

Abstract
The main source of mitochondrial DNA (mtDNA) damage is reactive oxygen species (ROS) generated during normal cellular metabolism. The main mtDNA lesions generated by ROS are base modifications, such as the ubiquitous 8-oxoguanine (8-oxoG) lesion; however, base loss and strand breaks may also occur. Many human diseases are associated with mtDNA mutations and thus maintaining mtDNA integrity is critical. All of these lesions are repaired primarily by the base excision repair (BER) pathway. It is now known that mammalian mitochondria have BER, which, similarly to nuclear BER, is catalyzed by DNA glycosylases, AP endonuclease, DNA polymerase (POLγ in mitochondria), and DNA ligase. This article outlines procedures for measuring oxidative damage formation and BER in mitochondria, including isolation of mitochondria from tissues and cells, protocols for measuring BER enzyme activities, gene-specific repair assays, chromatographic techniques, as well as current optimizations for detecting 8-oxoG lesions in cells by immunofluorescence. Throughout the assay descriptions we will include methodological considerations that may help optimize the assays in terms of resolution and repeatability.
Keywords: Mitochondria, Base excision repair, Incision activity, Reactive oxygen species
1. Introduction
Every eukaryotic cell contains hundreds to thousands of mitochondria, with each mitochondrion containing 2–10 copies of mtDNA [1]. The mtDNA encodes components of the electron transport chain (ETC), including 13 structural genes, 22 transfer RNAs and 2 ribosomal RNAs. The 13 polypeptides encoded by mtDNA are essential subunits of the ETC complexes; cells lacking mtDNA, or in some cases with mutations in mtDNA, lack aerobic metabolism and rely on anaerobic glycolysis for survival [2,3]. The mtDNA is associated with the inner mitochondrial membrane, in an environment containing high levels of ROS, produced by the nearby respiration machinery. ROS can damage not only mtDNA but also the proteins, lipids and RNA in mitochondria. It has been reported that mtDNA contain higher steady-state levels of oxidative DNA lesions compared to nuclear DNA (nDNA) [4,5]. Oxidative lesions produced by ROS lead to DNA mutations, particularly substitutions and deletions, which in turn can lead to mitochondrial dysfunction and cell death [6].
Possible cellular responses to oxidative mtDNA damage include autophagic elimination of mitochondria, complementation from the unaffected mtDNA molecules, and DNA repair of the lesion. Fig. 1 outlines the current repair pathways known to occur in nuclear and mitochondrial DNA. The mitochondrial BER pathway is partially characterized [7–12]. Recent evidence suggests that mismatch repair also occurs in mitochondria [13]. There is no evidence for nucleotide excision repair function in the mitochondria. This pathway removes bulky lesions from DNA in the nucleus. The presence of homologous replication (HR) activity within mammalian mtDNA extracts has been reported [14]. In addition, recombinational events have been detected in mtDNA from several animal species [15,16]. This suggests that HR is involved in mtDNA repair, but more data is needed to show whether it is common or if it only occurs under special circumstances.
Fig. 1.

Summary of DNA repair pathways characterized in the nucleus and mitochondria. All pathways are well characterized in the nucleus. There is no evidence for nucleotide excision repair or non homologous end joining (NHEJ) in the mitochondrial. There is has been homologous replication (HR) repair activity detected in the mitochondria of the cells of some animal species under some conditions, but it is not determined as to how commonly this pathway indeed occurs in mitochondria.
Damage to DNA, if not repaired, could lead to mutations during replication, and ultimately to disease. Mutations in mtDNA are associated with a number of hereditary diseases [17,18] and with carcinogenesis [19,20] and aging [21–24]. There are five distinct steps in BER, conserved in both the nucleus and mitochondria [7,9,25,26]. In brief, these five steps are: (1) excision of the damaged base by glycosylases, such as oxoguanine DNA glycosylase (OGG1) or uracil DNA glycosylase (UNG), (2) incision of the DNA backbone at the apurinic/apyrimidinic (AP) site by either AP endonuclease (e.g. APE1) or the DNA glycosylase-associated AP lyase, (3) processing of the 5′ and 3′ termini. The 5′ terminal deoxyribose phosphate (dRP) residue is removed by a polymerase; the 3′ residue (unsaturated aldehyde or phosphate) is removed by APE-1 or PNKP, (4) gap filling synthesis, by a polymerase. POLγ is the only polymerase present in the mitochondria, and thus has both polymerase, and dRP lyase activity, (5) ligation of the final nick, by a DNA ligase.
Thus, BER activity can be studied by individually examining any of these steps. For example, electrophoretic mobility shift assay and DNA cleavage assays are typically used to visualize the earlier steps in BER, whereas polymerase incorporation and ligation assays are used to examine the later steps.
Proper functioning of BER in both the nucleus and mitochondria is important in disease prevention and to sustain life, as illustrated by phenotypes seen in mice lacking BER genes. The removal of genes coding for key BER proteins, including XRCC1 [27], POLβ [28], APE1 [29,30], Fen1 [31] and DNA ligase 1 [32] leads to embryonic lethal in mice. More mild defects in these BER genes, such as point mutations or haploinsufficiency, are connected to high susceptibility to tumor formation [9]. Knockout of DNA glycosylases, such as oxoguanine DNA glycosylase (OGG1) or uracil DNA glycosylase (UNG), do not result in embryonic lethality, but do affect the level of DNA damage in the nDNA and mtDNA and are generally connected to elevated cancer susceptibility. Thus, it appears that the initial steps in the removal of lesions 8-oxoG and uracil, by OGG1 and UNG, respectively, have a level of protection from functional redundancy by way of other glycosylases [33–35].
Most of the repair enzymes in mitochondria are isoforms of nuclear enzymes, either generated by alternative splicing or alternative transcription initiation sites. In some cases, the counterpart proteins are identical, with signaling sequences that allow the protein to be targeted to both the nuclear and mitochondrial compartments [9,36,37]. As such, it is important for assays to distinguish the activities of mitochondrial enzymes from the activities of their nuclear counterparts. The human OGG1 gene produces two major isoforms generated by alternative splicing: αOGG1 localizes mainly to the nucleus, with relatively smaller amounts in the mitochondria, whereas βOGG1 localized exclusively to the mitochondria. The main activity of αOGG1 glycosylase is removal of oxidized purines, such 8oxoG. The βOGG1 protein does not have any apparent glycoslylase activity in vitro [38], and so an important ongoing study by our group and others is to determine the role of this mitochondrial isoform, using a variety of techniques, including those described here. Uracil DNA glycosylase (UNG) isoforms (mitochondrial UNG1, nuclear UNG2) are generated by alternative splicing and transcription from different positions in the UDG gene [39], each isoform carrying out uracil incision (as well as removal of oxidized cytosine derivatives) from mtDNA and nDNA, respectively. The mitochondrial form of apurinic/apyrimidinic endonuclease 1 (APE1) protein appears to be an N-terminal truncation of the full length nuclear APE1, which retains the apurinic/apyrimidinic site removal activity [40,41]. DNA polymerase γ (POLγ) is the only DNA polymerase present in mitochondria and has been shown to have both polymerase and dRp-lyase activity, catalyzing both reactions necessary for the repair synthesis step in BER [42].
In this article we describe techniques used to measure base excision repair activities. We describe several assay designs that have given us and other groups’ optimal specificity of mitochondrial activity measurements. To preclude any nuclear contamination in a mitochondrial activity assays it is paramount to isolate pure mitochondrial extracts without damage to the mitochondrial integrity and activities. Data suggests that isolation of mitochondria and purification of mtDNA result in potent induction of oxidative mtDNA damage [43]; in Fig. 2 it can be seen that the levels of oxidative stress-induced 8-oxoG detected in purified mtDNA can be much higher than that detected from crude extracts. Thus, it is important that the mtDNA isolation process does not lead to significant mtDNA oxidation, and thus unrealistically high estimates of oxidative lesions. Also, when measuring BER enzyme activities it is important to prevent contaminating nuclear activities on the specific substrate used, which can be ascertained by western blot analysis of specific markers (such as LaminB2 or mitochondrial transcription factor A to detect nuclear or mitochondrial contamination, respectively).
Fig. 2.

Endogenous oxidative damage is overestimated in mtDNA due to oxidative lesions generated from the mtDNA purification process. Fpg/Southern blot analysis was used to measure 8-oxoG levels in nDNA as well as in mtDNA with and without mitochondrial isolation; the crude homogenate and purified mtDNA was obtained from rat liver. Levels of DNA damage detected from nuclear and mitochondrial sequences in DNA isolated from crude homogenates were not significantly different. The endogenous levels based on DNA from isolated mitochondria were approximately three fold higher. Adpated from Anson et al. [43]
2. Isolation of Mitochondria
The key in isolating mitochondria for DNA repair assays is to minimize nuclear contamination while keeping the repair enzymes as active as possible. In contrast to isolation procedures for respiration measurements, the integrity of the respiratory complexes is secondary to getting a pure fraction. The tissue can thus be stored frozen before isolation procedures are performed, however it is important to remove and freeze the tissue as soon as possible after sacrificing the animals. Enzyme activity is protected by performing all steps on ice, performing centrifugations at 4°C and adding protease inhibitors to minimize digestion. Below we will go through methods we have used successfully in our lab for various tissues and cultured cells. For a more extensive review of isolation methods we recommend [44].
