

Abstract
The identification of host cell factors for virus replication holds great promise for the development of new antiviral therapies. Recently, high-throughput screening methods have emerged as powerful tools to identify candidate host factors for therapeutic intervention. The development of assay systems suitable for large-scale automated screening is of particular importance for novel viruses with high pathogenic potential for which limited biological information can be developed in a short period of time. This report presents a general enzymatic reporter system for the detection and characterization of multiple enveloped viruses that does not rely on engineering of the virus. Instead, reporter enzymes are incorporated into virus particles by targeting to lipid microdomains in producer cells. The approach allows a variety of human pathogenic enveloped viruses to be detected by sensitive, inexpensive and automatable enzymatic assays. Tagged viruses can be purified quickly and efficiently by a magnetic bead-based capture method. The method allows general detection of enveloped viruses without prior reference to their sequence.
Keywords: High-throughput screening, Alkaline phosphatase, Enveloped virus, Flag-tagged virus purification
1. Introduction
Enveloped viruses constitute a significant global health problem and potential bioterror hazard. This group of viruses includes human immunodeficiency virus (HIV), hepatitis C virus (HCV), influenza virus, members of the coronavirus family, and all of the high risk pathogenic viruses, including the arboviruses, filoviruses and poxviruses. All viruses require host cell factors for completion of their life cycle and the identification of these factors presents an opportunity to discover therapeutic targets that in principle should be less susceptible to virus evasion by mutation (Lama and Planelles, 2007). Genome-wide high-throughput screening has emerged as a powerful technique for identifying host factors that regulate virus replication and budding (Brass et al., 2008, Konig et al., 2008, Krishnan et al., 2008, Randall et al., 2007, Tai et al., 2009, Zhou et al., 2008). This is of particular interest with respect to developing effective methods to characterize novel or emerging viruses (Snowden, 2008). Although identification of specific viruses requires at least partial knowledge of the protein or DNA composition of the virus, improvements in sequencing technology have made it possible to discover novel viruses using microarray chips (Wang et al., 2002), cDNA amplification (van der Hoek et al., 2004) or next-generation sequencing methods (Briese et al., 2009, Zhang et al., 2006). However, few of these methods are compatible with high-throughput screening approaches.
Many known viruses have been engineered by the introduction of reporter sequences such as GFP or Luciferase in their genomes to provide activities that can be measured to provide a surrogate for viral replication (Hao et al., 2008, Konig et al., 2008). Alternatively, reporter virus particles can be produced by translational fusion of a specific virus component to a reporter gene such as chloramphenicol transferase (Sato et al., 1995, Wu et al., 1995, Yao et al., 1999), luciferase (Asokan et al., 2008, Kolokoltsov and Davey, 2004, McCarthy et al., 2006) or alkaline phosphatase (Iro et al., 2009). A similar concept is the incorporation of translational fusions with fluorescent proteins to observe trafficking of individual virions (Fritz et al., 2008, McDonald et al., 2002, Muller et al., 2004). However, in many cases the reporter gene interferes with infectivity of the virus (Furnes et al., 2008, Shih et al., 2004) and careful optimization is required for each individual virus construct. To develop more general labeled-virus assays, the reporter gene should be incorporated by a means common to a large group of viruses.
The membrane of enveloped viruses is derived entirely from the host cell, and contains both the lipid bilayer and some host cell membrane proteins (Brugger et al., 2006, Hammarstedt et al., 2000). It has been proposed that virus budding originates in lipid rafts, and as a consequence lipid raft-localized host cell proteins are highly enriched in viral membranes (Hammarstedt and Garoff, 2004, Hammarstedt et al., 2000, Manes et al., 2000, Ono and Freed, 2001). This property can be used to target enzymatic reporter activities to the virus membrane by modification of the reporter with a lipid raft anchor, a strategy that has previously been used for pseudotyping of virus particles (Pickl et al., 2001). The present report shows that host cells expressing GPI-anchored alkaline phosphatase can be used to detect diverse enveloped viruses as RSV, HSV1, HIV and human coronavirus 229E. The method thus allows the detection, isolation and characterization of a wide variety of known enveloped viruses and in principle could be applied to unknown or poorly characterized emerging viruses.
2. Materials and methods
2.1. Plasmids
For construction of AP::CD16, SEAP::APP pEAK12 (Schobel et al., 2006) was digested with XbaI and NotI and the PCR amplified, NheI and NotI tailed, GPI-anchor acceptor sequence of CD16b was inserted to create AP::CD16 pEAK12. A PCR fragment of placental alkaline phosphatase (PLAP, IMAGE clone, Open Biosystems) was subcloned into pEAK14 to generate pEAK14-PLAP. The leader peptide was replaced with the signal peptide of Gaussia princeps luciferase and the flag peptide sequence DDDDDYDK was inserted upstream of the signal peptide to generate a Flag-PLAP. Flag-PLAP was inserted in pMOWS (Ketteler et al., 2002), pEAK14, and pEAK14-PDG, a vector containing a fusion cassette of puromycin, dihydrofolate reductase (DHFR), and green fluorescent protein (GFP) linked by F2A and T2A self-cleaving peptide sequences to generate single peptides of each marker gene.
