

Abstract
The YopM protein of Yersinia sp. is a type III secreted effector that is required for virulence in murine models of infection. YopM has previously been shown to contain leucine-rich repeats (LRRs) and to interact with two host kinases, RSK1 and PRK2, although the consequence of these interactions is unknown. A series of YopM proteins missing different numbers of LRRs or a C-terminal domain were produced and used for in vitro binding reactions to map domains required for interaction with RSK1 and PRK2. A C-terminal domain of YopM (from LRR12 to the C terminus) was shown to be required for interaction with RSK1, while an internal portion encompassing LRR6 to LRR15 was shown to be required for interaction with PRK2. The virulence of a Yersinia pseudotuberculosis ΔyopM mutant in mice via an intravenous route of infection was significantly attenuated. At day 4 postinfection, there were significantly increased levels of gamma interferon and reduced levels of interleukin-18 (IL-18) and IL-10 in the serum of the ΔyopM-infected mice compared to that of mice infected with the wild type, suggesting that YopM action alters the balance of these key cytokines to promote virulence. The PRK2 and RSK1 interaction domains of YopM were both required for IL-10 induction in vivo, irrespective of splenic colonization levels. In an orogastric model of Y. pseudotuberculosis infection, a ΔyopM mutant was defective in dissemination from the intestine to the spleen and significantly reduced in virulence. In addition, Y. pseudotuberculosis mutants expressing YopM proteins unable to interact with either RSK1 (YopMΔ12-C) or PRK2 (YopMΔ6-15) were defective for virulence in this assay, indicating that both interaction domains are important for YopM to promote pathogenesis.
Yersinia pseudotuberculosis is one of three species of Yersinia that are pathogenic for humans. Like Yersinia enterocolitica, it typically causes a self-limiting gastroenteritis, although in rare cases, fatal septicemia may occur. In addition to these two species, Yersinia pestis is a recently evolved species that is the causative agent of plague. Yersinia pathogenesis is generally associated with the presence of a virulence plasmid, pYV (pCD1 in Y. pestis) that encodes a type III secretion system (T3SS) and a repertoire of T3SS effector proteins termed Yops.
The Yop proteins have been intensively studied, and the biochemical functions of most are known. The YopE protein is a Rho GTPase-activating protein with specificity for RhoA and Rac1 (6). YopT is a cysteine protease that cleaves both RhoA and Rac1, thereby preventing these proteins from remaining in a membrane-associated location (37). The YpkA protein is unusual in that it consists of two domains; the first is a serine/threonine protein kinase that phosphorylates Gαq, and the second is a C-terminal Rho GDP dissociation inhibitor that is able to inhibit the activity of RhoA and Rac1 (19, 32, 36). YopH is a potent tyrosine phosphatase that dephosphorylates multiple targets, including Fyn-binding protein and p130Cas, which are proteins involved in phagocytosis and cell signaling (5, 21). These four proteins (YopE, YopT, YpkA, and YopH) contribute to the antiphagocytic activity that Yersinia displays toward macrophages and neutrophils (48).
In addition, Yersinia species also secrete the YopJ acetyltransferase (YopP in Y. enterocolitica), which acetylates the normally phosphorylatable serine/threonine residues in the activation loop of mitogen-activated protein kinase (MAPK) kinase proteins, as well as in IKKβ (30, 31). This activity inhibits both MAPK and NF-κB signaling, leading to altered cytokine secretion and induction of apoptosis in infected macrophages (34, 50).
Another secreted effector protein of Yersinia spp. is YopM, the function of which remains enigmatic. The tertiary structure of Y. pestis YopM has been determined and consists of an N-terminal secretion signal followed by two α helices that serve to initiate the folding of the leucine-rich repeat (LRR) region that makes up the majority of the protein (17). Depending upon the Yersinia species examined, the YopM protein contains 13 to 21 LRRs. There is also a short, unstructured C-terminal domain that is highly conserved among all YopM isoforms. The protein itself has a twisted horseshoe-like structure in which the inside face of the horseshoe is believed to present a binding surface for eukaryotic proteins, as has been proposed for other LRR family proteins (26, 46).
While all other Yops have been shown to have enzymatic activity, the YopM protein is believed to be devoid of catalytic activity. Interestingly, YopM has been demonstrated by several groups to localize to the nuclei of both yeast and mammalian cells (4, 40, 41). In Saccharomyces cerevisiae, protein localization seems to depend on the vesicular trafficking system and both the N-terminal half and the C-terminal half of YopM were able to traffic to the nucleus, suggesting that the protein has more than one nuclear localization signal (41). In agreement with this, another group has reported that the C-terminal unstructured domain of YopM and the LRR1-to-LRR3 domain of the protein are able to independently target to the nuclei of both yeast and HEK293T cells (4). Although these studies have produced a general view of the importance of YopM nuclear localization, the consequence of this localization for virulence remains enigmatic.
Studies on the role of YopM in Y. pestis pathogenesis using mouse infection models have demonstrated a pronounced loss of virulence of yopM mutants. For example, whereas the wild-type KIM5 strain delivered intravenously had a 50% lethal dose (LD50) of 29 to 42 CFU, the yopM mutant had an LD50 of 3.4 × 105 to 9.8 × 105 CFU (22, 27). Results of studies in which several mutant versions of YopM were constructed suggested that some of the central LRRs are required for function, although the reason for the loss of virulence observed was thought to be an interaction with α-thrombin (22). The importance of YopM/α-thrombin interaction was refuted in a later publication (33). Somewhat paradoxically and in contrast to the first two publications cited, this study suggested that the deletion of yopM from Y. pestis did not result in a pronounced loss of virulence in an intravenous model of infection. In an orogastric infection study with a Y. enterocolitica O:8 strain, deletion of yopM resulted in complete loss of dissemination to the spleen and the liver by 5 days postinfection (45). However, in an intravenous challenge, the yopM mutant showed only a small but significant reduction in spleen and liver colonization, suggesting that YopM may be especially important for promoting bacterial dissemination. A recent publication has suggested that the YopM protein of Y. pestis CO92 is critical for virulence via the subcutaneous route of infection but is dispensable in a pneumonic model of plague (49).
