

Abstract
Gene knockout and knockdown methods were used to examine essentiality of pteridine reductase (PTR1) in pterin metabolism in the African trypanosome. Attempts to generate PTR1 null mutants in bloodstream form Trypanosoma brucei proved unsuccessful; despite integration of drug selectable markers at the target locus, the gene for PTR1 was either retained at the same locus or elsewhere in the genome. However, RNA interference (RNAi) resulted in complete knockdown of endogenous protein after 48 h, followed by cell death after 4 days. This lethal phenotype was reversed by expression of enzymatically active Leishmania major PTR1 in RNAi lines (oeRNAi) or by addition of tetrahydrobiopterin to cultures. Loss of PTR1 was associated with gross morphological changes due to a defect in cytokinesis, resulting in cells with multiple nuclei and kinetoplasts, as well as multiple detached flagella. Electron microscopy also revealed increased numbers of glycosomes, while immunofluorescence microscopy showed increased and more diffuse staining for glycosomal matrix enzymes, indicative of mis-localisation to the cytosol. Mis-localisation was confirmed by digitonin fractionation experiments. RNAi cell lines were markedly less virulent than wild-type parasites in mice and virulence was restored in the oeRNAi line. Thus, PTR1 may be a drug target for human African trypanosomiasis.
Introduction
Tetrahydrobiopterin (H4B) is an essential cofactor for various hydroxylation reactions catalysed by enzymes such as aromatic amino acid hydroxylases, glyceryl ether monooxygenases and NO synthases (Thony et al., 2000; Werner-Felmayer et al., 2002; Schmidt and Alp, 2007; Watschinger et al., 2009). In humans, H4B can be synthesized de novo from GTP or salvaged from dihydrobiopterin (H2B) via NADPH-dependent dihydrofolate reductase (DHFR, EC 1.5.13) (Nichol et al., 1983) or regenerated from quinonoid dihydrobiopterin, a by-product of hydroxylation reactions, via NAD(P)H-dependent 6,7-dihydropteridine reductase (DHPR; EC 1.5.1.34) (Armarego et al., 1983).
In contrast to humans, biological and genomic data indicate that trypanosomatids lack the ability to synthesize pterins de novo, and are totally dependent on salvage of extracellular pterins for growth (Bello et al., 1994; Cunningham and Beverley, 2001; Berriman et al., 2005; Ivens et al., 2005). The essential requirement for pterins was initially discovered in Crithidia fasciculata grown in a defined medium containing low amounts of folate (Kidder and Dutta, 1958; Kaufman, 1963); and our subsequent understanding of the uptake, salvage and functions of pterins in trypanosomatids has come principally from studies by the Beverley and Ouellette groups (Nare et al., 1997; Ouellette et al., 2002). In Leishmania spp., biopterin is taken up from the medium via the biopterin transporter BT1 (Kundig et al., 1999; Lemley et al., 1999) and then sequentially reduced to H2B and H4B by pteridine reductase 1 (PTR1; EC 1.5.1.33) (Bello et al., 1994). Previous studies on Leishmania ptr1- mutants suggested the presence of a residual pteridine reductase activity, provisionally assigned as PTR2. This activity is considered not to be due to quinonoid dihydropteridine reductase and the identity of PTR2 remains unknown (Lye et al., 2002). Some kinetic, inhibitor and structural properties have been reported for PTR1 from Leishmania major (Luba et al., 1998; Gourley et al., 2001; Schuttelkopf et al., 2005), Trypanosoma cruzi (Schormann et al., 2005) and Trypanosoma brucei (Dawson et al., 2006; Mpamhanga et al., 2009; Shanks et al., 2010). Gene knockout studies in L. major have demonstrated that PTR1 is essential for growth of the insect promastigote stage of the parasite, where growth of ptr1- null mutants could be restored with H2B or H4B, but not by dihydro- or tetrahydrofolate (Bello et al., 1994). These mutants showed increased formation of mammalian-infective metacyclic promastigote forms in stationary phase cultures and consequently gave larger lesions when injected into mice (Cunningham et al., 2001). This indicates that ptr1- amastigotes are viable in vivo, presumably due to abundant H4B in host macrophages and consequently that PTR1 is not a drug target on its own in L. major.
PTR1 has a secondary role in metabolism in being able to reduce dihydrofolate to tetrahydrofolate. Amplification of PTR1 in Leishmania spp. is one of the several mechanisms by which parasites acquire resistance to antifolates such as methotrexate (Callahan and Beverley, 1992; Papadopoulou et al., 1992; Bello et al., 1994) either by acting as a by-pass mechanism or by sequestering these DHFR inhibitors. Recent studies have demonstrated an intriguing link between unconjugated pterins and oxidative susceptibility in a variety of Leishmania PTR1 knockout and overexpressing cell lines (Moreira et al., 2009; Nare et al., 2009). However, the mechanism involved is not clear; neither is it known whether pterins have additional essential functions in these parasites.
In contrast to Leishmania spp. virtually nothing is known about pterin metabolism in African trypanosomes, parasites that occupy a completely different (extracellular) environment in the mammalian host. In this study we use genetic methods to examine the role of PTR1 in blood stream form T. brucei with respect to essentiality in vitro and infectivity in vivo. Our findings suggest that reduced pterins are required for glycosome and flagellar biogenesis and that loss of PTR1 activity cannot be compensated for by reduced pterins in plasma and extracellular fluids. Thus, unlike L. major, PTR1 is a potential drug target in the African trypanosome without reference to DHFR.
Results
Genotypic and phenotypic analysis of TbPTR1 knockdown in parasites
To assess the essentiality of PTR1, classical gene replacement methods were undertaken to try to produce a PTR1 null cell line of bloodstream trypanosomes. Consistent with genome sequence data for strain 927 (Berriman et al., 2005), Southern blot analysis confirmed that PTR1 is single copy per haploid genome in the ‘single marker’ bloodstream T. brucei 427 used in these studies (data not shown). This organism, subsequently referred to as wild-type (WT), constitutively expresses T7 RNA polymerase and the tetracycline repressor protein, and can be used to express other RNA constructs under the control of tetracycline (Wirtz et al., 1999). Replacement of one copy of PTR1 to generate a single knockout (SKO) could be readily achieved by transfection with a construct containing a drug resistance gene (either HYG or PAC) flanked by the 5′- and 3′-untranslated regions of PTR1, followed by drug selection with either hygromycin or puromycin, essentially as described (Sienkiewicz et al., 2008). SKO lines (PTR1/ptr1::HYG[SKOHYG] or PTR1/ptr1::PAC[SKOPAC]) were then subjected to a second round of transfection with the other knockout construct and selected for resistance to both drugs. Despite obtaining double drug-resistant trypanosomes in numerous independent experiments, we were unable to obtain clones in which both copies of PTR1 had been deleted. Southern blotting confirmed that integration of the drug-resistant genes had indeed occurred at the correct locus, but was associated with retention of an additional copy of PTR1 either at the same locus or elsewhere in the genome (see Fig. S1). In virulent strains of leishmania, such behaviour is typical of an essential gene (Cruz et al., 1993), but has not been reported for the African trypanosome. However, an increase in chromosomal copy number has been reported for trypanosomes selected for resistance to mycophenolic acid, an inhibitor of IMP dehydrogenase (Wilson et al., 1994). Attempts to generate conditional double knockout cell lines by introducing an ectopic copy of PTR1 (pLew100_TbPTR1) under tetracycline control were also unsuccessful.
In the light of these results, inducible RNA interference (RNAi) studies using the p2T7TiTAblue system were undertaken to deplete cells of PTR1 (Alibu et al., 2005). For the disruption of PTR1 function, a 403 bp region was ligated into the p2T7TiTAblue vector and a linearized construct transfected into WT cells by electroporation. Cells that had integrated the construct into the rDNA loci by homologous recombination were obtained by selection with hygromycin and then cloned. Generation of the correct RNAi clones was confirmed by Southern analysis (data not shown), then tetracycline was added to cell cultures to initiate the expression of PTR1-specific double-stranded RNA. The average cumulative growth with four different freshly derived clones is illustrated in Fig. 1A. In the absence of inducer these cells grew at essentially the same rate as WT cells. However, following induction of PTR1 dsRNA, growth ceased after 1 day, followed by a decline in cell numbers such that no motile parasites were visible after 4 days. Levels of PTR1 protein were diminished compared with WT in non-induced RNAi lines (Fig. 1A, inset) compared with the immunoglobulin heavy-chain-binding protein (BiP) used as a loading control. Densitometry of the PTR1 band relative to the BiP control band indicated that the non-induced RNAi line had 50% of WT protein levels; this decreased further to 9% 24 h after RNAi induction and was not detectable after 48 h. This RNAi phenotype was unstable; if the tetracycline-induction was repeated after a further 2–3 weeks in culture, cells either showed no growth defect with no knockdown of PTR1, or brief cytostasis followed by normal growth (Fig. S2). To confirm that the cidal effect of RNAi was due to loss of PTR1 activity, the effect of supplementation with H4B was examined on a freshly derived clone (Fig. 1B). Addition of 10 µM H4B to RNAi-induced cultures abolished the lethal growth phenotype.