2.1. Isolation of the mitochondrial fraction from liver, kidney and testis
These soft tissues can be isolated following the method in Croteau et al. [45]. Initially the tissue is transferred to a beaker containing ice cold buffer MSHE buffer (210 mM mannitol, 70 mM sucrose, 10 mM HEPES, 1 mM EGTA, 2 mM EDTA, 0.75 mM spermidine, 0.15 mM spermine, 2μg/mL leupeptin, 5mM DTT, 2 μM benzamidine, pH 7.4). After mincing with scissors the tissue is washed in MSHE buffer and transferred to a glass-teflon Potter-Elvehjem homogenizer and homogenized until a smooth homogenate is produced. This is followed by a low speed spin (LSS, 1000 g for 10 min) where unbroken cells and the nuclear fraction are precipitated. The supernatant is transferred to another tube and a high speed spin (HSS, 10000 g for 10 min) is performed. The resulting mitochondrial pellet is then washed with MSHE followed by another HSS (10000 g for 10 min) is carried out. The pellet is resuspended in 500 μL MSHE buffer and layered on top of a 50% percoll/50% 2 x MSHE gradient and spun at 50000 g. The mitochondrial fraction will appear as a band one third of the distance from the top of the centrifuge tube. The mitochondria are removed and washed free of percoll by the addition of 10 volumes of MSHE buffer. The pure mitochondria are precipitated in a 3000 g spin and mitochondrial enzymes can be further purified as described below.
2.2. Isolation of the mitochondrial fraction from brain tissue
The isolation can be performed as described in Karahalil et al. [46]. The tissue is added to ice cold MSHE buffer and minced with scissors. After an MSHE buffer wash, the tissue is homogenized and a LSS (1000 g for 10 min) is performed to precipitate the nuclear fraction. The supernatant contains the mitochondria. These are precipitated using a HSS (10000 g for 10 min). A portion of these mitochondria are contained in synaptosomal vesicles. Some authors suggests adding digitonin to disrupt the synaptosomal membrane [46–48] however this detergent has been shown to disrupt the outer mitochondrial membrane [49–52]. We have had success both with and without detergent. After a second HSS (10000 g for 10 min) the mitochondria can be further purified on a percoll gradient as described above.
2.3. Isolation of the mitochondrial fraction from skeletal muscle
Mitochondria in skeletal muscle are often isolated slightly differently than mitochondria from other tissues due to the morphological particularities of muscle tissue. Two main differences separate the procedure. First, the nuclear fraction is much smaller in skeletal muscle than many other tissues, thus nuclear contamination becomes less of a problem. Second, skeletal muscle tissue is organized in bundles of fibers bound together by different layers of connective tissue (endomysium, perimysium and epimysium) and mitochondria are organized very stringently in a crystallized pattern bound tightly to the cytoskeleton. Separation of subcellular compartments can be facilitated by the addition of a protease prior to [53], or during the homogenization [54]. We have found that adding 0.1 mg/mL nagarse (or a comparable protease) during homogenization to be especially useful when isolating mitochondria from heart or skeletal muscle.
The procedure can be performed as described in Scheibye-Knudsen and Quistorff [55] with minor modifications. We begin the procedure by placing the tissue in ice cold KCl-buffer (100 mM KCl, 50mM Tris-base, 5 mM Mg2SO4, 1 mM EDTA) and mincing it with scissors. The liquid is removed and 5 mL of ATP-buffer (KCl with 1 mM ATP, 0.5% BSA) is added. Tissue and buffer are transferred to a glass-teflon Potter-Elvehjem homogenizer and homogenized until a smooth homogenate is obtained. If nagarse has been added the homogenate is spun at 9500 g for 9 min, the supernatant is removed and the pellet is resuspended in ATP-buffer. A LSS (1000 g for 10 min) is performed and the supernatant is transferred to a new tube. A HSS (5400 g for 10 min) is performed and the pellet is resuspended and followed by another HSS (6700 g for 10 min). The resulting mitochondrial pellet can now be used for enzyme isolation as described below.
2.4. Isolation of the mitochondrial fraction from cultured cells
Isolation can be performed essentially as described in section 2.2 [56]. Depending on the cell type, 30 or more 150 mm tissue culture plates must be used for sufficient mitochondrial yield. Cells are washed in PBS and harvested in log-phase by scraping. The cells are precipitated by a LSS (500 g for 12 min) and 1 wet vol of MSHE buffer is then added. Using a dounce glass-glass homogenizer instead of a standard glass-teflon homogenizer appears to increase the yield from cultured cells. Since mechanical homogenization of cells is less efficient than tissue, 0.01% digitonin can be added to the MSHE buffer using a modified version of Greenawalts method [57], as follows: to check the homogenization efficiency a 10 μL aliquot of the homogenate is added to 20 μL trypan blue and inspected under microscope. At least 90% of the cells should take up the dye. If this is not the case another 0.01% of digitonin can be added and further homogenization can be performed. Several steps are sometimes necessary for complete homogenization. To ensure purity of the mitochondrial fraction it can occasionally be necessary to add a protease to digest cytosolic and nuclear proteins before the mitochondria are lysed. We have used subtilisin (1.4 U/mL) during the homogenization for this purpose [58]. After homogenization the remaining procedure can be performed as described in 2.1. At this point it is important to verify the purity of the mitochondrial preparation. We have used western blot to probe for nuclear contamination, often using the very abundant nuclear protein LaminB2 as a marker for nuclear contamination.
2.5. Preparation of mitochondrial extracts
After obtaining the purified mitochondrial fraction, a crude extract needs to be prepared for the in vitro assays. 100 μL of lysis buffer (10 mM tris-HCl pH 7.8, 400 mM KCl, 1 mM EDTA, 1 mM DTT, 20% glycerol, 0.1% NP-40, 0.25 mM PMSF, 1X protease inhibitor cocktail from Roche) is added to the mitochondrial pellet followed by 1.5 hr incubation at 4°C. The lysate is sonicated at 5 watts for 5 sec 5 times in ice, with 30 sec intervals, and spun at 130,000 g to precipitate contaminating DNA and membranes. The supernatant containing the purified proteins is dialyzed overnight against the dialysis buffer (25 mM HEPES, 100mM KCl, 1 mM EDTA, 17% glycerol, 12 mM MgCl2). After dialysis the lysate is spun at 16,000g for 10 min to precipitate any non-soluble fraction. Subsequently, the supernatant is aliquoted and transferred to new tubes, and then the protein concentration can be measured and assays can be performed. Such aliquots can be stored at −80°C for several months without significant loss of enzyme activity; however successive freeze-thaw cycles in the same aliquot should be avoided.
3. Measurement of BER enzyme activities
The BER pathway is carried out in four enzymatic steps, which are the damaged base release, the abasic site cleavage, the 3′ and 5′-end trimming and nucleotide insertion and the final ligation. Each of these activities can be measured separately, or the whole pathway can be measured directly using an incorporation assay. There are several ways of carrying out these measurements, but we have been using oligonucleotide-based assays for our measurements for over 10 years. One of the biggest advantages of the oligonucleotide-based assays is that one can design specific single lesion substrates, allowing for the specific probing of each enzymatic step individually. Table 1 [59] shows the oligonucleotide substrates we have used for the assays described below. Several companies now offer oligonucleotides with base modifications, such as 8-oxoG, uracil, 5-hydroxy-uracil and tetrahydrofuran, an abasic site analogue. We have had good success with oligonucleotides from Midland Certified Reagent Company, but have also used oligonucleotides from IDT and Invitrogen. It is important, however, that the oligonulceotides are extremely pure, and one can choose to have them gel-purified by the company, or perform the gel-purification in house, using standard protocols. We have been using radioactive-labeled substrates, and a protocol for labeling the oligonucleotides is presented below, but the development of new fluorescent labeled nucleotide analogues and the improvement in detection techniques allows for the adaptation of the assays to use fluorescence instead of radioactivity.
Table 1.
Names and sequences of oligonucleotides used in BER enzyme activity assays. Obtained from Weissman et al., [59].
| Assay | Name | Sequence |
|---|---|---|
| 8-Oxoguanine incision | OG | 5′-GAA CGA CTG T(OG)A CTT GAC TGC TAC TGA T 3′-CTT GCT GAC A C T GAA CTG ACG ATG ACT A |
| Uracil incision and BER synthesis incorporation | UU | 5′-ATA TAC CGC GG(U) CGG CCG ATC AAG CTT ATT 3′-TAT ATG GCG CC G GCC GGC TAG TTC GAA TAA |
| AP-site incision | AP | 5′-GAA CGA CTG T (F) A CTT GAC TGC TAC TGA T 3′-CTT GCT GAC A C T GAA CTG ACG ATG ACT A |
| Gap-filling | GAP | 5′-CTG CAG CTG ATG CGC OGT ACG GAT CCC CGG GTA C 3′-GAC GTC GAC TAC GCG GCA TGC CTA GGG GCC CAT G |
OG = 8-oxoguanine; F = tetrahydrofuran abasic site analog.