The Y. enterocolitica YopM protein has been shown to interact with two eukaryotic protein kinases, RSK1 and PRK2 (29). This interaction increases RSK1 kinase activity, which in turn activates the phosphorylation-dependent kinase activity of PRK2. The net effect of this interaction is increased kinase activity of these proteins toward a heterologous protein substrate. In addition, the Y. enterocolitica protein was shown to modify the phosphorylation pattern of RSK1 in Yersinia-infected J774A.1 cells. This has resulted in a model of YopM function in which these kinases would increase the phosphorylation of substrates of RSK1 and/or PRK2, thereby altering signaling via these proteins (29). The targets of this regulation have not, to date, been identified.
Although there has been a great deal of research examining the immune response to Yersinia, until recently there were few clues about what the role of YopM in this response may be. Kerschen et al. demonstrated that loss of yopM from Y. pestis results in increased recruitment of NK1.1+ cells and CD8+ T cells to the spleens of infected mice (25). However, a recent publication has suggested that this YopM-mediated NK1.1+ cell depletion is dispensable for virulence, as antibody-mediated ablation of NK1.1+ cells did not rescue the growth limitation of the ΔyopM strain in the spleens or livers of intravenously infected mice (49). Ye et al. presented evidence that neutrophils may be a target of YopM action, since depletion of neutrophils from mice increased the virulence of a Y. pestis yopM mutant in systemic plague (49).
In this work, we demonstrate that the LRR6 to LRR15 region (amino acid residues 176 to 379) of YopM is required for PRK2 binding. We also demonstrate that a C-terminal domain of the YopM protein (amino acid residues 299 to 409) is required for binding to RSK1. Deletion of either of these domains from YopM abrogates the virulence of Y. pseudotuberculosis via the orogastric route of infection. In addition, we obtained evidence that YopM's virulence function is associated with decreased production of gamma interferon (IFN-γ) and increased levels of interleukin-18 (IL-18) and IL-10 in the serum of Y. pseudotuberculosis-infected mice.
MATERIALS AND METHODS
Bacterial strains, plasmids, and primers.
Table 1 shows the strains and plasmids used in this study, while Table 2 shows the names and sequences of the primers used in this study. The Yersinia strains used in this study are derived from Y. pseudotuberculosis 32777, a serogroup O1 strain, and Y. pestis KIM5. KIM5 lacks the pgm locus and is exempt from select-agent guidelines. For construction of the Y. pseudotuberculosis 32777ΔyopM mutant, we used a variation of the FRT recombination-based system (15) We carried out a three-part PCR to produce a ΔyopM::npt fragment in which the neomycin phosphotransferase cassette is flanked by FRT sites and this cassette replaces codons 85 to 446 of the yopM gene. This construct was then cloned into the BamHI site of the suicide vector pSB890 and mobilized into Y. pseudotuberculosis by biparental mating with Escherichia coli S17-1λpir. Following integration and counterselection on sucrose, the npt gene was excised by conjugation of the pFLP2 plasmid into the 32777ΔyopM::npt strain, leaving behind a scar sequence. For the Y. pestis KIM5ΔyopM strain, the ΔyopM allele was constructed by single-overlap extension PCR producing a fragment containing ∼500 bp upstream of the +1 codon and ∼500 bp downstream of the yopM stop codon. This ΔyopM allele was then cloned into the BamHI site of the pSB890 cassette and mobilized into Y. pestis KIM5 by biparental mating with E. coli S17-1λpir, followed by positive selection for tetracycline resistance and negative selection for sucrose resistance. The Y. pestis KIM5ΔyopM strain was validated by PCR.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Reference(s) |
|---|---|---|
| Strains | ||
| Y. pseudotuberculosis 32777 | Serogroup O1 wild-type strain | 38 |
| Y. pseudotuberculosis 32777ΔyopM | ΔyopM; unmarked | This study |
| Y. pseudotuberculosis 32777ΔyopM + pyopMWT | Expresses full-length YopMWT from native promoter | This study |
| Y. pseudotuberculosis 32777ΔyopM + pyopMΔ6-15 | Expresses YopMΔ6-15 from native promoter | This study |