Fig. 1.
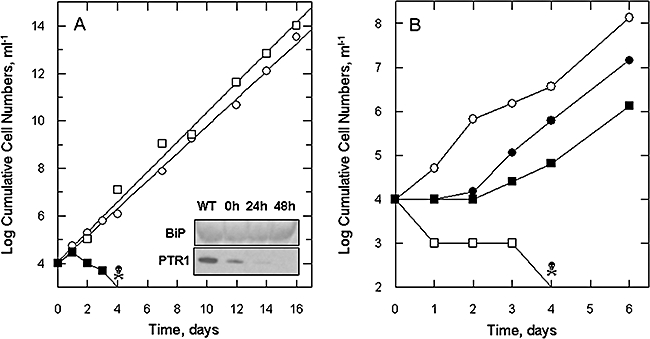
The effect of RNAi-dependent depletion of PTR1 on growth in the presence or absence of tetrahydrobiopterin. A. Effect of RNAi on cell growth. Four independent RNAi clones were analysed for growth defects following induction with tetracycline in the absence of tetrahydrobiopterin. WT (open circles) and RNAi cells (non-induced cells – open squares and induced cells – closed squares) were seeded at 1 × 104 cells ml−1 and growth was determined over a 2 week period. WT, non-induced and induced cells (1 × 106 cells per lane) were harvested at 0 (non-induced), 24 h and 48 h (induced) and processed for Western blot analysis. The blot was sequentially probed with anti-TbPTR1 (inset, lower panel) and anti-TbBIP (inset, upper panel) as loading control. B. Effect of tetrahydrobiopterin on cell growth. WT cells and a freshly derived RNAi clone were seeded into FDM or HMI9-T supplemented with or without 10 µM tetrahydrobiopterin at 1 × 104 cells ml−1 and growth monitored over 1 week. Symbols: WT (open circle; no additions), non-induced RNAi cells (closed circles) and tetracycline-induced cells plus or minus tetrahydrobiopterin (closed and open squares respectively). Data shown for FDM only; HMI9-T shows essentially the same effect.
Additional evidence of the essential requirement for PTR1 activity was obtained by introducing an inducible overexpressing ectopic copy of L. major PTR1 into the WT line (oeWT) prior to generation of TbPTR1 RNAi lines (oeRNAi). Due to the high GC content of the L. major gene, the sequence identity with the T. brucei RNAi construct is less than 50%, with no more than 13 nucleotide stretches of identity. Thus, LmPTR1 mRNA should not be susceptible to degradation, since assembly of an RNA-induced silencing complex requires formation of 20–26-mer double-stranded RNA (Ullu et al., 2004). Indeed, following tetracycline induction, oeRNAi cells showed no sign of any growth defect whatsoever (Fig. 2A). Western blotting with antisera raised against recombinant T. brucei PTR1 showed that TbPTR1 expression is undetectable following induction (Fig. 2B; TbPTR1 is ∼30 kDa and the 55 kDa cross-reacting band serves as an internal loading control). In contrast, Western blotting with a second antiserum specific to L. major PTR1 shows the opposite effect. LmPTR1 is visible as a ∼30 kDa band in non-induced (due to leaky nature of the promoter in pLew82) which increases following induction. PTR1 activity was measured in cell lysates to validate these results. As shown in Fig. 3, PTR1 activity is proportional to protein concentration for each cell line. WT cells have a specific activity of 22.9 ± 1.9 nmol min mg−1 and this is decreased to 8.0 ± 0.2 nmol min mg−1 in the SKO line, whereas this increases by 2.5- and 10-fold in RNA lines overexpressing LmPTR1 in the absence or presence of inducer (58.1 ± 9.3 and 231 ± 19 nmol min mg−1 respectively). These results demonstrate that LmPTR1 is enzymatically active when expressed in T. brucei and is able to rescue the lethal RNAi phenotype.
Fig. 2.
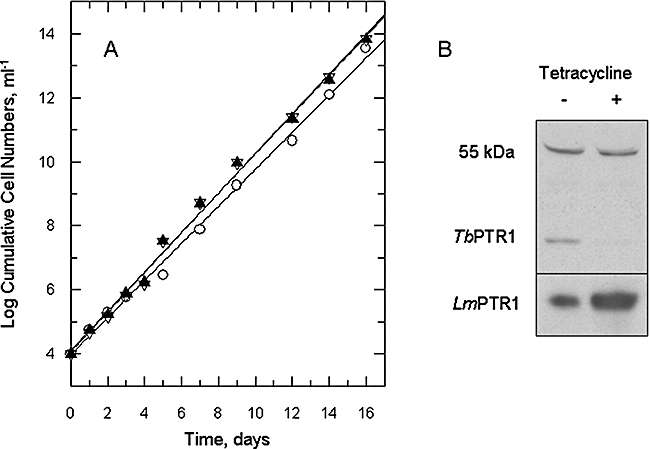
Rescue of RNAi growth defect by expression of LmPTR1. A. Cumulative growth of WT (open circles) non-induced oeRNAi cells (open inverted triangles) and tetracycline-induced oeRNAi cells (closed triangles). B. Western blot analysis demonstrating knockdown of TbPTR1 in oeRNAi cells. Non-induced (-) and 48 h tetracycline induced (+) cells were probed sequentially with anti-TbPTR1 and anti-LmPTR1. A 55 kDa non-specific band was detected with anti-TbPTR1, which was not effect by the knockdown of PTR1 and serves as an internal loading control (top panel), while LmPTR1 is present in both (bottom panel).
Fig. 3.
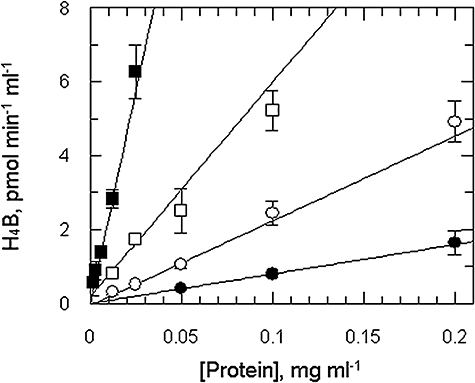
PTR1 enzyme activity in WT and transgenic T. brucei. Cell lysates were assayed for PTR1 activity at various protein concentrations as described in the materials and methods. WT, open circles; SKO, closed circles; oeRNAi minus tetracycline, open squares; oeRNAi plus tetracycline, closed squares.
Infectivity studies were undertaken in mice to examine the virulence of RNAi and oeRNAi transgenic trypanosomes. Wild-type and non-induced transgenic cells were inoculated into paired groups of mice, where one pair received doxycycline in their drinking water. Parasitaemia was monitored at intervals for 30 days; animals achieving a parasitaemia greater than 108 trypanosomes per millilitre were euthanized as previous studies had established that these levels were consistently lethal within the next 24 h. Mice infected with 105 WT cells were unable to survive longer than 3–4 days (Fig. 4), whereas mice infected with RNAi cells survived for much longer regardless of doxycycline treatment. The reduced virulence observed in the minus doxycycline group was probably due to leaky transcription of the p2T7 promoter, which lowers the baseline expression of PTR1 (Fig. 1A) and in some instances is associated with an initial lag in growth compared with WT (Fig. 1B). Six out of 10 mice treated with doxycycline failed to develop a parasitaemia and survived beyond 30 days. Infection with oeRNAi cells (with or without doxycycline) restored the virulence phenotype to that of WT cells.
Fig. 4.
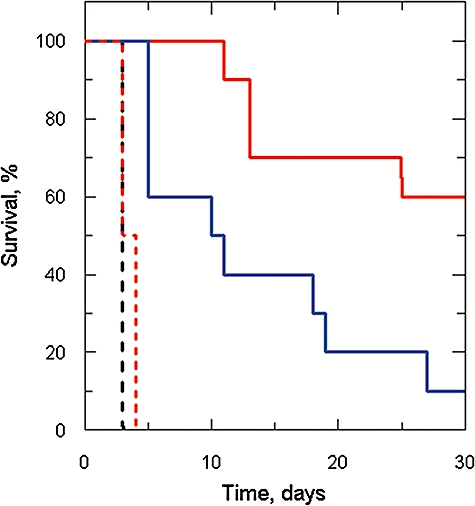
Virulence phenotype of PTR1 mutants in mice. Groups of five mice were infected with WT, non-induced cells for RNAi and oeRNAi cells and parasitaemia monitored at intervals over a 30 day period. The Kaplan-Meier survival graph shows the aggregated results of two independent experiments. Symbols: WT infection, no doxycycline (black dashed line); RNAi infection, no doxycycline (blue solid line); RNAi infection plus doxycycline (red solid line); oeRNAi infection, no doxycycline (red dashed line); oeRNAi infection with doxycycline (black dashed line, identical to WT infection).