2.2. Cell lines
Cell lines used were human embryonic kidney cells 293ET, Vero monkey kidney cells (ATCC, #CCL-81), lung fibroblast MRC5 cells (ATCC, #CCL-171) and the T cell line CEMx174-T2 (MT2; ATCC, #CRL-1992). All cell lines were maintained in DMEM supplemented with 10% calf serum (supplemented with iron), 0.25 μg/mL gentamycin and 50 μM β-mercaptoethanol. MT2 cells were maintained in IMEM with the same supplements. 293ET cells were transfected using calcium-phosphate precipitation as described (Ketteler et al., 2002). DHFR vectors were transfected in Vero cells using Nucleofection (Amaxa, Cologne, Germany) using Solution V and program V-01. Cells were selected in 6.0 μg/mL Puromycin. MRC5 cells were transduced with retroviral supernatants generated upon co-transfection of MoMLV Gag-Pol and VSV-G with pMOWS-FlagPLAP in 293ET cells. Stable AP::CD16 transfectants were established by electroporation (1.5 × 107 cells/400 medium, 950 μF, 400 V, 4 mm cuvettes) of 293 cells with 50 μg of SfiI linearized plasmid DNA and selection with puromycin (1 μg/mL, Sigma-Chemicals, St. Louis, USA).
2.3. Immunofluorescence analysis
For membrane staining, 50 μL of 293 or 293 AP::CD16 cells (1 × 107/mL) were incubated for 30 min at 4 °C with mouse–anti-human alkaline phosphatase-specific mAb (20 μg/mIgG2a, clone 8B6, Sigma-Chemicals), control mAb VIAP (negative control, IgG1, kindly provided by Dr. Otto Majdic, Vienna) or CD99 mAb 3B2/TA8 (positive control, IgG1, Caltag Laboratories, Burlingame, CA). Subsequently, cells were washed twice (1% BSA and 0.05% NaN3) followed by incubation with Oregon-green conjugated goat-anti-mouse IgG (Molecular Probes, Eugene, OR) at 4 °C for 30 min. After another wash cells were resuspended (200 μL PBS, 50 μL propidium iodide (0.5 μg/mL, Sigma-Chemicals) and subjected to flow cytometry (FACS Calibur, CellQuest software, Becton Dickinson, San Jose, CA). GPI-anchoring of AP::CD16 was determined by PI-PLC treatment with phosphatidylinositol specific phospholipase C from Bacillus thuringiensis (American Radiolabeled Chemicals, St. Louis, MO) according to the manufacturer's recommendations. Briefly, 293 AP::CD16 cells were washed in PBS w/o Ca2+ and Mg2+, resuspended at a concentration of 1 × 107 cells/mL in PBS. 100 mU of PI-PLC were added to 1 × 107 cells and incubated at 37 °C for 2 h. Subsequently cells were washed in PBS/1% BSA and subjected to surface staining for alkaline phosphatase (mAb 8B6), CD59 (mAb MEM-43/5), or CD99 (mAb 3B2/TA8). Percent release was calculated by relating the geometric mean expression of untreated cells to PI-PLC treated cells corrected by the geo-mean of a non-binding control mAb (VIAP).
2.4. Generation of MoMLV particles
293 or 293 AP::CD16 cells were transiently transfected by the calcium-phosphate precipitation method using 25 μg of plasmid DNA (either vector without insert, or bearing Gag-Pol). Supernatants were harvested after 48 h, cleared of cellular debris by filtration through 0.45 μm syringe filters (Millipore, Billerica, MA) and subjected to direct analyses (crude supernatant). Alternatively, membrane vesicles were concentrated by ultracentrifugation in a Beckman-Optima LE-80K centrifuge (Beckman Instruments, Palo Alto, CA), using a SW41 Ti rotor, at 100,000 × g for 1 h and were washed once in PBS. For determination of phosphatase activity, pellets were resuspended in culture medium at the original volume. This particulate fraction was analyzed and compared to cleared and crude supernatants. To destroy endogenous AP activity of 293 cells 1 mL aliquots of the various preparations were incubated at 56 °C for 15 min. Afterwards, 50 μL of the various preparations was added to 50 μL of p-NNP substrate buffer, incubated at 37 °C for 1 h and analyzed for substrate conversion at 405 nm (Thermomax Microplate Reader, Molecular Devices, Sunnyvale, CA, supported by the Softmax Pro software).
2.5. Animal viruses
Human respiratory syncytial virus (RSV; strain B WV/14617/’85; ATCC, #VR-1400), Aguacate virus (ATCC, #VR900), and human herpesvirus HSV1, strain F (ATCC, #VR-733) were propagated in Vero cells maintained in 2% calf serum (supplemented with iron) at 35 °C. Human coronavirus 229E (ATCC, #VR-740) was propagated in MRC5 cells (ATCC, #CCL-171) maintained with 2% calf serum (supplemented with iron) at 35 °C. Titer of viruses provided by ATCC was 104.5 TCID[50]/0.2 mL for HCoV 229E, 107.75 TCID[50]/0.2 mL for HSV1 and 104.25 TCID[50]/0.2 mL for RSV1. For all viruses, 100 μL of virus stocks were used for infection in 12-well plates. HSV1 and Aguacate virus infection was confirmed by rounding of cells and cytopathic effects. RSV was detected by the Light Diagnostics™ respiratory virus detection kit (Millipore, Billerica, MA) according to the manufacturer's instructions. HSV1 was detected by anti-HSV1 antibody (Sigma, St. Louis), HCoV 229E by anti-coronavirus 229E antiserum (Millipore, clone #401-4A).
