and hypertension in dialysis patients. Nephrol Dial Transplant. 2006;21:1729. doi: 10.1093/ndt/gfl099. [DOI] [PubMed] [Google Scholar]
- 308.Saraiva RM, Minhas KM, Zheng M, Pitz E, Treuer A, Gonzalez D, Schuleri KH, Vandegaer KM, Barouch LA, Hare JM. Reduced neuronal nitric oxide synthase expression contributes to cardiac oxidative stress and nitroso-redox imbalance in ob/ob mice. Nitric Oxide. 2006 doi: 10.1016/j.niox.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Sattar N, Wannamethee G, Sarwar N, Tchernova J, Cherry L, Wallace AM, Danesh J, Whincup PH. Adiponectin and coronary heart disease: a prospective study and meta-analysis. Circulation. 2006;114:623–629. doi: 10.1161/CIRCULATIONAHA.106.618918. [DOI] [PubMed] [Google Scholar]
- 310.Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE, Styles P, Radda GK, Neubauer S, Clarke K. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation. 2003;107:3040–3046. doi: 10.1161/01.CIR.0000072789.89096.10. [DOI] [PubMed] [Google Scholar]
- 311.Schmidt D, Salahudeen A. The obesity-survival paradox in hemodialysis patients: why do overweight hemodialysis patients live longer? Nutr Clin Pract. 2007;22:11–15. doi: 10.1177/011542650702200111. [DOI] [PubMed] [Google Scholar]
- 312.Schulze MB, Shai I, Rimm EB, Li T, Rifai N, Hu FB. Adiponectin and future coronary heart disease events among men with type 2 diabetes. Diabetes. 2005;54:534–539. doi: 10.2337/diabetes.54.2.534. [DOI] [PubMed] [Google Scholar]
- 313.Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol. 2002;283:H976–H982. doi: 10.1152/ajpheart.00088.2002. [DOI] [PubMed] [Google Scholar]
- 314.Seto S, Kapuku GK, Kawahara F, Suzuki S, Yano K. Influence of mild to moderate obesity on left ventricular stress filling pattern in hypertension. Hypertens Res. 1998;21:245–250. doi: 10.1291/hypres.21.245. [DOI] [PubMed] [Google Scholar]
- 315.Severson DL. Diabetic cardiomyopathy: recent evidence from mouse models of type 1 and type 2 diabetes. Can J Physiol Pharmacol. 2004;82:813–823. doi: 10.1139/y04-065. [DOI] [PubMed] [Google Scholar]
- 316.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18:1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 317.Sharma V, McNeill JH. The emerging roles of leptin and ghrelin in cardiovascular physiology and pathophysiology. Curr Vasc Pharmacol. 2005;3:169–180. doi: 10.2174/1570161053586868. [DOI] [PubMed] [Google Scholar]
- 318.Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. 2006;55:798–805. doi: 10.2337/diabetes.55.03.06.db05-1039. [DOI] [PubMed] [Google Scholar]
- 319.Shen X, Zheng S, Thongboonkerd V, Xu M, Pierce WM, Jr, Klein JB, Epstein PN. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am J Physiol Endocrinol Metab. 2004;287:E896–E905. doi: 10.1152/ajpendo.00047.2004. [DOI] [PubMed] [Google Scholar]
- 320.Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, Kumada M, Sato K, Schiekofer S, Ohashi K, Funahashi T, Colucci WS, Walsh K. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med. 2004;10:1384–1389. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–1103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Sidell RJ, Cole MA, Draper NJ, Desrois M, Buckingham RE, Clarke K. Thiazolidinedione treatment normalizes insulin resistance and ischemic injury in the Zucker fatty rat heart. Diabetes. 2002;51:1110–1117. doi: 10.2337/diabetes.51.4.1110. [DOI] [PubMed] [Google Scholar]
