

Abstract
The Met receptor tyrosine kinase is deregulated in a variety of cancers and is correlated with advanced stage and poor prognosis. Thus, Met has been identified as an attractive candidate for targeted therapy. We compared the tea polyphenol (−)-epigallocatechin-3-gallate (EGCG) and a specific Met inhibitor, SU11274, as suppressing agents of Met signaling in HCT116 human colon cancer cells. Treatment with hepatocyte growth factor increased phospho-Met levels, and this was inhibited in a concentration-dependent manner by EGCG and SU11274 (IC50 3.0 vs 0.05 µM, respectively). Downstream activation of Erk and Akt signaling pathways also was suppressed. Both compounds at a concentration of 5 µM lowered cell viability and proliferation, with EGCG being more effective than SU11274, and the invasion of colon cancer cells in Matrigel assays was strongly inhibited. These findings are discussed in the context of the pleiotropic effects of tea catechins, their tissue metabolite levels, and the potential to inhibit colon cancer metastasis and invasion.
Introduction
Met is a receptor tyrosine kinase (RTK), activated by the ligand hepatocyte growth factor (HGF). Met and HGF are required for embryonic development, but they can become deregulated in a variety of tumor types [1]. Met is a critical oncogene for tumor metastasis, facilitating cellular proliferation, invasion, and motility [2–6]. In human colorectal cancers, Met and HGF expression predicts tumor phenotype and propensity for metastasis, and is correlated with poor outcome [7]. Thus, Met and HGF are potential therapeutic targets for colorectal cancer.
SU11274 [(3Z)-N-(3-chlorophenyl)-3-({3,5-dimethyl-4-[(4-methylpiperazin-1-yl)carbonyl]-1H-pyrrol-2-yl}methylene)-N-methyl-2-oxoindoline-5-sulfonamide] is a Met inhibitor that induces G1 cell cycle arrest and apoptosis in cancer cells, interfering with phosphoinositide 3-kinase (PI3K) and other signaling pathways. Berthou et al. [8], observed that SU11274 differentially affects the kinase activity and downstream signaling of various mutant forms of Met. Whereas the variants M1268T and H1112Y were potently inhibited by SU11274, the mutants L1213V and Y1248H were resistant. The authors reported that “inhibition of the kinase altered cell proliferation, morphology and motility, while cells containing resistant mutants appeared unaffected by the compound” [8]. This has led to the suggestion that a combination approach might improve efficacy in the clinical setting, such as mTOR inhibitors in conjunction with cytotoxic chemotherapy [9].
Expanding upon the general idea of multiple mechanisms of attack on aberrant Met signaling, we recently undertook studies on Met activity in human colon cancer cells treated with green tea polyphenols. We observed that among the various catechins in green tea, (−)-epigallocatechin-3-gallate (EGCG) was the most effective inhibitor of Met [10], and that this occurred essentially independent of hydrogen peroxide [11]. The latter has been reported as a potential ‘artifact’ in some [12], but not all [13], cell culture studies that used tea polyphenols as test agents.
The work presented here sought to build upon our initial studies in colon cancer cells [10,11], as well as the findings of others indicating that tea polyphenols block Met activation in breast and hypopharyngeal cancer cells [14,15]. In this report, the specific goal was to examine downstream kinase pathways of Met, and the changes in cell growth and invasion following treatment with EGCG. It should be noted that EGCG also alters signaling via epidermal growth factor, platelet-derived growth factor, insulin-like growth factor 1, and vascular endothelial growth factor receptors [16–19]. EGCG inhibits the activities of cyclin-dependant kinases 2 and 4, and induces the expression of the Cdk inhibitors p21 and p27, leading to G1 arrest [19]. Using human HCT116 colon cancer cells, we compared EGCG and SU11274 as broad-spectrum and specific Met kinase inhibitors, respectively, and examined their downstream effects on PI3K and mitogen-activated protein kinase signaling (MAPK).
Experimental
Cell culture
HCT116 cells were obtained from American Type Tissue Collection (Manassas, VA) and maintained in McCoy's 5A media (Invitrogen, Carlsbad, CA) with 10% FBS and 1% penicillin/streptomycin. Cells were grown at 37°C with 5% CO2.
Cell treatments
Cells were plated at 1.5 × 105 cells in 12-well culture dishes and grown in serum containing media for 48 h. Cells were then incubated in serum-free media for 4 h. After serum starvation, cells were pretreated for 30 min with 5 µM EGCG (Sigma-Aldrich, MO) or 5 µM SU11274 (Calbiochem, San Diego, CA) followed by treatment with 30 ng/ml HGF (Calbiochem). We did not include catalase, because at the low concentrations of EGCG used here, effects on Met are essentially independent of H2O2 and the presence or absence of catalase [11].