2.6. Alkaline phosphatase
Secreted alkaline phosphatase was determined using the Phospha-Light™ secreted alkaline phosphatase reporter assay system (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. In all experiments, the supernatants were centrifuged at 10,700 × g prior to testing. Briefly, 50 μL of supernatant was mixed with 150 μL 1× Dilution buffer/1% Triton X-100 and incubated at 70 °C for 20 min. 50 μL of diluted supernatant was mixed with 50 μL of assay buffer and 50 μL of substrate solution before assaying in the TopCount luminescence plate reader.
2.7. Polyacrylamide gel electrophoresis and Western blotting
Samples were resolved onto 10% polyacrylamide gels (BioRad) and transferred to PVDF membranes (Millipore, Billerica, MA), stained with Ponceau Red (Sigma, St. Louis, MO), blocked with 2% BSA and incubated with AP specific mAb 8B6 (Sigma), MoMLV Gag specific mAb R187 (ATCC), anti-Flag M2 antibody (Sigma, St. Louis, MO), anti-HSV glycoprotein D (Sigma, St. Louis, MO) or anti-coronavirus (Millipore, Billerica, MA). After incubation with appropriate secondary antibodies (Jackson Immunoresearch, West Grove, PA), the bands were visualized with enhanced chemoluminescence (ECL) reagent.
2.8. Immunoprecipitation of virus particles
Viral supernatants were collected, anti-Flag M2 antibody (Sigma, St. Louis) and magnetic anti-mouse IgG coated Dynabeads® (Invitrogen) were added for 2 h at 4 °C, and 1/4 of the beads were used for re-infection of MT2 cells.
2.9. ELISA
ELISA for p24 (PerkinElmer Alliance Kit) was performed according to the manufacturer's instructions.
3. Results
3.1. Virus-like MLV particles incorporate membrane-anchored placental alkaline phosphatase
To evaluate the feasibility of establishing a general and quantitative detection method for enveloped viruses based on host-encoded reporter proteins targeted to lipid rafts (Hammarstedt and Garoff, 2004, Pickl et al., 2001), a number of candidate reporter systems were evaluated. One of these, placental alkaline phosphatase (AP) fused to CD16b GPI-anchor (AP::CD16b; Fig. 1A) proved suitable for the purpose. AP::CD16b was expressed on the cell surface of cells as shown by flow cytometry analysis using specific antibody against AP (Fig. 1B). Treatment with PI-PLC resulted in a reduction (67.7 ± 11.2%) of the amount of AP::CD16b on the cell surface, confirming that AP::CD16b is anchored by a glycosylphosphatidyl lipid. The release of AP activity into supernatant (SN) upon co-expression of MoMLV Gag-Pol was measured as a surrogate for virus infection. The MoMLV Gag protein has been shown to be sufficient to form virus-like particles in the absence of retroviral RNA or other structural proteins and thus serves as a good model system for membrane incorporation of host proteins (Mergener et al., 1992). In the absence of Gag-Pol, very little AP activity was detected in supernatant (Fig. 1C). In the presence of Gag-Pol, a 4-fold increase in AP activity in supernatant of cells expressing AP::CD16b was detected, indicating specific release of AP activity as a consequence of Gag-Pol expression (Fig. 1C). To determine, whether AP activity was associated with virus-like particles (VLP), AP activity in the soluble (Fig. 1D) and particulate fraction (Fig. 1E) after ultracentrifugation was measured. Very little AP activity was associated with the soluble fraction, while the majority of the activity was recovered in the particulate fraction (Fig. 1D and E). In the presence of Gag-Pol, a 10-fold increase of AP activity in the particulate fraction was detected (Fig. 1E). AP::CD16b was present at similar levels in cell lysates in the presence or absence of Gag-Pol. In contrast, full-length AP::CD16b was detected only in supernatants from cells expressing Gag-Pol, but not control cells (Fig. 1F). We conclude that AP::CD16b is expressed on the surface of virus-like particles and allows monitoring Gag-Pol induced particle formation.
Fig. 1.
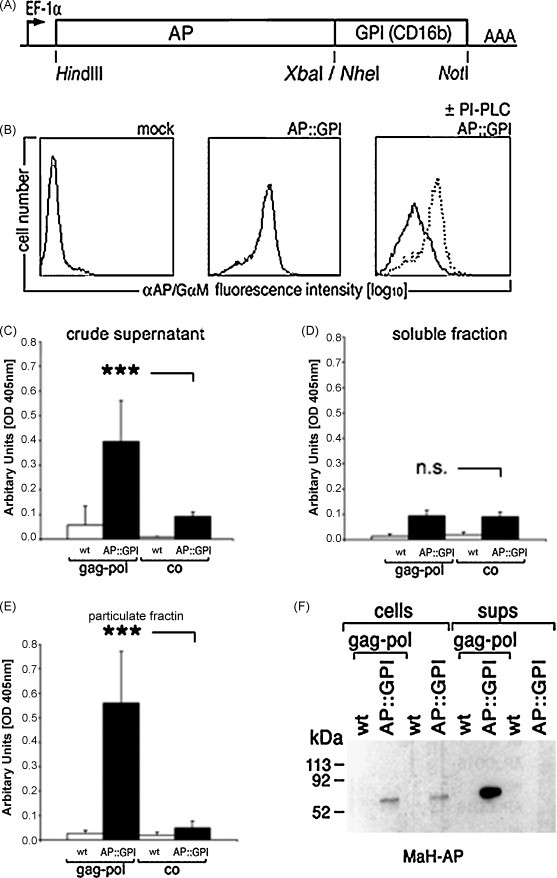
Incorporation of alkaline phosphatase activity in Gag-Pol derived virus-like particles. (A) Schematic representation of AP::CD16b. (B) Incubation of 293 AP::CD16 cells with phosphatidylinositol specific phospholipase C (PI-PLC) from Bacillus thuringiensis removes AP::CD16b from the cell surface. (C) Gag-Pol derived particles release alkaline phosphatase activity in SN. 293 wild type (wt) and AP::CD16 cells were transfected with MLV Gag-Pol, supernatants were harvested 72 h later, cleared of cellular debris by filtration and analyzed for alkaline phosphatase activity. (D) AP activity is not associated with the soluble fraction after ultracentrifugation of Gag-Pol derived virus-like particles. (E) AP activity is associated with the particulate fraction after ultracentrifugation of Gag-Pol derived virus-like particles. (F) AP-CD16b is detected in the supernatants (sups) of Gag-Pol expressing cells. Lysates of cells and vesicles concentrated from cell supernatants by ultracentrifugation were resolved by SDS-PAGE and probed with mouse–anti-human (MaH)AP-specific mAb. n.s., not significant; ***p < 0.001; Student's t-test, paired, two-tailed.