| Y. pseudotuberculosis 32777ΔyopM + pyopMΔ12-C | Expresses YopMΔ12-C from native promoter | This study |
| Y. pseudotuberculosis 32777ΔyopM + pyopMYPTB | Expresses YopMYPTB from native promoter | This study |
| Y. pestis KIM5 | Δpgm; Ampr | 47 |
| Y. pestis KIM5ΔyopM | ΔyopM; unmarked | This study |
| E. coli S17-1λpir | Tpr SmrhsdR pro recA RP4-2-Tc::Mu-Km::Tn7; lysogenized with λpir | 13, 50 |
| E. coli MACH1 | ΔrecA1398 endA1 tonA P80ΔlacM15ΔlacX74 hsdR | Invitrogen |
| E. coli BL21-AI | F−ompT hsdSBgal dcm araB:T7RNAP-tetA | Invitrogen |
| Plasmids | ||
| pSB890 | Suicide vector; Tetr | 34 |
| pFLP2 | Encodes Flp recombinase; sacB; Tetr | 23 |
| pKD4 | Contains npt cassette flanked by FRT sites; Kanr | 12 |
| pSB890-ΔyopMYP | Upstream and downstream regions of yopM in pSB890; Tetr | This study |
| pSB890-yopMYPTB:npt | yopM containing npt cassette flanked by FRT sites; Kanr; Tetr | This study |
| pENTR221-yopM | Full-length Y. pestis yopM open reading frame; Kanr | JCVIa |
| pDEST15 | Creates N-terminal GST fusions; Ampr | Invitrogen |
| pMMB67EH | Broad-host-range expression vector; low copy; Ampr | 18 |
| pDEST-yopMWT | N-terminal GST fusion to full-length YopM; Ampr | This study |
| pDEST-yopMΔ6-9 | Encodes N-terminal GST fusion to YopM lacking LRR6 to LRR9; Ampr | This study |
| pDEST-yopMΔ8-9 | Encodes N-terminal GST fusion to YopM lacking LRR8 and LRR9; Ampr | This study |
| pDEST-yopMΔ10-11 | Encodes N-terminal GST fusion to YopM lacking LRR10 and LRR11; Ampr | This study |
| pDEST-yopMΔ12-13 | Encodes N-terminal GST fusion to YopM lacking LRR12 and LRR13; Ampr | This study |
| pDEST-yopMΔ14-15 | Encodes N-terminal GST fusion to YopM lacking LRR14 and LRR15; Ampr | This study |
| pDEST-yopMΔ6-15 | Encodes N-terminal GST fusion to YopM lacking LRR6 to LRR15; Ampr | This study |
| pDEST-yopMΔ12-C | Encodes N-terminal GST fusion to YopM truncated after LRR11; Ampr | This study |
| pyopMYP | Full-length Y. pestis yopM gene and promoter in pMMB67EH | This study |
| pyopMΔ6-9 | yopMΔ6-9 gene and promoter in pMMB67EH | This study |
| pyopMΔ8-9 | yopMΔ8-9 gene and promoter in pMMB67EH | This study |
| pyopMΔ10-11 | yopMΔ10-11 gene and promoter in pMMB67EH | This study |
| pyopMΔ12-13 | yopMΔ12-13 gene and promoter in pMMB67EH | This study |
| pyopMΔ14-15 | yopMΔ14-15 gene and promoter in pMMB67EH | This study |
| pyopMΔ6-15 | yopMΔ6-15 gene and promoter in pMMB67EH | This study |
| pyopMΔ12-C | yopMΔ12-C gene and promoter in pMMB67EH | This study |
| pyopMYPTB | Full-length Y. pseudotuberculosis yopM gene and promoter in pMMB67EH | This study |
JCVI, J. Craig Venter Institute (Y. pestis gateway clone library, Pathogen Functional Genomics Resource Center, 2009).
TABLE 2.
Primers used in this study
| Primer name | Sequence (5′-3′) |
|---|---|
| yopMupsF | AAGAATTCAATAAAGCAGACTAACATTG |
| yopMupsR | TTCTGCCTCAACCGGCATCT |
| yopMyptbupsF | TTGAATTCGACTCACATTGAAGGATGCT |
| yopMF | ATGTTCATAAAATAAATCCAAGAAAT |
| yopMR | TTGGATCCCTACTCAAATACATCATCTTC |
| yopMYPTBR | CTACTCAAAAACATCATCTTC |
| yopMΔ1 | TTGGATCCAATAAAGCAGACTAACATT |
| yopMΔ2 | GAAGCAGCTCCAGCCTACACATTGAATGCCTTTCTGAAAAT |
| yopMΔ3 | ACTAAGGAGGATATTCATATGCTTCATTTAATCATCTTGC |
| yopMΔ4 | TTGGATCCGGGCCAACAATATCCAGT |
| yopMΔ5 | GTGTAGGCTGGAGCTGCTTC |
| yopMΔ6 | CATATGAATATCCTCCTTA |
| yopMΔLRR6-7F | AACTCGAGTCTATTGTTGCTGGTAATAAT |
| yopMΔLRR6-7R | AACTCGAGTGAAGGAGGTAAATCAGGTAG |
| yopMΔLRR8-9F | AACTCGAGGCACTTAATGTCAGATATAAT |
| yopMΔLRR8-9R | AACTCGAGTGAAAGAGGTAAATCAGGTAG |
| yopMΔLRR10-11F | AACTCGAGTATCTCAATGCATCCAGCAAT |
| yopMΔLRR10-11R | AACTCGAGGGAAGGGGGTAAATCGGGTAA |
| yopMΔLRR12-13F | AACTCGAGCGTTTAATCGCTTCATTTAAT |
| yopMΔLRR12-13R | AACTCGAGGTTTGGTGGCAATTCCGATAA |
| yopMΔLRR14-15F | AACTCGAGGATCTTCGGATGAACTCTGAA |
| yopMΔLRR14-15R | AACTCGAGGCGTGGAGGTAACGCTGGCAG |
In order to produce glutathione S-transferase (GST) fusion constructs containing internal-deletion alleles of yopM, we carried out inverse PCR using primer pair yopMΔLRR 6-7F and yopMΔLRR6-7R, yopMΔLRR8-9F and yopMΔLRR8-9R, yopMΔLRR10-11F and yopMΔLRR10-11R, yopMΔLRR12-13F and yopMΔLRR12-13R, yopMΔLRR14-15F and yopMΔLRR14-15R, or yopMΔLRR6-10R and yopMΔLRR11-15F on pDEST-yopMYP. The PCR product was then gel purified, SalI digested, and religated. It was then transformed into chemically competent DH5α cells (Invitrogen) and verified by sequencing.