Immunoblotting
Cells were placed in IP lysis buffer, vortexed, and centrifuged at 10,000 rpm for 5 min. The supernatant was collected and protein concentrations were determined by the bicinchoninic acid assay (Pierce, Rockford, IL). Proteins (10–20 µg) were separated by SDS-PAGE on a 4–12% bis-Tris gel (Novex, San Diego, CA) and transferred to nitrocellulose membrane (Invitrogen). Equal protein loading was confirmed by Amido Black staining and β-actin levels. The membrane was blocked for 1 h with Li-Cor Blocking Buffer (Li-Cor Biosciences, Lincoln, Nebraska), followed by overnight incubation with primary antibody at 4°C, and finally incubated for 1 h with goat anti-mouse secondary antibody conjugated with IRDye800 and goat anti-rabbit antibody conjugated with IRDye680 (Li-Cor Biosciences). Antibody dilutions were as follows: phospho-Met (Tyr1234/1235) 1:1000 (Cell Signaling Technology, Beverly, MA); total Met 1:1000 (Cell Signaling Technology); phospho-Akt 2 µg/ml (Upstate); total Akt 1:1000 (Cell Signaling Technology); phospho-Erk1/2 1:2000 (Cell Signaling Technology); total Erk1/2 1:1000 (Cell Signaling Technology); and β-actin 1:5000 (Sigma). Image acquisition and analysis were performed using the Odyssey® Infrared Imaging System (Li-Cor Biosciences).
Enzyme-linked immunosorbent assay (ELISA)
Cells were pretreated with EGCG or SU11274 for 30 min and then HGF (30 ng/ml) was added. Cells were incubated for an additional 15 min and lysed in RIPA buffer containing phosphatase inhibitors (Pierce). ELISA was performed as described in the instruction manual for STAR phospho-Met (Tyr1230/Tyr1234/Tyr1235) ELISA kit (Upstate). Concentration for 50% inhibition (IC50) was calculated using the sigmoidal dose-response method in GraphPad Prism 4 (GraphPad Software, La Jolla, CA).
Cell viability
Cells were incubated with 5 µM EGCG or 5 µM SU11274 for 30 min, and HGF (30 ng/ml) was then added to the culture media. Cells were collected at 0, 24, 48 and 72 h and stained with Guava ViaCount Reagent (Guava Technologies, Hayward, CA) for 5 min. The number of viable cells was determined by using the Guava ViaCount Assay on a Guava Personal Cell Analyzer.
Cell invasion assay
Matrigel (BD Transduction Laboratories) was diluted to 2 mg/ml in serum-free McCoy’s 5A media, and 50 µl were plated on 6.5 mm diameter Costar Transwell Inserts (VWR) and allowed to gel for 1 h at 37°C. Cells were diluted to 2 × 104 cells/ml in serum-free media with or without 5 µM EGCG or 5 µM SU11274 and plated in the top of the wells. Serum-free McCoy’s 5A with or without 30 ng/ml HGF was plated in the bottom of the wells. Cells were incubated for 24 h at 37°C in a 5% CO2 incubator. Matrigel and cells remaining on the top surface were removed with a cotton swab. Cells that migrated to the bottom of the insert were fixed in methanol and stained with ProLong Gold DAPI (Invitrogen) and counted using a Zeiss Axiovert 100 fluorescent microscope.
Statistics
Student’s t-test was used for pairwise comparisons. Additional analyses used a mixed model procedure with SPSS Statistics software (IBM Corp., Armonk, NY). P-values shown in the figures indicate significant differences from multiple comparisons, with Bonferroni correction.
Results
A side-by-side comparison of SU11274 and EGCG (Fig. 1A) examined their abilities to block Met activation in HCT116 colon cancer cells. Immunoblotting revealed little or no p-Met in the absence of HGF treatment and high levels of p-Met after HGF, with total Met expression remaining constant (Fig. 1B, left lanes). Both test compounds reduced the levels of p-Met in a concentration-dependent manner over the concentration range 0.05–10 µM (Fig. 1B). Quantification of p-Met in the corresponding cell lysates revealed SU11274 to be a more potent Met inhibitor than EGCG, with IC50 values of 0.05 µM and 3 µM, respectively (Fig 1C).