Morphological changes associated with PTR1 depletion
Severe morphological abnormalities could be observed in bloodstream trypanosomes by light and electron microscopy prior to cell death. After induction of RNAi for 24–48 h, cells showed reduced motility, with some fattening of the cell body and shortening of the flagellum compared with the control (non-induced) samples. After 72 h, Giemsa-stained preparations showed gross distension of the cell body with multiple nuclei, kinetoplasts and flagella.
To provide an insight into any changes in the cell cycle progression, DAPI-stained cells were analysed to determine the proportion of cells with one or more nucleus (N) and kinetoplast (K) from control, non-induced and tetracycline induced populations (Fig. 5). Non-induced samples were predominately 1K1N (92%), whereas tetracycline induction caused a dramatic time-dependent decrease in the number of 1K1N trypanosomes (28% at 72 h) with a corresponding increase in cells containing either multiple kinetoplasts plus multiple nuclei (MKMN) or anucleated zoids (1K0N). The number of cells with two kinetoplasts and one nucleus (2K1N) increased threefold from 6% to 19% by 48 h after induction, decreasing to 8% at 72 h. 2K2N did not vary markedly throughout the time course. These results suggest that most cells can undergo several rounds of mitosis, but not cytokinesis.
Fig. 5.
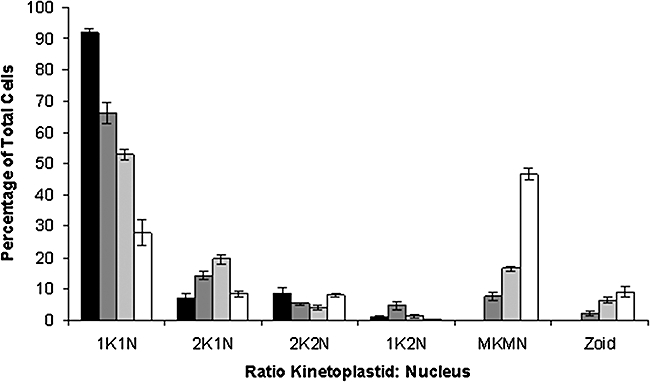
PTR1 depletion results in changes to cell cycle progression. Microscopic analysis of DAPI stained RNAi cells of non-induced (0 h) and induced (24, 48 and 72 h) populations analysed to determine the proportion of trypanosomes with different numbers of kinetoplasts (K) and nuclei (N). Percentage bar chart depicts changes in the K/N content per cell after tetracycline induction of RNAi. Approximately 300 parasites per population were analysed (n = 2, ± SD): non-induced control (black bars); 24 h induction (dark grey bars); 48 h induction (light grey bars) and 72 h induction (white bars).
Ultra-structural and surface alterations were examined by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) (Fig. 6). The most obvious aberrant feature present in the TEM sections of the RNAi-induced cells is the increased number of membrane bound electron-dense bodies (Fig. 6B and C) compared with the non-induced control (Fig. 6A). These round, sometimes sausage-shaped (Fig. 6C, inset) bodies are morphologically similar to glycosomes, an organelle related to peroxisomes (Opperdoes et al., 1984). Another distinct feature in these treated cells is the increased number of flagella present in the 72 h sections (Fig. 6B–E, arrowheads). Even though there are an aberrant number of flagella associated with the main body of the parasite, they appear to have the normal 9 + 2 arrangement of the microtubules of the axoneme and paraflagellar rod, suggesting that the structural organisation of the flagellum is unaffected by PTR1 depletion. Some sections also appear to have more than one flagellar pocket (Fig. 6B), as well as a number of autophagic vacuoles. Lastly, structures were evident in some sections, which appeared to be an internal non-membrane bound flagellum (Fig. 6B plus inset, starred), a phenotype previously observed in the knockdown of VSG (Sheader et al., 2005). Compared with a normal trypanosome (Fig. 6D, inset), tetracycline-induced trypanosomes show an increase in body size with some having sections of flagella detached from the body and lacking the undulating membrane (Fig. 6D, arrowheads). Multiple entangled flagella can be clearly identified in the SEM and DAPI- stained image (Fig. 6E and F respectively). Phase-contrast light microscopy of wet preparations showed that these grossly abnormal parasites are still viable, with multiple flagella moving in an uncoordinated fashion with detached flagella in some instances radiating from the central body in many directions. There is also considerable disruption of the parasite surface with what appears to be membrane disorganization and deterioration. The changes highlighted on the surface are probably an indication of the latter stages of the RNAi effect with cells that are close to death, as can be seen for the central parasite in Fig. 6D.
Fig. 6.
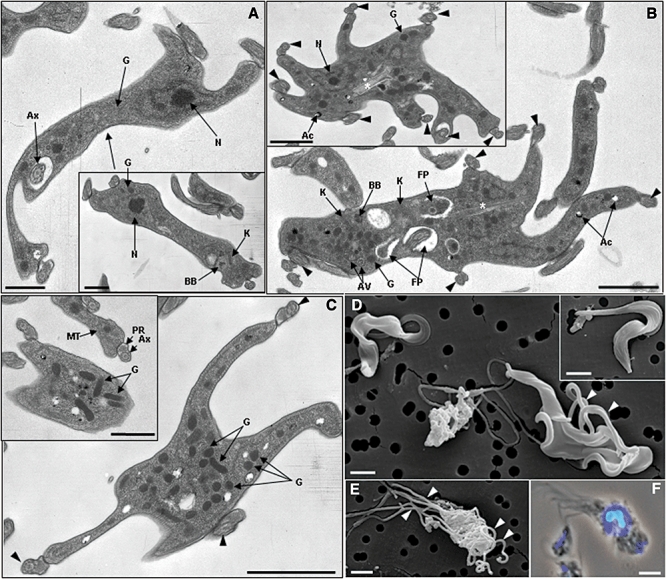
Morphology of PTR1-depleted cells by electron microscopy. A. TEM section of non-induced controls. B and C. TEM sections of 72-h induced cells depicting increased number of flagella and glycosomes. D. SEM images of 72-h induced cells, one with a normal (top left) and abnormal (bottom right) appearance compared with non-induced control (inset). E. Equivalent DAPI-stained 72-h induced cell with multiple flagella. Abbreviations used in TEM sections: nucleus (N); nuclear membrane (NM); basal bodies (BB); kinetoplast (K); flagellar rod (FR); flagellar pocket (FP); axonemes (Ax and black arrow heads); glycosomes (G); lysosomes (L); paraxial rod (PR); microtubules (MT); acidocalcisomes (Ac); autophagic vacuoles (AV) internal non-membrane bound flagellum (*). White arrow heads in SEM images highlight flagella. Black and white bars represent 200 nm.
The identity of the electron-dense organelles as glycosomes was verified by immuno-cytochemistry. TEM sections of WT, non-induced and induced samples at 72 h were labelled with an antibody to the glycosomal matrix enzyme marker, T. brucei glyceraldehyde phosphate dehydrogenase (GAPDH) and visualized with 10 nm protein-A gold particles (Fig. 7). In the non-induced samples, gold particles are exclusively localized to glycosomes confirming the specificity of the antibody reagent (Fig. 7A and B). In contrast, the induced samples show pronounced labelling of glycosomes with additional gold particles in the cytosol (Fig. 7C and D, arrows). The matrix of the elongated electron-dense structure in Fig. 7D is also heavily stained confirming that the sausage-shaped structures (Fig. 6C) are likely to be glycosomes.
Fig. 7.