For the 5′-radioactivity labeling of the substrates, the lesion-containing oligonucleotide (1 nmol) is 5′-32P-labeled by incubating with 3 μCi of [γ-32P] ATP (PerkinElmer, Boston, MA, USA) in the presence of 1 U of T4 polynucleotide kinase. Unincorporated free [γ-32P] ATP is removed from the reaction mixture using G25 desalting columns (GE Healthcare Corp., Piscataway, NJ, USA). The 32P-labeled oligonucleotide is then annealed to the complementary strands in the presence of 100 mM KCl by heating the samples at 90°C for 5 min and allowing them to slowly cool to room temperature. For the gap-filling assay and repair synthesis incorporation, unlabeled substrates are annealed as described above. It is important to check that the substrate has been annealed to completion, as several DNA glycosylases do not recognize single-stranded substrates.
3.1. DNA glycosylase assays
DNA glycosylases catalyze the first reaction in the BER pathway, releasing the damaged base through cleavage of the N-glycosyl bond that connects the base to the ribose in the nucleotide. These enzymes recognize specific base modifications, and as such have defined substrate specificity. All the known DNA glycosylases are classified into one of two classes, I and II, depending on the absence or presence of an associated abasic site lyase activity. The assays to measure their activity rely on their activity to cleave a single-lesion containing oligonucleotide at the site of the lesion, and vary slightly depending on whether they have associated lyase activity or not. The substrates are then resolved in a denaturing polyacrylamide gel, and the activity is calculated as the amount of cleaved substrate relative to the total amount of substrate in the lane.
3.1.1. OGG1 activity
OGG1 incision activity can be measured using oligonucleotide substrates containing single lesions recognized by this enzyme. 8-oxodG is one of the major in vivo substrates for this enzyme, and as such we have used substrates containing this modification. The 8-oxodG incision activity assay has been described in Souza-Pinto et al. [60]. After measuring the protein concentrations of the lysates, they are adjusted to the concentrations needed in each assay with the following buffer: 20 mM HEPES–KOH (pH 7.4), 1 mM EDTA, 100 mM KCl, 25% glycerol (v/v), 0.015% Triton X-100, 5 mM DTT and protease inhibitors. Incision reactions (20 μl volume) contain:
40 mM HEPES–KOH (pH7.4)
5 mM EDTA
1 mM DTT
75 mM KCl
10% glycerol
95 fmol of 32P-labeled duplex oligonucleotide
The reactions are initiated by adding the lysates, and the concentration of lysate needed will vary according to the source. We have found that mitochondria from animal tissues, such as liver and brain, have higher OGG1 activity than mitochondria from cultured cells, and thus require less lysate in the assay. Typically, OGG1 activity can be measured in the range of 10–100 μg protein per reaction, however, a concentration curve with increasing amounts of lysate must be performed to determine the lysate concentration in which the incision activity is linear to protein concentration. It is important to carefully calibrate the amount of protein for all the assays described here, for too much protein may result in non-specific degradation of the substrates.
The reactions are then incubated at 32°C for 12–16 h, at the end of which they are terminated by the addition of 1 μl each of the following, 5 mg/ml Proteinase K and 10% SDS, and incubated at 55°C for 30 min. The DNAs are then ethanol-precipitated by the addition of 1 μg of glycogen, 4 μl of 11 M ammonium acetate, and 63 μl ethanol, pelleted, dried and suspended in formamide dye (90% deionized formamide, 1 mM EDTA, 0.1% bromophenol blue, 0.1% xylene cyanol).
3.1.2. UDG activity
Uracil incision activity is measured using a 30-mer double-stranded oligonucleotide containing a single uracil (U) at position 12 (Table 1). Incision reactions (20 μl) contain:
70 mM HEPES–KOH (pH 7.4)
5 mM EDTA
1 mM DTT
75 mM NaCl
10% glycerol
50 fmoles of 32P-labeled duplex oligonucleotide
The reaction is also initiated by adding the lysate, and incubated for 1 h at 37°C. Because UDG activity is generally much higher than OGG1, much lower amounts of protein lysate are needed, usually within the range of 1–10 μg of protein; however, a protein concentration curve should also be performed to find the right concentration of lysate to be used. The reactions are then terminated and DNA processed as described for the measurement of OGG1 activity.
Because UDG does not have an associated AP-lyase activity, the product of the reaction is an abasic-site containing oligonucleotide which has to be converted into a single-strand break. We add 50 mM NaOH to the samples after they have been resuspended in the loading dye and incubate for 15 min at 75°C before loading into the gel.
3.1.3. Other DNA glycosylases
The activities of other DNA glycosylases, such as NTH and NEIL1, can be easily assayed using similar reactions and assay procedures. The substrates have to be chosen according to the substrate specificity of each enzyme. For example, we use a thymine-glycol-containing oligonucleotide to assay for NTH and a FapyGuanine-containing substrate for NEIL1, and the salt conditions may also need to be adjusted.
3.2. AP endonuclease assay
The next enzymatic step in the BER pathway is the cleavage of the abasic site resulting from the DNA glycosylase activity. In mammals, both in the nucleus and in the mitochondria, AP endonuclease 1 (APE1) is the major abasic endonuclease which catalyses this step. The assay is similar to the glycosylase incision assay, but the substrate now contains either a true abasic site, generated via a class I DNA glycosylase-catalyzed reaction or an abasic site analogue, such as tetrahydrofuran (THF). We prefer the latter, as those are efficiently recognized by APE1 and can be obtained from commercial sources. The substrate is 32P-labeled and annealed as described, with the small modification that the annealing is performed by heating up the samples to 75°C to avoid non-enzymatic cleavage at the abasic site.
APE1 incision activity is measured using a 28-mer oligonucleotide containing the THF analogue at position 11 (Table 1). The lysate samples are diluted in 10 mM HEPES–KOH (pH 7.4) containing 100 mM KCl. The reactions (10 μl) contain:
25 mM HEPES–KOH (pH 7.4)
25 mM KCl
0.1 mg/ml BSA
5 mM MgCl2
10% glycerol
0.05% Triton X-100
10 fmoles of 32P-labeled duplex oligonucleotide
The protein concentration range for the APE1 activity reactions is even lower than that of UDG, and we have worked with as little as 25 ng protein per reaction. The reactions are incubated at 37°C for 30 min, and terminated by the addition of formamide dye and heating at 90°C for 10 min. Samples are resolved, visualized and analyzed as described below.
3.3. Polymerase γ gap-filling assay
The next step in the BER pathway is the end-trimming and nucleotide insertion; in mitochondria, DNA Pol γ catalyses both activities. Until recently, it was believed that mitochondrial BER proceeded only through the short-patch BER sub-pathway, in which only one new nucleotide is incorporated, and this is the assay we describe here. Recently, however, several groups including ours [61] have shown that mitochondrial extracts also support long-patch BER, in which up to 6 nucleotides are incorporated through strand-displacement and which is dependent on the structure-specific endonuclease FEN1.
Single nucleotide gap-filling activity is measured using a non-labeled 34-mer duplex oligonucleotide containing a single gap at position 16 (Table 1); the assay measures the incorporation of a radioactive nucleotide during repair synthesis and the subsequent quantification of the amount of radioactivity incorporated in the full-length oligonucleotide substrate. In order to account for variations in sample loading in the gels, we recommend spiking the samples (after the incubations) with a 32P-labeled oligonucleotide of a different size than the substrate used in the assay, in order to normalize the radioactivity quantifications. The lysates are diluted in 10 mM Tris–HCl (pH 7.4) containing 100 mM KCl. The reactions (10 μl) contain:
50 mM Tris–HCl (pH 7.4)
50 mM KCl
1 mM DTT
5 mM MgCl2
5% glycerol
5 μM dCTP (Roche Applied Sciences, Indianapolis, IN, USA)
1 pmol of duplex gap oligonucleotide
4 μCi of α32P-dCTP (GE Healthcare Corp., Piscataway, NJ, USA)
The amount of protein used in these assays should also be calibrated for each sample, but we have had success with as little as 1 μg protein per reaction. Reactions are incubated at 37°C for 1 h and terminated by the addition of formamide dye and heating at 90°C for 10 min. Samples are resolved and visualized as described below.