As an alternative to the CD16b anchor, placental alkaline phosphatase with the natural GPI-anchor and an N-terminal Flag-tag (FPLAP) was tested for uptake by virus-like particles of MLV Gag-Pol. In addition, the GPI-anchor sequence was replaced with the transmembrane sequence of CD4 (AP::CD4) and compared to wild-type placental alkaline phosphatase with the natural GPI-anchor (Fig. 2A). In the presence of Gag-Pol, a 13.8-fold increase in alkaline phosphatase activity in the supernatant of 293ETN cells expressing FPLAP was observed, while AP::CD4 did not result in enhanced release of AP activity in the presence of Gag-Pol (Fig. 2B). Expression levels in cell lysates for FPLAP and AP::CD4 were not changed by expression of Gag-Pol (Fig. 2C). We therefore conclude that GPI-anchored alkaline phosphatase can be used to detect virus-like particles in the supernatant of transduced cells releasing viral particles.
Fig. 2.
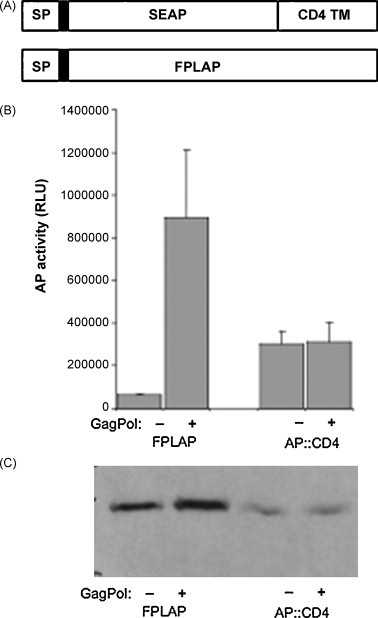
Incorporation of alkaline phosphatase activity in Gag-derived virus-like particles. (A) Schematic representation of AP::CD4 and FPLAP. The Flag-tag is indicated by a black box following the signal peptide (SP). TM, transmembrane domain. (B) 293ET cells were transiently co-transfected with MLV Gag-Pol and FPLAP or AP::CD4. Supernatants were harvested after 3 days and analyzed for alkaline phosphatase activity and compared to cells that were transfected with FPLAP constructs only. Results from three independent infections + standard deviation (SD) are shown. RLU, relative light units. (C) Cell lysates from A were analyzed by immunoblotting of FPLAP and AP::CD4 in the presence or absence of Gag-Pol using anti-Flag M2 antibody.
3.2. Generation of reporter cell lines to detect enveloped viruses
To investigate the potential use of the reporter system for detection of other enveloped viruses, stable reporter cell lines expressing FPLAP in combination with a PuromycinF2ADHFRT2AGFP cassette driven by the HSV1 TK promoter were generated. Vero cells are a good host for a variety of enveloped viruses such as Respiratory Syncytial Virus (RSV) and Herpes Simplex Virus (HSV). Vero cells have very little endogenous alkaline phosphatase activity and thus serve as a good reporter cell line. Vero cells were transfected by nucleofection (Amaxa) and selected with puromycin at 6.0 μg/mL. AP activity was measured in supernatants upon infection with respiratory syncytial virus (RSV1), Herpes Simplex Virus (HSV1) and Aguacate Virus (AV). Upon infection with RSV1, HSV1 and AV, 10.4 ± 0.2-fold, 9.0 ± 0.4-fold and 9.7 ± 3.3-fold increases in AP activity, respectively, were observed in supernatants compared to uninfected cells (Fig. 3A, C and E). Successful virus infection was confirmed with virus-specific assays. Infection with RSV was confirmed by immunofluorescence (Fig. 3B). An orange-red fluorescence specific for RSV was detected in infected Vero cells, but not uninfected cells. Immunoblotting with an anti-HSV1 antiserum recognizing glycoprotein D precursor showed a double band in infected cells but not in uninfected cells. Successful HSV1 infection was confirmed by cell rounding as early as 24 h and by cytopathic effects thereafter (Fig. 3D). Infection with AV resulted in cytopathic effects after 3 days in culture (Fig. 3F).
Fig. 3.