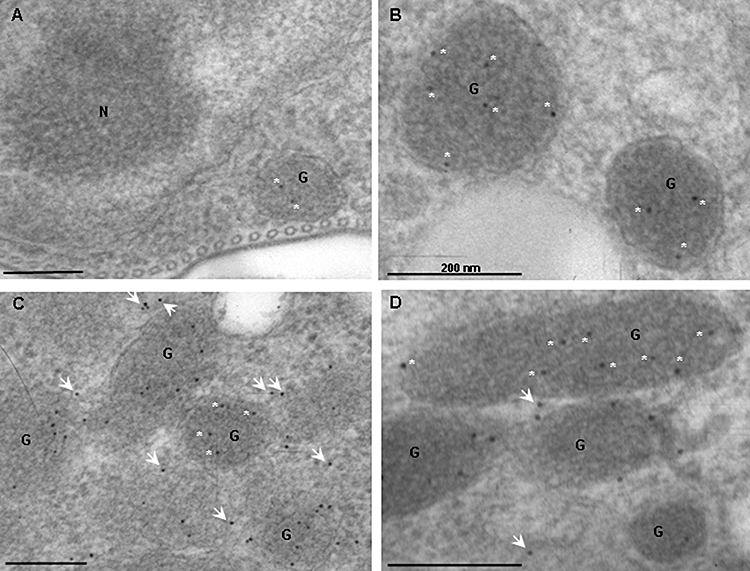
Localization and distribution of GAPDH by immuno-gold labelling. Thin-layer sections were labelled with anti-GAPDH and stained with protein A gold particles and examined by TEM. WT (A), RNAi non-induced (B) and induced (C–D) at 72 h. Abbreviations used: nucleus (N); glycosome (G) and white asterisks alongside black dots represent 10 nm gold particles present within glycosomes, while white arrows depict particles outside glycosomes. Scale bar represents 200 nm.
To verify whether there is an increase in the number of glycosomes following PTR1 depletion, immunofluorescence studies were undertaken using anti-GAPDH. Staining for GAPDH in the non-induced control is punctate in nature (Fig. 8A), which is more pronounced and diffuse following induction with tetracycline at 48 h. Staining increased to such a level that it covered almost the entire body of the parasite at 72 h. Some punctate staining is also visible along the long thin structures radiating out from the main body of these multinucleated and multikinetoplast parasites (Fig. 8C). These structures were confirmed to be detached flagella by staining with antibody to the paraflagellar rod protein, ROD1 (Fig. 9). The increase in glycosomal staining pattern illustrated with GAPDH following RNAi expression was confirmed using an alternative glycosomal matrix marker, aldolase (ALD). Induction at 48 h resulted in a partial diffuse staining into the cytoplasm; however, unlike GAPDH, no punctuate staining was visible in the flagella at 72 h.
Fig. 8.
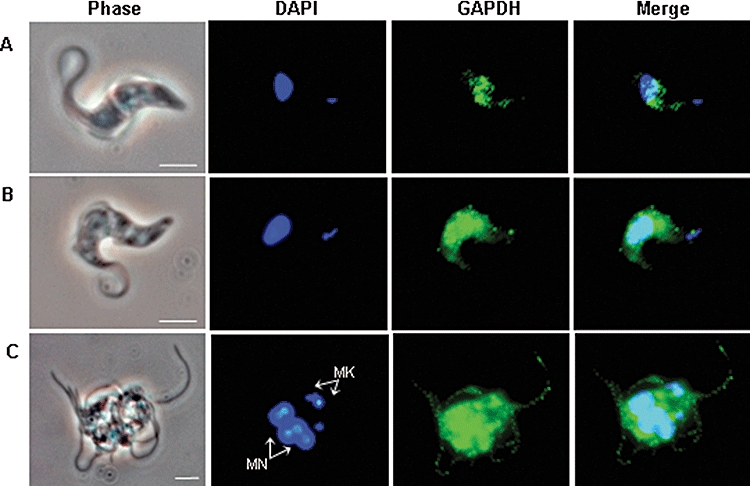
The effect of PTR1 depletion on the subcellular distribution of the glycosomal matrix enzyme, GAPDH by immunofluorescence microscopy. Non-induced (A), induced 48 h (B) and 72 h (C) RNAi cells were labelled with a glycosomal matrix marker, GAPDH (green) with staining of nuclear and kinetoplast DNA by DAPI (blue). MN, multinucleated, MK, multikinetoplastid. Scale bar represents 10 µm.
Fig. 9.
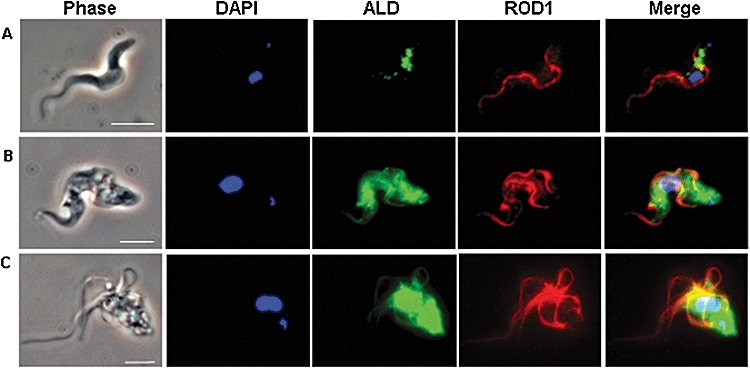
Immunolocalisation of aldolase and paraflagellar rod marker, ROD1 in RNAi depleted cells. Co-localization of aldolase (glycosomal marker, green) and ROD1 (paraflagellar rod marker, red) with DAPI (blue) on RNAi cells. Non-induced (A); induced 48 h (B); and 72 h (C) RNAi cells. MN, multinucleated, MK, multikinetoplastid, and scale bar represents 10 µm.
Subcellular distribution of PTR1 and glycosomal markers
Cytosolic and glycosomal fractions of bloodstream form T. brucei were prepared by ultracentrifugation, following digitonin treatment of WT and 48 h induced RNAi cells and analysed by Western blotting with antibodies to specific cytosolic and glycosomal markers (Fig. 10). PTR1 was recovered exclusively in the cytosolic fraction along with the cytosolic enzyme marker, enolase. Similar results were obtained for procyclic forms (Fig. S3). Reduced levels of PTR1 are evident in the cytosolic non-induced fraction of RNAi cells compared with WT, due to the leaky nature of p2T7TiTAblue RNAi vector (Alibu et al., 2005). Following RNAi induction for 48 h, PTR1 is below the limit of detection, whereas enolase and PEX13, a glycosomal membrane-bound protein, were unchanged. Densitometry of the band stained for the glycosomal matrix enzyme GAPDH indicated that the pellet fraction was unchanged whereas the amount in the cytosol was increased approximately twofold. Although analysis of 72 h induced samples was not possible due to lack of material, the increased amount of GAPDH observed 48 h after induction, supports the mis-localization of GAPDH to the cytosol illustrated in the immuno-gold labelling studies at 72 h (Fig. 7).
Fig. 10.
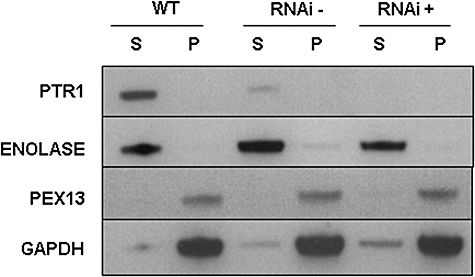
The effect of PTR1 depletion on the subcellular distribution of glycolytic enzymes. Digitonin fractionation of intact WT and RNAi cells grown for 48 h (in the absence ‘–’ and presence ‘+’ of tetracycline) were incubated with 150 µg ml−1 of digitonin and processed as described in Material and Methods. The release of PTR1, enolase, PEX13 and GAPDH from digitonin-treated cells was determined in supernatant (S) and pellet (P) fractions by Western blot.
Discussion
The gene knockout, RNAi knockdown and rescue studies reported here provide strong evidence that PTR1 is essential for the blood-stream form of the African trypanosome. Loss of PTR1 by RNAi is cytocidal rather than cytostatic, which is a distinct advantage from a drug discovery perspective. This lethal RNAi phenotype can be rescued by expression of functionally active L. major PTR1, or by supplementation with tetrahydrobiopterin. PTR1 levels were markedly reduced even in the absence of inducer due to poor regulation of the pLew82 vector. While this had no discernable effect on growth in rich medium in vitro, the virulence of the PTR1 RNAi T. brucei was considerably attenuated in mice. Further loss of virulence was evident when RNAi was induced, but some mice still succumbed to infection. This is most likely due to the instability of the RNAi machinery as previously reported (Chen et al., 2003). However, since supplementation with 10 µM tetrahydrobiopterin was able to rescue the trypanocidal effect in vitro, we cannot definitively exclude the possibility that the steady-state levels of tetrahydrobiopterin present within the animal host plasma (∼0.1 µM) are contributing to survival (Fukushima and Nixon, 1980; Yoshida et al., 2000). The fact that BH4 is extremely unstable in culture medium and that repeated daily addition of BH4 was found to be toxic to T. brucei could explain the failure to generate a double knockout line using BH4 as a nutritional rescue. Thus, ultimate proof that PTR1 is a suitable drug target on its own requires chemical validation using potent and specific drug-like inhibitors of PTR1 with appropriate pharmacological properties. Unfortunately, the currently available inhibitors do not meet these specifications (Cavazzuti et al., 2008; Mpamhanga et al., 2009; Shanks et al., 2010). Nonetheless, combined inhibition of dihydrofolate reductase and PTR1 remains an attractive chemotherapeutic strategy (Hardy et al., 1997; Cavazzuti et al., 2008; Tulloch et al., 2010).