- 323.Singh JP, Evans JC, Levy D, Larson MG, Freed LA, Fuller DL, Lehman B, Benjamin EJ. Prevalence and clinical determinants of mitral, tricuspid, aortic regurgitation (the Framingham Heart Study) Am J Cardiol. 1999;83:897–902. doi: 10.1016/s0002-9149(98)01064-9. [DOI] [PubMed] [Google Scholar]
- 324.Sjostrom L, Lindroos AK, Peltonen M, Torgerson J, Bouchard C, Carlsson B, Dahlgren S, Larsson B, Narbro K, Sjostrom CD, Sullivan M, Wedel H. Lifestyle, diabetes, cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med. 2004;351:2683–2693. doi: 10.1056/NEJMoa035622. [DOI] [PubMed] [Google Scholar]
- 325.Skilton MR, Celermajer DS. Endothelial dysfunction and arterial abnormalities in childhood obesity. Int J Obes. 2006;30:1041–1049. doi: 10.1038/sj.ijo.0803397. [DOI] [PubMed] [Google Scholar]
- 326.Smith CC, Mocanu MM, Davidson SM, Wynne AM, Simpkin JC, Yellon DM. Leptin, the obesity-associated hormone, exhibits direct cardioprotective effects. Br J Pharmacol. 2006;149:5–13. doi: 10.1038/sj.bjp.0706834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Sons HU, Hoffmann V. Epicardial fat cell size, fat distribution and fat infiltration of the right and left ventricle of the heart. Anat Anz. 1986;161:355–373. [PubMed] [Google Scholar]
- 328.Sowers JR. Obesity as a cardiovascular risk factor. Am J Med. 2003;115(Suppl 8A):37S–41S. doi: 10.1016/j.amjmed.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 329.St-Pierre AC, Cantin B, Bergeron J, Pirro M, Dagenais GR, Despres JP, Lamarche B. Inflammatory markers and long-term risk of ischemic heart disease in men A 13-year follow-up of the Quebec Cardiovascular Study. Atherosclerosis. 2005;182:315–321. doi: 10.1016/j.atherosclerosis.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 330.Su J, Zhang S, Tse J, Scholz PM, Weiss HR. Alterations in nitric oxide-cGMP pathway in ventricular myocytes from obese leptin-deficient mice. Am J Physiol Heart Circ Physiol. 2003;285:H2111–H2117. doi: 10.1152/ajpheart.00316.2003. [DOI] [PubMed] [Google Scholar]
- 331.Sukhija R, Aronow WS, Sandhu R, Kakar P, Maguire GP, Ahn C, Lehrman SG. Prevalence of left ventricular hypertrophy in persons with and without obstructive sleep apnea. Cardiol Rev. 2006;14:170–172. doi: 10.1097/01.crd.0000184455.52778.00. [DOI] [PubMed] [Google Scholar]
- 332.Suleiman M, Khatib R, Agmon Y, Mahamid R, Boulos M, Kapeliovich M, Levy Y, Beyar R, Markiewicz W, Hammerman H, Aronson D. Early inflammation and risk of long-term development of heart failure and mortality in survivors of acute myocardial infarction predictive role of C-reactive protein. J Am Coll Cardiol. 2006;47:962–968. doi: 10.1016/j.jacc.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 333.Suzuki K, Kostin S, Person V, Elsasser A, Schaper J. Time course of the apoptotic cascade and effects of caspase inhibitors in adult rat ventricular cardiomyocytes. J Mol Cell Cardiol. 2001;33:983–994. doi: 10.1006/jmcc.2001.1364. [DOI] [PubMed] [Google Scholar]
- 334.Svenson KL, Bogue MA, Peters LL. Invited review: identifying new mouse models of cardiovascular disease: a review of high-throughput screens of mutagenized and inbred strains. J Appl Physiol. 2003;94:1650–1659. doi: 10.1152/japplphysiol.01029.2003. [DOI] [PubMed] [Google Scholar]