Fig. 1.

EGCG and SU11274 inhibit Met activation in human colon cancer cells. (A) Chemical structures of SU11274 and EGCG. (B) HCT116 cells were serum-starved for 4 h and then treated with various concentrations of EGCG or SU11274 for 30 min. HGF (30 ng/ml) was added to the cell culture media, followed by incubation for an additional 15 min. Immunoblotting was performed on cell lysates using primary antibodies to phosphorylated-Met (p-Met), total Met, or β-actin (loading control). (C) Results from STAR phospho-Met ELISA kit (Upstate), using cell lysates from (B). Data (mean±SD, n=3) are representative of the findings from three separate experiments.
We next examined the downstream Met signaling pathways of Met kinase in time-course experiments. In the absence of EGCG, HGF induced a rapid increase in the phosphorylation of Met, and high p-Met levels were detected between 0.25–2 h, followed by a return to baseline (Fig. 2A). In the presence of 5 µM ECGC, the increase in p-Met was essentially abolished. Quantification of the immunoblots confirmed that the inhibition by EGCG was significant at all time points between 0.25–6 h (Fig. 2B, upper panel, p-Met normalized to total Met). In the absence of EGCG, HGF increased the levels of p-Akt at 0.25–2 h and p-Erk1/2 at 0.25–1 h (Fig. 2A). There was a trend towards inhibition following EGCG treatment, and this reached significance at 1 h for p-Akt/Akt, at 0.5, 1, and 6 h for p-Erk1/Erk1, and at 1–2 h for p-Erk2/Erk2 (Fig. 2B).
Fig. 2.
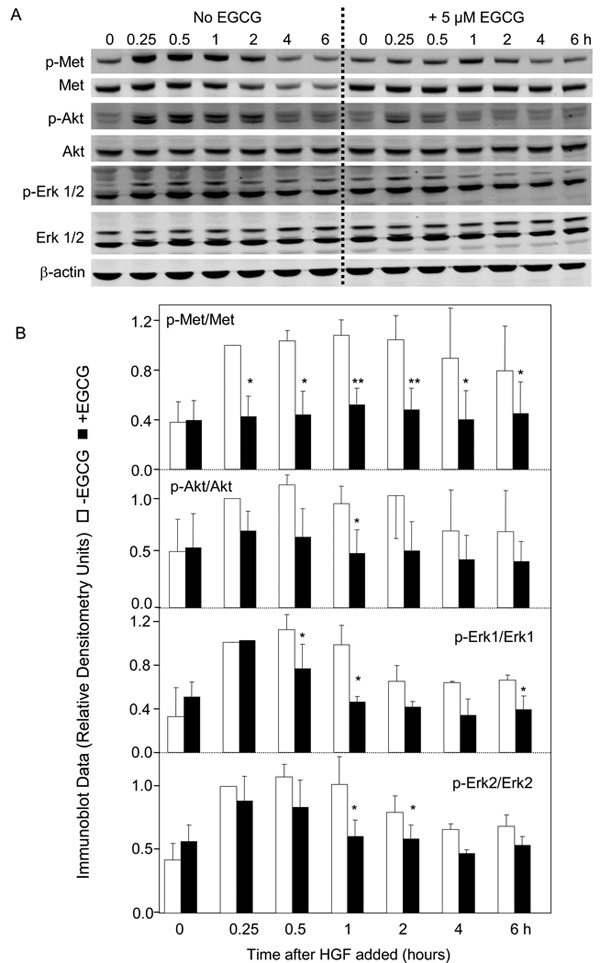

Inhibitory effects of EGCG on MAPK and PI3K signaling pathways. (A) HCT116 cells were serum-starved for 4 h and treated with or without 5 µM EGCG for 30 min. HGF (30 ng/ml) was added and incubations were continued for the times indicated. Immunoblotting was performed on cell lysates using primary antibodies to phosphorylated-Met (p-Met), total Met, phosphorylated-Akt (p-Akt), total Akt, phosphorylated-Erk (p-Erk), total Erk, or β-actin (loading control). (B) Quantification of immunoblots by densitometry, showing phosphorylated protein normalized to corresponding total protein. Open bars −EGCG, solid bars +EGCG. (C) Using the same approach as above, HCT116 cells were serum-starved for 4 h, treated for 30 min with 5 µM SU11274, 5 µM EGCG, or 5 µM SU11274 + 5 µM EGCG in combination, followed by HGF or media alone (-HGF). Immunoblots (not shown) were quantified from three separate experiments. In (B) and (C), data=mean±SD, n=3; *P<0.05; **P<0.01.