To put the ΔLRR alleles under the control of the native Y. pestis yopM promoter, the promoter was amplified using primers yopMupF and yopMupR. The promoter fragment (5 ng) was then mixed with the ΔLRR fragment (amplified by PCR using yopMF and yopMR) (5 ng) and then used as the template for single-overlap extension PCR with primers yopMupsF and yopMR. The product of this reaction was then gel cleaned, EcoRI-BamHI digested, and ligated into pMMB67EH. The C-terminal deletion allele of yopM was created by PCR amplification from Y. pestis virulence plasmid pCD1 using primers yopMupF and yopMΔ12-CR, gel purification, EcoRI-BamHI digestion, and ligation to pMMB67EH. The yopMYPTB coding region and promoter were PCR amplified by using primers yopMyptbupsF and yopMyptbR on the pYV virulence plasmid of Y. pseudotuberculosis 32777. The product obtained was then gel cleaned, EcoRI-BamHI digested, and ligated into pMMB67EH. All plasmid constructs were verified by DNA sequencing.
Analysis of YopM secretion by Yersinia.
Y. pseudotuberculosis strains 32777 and 32777ΔyopM and the 32777ΔyopM strain containing pyopMYP, pyopMYPTB, pyopMΔ6-15, or pyopMΔ12-C were grown overnight at 26°C. They were then diluted 1:40 in Luria-Bertani (LB) broth containing 20 mM sodium oxalate and 20 mM MgCl2 and grown for 1 h at 26°C before being temperature shifted to 37°C. The cultures were grown for 2 to 3 h, and the bacteria were removed by centrifugation. Trichloroacetic acid was added to 1 ml of culture supernatant to 10% (wt/vol), and the mixture was then incubated on ice for 1 h. The samples were centrifuged at 20,000 × g for 20 min, and the supernatant was removed. The protein pellets were washed once in acetone and then dried. The proteins were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and stained with GelCode Blue (Pierce, Rockford, IL). In parallel, a separate gel was blotted onto nitrocellulose before processing by immunoblotting with murine anti-YopM serum.
GST-YopM purification.
The full-length Y. pestis yopM gene from pENTR221-yopM (Y. pestis gateway clone library, Pathogen Functional Genomics Resource Center, J. Craig Venter Institute) was recombined into pDEST15 using Gateway LR Clonase II Enzyme Mix (Invitrogen, Carlsbad, CA). The deletion constructs were produced as described in the previous sections. These constructs were transformed into chemically competent E. coli BL21-AI (Invitrogen, Carlsbad, CA) and induced for 6 to 8 h at 18°C using 0.2% arabinose. This strain contains the T7 RNA polymerase gene under the control of the araBAD promoter. The induced cells were then collected by centrifugation and lysed using Lysonase and Bugbuster reagents according to the manufacturer's instructions (Novagen, Gibbstown, NJ) in the presence of Complete EDTA-free mini protease inhibitor cocktail (Roche, Indianapolis, IN). The supernatants were clarified by centrifugation and applied to 1 ml of GST-Bind Agarose (Novagen, Gibbstown, NJ) and allowed to bind for 1 h at 4°C. The beads were then washed and the protein eluted according to the manufacturer's instructions. The proteins were dialyzed against phosphate-buffered saline (PBS) for 8 h, and aliquots were frozen at −80°C until needed.
GST pull-down assays.
All GST pull-down assays were conducted using lysates of the human monocytic cell line THP-1. These lysates were prepared by resuspending cells at 107/ml in lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol) containing 1× EDTA-free Complete protease inhibitor (Roche) and three rounds of sonication for 10 s each at 4°C at a power setting of 3 using a microtip attachment on a Microson sonicator (Misonix, Farmingdale, NY). The lysates were then clarified by centrifugation at 10,000 × g for 5 min, aliquoted, and frozen at −20°C. The pull-downs were conducted as follows. Briefly, 20 μl of GST-Bind agarose beads (Novagen, Gibbstown, NJ) was incubated in 160 μl of lysate and 40 μl of 5× GST-Bind buffer (Novagen, Gibbstown, NJ) for 16 to 18 h at 4°C. The beads were then washed four times in 1 ml of GST-Bind/Wash buffer, boiled in Laemmli buffer, loaded directly onto an 8% polyacrylamide gel, and separated by SDS-PAGE.
Western blotting of YopM-interacting proteins.
Following separation, the proteins were transferred to nitrocellulose at 30 V for 16 to 18 h. The blots were blocked in 10 mM Tris-HCl (pH 7.4)-150 mM NaCl-0.1% Tween 20 (TBST) plus 5% skim milk powder for 1 h at room temperature. The blots were washed three times in TBST. Primary antibodies were as follows: rabbit polyclonal anti-PRK2 diluted 1:1,000 in TBST plus 5% bovine serum albumin (Abcam, Cambridge, MA) or rabbit polyclonal anti-RSK1 diluted 1:1,000 in TBST plus 5% bovine serum albumin (Abcam, Cambridge, MA). Primary antibody binding was allowed to occur for 16 to 18 h at 4°C. The primary-antibody-probed blots were washed three times in TBST, and then horseradish peroxidase-conjugated goat anti-rabbit IgG diluted 1:10,000 in TBST was allowed to bind for 1 h at 20°C. The blot was washed three times in TBST, and the blots were visualized using Western Lighting Enhanced Chemiluminescence reagent (Perkin-Elmer) and exposure to X-Omat Blue film (Kodak, Rochester, NY).
Mouse infections.