- 335.Sweeney G. Leptin signalling. Cell Signal. 2002;14:655–663. doi: 10.1016/s0898-6568(02)00006-2. [DOI] [PubMed] [Google Scholar]
- 336.Szczepaniak LS, Dobbins RL, Metzger GJ, Sartoni-D’Ambrosia G, Arbique D, Vongpatanasin W, Unger R, Victor RG. Myocardial triglycerides and systolic function in humans: in vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn Reson Med. 2003;49:417–423. doi: 10.1002/mrm.10372. [DOI] [PubMed] [Google Scholar]
- 337.Takahashi R, Okumura K, Asai T, Hirai T, Murakami H, Murakami R, Numaguchi Y, Matsui H, Ito M, Murohara T. Dietary fish oil attenuates cardiac hypertrophy in lipotoxic cardiomyopathy due to systemic carnitine deficiency. Cardiovasc Res. 2005;68:213–223. doi: 10.1016/j.cardiores.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 338.Takahashi T, Saegusa S, Sumino H, Nakahashi T, Iwai K, Morimoto S, Kanda T. Adiponectin replacement therapy attenuates myocardial damage in leptin-deficient mice with viral myocarditis. J Int Med Res. 2005;33:207–214. doi: 10.1177/147323000503300208. [DOI] [PubMed] [Google Scholar]
- 339.Takahashi T, Saegusa S, Sumino H, Nakahashi T, Iwai K, Morimoto S, Nojima T, Kanda T. Adiponectin, T-cadherin and tumour necrosis factor-alpha in damaged cardiomyocytes from autopsy specimens. J Int Med Res. 2005;33:236–244. doi: 10.1177/147323000503300212. [DOI] [PubMed] [Google Scholar]
- 340.Takahashi T, Yu F, Saegusa S, Sumino H, Nakahashi T, Iwai K, Morimoto S, Kurabayashi M, Kanda T. Impaired expression of cardiac adiponectin in leptin-deficient mice with viral myocarditis. Int Heart J. 2006;47:107–123. doi: 10.1536/ihj.47.107. [DOI] [PubMed] [Google Scholar]
- 341.Tanaka T, Ozaki K. Inflammation as a risk factor for myocardial infarction. J Hum Genet. 2006;51:595–604. doi: 10.1007/s10038-006-0411-8. [DOI] [PubMed] [Google Scholar]
- 342.Tandri H, Castillo E, Ferrari VA, Nasir K, Dalal D, Bomma C, Calkins H, Bluemke DA. Magnetic resonance imaging of arrhythmogenic right ventricular dysplasia: sensitivity, specificity, observer variability of fat detection versus functional analysis of the right ventricle. J Am Coll Cardiol. 2006;48:2277–2284. doi: 10.1016/j.jacc.2006.07.051. [DOI] [PubMed] [Google Scholar]
- 343.Tanko LB, Christiansen C. Can the obesity paradox be explained by the protective effects of peripheral adiposity? Arch Intern Med. 2005;165:1796–1798. doi: 10.1001/archinte.165.15.1796-b. [DOI] [PubMed] [Google Scholar]
- 344.Tansey DK, Aly Z, Sheppard MN. Fat in the right ventricle of the normal heart. Histopathology. 2005;46:98–104. doi: 10.1111/j.1365-2559.2005.02054.x. [DOI] [PubMed] [Google Scholar]
- 345.Teoh H, Strauss MH, Szmitko PE, Verma S. Adiponectin and myocardial infarction: a paradox or a paradigm? Eur Heart J. 2006;27:2266–2268. doi: 10.1093/eurheartj/ehl248. [DOI] [PubMed] [Google Scholar]
- 346.Thakker GD, Frangogiannis NG, Bujak M, Zymek P, Gaubatz JW, Reddy AK, Taffet G, Michael LH, Entman ML, Ballantyne CM. Effects of diet-induced obesity on inflammation and remodeling after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H2504–H2514. doi: 10.1152/ajpheart.00322.2006. [DOI] [PubMed] [Google Scholar]