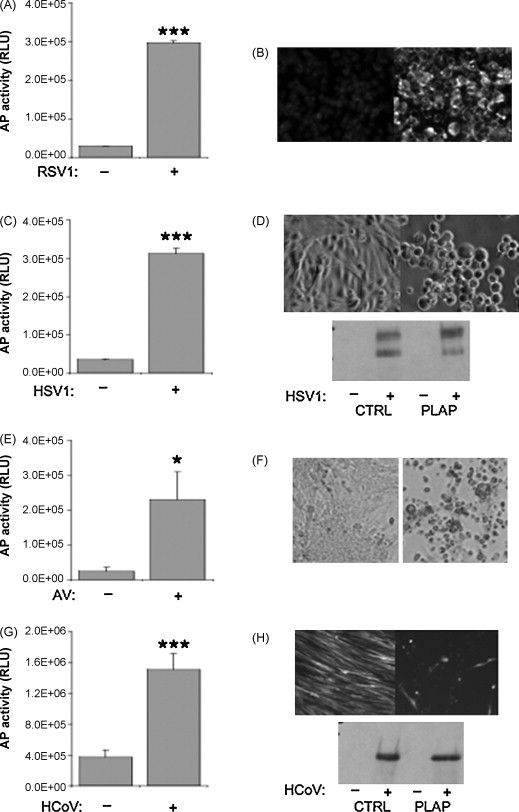
Alkaline phosphatase can be incorporated into multiple enveloped viruses. (A) Vero reporter cell lines were infected with Respiratory Syncytia Virus (RSV) and assayed for alkaline phosphatase (AP) activity in supernatants after 7 days. (B) The presence of RSV was confirmed with the respiratory detection kit (Chemicon) by orange-red fluorescence specific for RSV (lower panel). (C) Vero reporter cell lines were infected with Herpes Simplex Virus 1 (HSV1) and cultured for up to 3 days. Supernatants were collected, centrifuged and analyzed for alkaline phosphatase activity. (D) Vero cells were infected with HSV1 as in (C). After 24 h, cell rounding was observed as a hallmark of efficient infection with HSV1 by light microscopy in infected cells (lower panel) compared to uninfected cells (upper panel). The presence of HSV1 was confirmed by immunoblotting with antibodies raised against HSV1 (lower panel). (E) MRC5 cells transduced with GFP or FPLAP were infected with human coronavirus 229E in 2% serum at 35 °C. After 3 days, supernatants were analyzed for alkaline phosphatase activity. (F) Cytophathic effects were visible after 3 days in cells infected with HCoV 229E (lower panel) compared to the confluent layer of GFP-positive cells in uninfected MRC5 cells (upper panel). The cells were lysed and subjected to immunoblotting with anti-HCoV antibody (Chemicon, as recommended by ATCC) that is highly specific for the 229E sub-type of HCoV, but not for OC43. A single band was detected only in cells that were infected with HCoV 229E. Results from three independent infections + standard deviation (SD) are shown. RLU, relative light units. *p < 0.05; ***p < 0.001; Student's t-test, paired, two-tailed.
As an alternative host cell, MRC5 cells were transduced with FPLAP or GFP and a 4.1 ± 0.5-fold increase of AP activity in supernatant upon infection with human coronavirus 229E (HCoV) compared to uninfected cells (Fig. 3G) was detected. After 24 h, the majority of MRC5 cells was GFP-positive, indicating high transduction efficiencies. The cells were subsequently infected with HCoV 229E in 2% serum at 35 °C. After 3 days, cytopathic effects became visible (Fig. 3H), supernatants were harvested and AP activity was measured in SN. A 4.1 ± 0.5-fold increase of AP activity in supernatant upon infection with HCoV was observed compared to uninfected cells (Fig. 3G). No difference in AP activity in supernatant was detected in cells expressing GFP. To determine whether cells had been infected with HCoV 229E, the cells were lysed and subjected to immunoblotting with an anti-HCoV antibody that is highly specific for the 229E sub-type of HCoV, but not for OC43. A single band was detected in infected cells but not in uninfected cells (Fig. 3H). Thus, this system can be used to detect viruses of the coronavirus family in MRC5 cells.
To determine the sensitivity of this assay, serial dilutions of HSV1 (TCID50: 107.75 TCID[50]/0.2 mL) were used for infection of Vero cells. At 104-fold and 105-fold dilutions, a 5.52-fold and 2.78-fold increase in alkaline phosphatase activity compared to uninfected cells was detectable in supernatants, respectively (Table 1 ). Cytopathic effects were detectable at up to 104-fold dilution, but not at higher dilutions.
Table 1.
Detection of alkaline phosphatase and cytopathic effects in serial dilutions of HSV1.
| Dilution | 1/104 | 1/105 | 1/107 |
|---|---|---|---|
| Alkaline phosphatase (fold) | 5.52 ± 1.06 | 2.78 ± 0.46 | 0.97 ± 0.22 |
| Cytopathic effects | Yes | No | No |
Serial dilutions of HSV1 virus stock (107.75 TCID[50]/0.2 mL) were incubated with Vero cells expressing FPLAP. After 7 days, cells were analyzed for cytophathic effects and supernatants were tested for alkaline phosphatase activity.
3.3. Flag-tagged placental alkaline phosphatase can be incorporated in replicating HIV particles
To determine whether Flag-tagged PLAP can be incorporated in infectious HIV particles, FPLAP and HIV-GFP were co-expressed in 293ET cells. High levels of HIV expression were confirmed by fluorescence microscopy for expression of GFP (data not shown), which has been cloned in the nef open reading frame. Upon transfection of HIV-GFP and FPLAP in 293ET cells, a 4.2 ± 0.3-fold increase in AP activity in supernatant was detected compared to cells transfected with HIV-GFP only (Fig. 4A).