All experiments involving mice were carried out with the approval of the Stony Brook University Institutional Animal Care and Use Committee. For intravenous infections, Y. pestis strains were grown overnight in HI broth at 26°C, while Y. pseudotuberculosis was grown overnight in LB broth at 26°C. The following day, the cultures of Y. pestis were washed twice in PBS before being diluted to 1.4 × 103 to 1.7 × 103 CFU/ml, while Y. pseudotuberculosis cultures were diluted to 1.5 × 104 to 2.5 × 104 CFU/ml. A volume of 100 μl was delivered via tail vein injection into C57BL/6J mice (Jackson Labs, Bar Harbor, ME). Two groups of four mice were used for Y. pestis, while three groups of two to five mice were used for Y. pseudotuberculosis. Time to death was monitored for 14 days, at which point any surviving mice were euthanized. Any mice that were moribund at the time of observation were euthanized and scored as deceased.
Orogastric infections were carried out with Y. pseudotuberculosis strains as follows. C57BL/6J mice (Taconic, Hudson, NY) were fasted for 16 to 18 h with access to water ad libitum. Y. pseudotuberculosis strains were grown overnight in LB broth at 28°C. The cultures were washed twice in Hanks balanced salt solution and then resuspended to 2.5 × 1010 CFU/ml. The mice were infected with 200 μl of this culture (5 × 109 CFU) via a feeding needle attached to a 1-ml syringe. Time to death was monitored for 14 days, at which point any surviving mice were euthanized. Any mice that showed signs of disease (ruffled scruff, lethargy) at the time of observation were euthanized and scored as deceased.
Organ collection and processing for colonization, enzyme-linked immunosorbent assay (ELISA), and flow cytometry.
Infection for organ colonization/cytokine determination was carried out as described above, except that mice were euthanized by CO2 asphyxiation at 4 days postinfection. Blood was harvested by cardiac puncture, and the serum was isolated using Sarstedt S1.1 serum gel tubes (Sarstedt, Nümbrecht, Germany) according to the manufacturer's instructions. Spleens and mesenteric lymph nodes (MLN) were aseptically harvested in 5 ml of ice-cold PBS. The MLN were homogenized by use of a Stomacher lab blender, while the spleens were manually teased apart using two 18.5-gauge needles. The supernatants were serially diluted, and 100-μl aliquots of each were plated onto LB agar plates. Three Peyer's patches were harvested from each orogastrically infected mouse and homogenized by drawing through an 18.5-gauge needle 8 to 10 times. Serial dilutions of this homogenate were prepared, and 100-μl aliquots were plated on Yersinia selective medium agar plates.
Flow cytometry.
Erythrocytes were lysed by resuspending the washed splenocytes in 8 ml of lysis buffer (17 mM Tris HCl [pH 7.4], 144 mM NH4Cl) for 4 min. Lysis was stopped by adding 20 ml RPMI medium and removing the cells by centrifugation. The cells were filtered through a 70-μm nylon filter and then resuspended to 2 × 107/ml. Fifty microliters (1 × 106 cells) was blocked with FcBlock (αCD16/CD32) for 5 min at room temperature before staining with the following antibodies: Alexa Fluor 488-conjugated αLy6C (ABD Serotec), phycoerythrin-conjugated Ly6G (BD Pharmingen), and PerCP-Cy5.5-conjugated αCD11b (BD Pharmingen). Appropriate isotype controls were used to set gates, and the cells were sorted on a BD FACScalibur cell-sorting instrument. Analysis was done using WinList 5.0 software (Verity Software House, Topshem, ME).
ELISA analysis of mouse serum.
IL-10 ELISA kits were purchased from R&D Biosystems, Minneapolis, MN. IL-18 ELISA kits were purchased from MBL, Nagoya, Japan. Murine IFN-γ ELISA kits were purchased from Biolegend. Serum samples were diluted 1:10 and processed according to the manufacturer's instructions.
Statistical analysis.
Results of survival curve experiments were analyzed using log-rank testing and the GraphPad Prism software suite (GraphPad, La Jolla, CA). Changes in cytokine levels, CFU, or splenocyte recruitment were determined by using a Mann-Whitney test of significance.
RESULTS
YopM of Y. pestis interacts with RSK1 and PRK2.
In order to validate the previously described interactions of YopM with RSK1 and PRK2, we purified full-length YopM from Y. pestis as an N-terminal GST fusion protein. Equimolar amounts of both GST and GST-YopM were bound to glutathione Sepharose and washed before addition of the protein-coated beads to extracts of the human monocytic cell line THP-1. The beads were extensively washed before boiling in Laemmli buffer, separation on an 8% polyacrylamide gel, and blotting onto nitrocellulose. The blot was probed with monoclonal antibodies specific for RSK1 and PRK2. As shown in Fig. 1B and C, the GST-YopM protein exhibited interaction with RSK1 and PRK2 (lanes 5), while GST alone showed no interaction (lanes 3).
FIG. 1.

Interaction of YopM alleles with RSK1 and PRK2. (A) Representation of the YopM constructs used in this study. The black numbered boxes represent the LRRs, while the N-terminal dark gray boxes are the α-helical domains defined in reference 17. The C-terminal disordered region is represented as a pale gray box. (B and C) GST-YopM pulldown assays. GST alone or the GST-YopM constructs represented in panel A were purified and added in equimolar amounts to GST-Bind Sepharose beads. The protein-bound beads were then added to lysates of THP-1 monocytic cells, and the proteins were allowed to bind overnight before being removed and washed. Samples were processed for SDS-PAGE and Western blotting using antibodies against PRK2 or RSK1. Lane 1 shows a sample of the starting lysate. S, 10%, by volume, of the postbinding supernatant; B, GST-Bind bead-bound proteins; WT, wild type. Positions of molecular mass markers are shown on the left.
A C-terminal domain of YopM is required for interaction with RSK1, while LRR6 to LRR15 are required for interaction with PRK2.