- 347.Thim T, Bentzon JF, Kristiansen SB, Simonsen U, Andersen HL, Wassermann K, Falk E. Size of myocardial infarction induced by ischaemia/reperfusion is unaltered in rats with metabolic syndrome. Clin Sci. 2006;110:665–671. doi: 10.1042/CS20050326. [DOI] [PubMed] [Google Scholar]
- 348.Tilg H, Wolf AM. Adiponectin: a key fat-derived molecule regulating inflammation. Expert Opin Ther Targets. 2005;9:245–251. doi: 10.1517/14728222.9.2.245. [DOI] [PubMed] [Google Scholar]
- 349.Toblli JE, Cao G, DeRosa G, Di Gennaro F, Forcada P. Angiotensin-converting enzyme inhibition and angiogenesis in myocardium of obese Zucker rats. Am J Hypertens. 2004;17:172–180. doi: 10.1016/j.amjhyper.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 350.Toblli JE, Cao G, DeRosa G, Forcada P. Reduced cardiac expression of plasminogen activator inhibitor 1 and transforming growth factor beta1 in obese Zucker rats by perindopril. Heart. 2005;91:80–86. doi: 10.1136/hrt.2003.022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Todor A, Sharov VG, Tanhehco EJ, Silverman N, Bernabei A, Sabbah HN. Hypoxia-induced cleavage of caspase-3 and DFF45/ICAD in human failed cardiomyocytes. Am J Physiol Heart Circ Physiol. 2002;283:H990–H995. doi: 10.1152/ajpheart.01003.2001. [DOI] [PubMed] [Google Scholar]
- 352.Tsujino T, Kawasaki D, Masuyama T. Left ventricular diastolic dysfunction in diabetic patients: pathophysiology and therapeutic implications. Am J Cardiovasc Drugs. 2006;6:219–230. doi: 10.2165/00129784-200606040-00002. [DOI] [PubMed] [Google Scholar]
- 353.Turko IV, Li L, Aulak KS, Stuehr DJ, Chang JY, Murad F. Protein tyrosine nitration in the mitochondria from diabetic mouse heart. Implications to dysfunctional mitochondria in diabetes. J Biol Chem. 2003;278:33972–33977. doi: 10.1074/jbc.M303734200. [DOI] [PubMed] [Google Scholar]
- 354.Turko IV, Murad F. Quantitative protein profiling in heart mitochondria from diabetic rats. J Biol Chem. 2003;278:35844–35849. doi: 10.1074/jbc.M303139200. [DOI] [PubMed] [Google Scholar]
- 355.Turpin SM, Lancaster GI, Darby I, Febbraio MA, Watt MJ. Apoptosis in skeletal muscle myotubes is induced by ceramides and is positively related to insulin resistance. Am J Physiol Endocrinol Metab. 2006;291:E1341–E1350. doi: 10.1152/ajpendo.00095.2006. [DOI] [PubMed] [Google Scholar]
- 356.Unger RH. Hyperleptinemia: protecting the heart from lipid overload. Hypertension. 2005;45:1031–1034. doi: 10.1161/01.HYP.0000165683.09053.02. [DOI] [PubMed] [Google Scholar]
- 357.Van den Bergh A, Flameng W, Herijgers P. Type II diabetic mice exhibit contractile dysfunction but maintain cardiac output by favourable loading conditions. Eur J Heart Failure. 2006;8:777–783. doi: 10.1016/j.ejheart.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 358.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 359.Van Harmelen V, Elizalde M, Ariapart P, Bergstedt-Lindqvist S, Reynisdottir S, Hoffstedt J, Lundkvist I, Bringman S, Arner P. The association of human adipose angiotensinogen gene expression with abdominal fat distribution in obesity. Int J Obes Relat Metab Disord. 2000;24:673–678. doi: 10.1038/sj.ijo.0801217. [DOI] [PubMed] [Google Scholar]