3.4. Repair synthesis incorporation assay
The repair synthesis incorporation assays measures the activity of the BER pathway as a whole, in which the incorporation of a new, radioactive nucleotide into a substrate containing a damage base (we have used a uracil-containing substrate) is quantified. The repair synthesis reaction (10 μl) contains:
40 mM HEPES (pH 7.6)
0.1 mM EDTA
5 mM MgCl2
0.2 mg/ml BSA
20 mM KCl
1 mM DTT
40 mM phosphocreatine
100 μg/ml creatine phosphokinase
2 mM ATP
40 μM of each dATP, dTTP, dGTP and 4 μM of dCTP
0.8 μCi α32P-dCTP
3% glycerol
80 ng of double-strand U-containing oligonucleotide
The reaction is also initiated by the addition of the lysates, usually in the range of 10–50 μg protein per reaction. The reactions are incubated at 37°C for 3 h and terminated by adding 2.5 μg of proteinase K and 0.5 μl of 10% SDS and incubating at 55°C for 30 min. The DNA is then precipitated overnight at −20°C after addition of 1 μg glycogen, 4 μl of 11 M ammonium acetate, 63 μl of ethanol. Samples are centrifuged, dried, suspended in 10 μl of formamide loading dye and resolved and visualized as described below. The BER activity is quantified as 32P-dCTP signal strength of the product band relative to a control sample (relative activity = 1), after subtracting the background of a reaction without protein. It is important to note that this assay requires ATP, and thus an ATP-regenerating system (phosphocreatine/creatine kinase) is included in the reaction.
3.5. Electrophoresis and result analysis
All the assays described here require resolving the oligonucleotide substrates under denaturing conditions. We have used 20% polyacrylamide gels containing 7M urea, in 1X Tris-borate/EDTA (TBE) buffer. The acrylamide solution is prepared by diluting a commercially available acrylamide/bis-acrylamide solution (37:1) in H2O and adding 7M urea, from powder, and 0.1 vol of 10XTBE. After completely dissolving the urea, the solution is filtered. This stock can be stored up to 2 months in the refrigerator, protected from light.
For a better resolution, we have used 1mm gels of at least 16 cm length. The samples are loaded and the gels resolved in TBE buffer at 16W, for 1 h. The gels can be dried, but if the radioactive signal is strong enough, they can be exposed without drying. The signals are then acquired using a PhosphoImager (Molecular Dynamics) or equivalent scanner; we have found that the improved sensitivity of the newer versions, such as the Storm or Typhoon (GE Healthcare) allows for the use of smaller amounts of substrate and lysate. The radioactivity in the lanes is quantified using the ImageQuant software; incision activities are quantified as the amount of radioactivity in the product lane relative to the total radioactivity in the lane. Gap-filling and incorporation activity is calculated as the amount of radioactivity in the product lane, normalized by the control labeled substrate, relative to the incorporation of the control samples, which are set as 100% activity.
4. Gene-specific repair to assess mtDNA damage and repair
The gene-specific repair assay involves the generation of lesion-specific strand breaks within a restriction fragment of DNA, using damage-specific endonucleases such as T4 endonuclease V for cyclopyrimidine dimers, formamidopyrimidine DNA glycosylase (Fpg) for oxidized purines, or endonuclease III for oxidized pyrimidines. Quantification of lesion frequency can be done by either southern blotting after hybridization with a radioactive probe specific for the gene of interest, or more recent approaches involve using PCR primers flanking the gene region of interest. Both of these approaches are described below.
The gene-specific assays have the advantage that there is no need to isolate and purify mtDNA, and thus eliminates associated problems of variation, labor and oxidative damage over-estimation. One can also use this strategy to compare mtDNA and nDNA damage levels in the same sample. It also allows for flexibility in that different types of oxidative damage can be investigated.
4.1. Measurement of gene-specific repair using southern blotting
The Southern blot gene-specific repair assay is a reproducible and flexible method for measuring several kinds of DNA damage and associated repair kinetics [62,63]. The assay makes use of a lesion-specific endonuclease as a tool to nick a restriction DNA fragment at the site of damage (e.g. Fpg cutting at 8-oxoG lesions). In a denaturing agarose gel, the cleaved strand migrates further than the full length restriction fragment. The loss of the restriction fragment band on a southern blot due to the specific nicking within the fragment by the endonuclease is quantified against a control (Fig. 3). Since the radioactive probe in the Southern blot is gene-specific, only the damage of interest within a region of an investigated gene is measured. Fig. 4 shows examples of probes we have used and how their use in gene-specific repair assays demonstrates that mtDNA is repaired relatively efficiently after treatment with photosensitizer Ro-19-8022 (supplied by Roche, which generates 8-oxoG damage in nDNA and mtDNA) [64]. A random distribution of damage within the fragment is assumed and so allows the use of the Poisson equation in determining the average number of adducts per strand. Our focus has been on using this technique to measure oxidative lesions, especially 8-oxoG damage, but the assay can be applied to any type damage that can be converted to a single stand nick (converted either chemically or by using a repair enzyme) [65,66].
Fig. 3.
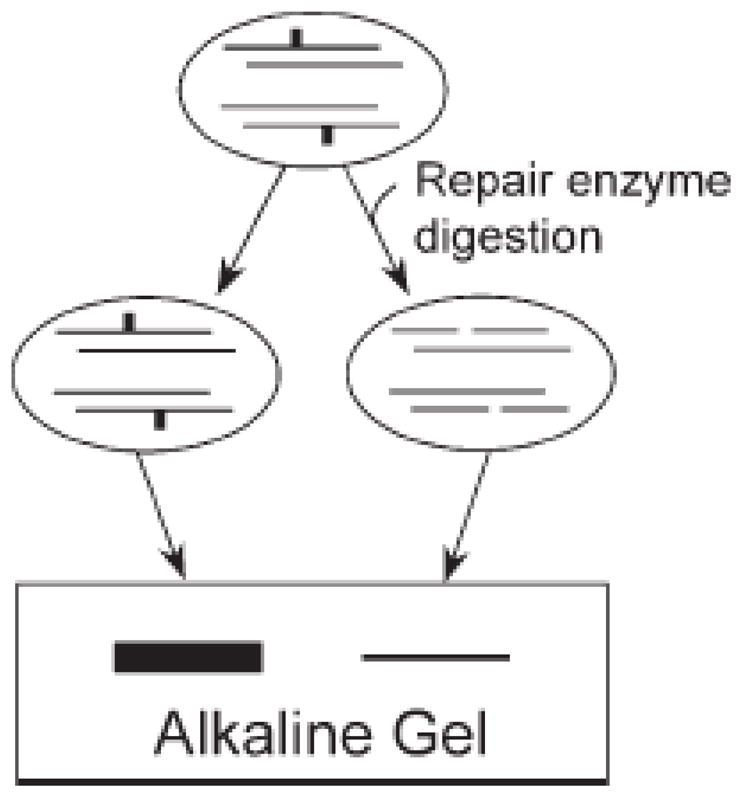
The principle behind the Southern blot gene-specific damage and repair assay. A DNA repair enzyme is used as a tool to nick a restricted DNA fragment at the site of damage. In a denaturing agarose gel, the cleaved strand migrates further than the full length restriction fragment. The band is detected by Southern blot. Obtained from Anson et al. [63].
Fig 4.


Use of gene-specific probes and Southern blotting to demonstrate efficient repair of 8-oxoG in mtDNA. (A) Regions of the Chinese hamster ovary (CHO) genome analyzed. K, KpnI. Horizontal bars below the gene-maps indicate the probes used. The DHFR gene was probed for by pMB5; the non-transcribed region was probed for by CS14; the rDNA was probed for by pABB which recognizes a 9 kb fragment containing the 5.8S and 28S region; the probing of the mtDNA by pCRII visualizes a 16 kb fragment. The map is based on the sequence known for guinea pig mitochondrial genome. (B) Repair of 8-oxoG in the genes probes for in part (A) was measured. KpnI digested genomic DNA (1 μg) from RO treated cells, and mock treated cells (control), was isolated at various time points after damage induction. Before Southern blot analysis DNA was either mock treated or treated with FPG (−/+). Representative autoradiograms of Southern blots are shown in the top panel, for the above probed genes. In the bottom panel quantification of the bands in the Southern blot is presented as percent repair of photoactivated RO induced 8-oxoG in the four different regions of the genome investigated. The data presented are the average +/− standard deviation from five biological experiments with several gels from each experiment. Adapted from Thorslund et al. [64].
The 8-oxoG modification is a significant lesion within the DNA of both the nucleus and mitochondria. We have found that photoactivated methylene blue (PA-MB) can be used to introduce 8-oxoG damage specifically in mtDNA, with minimal nDNA damage [63]. Therefore, one can use this method to study 8-oxoG damage and repair in mtDNA without mitochondrial isolation, a process know to induce oxidative DNA damage and give misrepresented results.
A detailed protocol is described by Bohr’s group [63]. The experiment should be set up in triplicate. For 8-oxoG, the suggested repair time points, after PA-MB treatment, are 0, 0.5, 1, 2, 4, 8 and 24 h. Three plates should serve as a negative control (not treated with MB or light) and three as MB control (not treated with light). Work with MB should be done in dim blue light. We find that safelights for photosensitive agents can generally be made with inexpensive colored plastic. In brief, the steps for this assay are as follows.