Fig. 4.
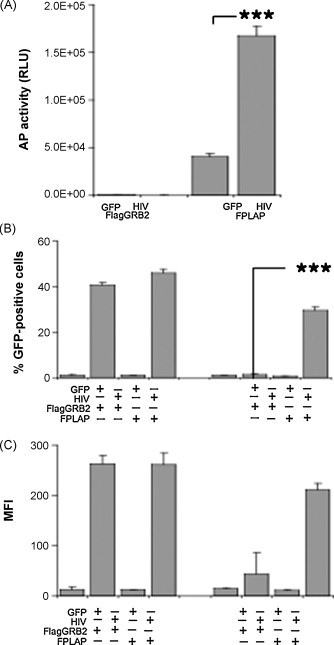
Incorporation of Flag-tagged alkaline phosphatase in replicating HIV. (A) 293ET cells were transfected with Flag-GRB2 (FGRB2) as control and FPLAP in combination with GFP or HIV. AP activity was determined after 2 days in SN. (B and C) Supernatants from 293ET cells expressing HIV and FPLAP or GFP were collected, pre-incubated with Flag M2 antibody and magnetic beads for 2 h at 4 °C and subsequently used for infection of CEMx174-T2 (MT2) cells. After 5 days, MT2 cells infected with plain supernatant or with captured virus were analyzed by flow cytometry for percentage of MT2 cells displaying green fluorescence (B) or mean fluorescence intensity (MFI) (C). Results from three independent infections + standard deviation (SD) are shown. RLU, relative light units. ***p < 0.001; Student's t-test, paired, two-tailed.
To explore whether this strategy could be used to purify unknown virus particles, supernatants of cells transfected with HIV and FPLAP were exposed to anti-Flag M2 antibody coupled to magnetic beads. CEMx174-T2 cells (MT2) were incubated with the magnetic beads or supernatants and analyzed by flow cytometry for GFP expression indicative of infection by HIV. Supernatants from cells transfected with HIV resulted in successful infection with 40–46% of cells displaying green fluorescence with high mean fluorescence intensities after 5 days (Fig. 4B and C). Coincubation of immunoadsorbed beads from cells expressing FPLAP and HIV resulted in successful infection of 30% MT2 cells displaying green fluorescence and a mean fluorescence intensity of 210.4 ± 13.4 (Fig. 4B and C), whereas similar coincubation with beads exposed to supernatant from cells expressing HIV and FGRB2 did not result in infection of MT2 cells. We conclude that FPLAP is incorporated into infectious virus particles and the Flag-tag is exposed on the virus surface in a way that permits the virus to be purified and delivered to recipient cells.
4. Discussion
High-throughput screening of RNAi or shRNA libraries enable the identification of host cell factors required for virus replication. Successful screening of several viruses for cellular host factors has been reported including HIV (Brass et al., 2008, Konig et al., 2008, Yeung et al., 2009, Zhou et al., 2008), HCV (Lupberger et al., 2008, Randall et al., 2007, Tai et al., 2009), West Nile virus (Krishnan et al., 2008) and Influenza virus (Hao et al., 2008). Targeting host cell factors for antiviral strategies may overcome virus resistance to conventional virus-targeted therapies that arises due to the high mutation rate of the viral genome. It would be interesting to identify host cell factors that are common to different pathogenic viruses, thus enabling “pan”-viral therapeutic targets. However, screening of multiple different viruses is difficult to achieve, largely because individual assays for each virus have to be established and optimized. The development of general virus detection methods would allow cross-virus screening. Prior to our study, a small molecule screen for SARS based on the observation of cytopathic effects was reported (Ivens et al., 2005). In principle, the detection of cytopathic effects is applicable to multiple viruses, but effects on general toxicity need to be controlled for. In addition, quantitation of cytopathic effects is much harder to achieve and less sensitive than enzymatic reporter systems. As such, cytopathic effects are largely a qualitative measure of general cytotoxicity rather than virus infectivity.
This study presents a host cell-based assay system for general detection of enveloped viruses based on lipid raft-mediated uptake of the reporter gene by the virus. The natural GPI-anchor of placental alkaline phosphatase incorporates effectively into the membrane of diverse enveloped virus particles. Other anchor sequences such as the extracellular domain of the low-affinity nerve growth factor receptor that has been used to funtionalize lentiviral particles (Nesbeth et al., 2006) may be used as alternative targeting motif. Several studies have demonstrated that the incorporation of reporter enzymes in the virus genome impacts budding and infectivity of the virus. The system described here is based on expression of the reporter in trans, thus enabling the detection of multiple viruses. The impact on budding, viral yields, potency and the immune profile when expressed in cis need to be addressed separately in future studies as these would be important aspects to address the suitability of this system for gene delivery and gene therapy approaches. Since the system described here does not modify integral virus components, but rather decorates the virus particle with the reporter enzyme, it has potentially a minimal effect on the virus structure itself. This is underscored by the demonstration that infectious HIV particles can be obtained by immuno-capture using anti-Flag antibodies.