Changes in Met, Erk, and Akt pathways also were examined following SU11274, EGCG, and EGCG plus SU11274 treatments. As expected, HGF alone increased significantly the levels of p-Met, p-Akt, and p-Erk1/2, and this increase was blocked by 5 µM SU11274 (Fig 2C). No additional inhibition was detected when 5 µM SU11274 was combined with 5 µM EGCG (data not shown).
As shown in Fig. 3A, both inhibitors reduced cell viability relative to the corresponding control, in the presence and absence of HGF, and significant differences between EGCG and SU11274 treatment groups were apparent at 48 h and 72 h. For example, in cells treated with HGF, cell viability at 72 h was decreased to 65% by EGCG and to 79% by SU11274 (**P<0.01, compare open and solid black bars, respectively, in Fig. 3A). EGCG generally was more effective in cells treated with HGF than –HGF (compare white bars at a given time-point, Fig 3A), but this difference was not apparent for SU11274.
Fig. 3.

Inhibition of cell viability and proliferation by EGCG and SU11274. (A) Viability of HCT116 cells treated with EGCG, SU11274, or vehicle control in the presence and absence of HGF. (B) Proliferation of HCT116 cells treated with EGCG, SU11274, or vehicle, in the presence and absence of HGF. Data shown in (A) and (B) are representative findings from three separate experiments (mean±SD, n=3); *P<0.05; **P<0.01; ***P<0.001.
Compared to the -HGF control, HGF treatment increased the overall cell number at 24, 48, and 72 h (square symbols in Fig. 3B, P<0.001 for all time-points). In the presence or absence of HGF, cell growth was suppressed markedly by SU11274 (circular symbols), and it was lowered relative to 0-h controls following EGCG treatment (triangles).
A matrigel assay was used to examine cell invasion in vitro (Fig 4A). The presence of HGF in the lower compartment increased the invasion of HCT116 cells, as expected (compare gray bars in Fig. 4B, ***P<0.001). Following HGF treatment, EGCG and SU11274 decreased the cell invasion to 32% and 18%, respectively (**P<0.01 for both treatment groups vs control), but under these conditions there was no significant difference between EGCG and SU11274 groups (compare white vs black bars in Fig. 4B). In the absence of HGF, however, SU11274 was more effective at reducing cell invasion compared to EGCG (*P<0.05, black vs green bars).
Fig. 4.

Suppression of invasion by ECGC and SU11274. (A) Representative images showing invasion of HCT116 cells through matrigel-coated Transwell inserts, under the assays conditions indicated. (B) Quantification of three independent in vitro invasion assays (mean±SD, n=3). *P<0.05; **P<0.01; n/s = not significant.
Discussion
The Met receptor tyrosine kinase is considered an important prognostic factor for metastasis, tumor stage, and reduced survival [7]. We compared the effects of two Met inhibitors in HCT116 human colon cancer cells. SU11274 is a specific Met inhibitor [8], whereas EGCG most likely acts on multiple RTKs [16–19]. The results were consistent with a potent selective Met inhibitor being more effective than a widely consumed tea catechin in suppressing Met activation. SU11274 had a lower IC50 value than EGCG in reducing p-Met levels in HCT116 cells, and SU11274 was more effective than EGCG at inhibiting Akt and Erk pathways. Met activation results in increased MAPK and PI3K signaling in breast epithelial cells, and these pathways have been strongly implicated in HGF-induced cellular invasion [14].
Interestingly, 5 µM EGCG had a greater inhibitory effect on cell viability than the same concentration of SU11274, in both HGF-treated and HGF-untreated cells. EGCG also was more effective than SU11274 at decreasing proliferation of HCT116 cells over a 72-h period. However, in a matrigel invasion assay minus HGF treatment, SU11274 was slightly more effective than EGCG at decreasing the number of invading cells, whereas in the presence of HGF both compounds were equally effective at suppressing invasion. Our interpretation of these findings is that SU11274 acts more effectively on the specific kinase pathways studied, but additional chemopreventive mechanisms of EGCG [13–16] likely result in greater overall inhibition of cell viability and proliferation. Both test compounds nonetheless strongly suppressed HGF-mediated invasion in vitro.