- 360.Vasan RS, Larson MG, Benjamin EJ, Evans JC, Reiss CK, Levy D. Congestive heart failure in subjects with normal versus reduced left ventricular ejection fraction: prevalence and mortality in a population-based cohort. J Am Coll Cardiol. 1999;33:1948–1955. doi: 10.1016/s0735-1097(99)00118-7. [DOI] [PubMed] [Google Scholar]
- 361.Verreth W, De Keyzer D, Pelat M, Verhamme P, Ganame J, Bielicki JK, Mertens A, Quarck R, Benhabiles N, Marguerie G, Mackness B, Mackness M, Ninio E, Herregods MC, Balligand JL, Holvoet P. Weight-loss-associated induction of peroxisome proliferator-activated receptor-alpha and peroxisome proliferator-activated receptor-gamma correlate with reduced atherosclerosis and improved cardiovascular function in obese insulin-resistant mice. Circulation. 2004;110:3259–3269. doi: 10.1161/01.CIR.0000147614.85888.7A. [DOI] [PubMed] [Google Scholar]
- 362.Verreth W, Ganame J, Mertens A, Bernar H, Herregods MC, Holvoet P. Peroxisome proliferator-activated receptor-alpha-, gamma-agonist improves insulin sensitivity and prevents loss of left ventricular function in obese dyslipidemic mice. Arterioscler Thromb Vasc Biol. 2006;26:922–928. doi: 10.1161/01.ATV.0000207318.42066.bb. [DOI] [PubMed] [Google Scholar]
- 363.Vikramadithyan RK, Hirata K, Yagyu H, Hu Y, Augustus A, Homma S, Goldberg IJ. Peroxisome proliferator-activated receptor agonists modulate heart function in transgenic mice with lipotoxic cardiomyopathy. J Pharmacol Exp Ther. 2005;313:586–593. doi: 10.1124/jpet.104.080259. [DOI] [PubMed] [Google Scholar]
- 364.Villa E, Rabano A, Albarran OG, Ruilope LM, Garcia-Robles R. Effects of chronic combined treatment with captopril and pravastatin on the progression of insulin resistance and cardiovascular alterations in an experimental model of obesity in dogs. Am J Hypertens. 1998;11:844–851. doi: 10.1016/s0895-7061(98)00053-3. [DOI] [PubMed] [Google Scholar]
- 365.Villareal DT, Miller BV, 3rd, Banks M, Fontana L, Sinacore DR, Klein S. Effect of lifestyle intervention on metabolic coronary heart disease risk factors in obese older adults. Am J Clin Nutr. 2006;84:1317–1323. doi: 10.1093/ajcn/84.6.1317. [DOI] [PubMed] [Google Scholar]
- 366.Von Eynatten M, Hamann A, Twardella D, Nawroth PP, Brenner H, Rothenbacher D. Relationship of adiponectin with markers of systemic inflammation, atherogenic dyslipidemia, heart failure in patients with coronary heart disease. Clin Chem. 2006;52:853–859. doi: 10.1373/clinchem.2005.060509. [DOI] [PubMed] [Google Scholar]
- 367.Von Haehling S, Doehner W, Anker SD. Obesity and the heart a weighty issue. J Am Coll Cardiol. 2006;47:2274–2276. doi: 10.1016/j.jacc.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 368.Wang J, Zhen L, Klug MG, Wood D, Wu X, Mizrahi J. Involvement of caspase 3- and 8-like proteases in ceramide-induced apoptosis of cardiomyocytes. J Card Failure. 2000;6:243–249. doi: 10.1054/jcaf.2000.9502. [DOI] [PubMed] [Google Scholar]
- 369.Wang P, Chatham JC. Onset of diabetes in Zucker diabetic fatty (ZDF) rats leads to improved recovery of function after ischemia in the isolated perfused heart. Am J Physiol Endocrinol Metab. 2004;286:E725–E736. doi: 10.1152/ajpendo.00295.2003. [DOI] [PubMed] [Google Scholar]
- 370.Wang P, Lloyd SG, Zeng H, Bonen A, Chatham JC. Impact of altered substrate utilization on cardiac function in isolated hearts from Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol. 2005;288:H2102–H2110. doi: 10.1152/ajpheart.00935.2004. [DOI] [PubMed] [Google Scholar]