The general virus detection system described here offers several advantages. First, it allows the detection of multiple enveloped viruses that bud from the plasma membrane. Second, the assay is amenable to high-throughput screening and can be used for multiple enveloped viruses. Third, a magnetic purification scheme is described that enables the recovery of infectious virus for further analysis. Thus, this system enables detection and subsequent analysis of enveloped viruses without prior reference to their composition. One limitation of this system is cell culture adaptation as a pre-requisite for virus screening. Vero cells have proven as suitable host for multiple cell culture adapted viruses, but may not be suitable for all enveloped viruses. In such cases, the use of alternate hosts such as MRC5 or MDCK cells can be explored. We propose that this tool will be highly useful as a research tool for characterization of known and novel enveloped viruses.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
R.K. was supported by the Deutsche Forschungsgemeinschaft (Ke904/2-1). W.F.P. and A.N. were supported by the Austrian Science Foundation (SFB F1816). We thank Slim Sassi and Zahedi Mujawar for critically reading the manuscript.
References
- Asokan A., Johnson J.S., Li C., Samulski R.J. Bioluminescent virion shells: new tools for quantitation of AAV vector dynamics in cells and live animals. Gene Ther. 2008;15:1618–1622. doi: 10.1038/gt.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass A.L., Dykxhoorn D.M., Benita Y., Yan N., Engelman A., Xavier R.J., Lieberman J., Elledge S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- Briese T., Paweska J.T., McMullan L.K., Hutchison S.K., Street C., Palacios G., Khristova M.L., Weyer J., Swanepoel R., Egholm M., Nichol S.T., Lipkin W.I. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 2009;5:e1000455. doi: 10.1371/journal.ppat.1000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugger B., Glass B., Haberkant P., Leibrecht I., Wieland F.T., Krausslich H.G. The HIV lipidome: a raft with an unusual composition. Proc. Natl. Acad. Sci. U.S.A. 2006;103:2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz J.V., Didier P., Clamme J.-P., Schaub E., Muriaux D., Cabanne C., Morellet N., Bouaziz S., Darlix J.-L., Mely Y., de Rocquigny H. Direct Vpr–Vpr interaction in cells monitored by two photon fluorescence correlation spectroscopy and fluorescence lifetime imaging. Retrovirology. 2008;5:87. doi: 10.1186/1742-4690-5-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnes C., Andresen V., Szilvay A.M. Functional rescue of an oligomerization-defective HIV-1 Rev mutant by fusion with an oligomeric tag. Arch. Virol. 2008;153:357–362. doi: 10.1007/s00705-007-1095-x. [DOI] [PubMed] [Google Scholar]
- Hammarstedt M., Garoff H. Passive and active inclusion of host proteins in human immunodeficiency virus type 1 gag particles during budding at the plasma membrane. J. Virol. 2004;78:5686–5697. doi: 10.1128/JVI.78.11.5686-5697.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarstedt M., Wallengren K., Pedersen K.W., Roos N., Garoff H. Minimal exclusion of plasma membrane proteins during retroviral envelope formation. Proc. Natl. Acad. Sci. U.S.A. 2000;97:7527–7532. doi: 10.1073/pnas.120051597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L., Sakurai A., Watanabe T., Sorensen E., Nidom C.A., Newton M.A., Ahlquist P., Kawaoka Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454:890–893. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iro M., Witteveldt J., Angus A.G.N., Woerz I., Kaul A., Bartenschlager R., Patel A.H. A reporter cell line for rapid and sensitive evaluation of hepatitis C virus infectivity and replication. Antivir. Res. 2009;83:148–155. doi: 10.1016/j.antiviral.2009.04.007. [DOI] [PubMed] [Google Scholar]
- Ivens T., Van den Eynde C., Van Acker K., Nijs E., Dams G., Bettens E., Ohagen A., Pauwels R., Hertogs K. Development of a homogeneous screening assay for automated detection of antiviral agents active against severe acute respiratory syndrome-associated coronavirus. J. Virol. Methods. 2005;129:56–63. doi: 10.1016/j.jviromet.2005.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketteler R., Glaser S., Sandra O., Martens U.M., Klingmuller U. Enhanced transgene expression in primitive hematopoietic progenitor cells and embryonic stem cells efficiently transduced by optimized retroviral hybrid vectors. Gene Ther. 2002;9:477–487. doi: 10.1038/sj.gt.3301653. [DOI] [PubMed] [Google Scholar]
- Kolokoltsov A.A., Davey R.A. Rapid and sensitive detection of retrovirus entry by using a novel luciferase-based content-mixing assay. J. Virol. 2004;78:5124–5132. doi: 10.1128/JVI.78.10.5124-5132.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig R., Zhou Y., Elleder D., Diamond T.L., Bonamy G.M., Irelan J.T., Chiang C.Y., Tu B.P., De Jesus P.D., Lilley C.E., Seidel S., Opaluch A.M., Caldwell J.S., Weitzman M.D., Kuhen K.L., Bandyopadhyay S., Ideker T., Orth A.P., Miraglia L.J., Bushman F.D., Young J.A., Chanda S.K. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan M.N., Ng A., Sukumaran B., Gilfoy F.D., Uchil P.D., Sultana H., Brass A.L., Adametz R., Tsui M., Qian F., Montgomery R.R., Lev S., Mason P.W., Koski R.A., Elledge S.J., Xavier R.J., Agaisse H., Fikrig E. RNA interference screen for human genes associated with West Nile virus infection. Nature. 2008;455:242–245. doi: 10.1038/nature07207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lama J., Planelles V. Host factors influencing susceptibility to HIV infection and AIDS progression. Retrovirology. 