(1) Incubation of cells with MB, followed by photoactivation of the MB
Use a 15 cm plate, cells at approx 80% confluence, washed with DPBS (+ Ca, + Mg) between each step, and a 300-W tungsten bulb that is placed18 cm from the dish. Before treating these cells, one must first establish the sensitivity of the untested cells to photoactivated MB, both in terms of survival and the level of damage induced, on cells in a 96 well dish. This is described in [63]; the concentration of MB, time of treatment and level of light exposure should be between 20–200 μM, 0.5–2 h, and 2–6 min, respectively; the treatment conditions should results in minimal cell death and the level of damage should be 1–1.5 lesions/fragment. The average number of lesions can estimated from the average incisions for each sample, which is calculated from band intensities on the southern, as described in [63] and “Data Analysis (part 5)” below.
(2) Isolate DNA from the cells and subject to enzyme restriction
The DNA is isolated then cut with a restriction enzyme. For the mtDNA, single-cutter restriction enzymes will generate a single, linear fragment, which allows for accurate quantification. PvuII, for example, is useful to linearize human mtDNA. The DNA must be fully restricted prior to proceeding, and this can be quickly tested in a 0.8% agarose minigel. Purified DNA should be prepared and restricted as a positive control and to optimize reaction conditions to give complete cleavage.
(3) Treat DNA with Fpg
The concentration of Fpg and reaction time used should be based on optimal enzyme:DNA ratios, determined in optimization experiments on purified DNA, as follows: Serial dilution of Fpg are made in 1X reaction buffer (100mM Tris-HCl pH 7.5–8.0, 100 mM KCl, 2mM EDTA) at 0.01, 0.1, 1, 10 ng/μl and added to tubes containing damaged and undamaged DNA. Use 15μl DNA preparation per tube at 200 μg/ml 1X reaction buffer; add 5 μl of Fpg per tube, incubate in 37°C water bath for each Fpg dilution for 10, 20, 40 and 80 min. The samples are run on an alkaline gel and transferred for use in southern blotting, using mitochondrial probes (see below). Make a note the highest enzyme:DNA ratio at which little or no cutting that was seen in the control (undamaged) DNA after 20 minutes; this is the highest enzyme:DNA ratio that will be used. Also, note the lowest enzyme:DNA ratio and reaction time where specific incision [number of incisions (see above) observed in undamaged DNA subtracted from number of incisions seen in damaged (MB-treated) DNA] had reached a plateau. At plateau, the number of incisions now corresponds to actual number of lesions and thus the reaction had gone to completion.
The above determined optimal enzyme:DNA ratio and reaction time can now be used in the DNA isolated from photoactivated MB treated cells. Use 2.2 μg of DNA in 33 μl of 1 x reaction buffer for each sample; 15 μl of this solution goes into each of two tubes- one for Fpg treatment (tube containing 5 μl of the predetermined Fpg dilution) and one for 1 X reaction buffer only control (tube containing 5 μl 1 x reaction buffer). Samples are placed in the 37°C water bath and incubated for the pre-determined time. The reactions are stopped at the appropriate times by the addition of 2.2 μl of 10X alkaline loading buffer [25% Ficoll, 10 mM EDTA, 0.025% (w/v) bromocresol purple, 0.5 M NaOH (NaOH is not added until the day of use)], mixed, and incubated at 37°C for 15 minutes to fully denature the DNA. At that time, an aliquot of HindIII-digested λ DNA molecular weight marker should be prepared and denatured in the same way. Southern blotting is then carried out [63].
(4) Run alkaline gel, transfer and perform Southern blotting
The techniques used here are standard and described in [63]. For a 20 kb gene of interest use 0.6% (w/v) gel, and increase it by about 0.2% for each 5 kb decrease in fragment size; thus, for a linearized mammalian mitochondrial genome, a 0.75% gel is used. For transfer we use the PosiBlot apparatus (Strategene, La Jolla, CA), following the manufacturer’s instructions. The riboprobe against the gene of interest is prepared using any appropriate kit, and following the manufacturer’s instructions. The use of a riboprobe is recommended because it allows for the possibility of strand-specific annealing if desired and for effective stripping of the membrane. The riboprobe template plasmids used by our laboratory were prepared by cloning the mitochondrial sequences amplified by PCR into the PCRII vector (Invitrogen, Carlsbad, CA) between the T7 and SP6 promoter sequences. The sequences were from 652 to 3226 and from 5902 to 7433 of the mitochondrial genome [63,67].
(5) Data analysis
Quantification of the band intensities is ideally carried out using Phosphorimager (GE HealthCare, Inc., Sunnyvale, CA), due to its extended linear range. The average number of incisions for each sample is calculated using the Poisson distribution as follows: -ln (band intensity in a minus enzyme lane/band intensity in an enzyme-treated lane) [63].
4.2. Measurement of gene-specific repair using quantitative PCR
An alternative means to the Southern blotting method for measuring gene specific repair of the nuclear and mitochondrial genomes is to use quantitative PCR (QPCR) [68,69]. In this case, measurement of damage formation and repair is based on amplification of long DNA targets. The principle of this assay is that many lesions types block the progression of DNA polymerase on a long template, resulting in decreased amplification of the target DNA. The repair of DNA lesions is quantified by following restoration of amplification of the target DNA over time, after removal of the damaging agent. The amount of DNA is quantified by comparing the level of PCR amplification in the treated cells relative to control (untreated) cells, and expressed as the number of lesions, due to the damage, per 10 kb (assuming a Poisson distribution of the lesions on the template) [70,71]. We will not present here a protocol but instead refer to [68,69] for details.
An advantage of this technique over the Southern blotting method (and other methods such as HPLC), is that only small nanogram quantities of DNA are required. In addition, the amplification of long DNA targets makes this technique sensitive [72], and thus a small number of lesions can be detected (as low as 1 lesion per 100,000 nucleotides). However, a major disadvantage of this QPCR technique is that only lesions that block or slow down the amplifying DNA polymerase can be measured. Single or double strand breaks are easily detected while other lesions, such as the ubiquitous oxidative lesion 8-oxoG, do not significantly stop polymerase progression and therefore are not detected with high efficiency. Since our group has a major interest in oxidative damage and base excision repair of which 8-oxoG is a major lesion, we have preferred the Southern blotting strategy. Moreover, the QPCR technique does not give information on the nature of the lesion; whereas the Southern blotting strategy makes use of lesion specific enzymes to produce DNA breaks and so has a great deal of specificity in this respect.
5. Detection of 8-oxoG by chromatographic methods and comparison to Fpg-based methods
Common analytical methods to measure oxidative DNA damage in the nucleus and mitochondria involve detection of modified nucleotides in the DNA by chromatography; the most commonly measured marker of oxidative DNA damage is 8-oxoG. These chromatographic techniques include high performance liquid chromatography with electrochemical detection (HPLC-EC) [5,73,74], gas chromatography coupled to mass spectroscopy (GC-MS) [75,76] and HPLC-MS/MS [77]. The details of these procedures, as well as advantages and limitations, are described more fully in the associated references above. Published estimates of the in vivo levels of 8-oxoG in normal human cells vary widely, due mainly to spurious levels of oxidation generated during sample preparation. Protocols have been revised to alleviate this problem, and it is now standard practice to include antioxidants and metal chelators during sample preparation. The European Standards Committee on Oxidative DNA Damage (ESCODD) has compiled data from a number of different laboratories to compare the strategies used to measure 8-oxoG levels [78–80]. Laboratories using HPLC-EC were able to measure induced DNA damage (8-oxoG; induced using a photosensitizer, Ro-19-8022) with high accuracy (dose response gradients were measured). GC-MS and HPLC-MS/MS did not convincingly give accurate dose response values.
All these chromatographic techniques are still prone to variable measured levels of background 8-oxoG, due to DNA oxidation during sample preparation. These chromatographic techniques were compared to Fpg-based methods, which use Fpg to convert 8-oxoG to single strand breaks, which are then measured by alkaline unwinding [81–84], alkaline elution [85,86] or the comet assay [87–89]. The Fpg-based methods seem to be less prone to spurious oxidation. They can be used quantitatively but require careful calibration and standardization. Of these Fpg methods, alkaline unwinding is the most suited for measurement of mtDNA damage and repair, in cells exposed to oxidative damage. After treatment of purified mtDNA with Fpg to convert oxidative DNA base lesions into strand breaks, the subsequent relaxation of the supercoiled mtDNA is used as a measure of extent of lesions that were present. In the case of the comet assay, cells are embedded on a thin layer of agarose on microscope slides, lysed by detergent and high salt to produce a nucleoid of DNA, and then electrophoresed. The comet assay, at present however cannot detect the low levels of mtDNA at the single cell level (comet tail from mtDNA cannot be detected). The alkaline elution technique also is not appropriate for mtDNA lesion measurements since it does not typically involve purifying mtDNA; this technique involves lysing cells on a membrane filter leaving the total DNA embedded on the filter, followed by treatment with the repair endonuclease (such as Fpg) such that the amount of DNA eluted through the filter is proportional to the amount of single strand breaks generated by Fpg acting on oxidative lesions.