- 371.Wang TJ, Parise H, Levy D, D’Agostino RB, Sr, Wolf PA, Vasan RS, Benjamin EJ. Obesity and the risk of new-onset atrial fibrillation. J Am Med Assoc. 2004;292:2471–2477. doi: 10.1001/jama.292.20.2471. [DOI] [PubMed] [Google Scholar]
- 372.Wannamethee SG, Tchernova J, Whincup P, Lowe GD, Rumley A, Brown K, Cherry L, Sattar N. Associations of adiponectin with metabolic and vascular risk parameters in the British Regional Heart Study reveal stronger links to insulin resistance-related than to coronory heart disease risk-related parameters. Int J Obes. 2007;31:1089–1098. doi: 10.1038/sj.ijo.0803544. [DOI] [PubMed] [Google Scholar]
- 373.Weiss JW, Remsburg S, Garpestad E, Ringler J, Sparrow D, Parker JA. Hemodynamic consequences of obstructive sleep apnea. Sleep. 1996;19:388–397. doi: 10.1093/sleep/19.5.388. [DOI] [PubMed] [Google Scholar]
- 374.Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:1497–1504. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Wikstrand J, Pettersson P, Bjorntorp P. Body fat distribution and left ventricular morphology and function in obese females. J Hypertens. 1993;11:1259–1266. [PubMed] [Google Scholar]
- 376.Wilhelmsen L, Rosengren A, Eriksson H, Lappas G. Heart failure in the general population of men—morbidity, risk factors and prognosis. J Intern Med. 2001;249:253–261. doi: 10.1046/j.1365-2796.2001.00801.x. [DOI] [PubMed] [Google Scholar]
- 377.Wong CY, O’Moore-Sullivan T, Leano R, Byrne N, Beller E, Marwick TH. Alterations of left ventricular myocardial characteristics associated with obesity. Circulation. 2004;110:3081–3087. doi: 10.1161/01.CIR.0000147184.13872.0F. [DOI] [PubMed] [Google Scholar]
- 378.Wong CY, O’Moore-Sullivan T, Leano R, Hukins C, Jenkins C, Marwick TH. Association of subclinical right ventricular dysfunction with obesity. J Am Coll Cardiol. 2006;47:611–616. doi: 10.1016/j.jacc.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 379.Xu FP, Chen MS, Wang YZ, Yi Q, Lin SB, Chen AF, Luo JD. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation. 2004;110:1269–1275. doi: 10.1161/01.CIR.0000140766.52771.6D. [DOI] [PubMed] [Google Scholar]
- 380.Xu Y, Gen M, Lu L, Fox J, Weiss SO, Brown RD, Perlov D, Ahmad H, Zhu P, Greyson C, Long CS, Schwartz GG. PPAR-gamma activation fails to provide myocardial protection in ischemia and reperfusion in pigs. Am J Physiol Heart Circ Physiol. 2005;288:H1314–H1323. doi: 10.1152/ajpheart.00618.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, Bensadoun A, Homma S, Goldberg IJ. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest. 2003;111:419–426. doi: 10.1172/JCI16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100:1208–1217. doi: 10.1161/01.RES.0000264104.25265.b6. [DOI] [PubMed] [Google Scholar]
- 383.Yarbrough WM, Mukherjee R, Escobar GP, Sample JA, McLean JE, Dowdy KB, Hendrick JW, Gibson WC, Hardin AE, Mingoia JT, White PC, Stiko A, Armstrong RC, Crawford FA, Spinale FG. Pharmacologic inhibition of intracellular caspases after myocardial infarction attenuates left ventricular remodeling: a potentially novel pathway. J Thorac Cardiovasc Surg. 2003;126:1892–1899. doi: 10.1016/j.jtcvs.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 384.Yarbrough WM, Mukherjee R, Squires CE, Reese ES, Leiser JS, Stroud RE, Sample JA, Hendrick JW, Mingoia JT, McLean JE, Hardin AE, Dowdy KB, Spinale FG. Caspase inhibition attenuates contractile dysfunction following cardioplegic arrest and rewarming in the setting of left ventricular failure. J Cardiovasc Pharmacol. 2004;44:645–650. doi: 10.1097/00005344-200412000-00004. [DOI] [PubMed] [Google Scholar]