In order to determine the domains of YopM required for interaction with each of these partner proteins, we constructed a series of deletion mutant proteins. The characteristics of the mutant YopM proteins tested are shown in Fig. 1A. As shown in Fig. 1C, the deletion of the C terminus through LRR12 resulted in complete loss of interaction with RSK1, but this protein still exhibited the ability to interact with PRK2 (lane 17). Deletion of LRR10 and LRR11 or LRR14 and LRR15 resulted in a slightly reduced interaction with PRK2 compared to that of full-length YopM, but the interaction of these mutant proteins with RSK1 was unaffected (Fig. 1B, lanes 11 and 15, respectively, and C, lanes 7 and 11, respectively). Deletion of either LRR6 and LRR7 or LRR6 to LRR9 did not affect the binding of YopM to either RSK1 or PRK2 (Fig. 1B). Larger deletions encompassing the entire LRR domain or LRR6 to LRR15 showed a profoundly reduced interaction with PRK2, but the interaction with RSK1 was still maintained (Fig. 1C, lanes 13 and 15, respectively). This demonstrated that the interaction of YopM with RSK1 and with PRK2 requires different regions of the YopM protein.
Phenotypic characterization of Y. pestis and Y. pseudotuberculosis yopM mutants in mouse infection models.
To investigate the role of YopM in Yersinia pathogenesis, we constructed yopM deletion mutants of Y. pestis KIM5 and Y. pseudotuberculosis 32777. The mutants were confirmed to lack yopM sequences by PCR, and to be defective for YopM production and secretion by SDS-PAGE and GelCode Blue staining or Western blotting (see Fig. 6A; data not shown). C57BL/6J mice were infected with the resulting strains or the wild-type controls, followed by analysis of time to death and bacterial colonization of lymphoid organs. In the intravenous model of infection, we did not observe any difference in the time to death between Y. pestis KIM5 and the isogenic KIM5ΔyopM mutant (Fig. 2A). In contrast, the Y. pseudotuberculosis ΔyopM mutant exhibited a clear defect in the ability to cause lethal disease via this model (P = 0.024 by log-rank test) (Fig. 2B). In addition to the difference in kinetics of killing, the splenic bacterial colonization levels in the mice infected via the intravenous route with wild-type and ΔyopM mutant Y. pseudotuberculosis were significantly different (P = 0.050 by Mann-Whitney test) at day 4 postinfection (Fig. 3A). Infection with the 32777ΔyopM mutant via the orogastric route resulted in an even more profound loss of virulence, with 12/12 mice surviving to 14 days postinfection (see Fig. 6B). In contrast, only 1/13 mice survived oral infection with 32777 to day 14 with a mean time to death of 7 days (see Fig. 6B). Following orogastric infection with the ΔyopM mutant, there was no statistically significant difference in the colonization of the Peyer's patches (Fig. 3B) but dissemination to the MLN and spleen was compromised in the 32777ΔyopM-infected mice (P = 0.050 and P < 0.0001 by Mann-Whitney test, respectively; Fig. 3C and D).
FIG. 6.

Phenotypic characterization of Y. pseudotuberculosis yopM mutants by secretion assay and mouse virulence assay. (A) The following strains were grown under T3SS-inducing conditions as described in Materials and Methods: 32777 (lanes 1 and 7), 32777ΔyopM (lanes 2 and 8), 32777ΔyopM + pyopMYP (lanes 3 and 9), 32777ΔyopM + pyopMΔ6-15 (lanes 4 and 10), 32777ΔyopM + pyopMΔ12-C (lanes 5 and 11), and 32777ΔyopM + pyopMYPTB (lanes 6 and 12). Secreted proteins were analyzed by SDS-PAGE, followed by GelCode Blue staining (left), or a duplicate gel was blotted onto nitrocellulose and probed with mouse antiserum raised against GST-YopM (right). The values to the left are molecular sizes in kilodaltons. (B) Time-to-death analysis of orogastric Y. pseudotuberculosis infections. Groups of three to five mice were infected by oral gavage with 5 × 109 CFU of Y. pseudotuberculosis, and time to death was monitored for 14 days. Results are pooled from two to four independent experiments (32777, n = 13; 32777ΔyopM, n = 12; 32777ΔyopM + pyopMWT, n = 13; 32777ΔyopM + pyopMΔ6-15, n = 11; 32777ΔyopM + pyopMΔ12-C, n = 12; 32777ΔyopM + pyopMYPTB, n = 11).
FIG. 2.

Time-to-death analysis of intravenous Yersinia infections. (A) About 150 CFU of Y. pestis KIM5 or Y. pestis KIM5ΔyopM were delivered intravenously via tail vein injection to groups of four C57BL/6J mice each. Time to death was monitored for 14 days. Results are pooled from two independent experiments (n = 8). (B) Time-to-death analysis of intravenous Y. pseudotuberculosis infection. About 1,500 to 2,500 CFU of Y. pseudotuberculosis 32777 or Y. pseudotuberculosis 32777ΔyopM were delivered to groups of C57BL/6J mice, and survival was monitored for 21 days. Results are pooled from two or three independent experiments with 2 to 4 mice per group (n = 10 for 32777; n = 9 for 32777ΔyopM).
FIG. 3.

Organ colonization assays of C57BL/6J mice at 4 days postinfection. (A) Splenic colonization following intravenous Y. pseudotuberculosis 32777 or Y. pseudotuberculosis 32777ΔyopM infection. (B) Peyer's patch colonization following orogastric Y. pseudotuberculosis infection. (C) MLN colonization following orogastric Y. pseudotuberculosis infection. (D) Splenic colonization following orogastric Y. pseudotuberculosis infection. Each filled circle represents the log10 CFU per organ recovered from a single mouse. The horizontal line represents the mean colonization level, while the dashed line is the detection limit for each organ. Results are pooled from two to four independent experiments.