Our morphological data illustrate that knockdown of PTR1 results in cells that are unable to divide normally, although they can still undergo nuclear division and kinetoplast segregation. Formation of multiflagellated parasites has also been reported in a number of other RNAi studies dissecting organelle biogenesis including: depletion of trypanin, a component of the dynein regulatory complex of the flagellum (Ralston and Hill, 2006); depletion of centrin1, a component associated with the basal bodies and responsible for segregation of the linked organelles: flagellum, kinetoplasts and Golgi (Selvapandiyan et al., 2007); depletion of the aurora-like kinase AUK1, a chromosome passenger protein (Li and Wang, 2006; Jetton et al., 2009); and depletion of CAP5.5 resulting in defects in microtubule organization (Olego-Fernandez et al., 2009). However, none of these studies have observed the glycosomal abnormalities reported here. Further work is required to determine whether other RNAi mutants with cytokinesis defects might show similar glycosomal abnormalities.
Knockdown of PTR1 also affects glycosomal numbers and their morphology, as judged by TEM. PTR1 knockdown also causes partial mislocalisation to the cytosol of the glycosomal matrix enzymes, GAPDH and aldolase, as seen in the immunofluorescence and digitonin fractionation experiments. In our view, mislocalization could be due to either defective import into the glycosome or due to loss of integrity of the glycosomal membrane. However, similar effects observed in studies on the functional roles of peroxins have been attributed to defective import. Peroxins (PEX) are a large family of peroxisomal proteins which are involved in glycosomal biogenesis via the import of newly synthesized proteins from the cytosol (Parsons et al., 2001). Cellular knockdown by RNA interference of PEX 13 (Furuya et al., 2002; Verplaetse et al., 2009), GIM5 (a PEX11-related protein) (Furuya et al., 2002; Moyersoen et al., 2003; Voncken et al., 2003; Verplaetse et al., 2009), PEX14 (Moyersoen et al., 2003), PEX19 (Banerjee et al., 2005); and PEX6, PEX10 and PEX12 (Krazy and Michels, 2006) all resulted in the partial mislocalization of glycosomal matrix enzymes to the cytosol. Overexpression of PEX11 led to an increased number of glycosomes which were elongated and clustered (Lorenz et al., 1998), whereas knockdown of PEX19 resulted in smaller numbers of larger glycosomes (Banerjee et al., 2005). However, none of these studies reported defects in cytokinesis that we observed here.
The discovery that TbPTR1 is present within the cytosol in bloodstream (Fig. 10) and procyclic forms (Fig. S3) refutes the predicted glycosomal location for PTR1, based on the PST1 consensus sequence -VHA at its C-terminus (Opperdoes and Szikora, 2006). This is consistent with the failure to detect PTR1 by a proteomic analysis of purified T. brucei glycosomes (Vertommen et al., 2008) and agrees with the cytosolic location of a GFP-PTR1 chimera in L. donovani (Kumar et al., 2007). Likewise, both trypanothione reductase and trypanothione peroxidase from T. brucei also contain putative glycosomal targeting sequences, yet the former is clearly cytosolic (Smith et al., 1991) and the latter is located in the cytosol and mitochondria (Schlecker et al., 2005). This underlines the need for experimental verification of any in silico bioinformatic predictions (Wanders and Waterham, 2006).
PTR1 clearly has pleiotropic effects on cytokinesis, flagellar morphology and glycosome biogenesis. The challenging question is what is the underlying biochemical mechanism? Of the two known functions for PTR1, the maintenance of biopterin in its tetrahydro-form seems more likely than the salvage of folate to dihydrofolate, since the growth phenotype can be rescued by supplementation with H4B in high- or low-folate media containing thymidine. Since the functions of pterins remain to be elucidated in T. brucei one can only speculate what these functions might be. For example, part of the ether lipid biosynthetic pathway is present in mammalian peroxisomes cells (Kaufman et al., 1990) and in trypanosomatid glycosomes (Lux et al., 2000; Parsons et al., 2001; Michels et al., 2006). To date, the first three enzymes of this pathway have been identified in trypanosomatids by fractionation, bioinformatic and proteomic based analysis (Heise and Opperdoes, 1997; Opperdoes and Szikora, 2006; Vertommen et al., 2008), but none of these are known to be pterin-dependent. However, BH4 is involved in ether lipid catabolism in mammalian cells, where it serves as an essential cofactor for the cleavage of ether lipids via glyceryl ether monooxygenase (EC 1.14.16.5) located in the microsomal compartment (Watschinger et al., 2009). The ultimate products of ether lipid cleavage are glycerol and a fatty alcohol. Possibly, these fatty alcohols could serve in the subsequent ether lipid synthesis in the glycosome. However, the fact that oxidative cleavage of ether lipids in L. donovani was found to be dependent on NADPH rather than tetrahydrobiopterin (Ma et al., 1996) tends to argue against this hypothesis. Tetrahydrobiopterin also plays a role in defence against oxidative and nitrosative stress in mammalian cells (Mogi et al., 1999; Ozkor and Quyyumi, 2008) and recent studies in PTR1 knockout and overexpressing lines suggest a similar role for Leishmania spp. (Moreira et al., 2009; Nare et al., 2009).
In conclusion, our data strongly suggest that PTR1 is essential for blood stream T. brucei for growth and survival in vitro and in the animal host. The phenotypic abnormalities uncovered in this investigation suggest a potential role for PTR1 in the biogenesis of the glycosome and the flagellum. Further studies are underway in our laboratory to investigate these and other possibilities.
Experimental procedures
Trypanosome culture
Trypanosoma brucei bloodstream form ‘single marker’ S427 (T7RPOL TETR NEO) and transfected cells were cultured as previously described (Wirtz et al., 1999) and (Alibu et al., 2005). Folate deficient media (FDM) and growth conditions used in the study were as previously described (Sienkiewicz et al., 2008). Tetrahydrobiopterin was purchased from B. Shircks Laboratories (Jona, Switzerland) and 10 mM stock was prepared in degassed 0.1 M HCl.
Cloning and generation of transgenic cell lines
All constructs made were sequenced and prepared for electroporation using QIAprep Miniprep Plasmid Kit (Qiagen). DNA constructs were prepared for electroporation into trypanosomes and cloned with all procedures performed as previously described (Wirtz et al., 1999; Sienkiewicz et al., 2008). Primers used in this study to generate the RNAi and overexpressor constructs (Table S1) are based on the GeneDB sequences for T. brucei PTR1 (Tb927.8.2210, 927 strain) and L. major PTR1 (LmjF23.0270, Friedlin strain). For the disruption of PTR1 function, a 403 bp region (nucleotide position 399–801; http://trypanofan.path.cam.ac.uk/software/RNAit.html) was directly ligated into the p2T7TiTAblue vector (http://trypanofan.path.cam.ac.uk/trypanofan/vector/; a generous gift from David Horn), linearized by digestion with NotI and transfected by electroporation to generate RNAi cells under hygromycin selection (4 µg ml−1). An overexpressor construct (pLew82), containing the L. major PTR1 ORF was also made for generation of an overexpressor WT (oeWT) under phleomycin selection (2.5 µg ml−1). The oeWT cells generated were used for the reintroduction of the p2T7TiTAblue RNAi construct to generate oeRNAi cells. All tetracycline induction experiments were initiated at 1 × 104 cells ml−1 and tetracycline added daily at 1 µg ml−1.
DNA analysis
The WT and RNAi genomic DNA (5 µg) was digested with the restriction enzyme StuI and blots were subsequently probed with TbPTR1 ORF as described previously (Sienkiewicz et al., 2008). Southern blot analysis was used to confirm the successful integration of the selectable gene markers into WT cells (see Supporting information).
Western blot analysis
The WT, RNAi (0, 24 and 48 h) and oeRNAi (48 h) cells were centrifuged at 20 000 g for 5 min at 4°C. The cell pellets (1 × 106 parasites per sample) were resuspended in SDS Laemmli buffer, boiled for 5 min and micro-centrifuged as above. The supernatants were then separated by SDS-PAGE and subsequently transferred onto nitrocellulose for immunoblotting as previously described (Sienkiewicz et al., 2008). The immunoblot for the RNAi set was sequentially probed with polyclonal antisera raised against T. brucei PTR1 (1:500 dilution) and BiP (1:10 000), in conjunction with a secondary rabbit anti-rat HRP-conjugated antibody (1:5 000, Dako). The oeRNAi samples were sequentially probed with T. brucei PTR1 antisera and L. major PTR1 (1:10 000), in conjunction with secondary rabbit anti-rat and goat anti-rabbit HRP-conjugated antibodies (1:5000 and 1:10 000 respectively) and the immunoblots visualized with the ECL plus (enhanced chemiluminescence) system from GE Healthcare.