6. Immunofluorescent detection of 8-oxoG mtDNA damage and repair using 8-oxoG-specific antibodies
The use of immunofluorescence methods to detect 8-oxoG eliminates the need for mitochondrial isolation and the associated problems of spurious oxidation during preparation. It also allows for a direct look at the in situ localization (e.g. nuclear or mitochondrial) and kinetics of formation and repair of this oxidative lesion at the individual cell level. Several research groups and companies have now developed antibodies for detection of 8-oxoG in nuclear or mitochondrial DNA. One approach in this regard is to produce the immunogens against 8-oxoG lesions by conjugating synthesized 8-oxoG to serum albumin [90–92]. Another strategy describes the generation of a mouse recombinant Fab, generated by repertoire cloning and combinatorial phage display [93]. Alternatively, Struthers et al. [94] demonstrated that avidin-FITC conjugates can be used in fluorescent detection of 8-oxoG lesions after demonstrating that avidin binds 8-oxoG with high specificity. We have used the 8-oxoG antibody developed by Trevigen, Inc (Clone 2E2) to study repair of 8-oxoG lesions mammalian cells [95]; in support of the specificity of this antibody for 8-oxoG lesions we have preincubated fixed cells with Fpg, which resulted in a significant loss of signal (unpublished data). Trevigen’s 8-oxoG antibody has also been used by Kong et al. [96] to study repair of nuclear 8-oxoG lesions in laser treated mammalian cells. Another commercial 8-oxoG antibody (Clone 45.1 from Nikken SEIL Co., Ltd) has been validated by Ohno et al. [97], for detection of 8-oxoG in both nDNA and mtDNA. Despite the above advances we still remain skeptical about the specificity of the 8-oxoG antibody; we believe there are still considerable background problems, and that use of this strategy should still be supplemented by other approaches to reach reliable conclusions.
Different strategies have been applied for detecting 8-oxoG in mtDNA as apposed to nDNA. Soultanakis et al. [93] demonstrated simultaneous staining of nuclear and mitochondrial 8-oxoG in cultures cells, using confocal microscopy combined with mitotracker labeling to discriminate the subcellular compartments. Ohno et al. [97] used conventional fluorescence microscopy, and found that they can discriminate 8-oxoG staining in mtDNA from that in nDNA based on the difference in denaturing conditions required (likely because the differences in DNA packaging): for detection in mtDNA, pretreatment with NaOH was ideal, whereas denaturing with HCl resulted in a nDNA signal. Based on the above strategies we describe here a consensus protocol for detection and quantification of mtDNA 8-oxoG damage and repair. (1) Cells growing in covered slides are treated with oxidative damaging agent, such as hydrogen peroxide (e.g. 500μM for 30 minutes) or menadione (e.g. 50μM for 30 min), in serum free medium. (2) Then the medium is removed, replaced with complete medium, and incubated for repair times 0, 2, 4, 12, 24 h. Also include an untreated control slide. (3) After washing three times with cold PBS, the slides are fixed with cold methanol (we have also used 1:1 methanol-acetone) for 20 min at −20°C. (4) After rinsing slides in room temperature (RT) PBS two times, the slides are treated with RNase solution for 1 h at 37°C. Complete digestion of RNA in the fixed cells is required to obtain a clear immunofluorescent signal. We have used 100 μg/ml of RNAase in Tris buffer (pH 7.5). (5) At this point one can perform a few 3 min washes in 1% Triton X-100 (in PBS), for cellular permeabilization, if desired (see [97]), then wash and replace with PBS. (6) To confirm specificity of the 8-oxoG antibody, one can now incubate the slides with Fpg (we have used 1 U/μl in FLARE buffer (Trevigen) and) or MutM (10 μg/ml) or Dnase I (1 U/μl) for 1 hour at 37°C (see [97]). The final 8-oxoG signal should be significantly reduced in these slides. (7) After washing again with PBS the slides should be treated with proteinase K (we used10 μg/ml for 7 min at RT) to prevent cross reactivity of the 8-oxoG antibody to protein. (8) If the cells are to be treated with NaOH in the next step (i.e. for immunodetection 8-oxoG in mtDNA) then it is suggested to refix the cells in 4% paraformaldehyde (in PBS) at this point (10 minutes at RT). (9) When examining for nDNA 8-oxoG signal pattern, the slides are washed with PBS and treated with 4 N HCl for 7 minutes at RT to denature the DNA. As mentioned above, it is recommended to instead use NaOH (50 mM in 50% ethanol for 7 minutes at RT) if mtDNA 8-oxoG signal pattern is the goal.
From this point, standard blocking and addition of primary antibody and secondary antibody procedures are used for both nuclear and mitochondrial DNA investigation. In [95] we blocked overnight at 4°C with 10% goat serum (in PBS); we used the Trevigen 8-oxoG antibody at 1:250 dilution (in 10% goat serum-PBS for 1 hour at 37°C) and the AlexaFluor 488 anti-mouse (Invitrogen) secondary antibody at 1:1000 dilution for 1 hour at 37°C in the dark. The slides were then mounted with DAPI-containing Vectasheild (Vector Laboratories) and analyzed with a Zeiss Axiovert 200M microscope [95]. Negative controls consisted of secondary antibody-only and normal mouse IgG (since the Trevigen 8-oxoG antibody in made in mouse) in place of the primary antibody.
7. Conclusion
There is no single standard consensus method for inducing and measuring oxidative damage in mitochondria. In fact, comparison data on oxidative damage between different methods [78,98] showed great variation in damage levels. In fact, reported data on the extent of oxidative DNA damage in both nucleus and mitochondria varies widely, even within a single method [80,99,100]. Our descriptions here of various techniques for identifying and quantifying various oxidative lesions in mtDNA has endeavored to give strategies to mitigate these problems by way of optimal purity, minimal damage, and appropriate control and verification.
Many of the techniques described above require isolation of mitochondria and purification of mtDNA. These manipulations lead to unintentional DNA oxidation, and therefore likely overestimation of DNA damage. Also, large amount of cells are often necessary to obtain sufficient amounts of mitochondria or mtDNA, specifically in the case of the in vitro enzyme assays. The gene specific repair techniques do not involve mitochondria/mtDNA isolation; in the case of the PCR based gene specific repair assay, detection of 8-oxoG lesions is limited (and thus so is BER investigation) since 8-oxoG does not block or slow down the DNA polymerase significantly. The imaging techniques also avoid the mitochondria/mtDNA isolation problems, and offer particular qualitative insight into localization of the oxidative damage. However, this method is not quantitative and the cellular localization may be disturbed by the fixation, permeabilization and acid (or base) treatment, which may alter structural morphology and distribution of the 8-oxoG [101]. In addition, specificity of the 8-oxoG antibody may be in question, since DNA is a poor immunogen [101]; repair enzymes with lesion-cutting specificity (such as Fpg at 8-oxoG lesions) combined with mitochondrial stains are critical in verifying the specificity of the fluorescent signal obtained.
The in vitro techniques (section 2 and 3) are very quantitative and lesion-specific, however the biological relevance of the results has to be considered. While these assays accurately measure reaction kinetic parameters, in the cell other factors which are not represented in vitro may significantly alter the efficiency of the overall pathway. Moreover, aspects such as cellular localization and variations in enzyme levels with the cell cycle are not easily taken into account. The in vivo strategies must involve strict controls, validation and optimizations to be quantitatively relevant. For optimal qualitative and quantitative data on repair of oxidative lesions by BER, it is recommended that a combination of the highly quantitative in vitro assays with one or more cell based techniques be performed to provide information on both amount and localization of DNA damage and repair in a particular sample.