In this investigation, EGCG had an IC50 value of 3 µM for inhibition of Met activation, which is noteworthy given that a peak plasma concentration of 7.5 µM EGCG has been detected in humans after pharmacological oral dosing [20]. These concentrations of EGCG and other tea catechins also might be feasible in the gastrointestinal tract following oral tea intake, despite extensive methylation, glucuronidation, and sulfation, or conversion to valerolactone breakdown products [21–23]. Indeed, Stalmach et al. [24] recently reported that following green tea consumption by ileostomy patients, substantial quantities of flavan-3-ols passed into the small intestine, and would be available to the large intestine at levels exceeding those employed in the current investigation. Given this fact, it will be of great interest to ascertain whether catechin metabolites and breakdown products generated in the large intestine have improved efficacy towards suppression of Met signaling, cell proliferation, and invasion in patients with colon cancer or other intestinal pathologies.
Acknowledgments
We thank Dr. Donald Jump of the Department of Nutrition and Exercise Sciences for access to the Li-Cor instrument, and Dr. Jeffrey Greenwood and Dr. Hyo Sang Jang of the Cell Imaging and Analysis Core of the Environmental Health Sciences Center for assistance with invasion assays, Dr. Praveen Rajendran for assistance with the calculation of IC50 values for EGCG and SU11274 with the Met receptor and Hui Nian for performing statistical analysis of cell proliferation data. This work was supported in part by NIEHS Center grant P30 ES00210 and by Program Project P01 CA090890 from the US National Cancer Institute.
Abbreviations used
- RTK
receptor tyrosine kinase
- HGF
hepatocyte growth factor
- SU11274
[(3Z)-N-(3-chlorophenyl)-3-({3,5-dimethyl-4-[(4-methylpiperazin-1-yl)carbonyl]-1H-pyrrol-2-yl}methylene)-N-methyl-2-oxoindoline-5-sulfonamide]
- PI3K
phosphoinositide 3-kinase
- EGCG
(−)-epigallocatechin-3-gallate
- MAPK
mitogen-activated protein kinase
- ELISA
enzyme-linked immunosorbent assay
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Naran S, Zhang X, Hughes SJ. Inhibition of HGF/MET as therapy for malignancy. Expert Opin. Ther. Targets. 2009;13:569–581. doi: 10.1517/14728220902853917. [DOI] [PubMed] [Google Scholar]
- 2.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 3.Christensen JG, Burrows J, Salgia R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005;225:1–26. doi: 10.1016/j.canlet.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 4.Fujita S, Sugano K. Expression of c-met proto-oncogene in primary colorectal cancer and liver metastases. Jpn. J. Clin. Oncol. 1997;27:378–383. doi: 10.1093/jjco/27.6.378. [DOI] [PubMed] [Google Scholar]
- 5.Jeffers M, Rong S, Woude GF. Hepatocyte growth factor/scatter factor-Met signaling in tumorigenicity and invasion/metastasis. J. Mol. Med. 1996;74:505–513. doi: 10.1007/BF00204976. [DOI] [PubMed] [Google Scholar]
- 6.Jeffers M, Schmidt L, Nakaigawa N, Webb CP, Weirich G, Kishida T, Zbar B, Vande Woude GF. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc. Natl. Acad. Sci. USA. 1997;94:11445–11450. doi: 10.1073/pnas.94.21.11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kammula US, Kuntz EJ, Francone TD, Zeng Z, Shia J, Landmann RG, Paty PB, Weiser MR. Molecular co-expression of the c-Met oncogene and hepatocyte growth factor in primary colon cancer predicts tumor stage and clinical outcome. Cancer Lett. 2007;248:219–228. doi: 10.1016/j.canlet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Berthou S, Aebersold DM, Schmidt LS, Stroka D, Heigl C, Streit B, Stalder D, Gruber G, Liang C, Howlett AR, Candinas D, Greiner RH, Lipson KE, Zimmer Y. The Met kinase inhibitor SU11274 exhibits a selective inhibition pattern toward different receptor mutated variants. Oncogene. 2004;23:5387–5393. doi: 10.1038/sj.onc.1207691. [DOI] [PubMed] [Google Scholar]