Comparison of murine immune responses to wild-type and ΔyopM mutant Y. pseudotuberculosis.
The yopM mutant of Y. pseudotuberculosis showed a slight defect in splenic colonization at day 4 following intravenous infection (average of 6.6 × 106 CFU/spleen for Y. pseudotuberculosis 32777 versus 1.2 × 106 CFU/spleen for the ΔyopM mutant strain) (Fig. 3A). However, the ΔyopM mutant was significantly attenuated in this model when the mean time to death was measured (Fig. 2B). To examine early immune responses that might be altered by YopM function, resulting in increased virulence of the wild-type strain over time, mice were infected by the intravenous route. At day 4 postinfection, spleens and blood samples were collected from euthanized mice. Spleen cells were analyzed by flow cytometry to characterize the populations of neutrophils (CD11b+ Ly6G+; Fig. 4 B), inflammatory monocytes (CD11b+ Ly6C+; Fig. 4C), or natural killer (NK) cells (NK1.1+; Fig. 4A). Although we did not observe any significant differences in the recruitment of neutrophils or of NK1.1+ cells in the 32777-infected mice versus the 32777ΔyopM-infected mice, there were significant changes in the recruitment of inflammatory monocytes. In the 32777-infected mice, there was a significantly increased percentage of CD11b+ Ly6C+ inflammatory monocytes (10.1% ± 0.6% in 32777-infected mice versus 7.9% ± 0.6% in 32777ΔyopM-infected mice). We reasoned that this differential monocyte recruitment may reflect alterations in cytokine production due to the yopM status of the infecting strain.
FIG. 4.
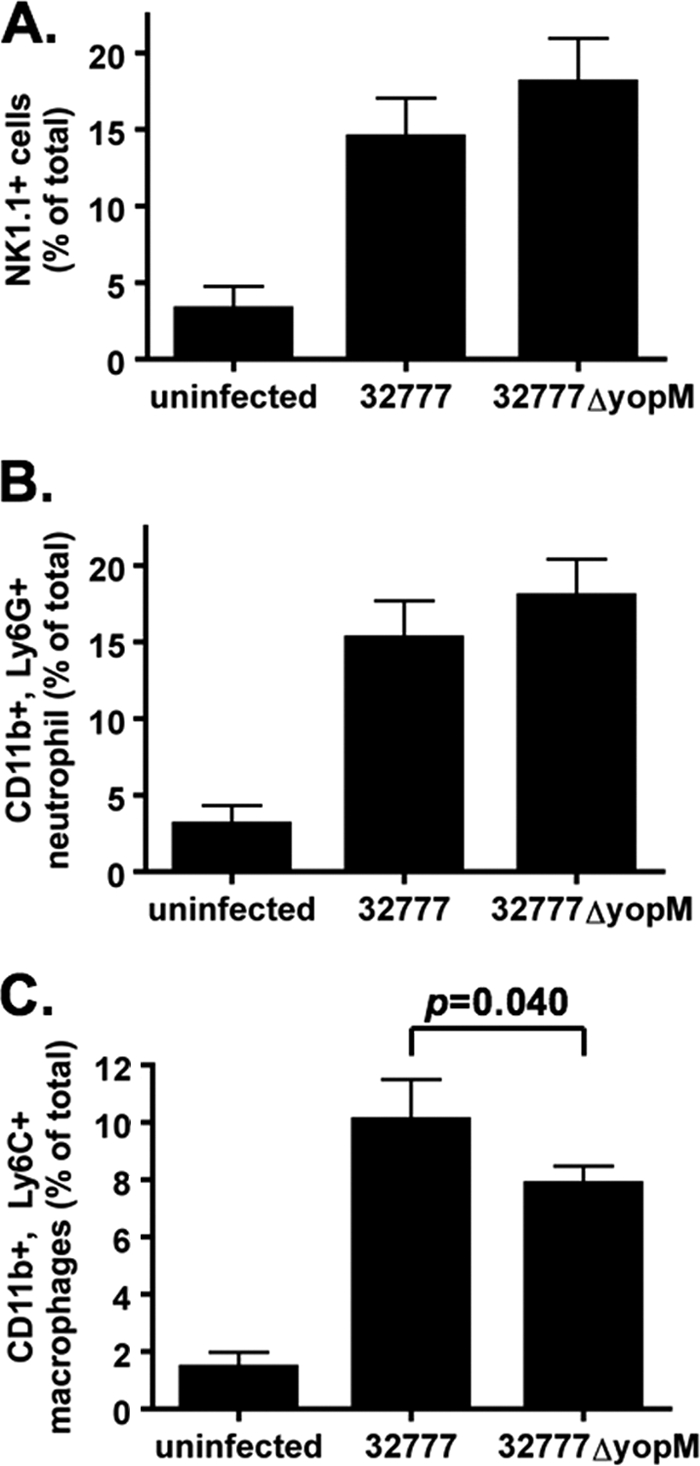
Fluorescence-activated cell sorter analysis of immune cells in the spleens of infected mice at 4 days postinfection. (A) NK1.1+ cells. (B) CD11b+ Ly6G+ neutrophils. (C) CD11b+ Ly6C+ inflammatory monocytes (macrophages). Results show percentages of total spleen cells averaged from 3 independent experiments with 3 or 4 mice in each experiment, except for “uninfected,” which represents 3 mice from 1 experiment.