Acknowledgments
We thank Dr. Chandrika Canugovi and Dr. Mahesh Ramamoorthy for editing the paper. Support was provided by funds from the National Institute of Health, National Institute on Aging Intramural Research Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moraes CT. Trends Genet. 2001;17:199–205. doi: 10.1016/s0168-9525(01)02238-7. [DOI] [PubMed] [Google Scholar]
- 2.Bollmann FM. Rejuvenation Res. 2007;10:327–333. doi: 10.1089/rej.2006.0527. [DOI] [PubMed] [Google Scholar]
- 3.Desjardins P, de Muys JM, Morais R. Somat Cell Mol Genet. 1986;12:133–139. doi: 10.1007/BF01560660. [DOI] [PubMed] [Google Scholar]
- 4.Croteau DL, Bohr VA. J Biol Chem. 1997;272:25409–25412. doi: 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- 5.Hamilton ML, Guo Z, Fuller CD, Van RH, Ward WF, Austad SN, Troyer DA, Thompson I, Richardson A. Nucleic Acids Res. 2001;29:2117–2126. doi: 10.1093/nar/29.10.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN, Turnbull DM. Nat Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- 7.de Souza-Pinto NC, Wilson DM, III, Stevnsner TV, Bohr VA. DNA Repair (Amst) 2008;7:1098–1109. doi: 10.1016/j.dnarep.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LeDoux SP, Wilson GL, Beecham EJ, Stevnsner T, Wassermann K, Bohr VA. Carcinogenesis. 1992;13:1967–1973. doi: 10.1093/carcin/13.11.1967. [DOI] [PubMed] [Google Scholar]
- 9.Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Carcinogenesis. 2009;30:2–10. doi: 10.1093/carcin/bgn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nyaga SG, Bohr VA. Methods Mol Biol. 2002;197:227–244. doi: 10.1385/1-59259-284-8:227. [DOI] [PubMed] [Google Scholar]
- 11.Pinz KG, Bogenhagen DF. Mol Cell Biol. 1998;18:1257–1265. doi: 10.1128/mcb.18.3.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stuart JA, Mayard S, Hashiguchi K, Souza-Pinto NC, Bohr VA. Nucleic Acids Res. 2005;33:3722–3732. doi: 10.1093/nar/gki683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Souza-Pinto NC, Mason PA, Hashiguchi K, Weissman L, Tian J, Guay D, Lebel M, Stevnsner TV, Rasmussen LJ, Bohr VA. DNA Repair (Amst) 2009;8:704–719. doi: 10.1016/j.dnarep.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thyagarajan B, Padua RA, Campbell C. J Biol Chem. 1996;271:27536–27543. doi: 10.1074/jbc.271.44.27536. [DOI] [PubMed] [Google Scholar]
- 15.Ladoukakis ED, Zouros E. Mol Biol Evol. 2001;18:1168–1175. doi: 10.1093/oxfordjournals.molbev.a003904. [DOI] [PubMed] [Google Scholar]
- 16.Tsaousis AD, Martin DP, Ladoukakis ED, Posada D, Zouros E. Mol Biol Evol. 2005;22:925–933. doi: 10.1093/molbev/msi084. [DOI] [PubMed] [Google Scholar]
- 17.Schon EA. Trends Biochem Sci. 2000;25:555–560. doi: 10.1016/s0968-0004(00)01688-1. [DOI] [PubMed] [Google Scholar]
- 18.Wallace DC. J Bioenerg Biomembr. 1994;26:241–250. doi: 10.1007/BF00763096. [DOI] [PubMed] [Google Scholar]
- 19.Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, Jen J, Sidransky D. Science. 2000;287:2017–2019. doi: 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- 20.Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B. Nat Genet. 1998;20:291–293. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- 21.Salvioli S, Capri M, Santoro A, Raule N, Sevini F, Lukas S, Lanzarini C, Monti D, Passarino G, Rose G, De BG, Franceschi C. Biotechnol J. 2008;3:740–749. doi: 10.1002/biot.200800046. [DOI] [PubMed] [Google Scholar]
- 22.Tonska K, Solyga A, Bartnik E. J Appl Genet. 2009;50:55–62. doi: 10.1007/BF03195653. [DOI] [PubMed] [Google Scholar]
- 23.Wallace DC. Novartis Found Symp. 2001;235:247–263. doi: 10.1002/0470868694.ch20. [DOI] [PubMed] [Google Scholar]
- 24.Cortopassi GA, Shibata D, Soong NW, Arnheim N. Proc Natl Acad Sci U S A. 1992;89:7370–7374. doi: 10.1073/pnas.89.16.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dianov GL, Souza-Pinto N, Nyaga SG, Thybo T, Stevnsner T, Bohr VA. Prog Nucleic Acid Res Mol Biol. 2001;68:285–297. doi: 10.1016/s0079-6603(01)68107-8. [DOI] [PubMed] [Google Scholar]
- 26.Robertson AB, Klungland A, Rognes T, Leiros I. Cell Mol Life Sci. 2009;66:981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tebbs RS, Flannery ML, Meneses JJ, Hartmann A, Tucker JD, Thompson LH, Cleaver JE, Pedersen RA. Dev Biol. 1999;208:513–529. doi: 10.1006/dbio.1999.9232. [DOI] [PubMed] [Google Scholar]
- 28.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 29.Meira LB, Devaraj S, Kisby GE, Burns DK, Daniel RL, Hammer RE, Grundy S, Jialal I, Friedberg EC. Cancer Res. 2001;61:5552–5557. [PubMed] [Google Scholar]
- 30.Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. Proc Natl Acad Sci U S A. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larsen E, Gran C, Saether BE, Seeberg E, Klungland A. Mol Cell Biol. 2003;23:5346–5353. doi: 10.1128/MCB.23.15.5346-5353.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bentley D, Selfridge J, Millar JK, Samuel K, Hole N, Ansell JD, Melton DW. Nat Genet. 1996;13:489–491. doi: 10.1038/ng0896-489. [DOI] [PubMed] [Google Scholar]
- 33.de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, Bohr VA. Cancer Res. 2001;61:5378–5381. [PubMed] [Google Scholar]
- 34.Endres M, Biniszkiewicz D, Sobol RW, Harms C, Ahmadi M, Lipski A, Katchanov J, Mergenthaler P, Dirnagl U, Wilson SH, Meisel A, Jaenisch R. J Clin Invest. 2004;113:1711–1721. doi: 10.1172/JCI20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie Y, Yang H, Cunanan C, Okamoto K, Shibata D, Pan J, Barnes DE, Lindahl T, McIlhatton M, Fishel R, Miller JH. Cancer Res. 2004;64:3096–3102. doi: 10.1158/0008-5472.can-03-3834. [DOI] [PubMed] [Google Scholar]
- 36.Larsen NB, Rasmussen M, Rasmussen LJ. Mitochondrion. 2005;5:89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 37.Nakabeppu Y. Prog Nucleic Acid Res Mol Biol. 2001;68:75–94. doi: 10.1016/s0079-6603(01)68091-7. [DOI] [PubMed] [Google Scholar]
- 38.Hashiguchi K, Stuart JA, de Souza-Pinto NC, Bohr VA. Nucleic Acids Res. 2004;32:5596–5608. doi: 10.1093/nar/gkh863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, Krokan HE. Nucleic Acids Res. 1997;25:750–755. doi: 10.1093/nar/25.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chattopadhyay R, Wiederhold L, Szczesny B, Boldogh I, Hazra TK, Izumi T, Mitra S. Nucleic Acids Res. 2006;34:2067–2076. doi: 10.1093/nar/gkl177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitra S, Izumi T, Boldogh I, Bhakat KK, Chattopadhyay R, Szczesny B. DNA Repair (Amst) 2007;6:461–469. doi: 10.1016/j.dnarep.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 42.Longley MJ, Prasad R, Srivastava DK, Wilson SH, Copeland WC. Proc Natl Acad Sci U S A. 1998;95:12244–12248. doi: 10.1073/pnas.95.21.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anson RM, Hudson E, Bohr VA. FASEB J. 2000;14:355–360. doi: 10.1096/fasebj.14.2.355. [DOI] [PubMed] [Google Scholar]
- 44.Pallotti F, Lenaz G. Methods Cell Biol. 2007;80:3–44. doi: 10.1016/S0091-679X(06)80001-4. [DOI] [PubMed] [Google Scholar]
- 45.Croteau DL, ap Rhys CM, Hudson EK, Dianov GL, Hansford RG, Bohr VA. J Biol Chem. 1997;272:27338–27344. doi: 10.1074/jbc.272.43.27338. [DOI] [PubMed] [Google Scholar]
- 46.Karahalil B, Hogue BA, de Souza-Pinto NC, Bohr VA. FASEB J. 2002;16:1895–1902. doi: 10.1096/fj.02-0463com. [DOI] [PubMed] [Google Scholar]
- 47.Battino M, Gorini A, Villa RF, Genova ML, Bovina C, Sassi S, Littarru GP, Lenaz G. Mech Ageing Dev. 1995;78:173–187. doi: 10.1016/0047-6374(94)01535-t. [DOI] [PubMed] [Google Scholar]
- 48.Peterson C, Nicholls DG, Gibson GE. J Neurochem. 1985;45:1779–1790. doi: 10.1111/j.1471-4159.1985.tb10534.x. [DOI] [PubMed] [Google Scholar]
- 49.Derdak Z, Garcia TA, Baffy G. Methods Mol Biol. 2009;559:205–217. doi: 10.1007/978-1-60327-017-5_15. [DOI] [PubMed] [Google Scholar]