- 9.Toschi L, Jänne PA. Single-agent and combination therapeutic strategies to inhibit hepatocyte growth factor/MET signaling in cancer. Clin. Cancer Res. 2008;14:5941–5946. doi: 10.1158/1078-0432.CCR-08-0071. [DOI] [PubMed] [Google Scholar]
- 10.Larsen CA, Bisson WH, Dashwood RH. Tea catechins inhibit hepatocyte growth factor receptor (MET kinase) activity in human colon cancer cells: kinetic and molecular docking studies. J. Med. Chem. 2009;52:6543–6545. doi: 10.1021/jm901330e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsen CA, Dashwood RH. Suppression of Met activation in human colon cancer cells treated with (−)-epigallocatechin-3-gallate: minor role of hydrogen peroxide. Biochem. Biophys. Res. Commun. 2009;389:527–530. doi: 10.1016/j.bbrc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long LH, Clement MV, Halliwell B. Artifacts in cell culture: rapid generation of hydrogen peroxide on addition of (−)-epigallocatechin, (−)-epigallocatechin gallate, (+)-catechin, and quercetin to commonly-used cell culture media. Biochem. Biophys. Res. Commun. 2000;273:50–53. doi: 10.1006/bbrc.2000.2895. [DOI] [PubMed] [Google Scholar]
- 13.Dashwood WM, Orner GA, Dashwood RH. Inhibition of beta-catechin/Tcf activity by white tea, green tea, and epigallocatechin-3-gallate (EGCG): minor contribution of H2O2 at physiologically relevant EGCG concentrations. Biochem. Biophys. Res. Commun. 2002;296:584–588. doi: 10.1016/s0006-291x(02)00914-2. [DOI] [PubMed] [Google Scholar]
- 14.Bigelow RL, Cardelli JA. The green tea catechins, (−)-Epigallocatechin-3-gallate (EGCG) and (−)-Epicatechin-3-gallate (ECG), inhibit HGF/Met signaling in immortalized and tumorigenic breast epithelial cells. Oncogene. 2006;25:1922–1930. doi: 10.1038/sj.onc.1209227. [DOI] [PubMed] [Google Scholar]
- 15.Lim YC, Park HY, Hwang HS, Kang SU, Pyun JH, Lee MH, Choi EC, Kim CH. (−)-Epigallocatechin-3-gallate (EGCG) inhibits HGF-induced invasion and metastasis in hypopharyngeal carcinoma cells. Cancer Lett. 2008;271:140–152. doi: 10.1016/j.canlet.2008.05.048. [DOI] [PubMed] [Google Scholar]
- 16.Khan N, Mukhtar H. Multitargeted therapy of cancer by green tea polyphenols. Cancer Lett. 2008;269:269–280. doi: 10.1016/j.canlet.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimizu M, Weinstein IB. Modulation of signal transduction by tea catechins and related phytochemicals. Mutat. Res. 2005;591:147–160. doi: 10.1016/j.mrfmmm.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Teillet F, Boumendjel A, Boutonnat J, Ronot X. Flavonoids as RTK inhibitors and potential anticancer agents. Med. Res. Rev. 2008;28:715–745. doi: 10.1002/med.20122. [DOI] [PubMed] [Google Scholar]
- 19.Lin JK, Liang YC, Lin-Shiau SY. Cancer chemoprevention by tea polyphenols through mitotic signal transduction blockade. Biochem. Pharmacol. 1999;58:911–915. doi: 10.1016/s0006-2952(99)00112-4. [DOI] [PubMed] [Google Scholar]
- 20.Yang CS, Sang S, Lambert JD, Lee MJ. Bioavailability issues in studying the health effects of plant polyphenolic compounds. Mol. Nutr. Food Res. 2008;52 Suppl 1:S139–S151. doi: 10.1002/mnfr.200700234. [DOI] [PubMed] [Google Scholar]
- 21.Chow HH, Cai Y, Hakim IA, Crowell JA, Shahi F, Brooks CA, Dorr RT, Hara Y, Alberts DS. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin. Cancer Res. 2003;9:3312–3319. [PubMed] [Google Scholar]
- 22.Auger C, Mullen W, Hara Y, Crozier A. Bioavailability of polyphenon E flavin-3-ols in humans with an ileostomy. J. Nutr. 2008;138:1535S–1542S. doi: 10.1093/jn/138.8.1535S. [DOI] [PubMed] [Google Scholar]
- 23.Raab T, Barron D, Vera FA, Crespy V, Oliveira M, Williamson G. Catechin glucosides: occurrence, synthesis, and stability. J. Agric. Food Chem. 2010 Jan 29; doi: 10.1021/jf9034095. [epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 24.Stalmach A, Mullen W, Steiling H, Williamson G, Lean ME, Crozier A. Absorption, metabolism, efflux and excretion of green tea flavan-3-ols in humans with an ileostomy. Mol. Nutr. Food Res. 2010;54:323–334. doi: 10.1002/mnfr.200900194. [DOI] [PubMed] [Google Scholar]