- 385.Yndestad A, Damas JK, Oie E, Ueland T, Gullestad L, Aukrust P. Systemic inflammation in heart failure—the whys and where-fores. Heart Fail Rev. 2006;11:83–92. doi: 10.1007/s10741-006-9196-2. [DOI] [PubMed] [Google Scholar]
- 386.Yokoyama M, Yagyu H, Hu Y, Seo T, Hirata K, Homma S, Goldberg IJ. Apolipoprotein B production reduces lipotoxic cardiomyopathy: studies in heart-specific lipoprotein lipase transgenic mouse. J Biol Chem. 2004;279:4204–4211. doi: 10.1074/jbc.M311995200. [DOI] [PubMed] [Google Scholar]
- 387.Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, Taegtmeyer H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes. 2002;51:2587–2595. doi: 10.2337/diabetes.51.8.2587. [DOI] [PubMed] [Google Scholar]
- 388.Yue P, Arai T, Terashima M, Sheikh AY, Cao F, Charo D, Hoyt G, Robbins RC, Ashley EA, Wu J, Yang PC, Tsao PS. Magnetic resonance imaging of progressive cardiomyopathic changes in the db/db mouse. Am J Physiol Heart Circ Physiol. 2007;292:H2106–H2118. doi: 10.1152/ajpheart.00856.2006. [DOI] [PubMed] [Google Scholar]
- 389.Yue TL, Bao W, Gu JL, Cui J, Tao L, Ma XL, Ohlstein EH, Jucker BM. Rosiglitazone treatment in Zucker diabetic fatty rats is associated with ameliorated cardiac insulin resistance and protection from ischemia/reperfusion-induced myocardial injury. Diabetes. 2005;54:554–562. doi: 10.2337/diabetes.54.2.554. [DOI] [PubMed] [Google Scholar]
- 390.Yue TL, Chen J, Bao W, Narayanan PK, Bril A, Jiang W, Lysko PG, Gu JL, Boyce R, Zimmerman DM, Hart TK, Buckingham RE, Ohlstein EH. In vivo myocardial protection from ischemia/reperfusion injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2001;104:2588–2594. doi: 10.1161/hc4601.099403. [DOI] [PubMed] [Google Scholar]
- 391.Zaman AK, Fujii S, Goto D, Furumoto T, Mishima T, Nakai Y, Dong J, Imagawa S, Sobel BE, Kitabatake A. Salutary effects of attenuation of angiotensin II on coronary perivascular fibrosis associated with insulin resistance and obesity. J Mol Cell Cardiol. 2004;37:525–535. doi: 10.1016/j.yjmcc.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 392.Zaman AK, Fujii S, Sawa H, Goto D, Ishimori N, Watano K, Kaneko T, Furumoto T, Sugawara T, Sakuma I, Kitabatake A, Sobel BE. Angiotensin-converting enzyme inhibition attenuates hypofibrinolysis and reduces cardiac perivascular fibrosis in genetically obese diabetic mice. Circulation. 2001;103:3123–3128. doi: 10.1161/01.cir.103.25.3123. [DOI] [PubMed] [Google Scholar]
- 393.Zannad F, Gille B, Grentzinger A, Bruntz JF, Hammadi M, Boivin JM, Hanotin C, Igau B, Drouin P. Effects of sibutramine on ventricular dimensions and heart valves in obese patients during weight reduction. Am Heart J. 2002;144:508–515. doi: 10.1067/mhj.2002.124403. [DOI] [PubMed] [Google Scholar]
- 394.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci USA. 2000;97:1784–1789. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]