- 50.Molina M, Segura JA, Aledo JC, Medina MA, Nunez dC, Marquez IJ. Biochem J. 1995;308(Pt 2):629–633. doi: 10.1042/bj3080629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han D, Williams E, Cadenas E. Biochem J. 2001;353:411–416. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Q, Lam PY, Han D, Cadenas E. J Neurochem. 2008;104:325–335. doi: 10.1111/j.1471-4159.2007.04957.x. [DOI] [PubMed] [Google Scholar]
- 53.Rasmussen HN, Andersen AJ, Rasmussen UF. Anal Biochem. 1997;252:153–159. doi: 10.1006/abio.1997.2304. [DOI] [PubMed] [Google Scholar]
- 54.Stuart JA, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. FASEB J. 2004;18:595–597. doi: 10.1096/fj.03-0890fje. [DOI] [PubMed] [Google Scholar]
- 55.Scheibye-Knudsen M, Quistorff B. Eur J Appl Physiol. 2009;105:279–287. doi: 10.1007/s00421-008-0901-9. [DOI] [PubMed] [Google Scholar]
- 56.Stuart JA, Hashiguchi K, Wilson DM, III, Copeland WC, Souza-Pinto NC, Bohr VA. Nucleic Acids Res. 2004;32:2181–2192. doi: 10.1093/nar/gkh533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Greenawalt JW. Methods Enzymol. 1974;31:310–323. doi: 10.1016/0076-6879(74)31033-6. [DOI] [PubMed] [Google Scholar]
- 58.Gredilla R, Garm C, Holm R, Bohr VA, Stevnsner T. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weissman L, Jo DG, Sorensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, Bohr VA. Nucleic Acids Res. 2007;35:5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Souza-Pinto NC, Croteau DL, Hudson EK, Hansford RG, Bohr VA. Nucleic Acids Res. 1999;27:1935–1942. doi: 10.1093/nar/27.8.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu P, Qian L, Sung JS, de Souza-Pinto NC, Zheng L, Bogenhagen DF, Bohr VA, Wilson DM, III, Shen B, Demple B. Mol Cell Biol. 2008;28:4975–4987. doi: 10.1128/MCB.00457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bohr VA. Carcinogenesis. 1991;12:1983–1992. doi: 10.1093/carcin/12.11.1983. [DOI] [PubMed] [Google Scholar]
- 63.Anson RM, Mason PA, Bohr VA. Methods Mol Biol. 2006;314:155–181. doi: 10.1385/1-59259-973-7:155. [DOI] [PubMed] [Google Scholar]
- 64.Thorslund T, Sunesen M, Bohr VA, Stevnsner T. DNA Repair (Amst) 2002;1:261–273. doi: 10.1016/s1568-7864(02)00003-4. [DOI] [PubMed] [Google Scholar]
- 65.Bohr VA. IARC Sci Publ. 1994:361–369. [PubMed] [Google Scholar]
- 66.Bohr VA, Anson RM. Mutat Res. 1995;338:25–34. doi: 10.1016/0921-8734(95)00008-t. [DOI] [PubMed] [Google Scholar]
- 67.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 68.Kovalenko OA, Santos JH. Curr Protoc Hum Genet. 2009;Chapter 19(Unit) doi: 10.1002/0471142905.hg1901s62. [DOI] [PubMed] [Google Scholar]
- 69.Santos JH, Meyer JN, Mandavilli BS, Van HB. Methods Mol Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- 70.Santos JH, Meyer JN, Skorvaga M, Annab LA, Van HB. Aging Cell. 2004;3:399–411. doi: 10.1111/j.1474-9728.2004.00124.x. [DOI] [PubMed] [Google Scholar]
- 71.yala-Torres S, Chen Y, Svoboda T, Rosenblatt J, Van HB. Methods. 2000;22:135–147. doi: 10.1006/meth.2000.1054. [DOI] [PubMed] [Google Scholar]
- 72.Van HB, Chen Y, Nicklas JA, Rainville IR, O’Neill JP. Mutat Res. 1998;403:171–175. doi: 10.1016/s0027-5107(98)00076-1. [DOI] [PubMed] [Google Scholar]
- 73.Helbock HJ, Beckman KB, Shigenaga MK, Walter PB, Woodall AA, Yeo HC, Ames BN. Proc Natl Acad Sci U S A. 1998;95:288–293. doi: 10.1073/pnas.95.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hudson EK, Hogue BA, Souza-Pinto NC, Croteau DL, Anson RM, Bohr VA, Hansford RG. Free Radic Res. 1998;29:573–579. doi: 10.1080/10715769800300611. [DOI] [PubMed] [Google Scholar]
- 75.Halliwell B, Dizdaroglu M. Free Radic Res Commun. 1992;16:75–87. doi: 10.3109/10715769209049161. [DOI] [PubMed] [Google Scholar]
- 76.Jaruga P, Speina E, Gackowski D, Tudek B, Olinski R. Nucleic Acids Res. 2000;28:E16-. doi: 10.1093/nar/28.6.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dizdaroglu M, Jaruga P, Rodriguez H. Nucleic Acids Res. 2001;29:E12-. doi: 10.1093/nar/29.3.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Free Radic Biol Med. 2003;34:1089–1099. doi: 10.1016/s0891-5849(03)00041-8. [DOI] [PubMed] [Google Scholar]
- 79.Collins AR, Cadet J, Moller L, Poulsen HE, Vina J. Arch Biochem Biophys. 2004;423:57–65. doi: 10.1016/j.abb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 80.Valavanidis A, Vlachogianni T, Fiotakis C. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009;27:120–139. doi: 10.1080/10590500902885684. [DOI] [PubMed] [Google Scholar]
- 81.Hartwig A, Dally H, Schlepegrell R. Toxicol Lett. 1996;88:85–90. doi: 10.1016/0378-4274(96)03722-8. [DOI] [PubMed] [Google Scholar]
- 82.Moreno-Villanueva M, Pfeiffer R, Sindlinger T, Leake A, Muller M, Kirkwood TB, Burkle A. BMC Biotechnol. 2009;9:39-. doi: 10.1186/1472-6750-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trapp C, McCullough AK, Epe B. Mutat Res. 2007;625:155–163. doi: 10.1016/j.mrfmmm.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 84.Wenzel P, Schuhmacher S, Kienhofer J, Muller J, Hortmann M, Oelze M, Schulz E, Treiber N, Kawamoto T, Scharffetter-Kochanek K, Munzel T, Burkle A, Bachschmid MM, Daiber A. Cardiovasc Res. 2008;80:280–289. doi: 10.1093/cvr/cvn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Osterod M, Larsen E, Le PF, Hengstler JG, Van Der Horst GT, Boiteux S, Klungland A, Epe B. Oncogene. 2002;21:8232–8239. doi: 10.1038/sj.onc.1206027. [DOI] [PubMed] [Google Scholar]
- 86.Pflaum M, Will O, Mahler HC, Epe B. Free Radic Res. 1998;29:585–594. doi: 10.1080/10715769800300631. [DOI] [PubMed] [Google Scholar]
- 87.Collins AR, Duthie SJ, Dobson VL. Carcinogenesis. 1993;14:1733–1735. doi: 10.1093/carcin/14.9.1733. [DOI] [PubMed] [Google Scholar]
- 88.Maynard S, Swistowska AM, Lee JW, Liu Y, Liu ST, Da Cruz AB, Rao M, de Souza-Pinto NC, Zeng X, Bohr VA. Stem Cells. 2008;26:2266–2274. doi: 10.1634/stemcells.2007-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Singh NP, McCoy MT, Tice RR, Schneider EL. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 90.Ide H, Kow YW, Chen BX, Erlanger BF, Wallace SS. Cell Biol Toxicol. 1997;13:405–417. doi: 10.1023/a:1007467726635. [DOI] [PubMed] [Google Scholar]
- 91.Yarborough A, Zhang YJ, Hsu TM, Santella RM. Cancer Res. 1996;56:683–688. [PubMed] [Google Scholar]
- 92.Yin B, Whyatt RM, Perera FP, Randall MC, Cooper TB, Santella RM. Free Radic Biol Med. 1995;18:1023–1032. doi: 10.1016/0891-5849(95)00003-g. [DOI] [PubMed] [Google Scholar]
- 93.Soultanakis RP, Melamede RJ, Bespalov IA, Wallace SS, Beckman KB, Ames BN, Taatjes DJ, Janssen-Heininger YM. Free Radic Biol Med. 2000;28:987–998. doi: 10.1016/s0891-5849(00)00185-4. [DOI] [PubMed] [Google Scholar]
- 94.Struthers L, Patel R, Clark J, Thomas S. Anal Biochem. 1998;255:20–31. doi: 10.1006/abio.1997.2354. [DOI] [PubMed] [Google Scholar]
- 95.de Souza-Pinto NC, Maynard S, Hashiguchi K, Hu J, Muftuoglu M, Bohr VA. Mol Cell Biol. 2009;29:4441–4454. doi: 10.1128/MCB.00265-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kong X, Mohanty SK, Stephens J, Heale JT, Gomez-Godinez V, Shi LZ, Kim JS, Yokomori K, Berns MW. Nucleic Acids Res. 2009;37:e68-. doi: 10.1093/nar/gkp221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ohno M, Oka S, Nakabeppu Y. Methods Mol Biol. 2009;554:199–212. doi: 10.1007/978-1-59745-521-3_13. [DOI] [PubMed] [Google Scholar]
- 98.Carcinogenesis. 2002;23:2129–2133. doi: 10.1093/carcin/23.12.2129. [DOI] [PubMed] [Google Scholar]
- 99.Beckman KB, Ames BN. Methods Enzymol. 1996;264:442–453. doi: 10.1016/s0076-6879(96)64040-3. [DOI] [PubMed] [Google Scholar]
- 100.Beckman KB, Ames BN. Mutat Res. 1999;424:51–58. doi: 10.1016/s0027-5107(99)00007-x. [DOI] [PubMed] [Google Scholar]
- 101.Persinger RL, Melamede R, Bespalov I, Wallace S, Taatjes DJ, Janssen-Heininger Y. Exp Gerontol. 2001;36:1483–1494. doi: 10.1016/s0531-5565(01)00134-6. [DOI] [PubMed] [Google Scholar]