2007;4:52. doi: 10.1186/1742-4690-4-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupberger J., Brino L., Baumert T.F. RNAi: a powerful tool to unravel hepatitis C virus-host interactions within the infectious life cycle. J. Hepatol. 2008;48:523–525. doi: 10.1016/j.jhep.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Manes S., del Real G., Lacalle R.A., Lucas P., Gomez-Mouton C., Sanchez-Palomino S., Delgado R., Alcami J., Mira E., Martinez A.C. Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep. 2000;1:190–196. doi: 10.1093/embo-reports/kvd025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S.E., Licata J.M., Harty R.N. A luciferase-based budding assay for Ebola virus. J. Virol. Methods. 2006;137:115–119. doi: 10.1016/j.jviromet.2006.06.007. [DOI] [PubMed] [Google Scholar]
- McDonald D., Vodicka M.A., Lucero G., Svitkina T.M., Borisy G.G., Emerman M., Hope T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002;159:441–452. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergener K., Facke M., Welker R., Brinkmann V., Gelderblom H.R., Krausslich H.G. Analysis of HIV particle formation using transient expression of subviral constructs in mammalian cells. Virology. 1992;186:25–39. doi: 10.1016/0042-6822(92)90058-w. [DOI] [PubMed] [Google Scholar]
- Muller B., Daecke J., Fackler O.T., Dittmar M.T., Zentgraf H., Krausslich H.G. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J. Virol. 2004;78:10803–10813. doi: 10.1128/JVI.78.19.10803-10813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbeth D., Williams S.L., Chan L., Brain T., Slater N.K., Farzaneh F., Darling D. Metabolic biotinylation of lentiviral pseudotypes for scalable paramagnetic microparticle-dependent manipulation. Mol. Ther. 2006;13:814–822. doi: 10.1016/j.ymthe.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Ono A., Freed E.O. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. U.S.A. 2001;98:13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickl W.F., Pimentel-Muinos F.X., Seed B. Lipid rafts and pseudotyping. J. Virol. 2001;75:7175–7183. doi: 10.1128/JVI.75.15.7175-7183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall G., Panis M., Cooper J.D., Tellinghuisen T.L., Sukhodolets K.E., Pfeffer S., Landthaler M., Landgraf P., Kan S., Lindenbach B.D., Chien M., Weir D.B., Russo J.J., Ju J., Brownstein M.J., Sheridan R., Sander C., Zavolan M., Tuschl T., Rice C.M. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A., Isaka Y., Kodama M., Yoshimoto J., Kawauchi S., Kuwata T., Adachi A., Hayami M., Yoshi O., Fujiwara T. Targeting of chrolamphenicol acetyltransferase to human immunodeficiency virus particles via Vpr and Vpx. Microbiol. Immunol. 1995;39:1015–1019. doi: 10.1111/j.1348-0421.1995.tb03293.x. [DOI] [PubMed] [Google Scholar]
- Schobel S., Neumann S., Seed B., Lichtenthaler S.F. Expression cloning screen for modifiers of amyloid precursor protein shedding. Int. J. Dev. Neurosci. 2006;24:141–148. doi: 10.1016/j.ijdevneu.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Shih K.-N., Chuang Y.-T., Liu H., Lo S.J. Hepatitis D virus RNA editing is inhibited by a GFP fusion protein containing a C-terminally deleted delta antigen. J. Gen. Virol. 2004;85:947–957. doi: 10.1099/vir.0.19661-0. [DOI] [PubMed] [Google Scholar]
- Snowden F.M. Emerging and reemerging diseases: a historical perspective. Immunol. Rev. 2008;225:9–26. doi: 10.1111/j.1600-065X.2008.00677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai A.W., Benita Y., Peng L.F., Kim S.-S., Sakamoto N., Xavier R.J., Chung R.T. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe. 2009;5:298–307. doi: 10.1016/j.chom.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L., Pyrc K., Jebbink M.F., Vermeulen-Oost W., Berkhout R.J., Wolthers K.C., Wertheim-van Dillen P.M., Kaandorp J., Spaargaren J., Berkhout B. Identification of a new human coronavirus. Nat. Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Coscoy L., Zylberberg M., Avila P.C., Boushey H.A., Ganem D., DeRisi J.L. Microarray-based detection and genotyping of viral pathogens. Proc. Natl. Acad. Sci. U.S.A. 2002;99:15687–15692. doi: 10.1073/pnas.242579699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Liu H., Xiao H., Kim J., Seshaiah P., Natsoulis G., Boeke J.D., Hahn B.H., Kappes J.C. Targeting foreign proteins to human immunodeficiency virus particles via fusion with Vpr and Vpx. J. Virol. 1995;69:3389–3398. doi: 10.1128/jvi.69.6.3389-3398.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.J., Kobinger G., Dandache S., Rougeau N., Cohen E. HIV-1 Vpr-chloramphenicol acetyltransferase fusion proteins: sequence requirement for virion incorporation and analysis of antiviral effect. Gene Ther. 1999;6:1590–1599. doi: 10.1038/sj.gt.3300988. [DOI] [PubMed] [Google Scholar]
- Yeung M.L., Houzet L., Yedavalli V.S., Jeang K.T. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 2009;284:19463–19473. doi: 10.1074/jbc.M109.010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Breitbart M., Lee W.H., Run J.Q., Wei C.L., Soh S.W., Hibberd M.L., Liu E.T., Rohwer F., Ruan Y. RNA viral community in human feces: prevalence of plant pathogenic viruses. PLoS Biol. 2006;4:e3. doi: 10.1371/journal.pbio.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Xu M., Huang Q., Gates A.T., Zhang X.D., Castle J.C., Stec E., Ferrer M., Strulovici B., Hazuda D.J., Espeseth A.S. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]