Enzyme assays
Lysates of T. brucei were prepared as previously described (Vickers and Fairlamb, 2004). To ensure adequate extraction of parasites, trypanothione reductase activity was measured in clarified lysates as previously described (Jockers-Scherubl et al., 1989). Assays for PTR1 activity in clarified lysates were carried out at 22°C in 50 mM HEPES (pH 7.4), containing 0.1% (v/v) triton X-100, adjusted to an ionic strength of 100 mM with KCl. Lysates were preincubated with 100 µM NADPH cofactor for 2 min and reactions were initiated by the addition of 25 nM H2B, in a final volume of 200 µl. Enzymatic reactions were further incubated for 1 min, before aliquots (100 µl) were removed and oxidized with iodine under alkaline conditions and analysed by HPLC as previously described (Shanks et al., 2010). Protein was measured using Coomassie blue using bovine serum albumin as standard (Bradford, 1976).
Morphological analysis
RNAi cells (grown in the presence and absence of tetracycline) were fixed with methanol or 4% (v/v) paraformaldehyde and stained with either Giemsa or 4′,6-diamidino-2-phenylindole (Slowfade Gold antifade reagent with DAPI, Invitrogen). Images were captured on a Zeiss Axiovert 200 M fluorescence and light microscope using Zeiss AxioVision Software. Approximately 300 parasites per population were assessed for changes in N:K ratio and data presented as the mean of two independent knockdown experiments. For electron microscopy, cells were fixed for 24 h in 2% (v/v) glutaraldehyde and 4% (v/v) paraformaldehyde in 0.2 M PIPES, pH 7.2 and prepared for both SEM and TEM as previously described (Sienkiewicz et al., 2008).
Immuno-gold labelling of TEM sections
For immunoelectron microscopy the labelling and staining procedure was carried out as essentially described (Sandilands et al., 2004; Sienkiewicz et al., 2008). Briefly, samples previously embedded in Durcupan resin were etched with 1% (v/v) periodic acid and the osmium removed by 2% (w/v) sodium periodate. The sections were then probed with anti-GAPDH (1: 50 dilution) antibody and subsequently incubated with Protein A labelled with 10 nm gold particles (1:50 dilution, BB International) to visualize the primary antibody. The sections were stained with 3% (v/v) aqueous uranyl acetate and Reynold's lead citrate prior to examination with a JEOl-1200 EX TEM.
Immunofluorescence
Cells were fixed with 4% (v/v) paraformaldehyde onto poly-L-lysine coated chamber slides and probed with antisera raised against T. brucei GAPDH (rabbit antiserum, 1:2500), ALD (rabbit antiserum, 1:6000) and ROD1 (monoclonal antiserum, neat), generous gifts from Paul Michels and Keith Gull, as previously described (Woods et al., 1989; Verplaetse et al., 2009). Combination of secondary Alexafluor 488 and 555 conjugated ant-rabbit or anti-mouse immunoglobulins were used (both 1:800 dilution, Molecular Probes) and slides were counter-stained with DAPI prior to analysis. Images were captured using FITC, Texas Red and DAPI filters as previously described. Property parameters were set on the AxioVision microscope software to optimize the intensity of the fluorescent stains standardized against the non-induced controls and these values were retained for all images captured. Images shown are representative of three independent experiments.
Separation of cytosolic and glycosomal rich proteins by digitonin permeabilization
Digitonin permeabilization experiments were undertaken to separate cytosolic and glycosomal rich proteins from WT and RNAi cells (grown in the absence and presence of tetracycline for 48 h), as previously described (Voncken et al., 2003). Briefly, live cells were washed once in STEN (250 mM sucrose, 25 mM Tri-HCl, 1 mM EDTA, 150 mM NaCl, pH 7.4), pelleted and resuspended in STEN containing 150 µg ml−1 digitonin (at a final concentration 5 × 108 cells ml−1) which was subsequently incubated for 1 h on ice. The digitonin treated cells were then fractionated by centrifugation (2 min, 20 000 g, at 4°C) resulting in an enriched cytosolic (s, supernatant) and glycosomal (p, pellet) fractions. Approximately 5 × 106 fractionated parasites were processed for Western blot analysis and sequentially probed with the following antibodies raised against T. brucei PTR1 (1:500), rabbit antisera raised against PEX13 (1:20 000), enolase (1:100 000) and GAPDH (1:100 000). Blots were subsequently incubated with secondary anti-rat (1:5000 dilution) and anti-rabbit HRP-conjugated antiserum (1:15 000) and visualized as previously described.
In vivo studies
All cell lines (WT, RNAi, oeRNAi) were cultured for 24 h in the absence of selectable drugs before female National Medical Research Institute (NMRI) mice (5 per group) were infected with a single intraperitoneal injection of 105 parasites in 0.2 ml glucose saline as previously described (Sienkiewicz et al., 2008). For the RNAi and oeRNAi cells, a single inoculum of non-induced cells were infected into two sets of mice with one group being given 2.5 µg ml−1 doxycycline in their drinking water (starting a week prior to infection and continuing throughout the 30 day period). Animals were fed ad libitum on standard chow (Diet 3, SAS, Edinburgh). Parasitaemia levels were monitored throughout the 30 day experiment (Sienkiewicz et al., 2008). All animal experiments undertaken were carried out following local ethical review and under UK regulatory licensing in accordance with the European Communities Council Directive (86/609/EEC).
Acknowledgments
We would like to thank Professor Steve Beverley (Washington University School of Medicine) and Professor Paul Michels (Université Catholique de Louvain), for helpful discussions, useful advice and encouragement; Professor George Cross and Dr Simone Leal (Rockefeller University, New York, USA) for the provision of transgenic cell lines; Professor David Horn (London School of Hygiene and Tropical Medicine) for the p2T7TiTAblue vector; Professor Steve Beverley, Professor James Bangs (University of Wisconsin), Professor Paul Michels and Professor Keith Gull (University of Oxford) for all their kind gifts of antisera and helpful insights into trypanosome biology; Dr Lucia Güther for many helpful discussions and reagents; Dr Susan Wyllie for critical analysis of the manuscript; and Mr Martin Keirens from the CHIPs facility for help and advice with electron microscopy. This study was supported by funding from the Wellcome Trust (WT079838 and WT WT083481).
References
- Alibu VP, Storm L, Haile S, Clayton C, Horn D. A doubly inducible system for RNA interference and rapid RNAi plasmid construction in Trypanosoma brucei. Mol Biochem Parasitol. 2005;139:75–82. doi: 10.1016/j.molbiopara.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Armarego WLF, Randles D, Taguchi H. Km and kcat values for [6,6,7,7-2H]7,8(6H)-dihydropterin and 2,6-diamino-5-iminopyrimidin-4-one with dihydropteridine reductase. Biochem J. 1983;211:357–361. doi: 10.1042/bj2110357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee SK, Kessler PS, Saveria T, Parsons M. Identification of trypanosomatid PEX19: functional characterization reveals impact on cell growth and glycosome size and number. Mol Biochem Parasitol. 2005;142:47–55. doi: 10.1016/j.molbiopara.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Bello AR, Nare B, Freedman D, Hardy L, Beverley SM. PTR1: a reductase mediating salvage of oxidized pteridines and methotrexate resistance in the protozoan parasite Leishmania major. Proc Natl Acad Sci USA. 1994;91:11442–11446. doi: 10.1073/pnas.91.24.11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, et al. The Genome of the African Trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Callahan HL, Beverley SM. A member of the aldoketo reductase family confers methotrexate resistance in Leishmania. J Biol Chem. 1992;267:24165–24168. [PubMed] [Google Scholar]
- Cavazzuti A, Paglietti G, Hunter WN, Gamarro F, Piras S, Loriga M, et al. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc Natl Acad Sci USA. 2008;105:1448–1453. doi: 10.1073/pnas.0704384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YL, Hung CH, Burderer T, Lee GSM. Development of RNA interference revertants in Trypanosoma brucei cell lines generated with a double stranded RNA expression construct driven by two opposing promoters. Mol Biochem Parasitol. 2003;126:275–279. doi: 10.1016/s0166-6851(02)00276-1. [DOI] [PubMed] [Google Scholar]
- Cruz AK, Titus R, Beverley SM. Plasticity in chromosome number and testing of essential genes in Leishmania by targeting. Proc Natl Acad Sci USA. 1993;90:1599–1603. doi: 10.1073/pnas.90.4.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham ML, Beverley SM. Pteridine salvage throughout the Leishmania infectious cycle: implications for antifolate chemotherapy. Mol Biochem Parasitol. 2001;113:199–213. doi: 10.1016/s0166-6851(01)00213-4. [DOI] [PubMed] [Google Scholar]
- Cunningham ML, Titus RG, Turco SJ, Beverley SM. Regulation of differentiation to the infective stage of the protozoan parasite Leishmania major by tetrahydrobiopterin. Science. 2001;292:285–287. doi: 10.1126/science.1057740. [DOI] [PubMed] [Google Scholar]
- Dawson A, Gibellini F, Sienkiewicz N, Tulloch LB, Fyfe PK, McLuskey K, et al. Structure and reactivity of Trypanosoma brucei pteridine reductase: inhibition by the archetypal antifolate methotrexate. Mol Microbiol. 2006;61:1457–1468. doi: 10.1111/j.1365-2958.2006.05332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima T, Nixon JC. Analysis of reduced forms of biopterin in biological tissues and fluids. Anal Biochem. 1980;102:176–188. doi: 10.1016/0003-2697(80)90336-x. [DOI] [PubMed] [Google Scholar]
- Furuya T, Kessler P, Jardim A, Schnaufer A, Crudder C, Parsons M. Glucose is toxic to glycosome-deficient trypanosomes. Proc Natl Acad Sci USA. 2002;99:14177–14182. doi: 10.1073/pnas.222454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley DG, Schuttelkopf AW, Leonard GA, Luba J, Hardy LW, Beverley SM, Hunter WN. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat Struct Biol. 2001;8:521–525. doi: 10.1038/88584. [DOI] [PubMed] [Google Scholar]
- Hardy LW, Matthews W, Nare B, Beverley SM. Biochemical and genetic tests for inhibitors of Leishmania pteridine pathways. Exp Parasitol. 1997;87:157–169. [PubMed] [Google Scholar]
- Heise N, Opperdoes FR. The dihydroxyacetonephosphate pathway for biosynthesis of ether lipids in Leishmania mexicana promastigotes. Mol Biochem Parasitol. 1997;89:61–72. doi: 10.1016/s0166-6851(97)00101-1. [DOI] [PubMed] [Google Scholar]
- Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetton N, Rothberg KG, Hubbard JG, Wise J, Li Y, Ball HL, Ruben L. The cell cycle as a therapeutic target against Trypanosoma brucei: hesperadin inhibits Aurora kinase-1 and blocks mitotic progression in bloodstream forms. Mol Microbiol. 2009;72:442–458. doi: 10.1111/j.1365-2958.2009.06657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockers-Scherubl MC, Schirmer RH, Krauth-Siegel RL. Trypanothione reductase from Trypanosoma cruzi: catalytic properties of the enzyme and inhibition studies with trypanocidal compounds. Eur J Biochem. 1989;180:267–272. doi: 10.1111/j.1432-1033.1989.tb14643.x. [DOI] [PubMed] [Google Scholar]
- Kaufman S. The structure of the phenylalanine-hydroxylation cofactor. Proc Natl Acad Sci USA. 1963;50:1085–1093. doi: 10.1073/pnas.50.6.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman S, Pollock RJ, Summer GK, Das AK, Hajra AK. Dependence of an alkyl glycol-ether monooxygenase activity upon tetrahydropterins. Biochim Biophys Acta. 1990;1040:19–27. doi: 10.1016/0167-4838(90)90141-2. [DOI] [PubMed] [Google Scholar]
- Kidder GW, Dutta BN. The growth and nutrition of Crithidia fasciulata. J Gen Microbiol. 1958;18:621–638. doi: 10.1099/00221287-18-3-621. [DOI] [PubMed] [Google Scholar]
- Krazy H, Michels PAM. Identification and characterization of three peroxins – PEX6, PEX10 and PEX12 – involved in glycosome biogenesis in Trypanosoma brucei. Biochim Biophys Acta. 2006;1763:6–17. doi: 10.1016/j.bbamcr.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Kumar P, Sundar S, Singh N. Degradation of pteridine reductase 1 (PTR1) enzyme during growth phase in the protozoan parasite Leishmania donovani. Exp Parasitol. 2007;116:182–189. doi: 10.1016/j.exppara.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Kundig C, Haimeur A, Legare D, Papadopoulou B, Ouellette M. Increased transport of pteridines compensates for mutations in the high affinity folate transporter and contributes to methotrexate resistance in the protozoan parasite Leishmania tarentolae. EMBO J. 1999;18:2342–2351. doi: 10.1093/emboj/18.9.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemley C, Yan SF, Dole VS, Madhubala R, Cunningham ML, Beverley SM, et al. The Leishmania donovani LD1 locus gene ORFG encodes a biopterin transporter (BT1) Mol Biochem Parasitol. 1999;104:93–105. doi: 10.1016/s0166-6851(99)00132-2. [DOI] [PubMed] [Google Scholar]
- Li ZY, Wang CC. Changing roles of aurora-B kinase in two life cycle stages of Trypanosoma brucei. Eukaryotic Cell. 2006;5:1026–1035. doi: 10.1128/EC.00129-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz P, Maier AG, Baumgart E, Erdmann R, Clayton C. Elongation and clustering of glycosomes in Trypanosoma brucei overexpressing the glycosomal Pex11p. EMBO J. 1998;17:3542–3555. doi: 10.1093/emboj/17.13.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luba J, Nare B, Liang PH, Anderson KS, Beverley SM, Hardy LW. Leishmania major pteridine reductase 1 belongs to the short chain dehydrogenase family: stereochemical and kinetic evidence. Biochemistry. 1998;37:4093–4104. doi: 10.1021/bi972693a. [DOI] [PubMed] [Google Scholar]
- Lux H, Heise N, Klenner T, Hart D, Opperdoes FR. Ether-lipid (alkyl-phospholipid) metabolism and the mechanism of action of ether-lipid analogues in Leishmania. Mol Biochem Parasitol. 2000;111:1–14. doi: 10.1016/s0166-6851(00)00278-4. [DOI] [PubMed] [Google Scholar]
- Lye LF, Cunningham ML, Beverley SM. Characterization of quinonoid-dihydropteridine reductase (QDPR) from the lower eukaryote Leishmania major. J Biol Chem. 2002;277:38245–38253. doi: 10.1074/jbc.M206543200. [DOI] [PubMed] [Google Scholar]
- Ma DQ, Beverley SM, Turco SJ. Leishmania donovani possess a NADPH-dependent alkylglycerol cleavage enzyme. Biochem Biophys Res Commun. 1996;227:885–889. doi: 10.1006/bbrc.1996.1600. [DOI] [PubMed] [Google Scholar]
- Michels PAM, Bringaud F, Herman M, Hannaert V. Metabolic functions of glycosornes in trypanosomatids. Biochim Biophys Acta. 2006;1763:1463–1477. doi: 10.1016/j.bbamcr.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Mogi M, Kinpara K, Kondo A, Togari A. Involvement of nitric oxide and biopterin in proinflammatory cytokine-induced apoptotic cell death in mouse osteoblastic cell line MC3T3-E1. Biochem Pharmacol. 1999;58:649–654. doi: 10.1016/s0006-2952(99)00131-8. [DOI] [PubMed] [Google Scholar]
- Moreira W, Leblanc E, Ouellette M. The role of reduced pterins in resistance to reactive oxygen and nitrogen intermediates in the protozoan parasite Leishmania. Free Radic Biol Med. 2009;46:367–375. doi: 10.1016/j.freeradbiomed.2008.10.034. [DOI] [PubMed] [Google Scholar]
- Moyersoen J, Choe J, Kumar A, Voncken FGJ, Hol WGJ, Michels PAM. Characterization of Trypanosoma brucei PEX14 and its role in the import of glycosomal matrix proteins. Eur J Biochem. 2003;270:2059–2067. doi: 10.1046/j.1432-1033.2003.03582.x. [DOI] [PubMed] [Google Scholar]
- Mpamhanga CP, Spinks D, Tulloch LB, Shanks EJ, Robinson DA, Collie IT, et al. One scaffold, three binding modes: novel and selective pteridine reductase 1 Inhibitors derived from fragment hits discovered by virtual screening. J Med Chem. 2009;52:4454–4465. doi: 10.1021/jm900414x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nare B, Luba J, Hardy LW, Beverley SM. New approaches to Leishmania chemotherapy: pteridine reductase 1 (PTR1) as a target and modulator of antifolate sensitivity. Parasitol. 1997;114:S101–S110. [PubMed] [Google Scholar]
- Nare B, Garraway LA, Vickers TJ, Beverley SM. PTR1-dependent synthesis of tetrahydrobiopterin contributes to oxidant susceptibility in the trypanosomatid protozoan parasite Leishmania major. Curr Genet. 2009;55:287–299. doi: 10.1007/s00294-009-0244-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichol CA, Lee CL, Edelstein MP, Chao JY, Duch DS. Biosynthesis of tetrahydrobiopterin by de novo and salvage pathways in adrenal-medulla extracts, mammalian-cell cultures, and rat-brain in vivo. Proc Natl Acad Sci USA. 1983;80:1546–1550. doi: 10.1073/pnas.80.6.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olego-Fernandez S, Vaughan S, Shaw MK, Gull K, Ginger ML. Cell morphogenesis of Trypanosoma brucei requires the paralogous, differentially expressed calpain-related proteins CAP5.5 and CAP5.5V. Protist. 2009;160:576–590. doi: 10.1016/j.protis.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Opperdoes FR, Szikora JP. In silico prediction of the glycosomal enzymes of Leishmania major and trypanosomes. Mol Biochem Parasitol. 2006;147:193–206. doi: 10.1016/j.molbiopara.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Opperdoes FR, Baudhuin P, Coppens I, de Roe C, Edwards SW, Weijers PJ, Misset O. Purification, morphometric analysis, and characterization of the glycosomes (microbodies) of the protozoan hemoflagellate Trypanosoma brucei. J Cell Biol. 1984;98:1178–1184. doi: 10.1083/jcb.98.4.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellette M, Drummelsmith J, El Fadili A, Kundig C, Richard D, Roy G. Pterin transport and metabolism in Leishmania and related trypanosomatid parasites. Int J Parasitol. 2002;32:385–398. doi: 10.1016/s0020-7519(01)00346-0. [DOI] [PubMed] [Google Scholar]
- Ozkor MA, Quyyumi AA. Tetrahydrobiopterin. Curr Hypertens Rep. 2008;10:58–64. doi: 10.1007/s11906-008-0012-3. [DOI] [PubMed] [Google Scholar]
- Papadopoulou B, Roy G, Ouellette M. A novel antifolate resistance gene on the amplified H circle of Leishmania. EMBO J. 1992;11:3601–3608. doi: 10.1002/j.1460-2075.1992.tb05444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M, Furuya T, Pal S, Kessler P. Biogenesis and function of peroxisomes and glycosomes. Mol Biochem Parasitol. 2001;115:19–28. doi: 10.1016/s0166-6851(01)00261-4. [DOI] [PubMed] [Google Scholar]
- Ralston KS, Hill KL. Trypanin, a component of the flagellar dynein regulatory complex, is essential in bloodstream form African trypanosomes. PLoS Pathog. 2006;2:873–882. doi: 10.1371/journal.ppat.0020101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandilands A, Wang X, Hutcheson AM, James J, Prescott AR, Wegener A, et al. Bfsp2 mutation found in mouse 129 strains causes the loss of CP49 and induces vimentin-dependent changes in the lens fibre cell cytoskeleton. Exp Eye Res. 2004;78:109–123. doi: 10.1016/j.exer.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Schlecker T, Schmidt A, Dirdjaja N, Voncken F, Clayton C, Krauth-Siegel RL. Substrate specificity, localization, and essential role of the glutathione peroxidase-type tryparedoxin peroxidases in Trypanosoma brucei. J Biol Chem. 2005;280:14385–14394. doi: 10.1074/jbc.M413338200. [DOI] [PubMed] [Google Scholar]
- Schmidt TS, Alp NJ. Mechanisms for the role of tetrahydrobiopterin in endothelial function and vascular disease. Clin Sci. 2007;113:47–63. doi: 10.1042/CS20070108. [DOI] [PubMed] [Google Scholar]
- Schormann N, Pal B, Senkovich O, Carson M, Howard A, Smith C, et al. Crystal structure of Trypanosoma cruzi pteridine reductase 2 in complex with a substrate and an inhibitor. J Struct Biol. 2005;152:64–75. doi: 10.1016/j.jsb.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf AW, Hardy LW, Beverley SM, Hunter WN. Structures of Leishmania major pteridine reductase complexes reveal the active site features important for ligand binding and to guide inhibitor design. J Mol Biol. 2005;352:105–116. doi: 10.1016/j.jmb.2005.06.076. [DOI] [PubMed] [Google Scholar]
- Selvapandiyan A, Kumar P, Morris JC, Salisbury JL, Wang CC, Nakhasi HL. Centrin1 is required for organelle segregation and cytokinesis in Trypanosoma brucei. Mol Biol Cell. 2007;18:3290–3301. doi: 10.1091/mbc.E07-01-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks EJ, Ong HB, Robinson DA, Thompson S, Sienkiewicz N, Fairlamb AH, Frearson JA. Development and validation of a cytochrome c coupled assay for pteridine reductase 1 and dihydrofolate reductase. Anal Biochem. 2010;396:194–203. doi: 10.1016/j.ab.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheader K, Vaughan S, Minchin J, Hughes K, Gull K, Rudenko G. Variant surface glycoprotein RNA interference triggers a precytokinesis cell cycle arrest in African trypanosomes. Proc Natl Acad Sci USA. 2005;102:8716–8721. doi: 10.1073/pnas.0501886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienkiewicz N, Jaroslawski S, Wyllie S, Fairlamb AH. Chemical and genetic validation of dihydrofolate reductase-thymidylate synthase as a drug target in African trypanosomes. Mol Microbiol. 2008;69:520–533. doi: 10.1111/j.1365-2958.2008.06305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K, Opperdoes FR, Fairlamb AH. Subcellular distribution of trypanothione reductase in bloodstream and procyclic forms of Trypanosoma brucei. Mol Biochem Parasitol. 1991;48:109–112. doi: 10.1016/0166-6851(91)90170-b. [DOI] [PubMed] [Google Scholar]
- Thony B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347:1–16. [PMC free article] [PubMed] [Google Scholar]
- Tulloch LB, Martini VP, Iulek J, Huggan JK, Lee JH, Gibson CL, et al. Structure-based design of pteridine reductase inhibitors targeting African sleeping sickness and the Leishmaniases. J Med Chem. 2010;53:221–229. doi: 10.1021/jm901059x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullu E, Tschudi C, Chakraborty T. RNA interference in protozoan parasites. Cell Microbiol. 2004;6:509–519. doi: 10.1111/j.1462-5822.2004.00399.x. [DOI] [PubMed] [Google Scholar]
- Verplaetse E, Rigden DJ, Michels PAM. Identification, characterization and essentiality of the unusual peroxin 13 from Trypanosoma brucei. Biochim Biophys Acta. 2009;1793:516–527. doi: 10.1016/j.bbamcr.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Vertommen D, Van Roy J, Szikora JP, Rider MH, Michels PAM, Opperdoes FR. Differential expression of glycosomal and mitochondrial proteins in the two major life-cycle stages of Trypanosoma brucei. Mol Biochem Parasitol. 2008;158:189–201. doi: 10.1016/j.molbiopara.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Vickers TJ, Fairlamb AH. Trypanothione S-transferase activity in a trypanosomatid ribosomal elongation factor 1B. J Biol Chem. 2004;279:27246–27256. doi: 10.1074/jbc.M311039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voncken F, Van Hellemond JJ, Pfisterer I, Maier A, Hillmer S, Clayton C. Depletion of GIM5 causes cellular fragility, a decreased glycosome number, and reduced levels of ether-linked phospholipids in trypanosomes. J Biol Chem. 2003;278:35299–35310. doi: 10.1074/jbc.M301811200. [DOI] [PubMed] [Google Scholar]
- Wanders RJA, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329. [DOI] [PubMed] [Google Scholar]
- Watschinger K, Keller MA, Hermetter A, Golderer G, Werner-Felmayer G, Werner ER. Glyceryl ether monooxygenase resembles aromatic amino acid hydroxylases in metal ion and tetrahydrobiopterin dependence. Biol Chem. 2009;390:3–10. doi: 10.1515/BC.2009.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner-Felmayer G, Golderer G, Werner ER. Tetrahydrobiopterin biosynthesis, utilization and pharmacological effects. Curr Drug Metab. 2002;3:159–173. doi: 10.2174/1389200024605073. [DOI] [PubMed] [Google Scholar]
- Wilson K, Berens RL, Sifri CD, Ullman B. Amplification of the inosinate dehydrogenase gene in Trypanosoma brucei gambiense due to an increase in chromosome copy number. J Biol Chem. 1994;269:28979–28987. [PubMed] [Google Scholar]
- Wirtz E, Leal S, Ochatt C, Cross GAM. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- Woods A, Sherwin T, Sasse R, Macrae TH, Baines AJ, Gull K. Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J Cell Sci. 1989;93:491–500. doi: 10.1242/jcs.93.3.491. [DOI] [PubMed] [Google Scholar]
- Yoshida YI, Eda S, Masada M. Alterations of tetrahydrobiopterin biosynthesis and pteridine levels in mouse tissues during growth and aging. Brain Dev. 2000:S45–S49. doi: 10.1016/s0387-7604(00)00144-3. [DOI] [PubMed] [Google Scholar]