To measure levels of cytokines at day 4 postinfection, serum was obtained from the infected mice and cytokine concentrations were determined using ELISA kits for mouse IFN-γ (Fig. 5A), IL-18 (Fig. 5B), and IL-10 (Fig. 5C). In these infections, the Y. pseudotuberculosis 32777ΔyopM-infected mice produced significantly higher (P = 0.028 by Mann-Whitney test) levels of IFN-γ (5.74 ± 0.77 ng/ml) than the 32777-infected mice (3.02 ± 0.55 ng/ml). In contrast, the IL-18 results show that there were significantly higher (P < 0.004 by Mann-Whitney test) concentrations of this cytokine in the 32777-infected mice (4.89 ± 0.87 ng/ml) than in the 32777ΔyopM-infected mice (1.20 ± 0.22 ng/ml). Similarly, the IL-10 ELISA demonstrated that the 32777-infected mice produced significantly higher levels of IL-10 (381 ± 84 pg/ml) than the 32777ΔyopM-infected mice (65 ± 15 pg/ml; P < 0.001). It is worth noting that the concentrations of IL-10 detected in the 32777ΔyopM-infected mice are below the lowest dilution standard provided with the ELISA kit and are therefore all extrapolated values.
FIG. 5.
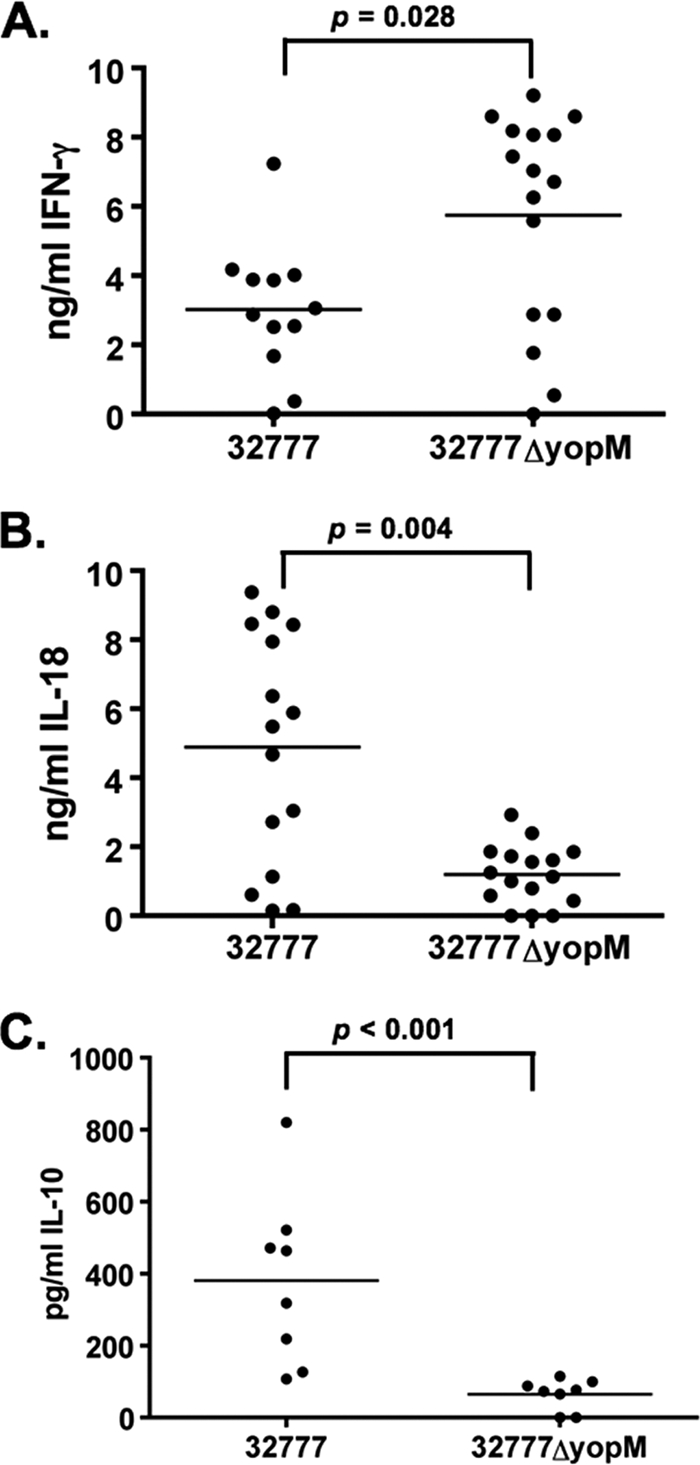
Cytokine levels in the serum of C57BL/6J mice at 4 days postinfection. (A) IFN-γ, (B) IL-18, and (C) IL-10 concentrations were determined by ELISA. Each filled circle represents the concentration of cytokine present in the serum from a single mouse. The horizontal line is the mean concentration from all mice. Results shown are the summary of 4 experiments with 2 to 4 mice per group (A and B) or 2 experiments with 4 mice per group (C).
Both the RSK1 and PRK2 interaction domains of YopM are required for virulence.
In order to determine the contribution of the RSK1 and PRK2 interaction domains of YopM to virulence, complementation analysis of the Y. pseudotuberculosis 32777ΔyopM mutant was performed. The mutant alleles yopMΔ6-15 and yopMΔ12-C were placed under the control of the native yopM promoter and inserted into the low-copy-number pMMB67EH vector. Full-length yopM sequences from Y. pestis (yopMYP) and Y. pseudotuberculosis (yopMYPTB) were similarly placed into the same plasmid as positive controls. These vectors were introduced into Y. pseudotuberculosis ΔyopM, and the resulting strains were tested for secretion of YopM by SDS-PAGE, followed by GelCode Blue staining and Western blotting. As shown in Fig. 6A, the YopM protein of 32777 at ∼60 kDa (lanes 1 and 7) is not found in the 32777ΔyopM mutant (lanes 2 and 8). Secretion of YopM proteins encoded by pyopMYP (lanes 3 and 9), pyopMΔ6-15 (lanes 4 and 10), pyopMΔ12-C (lane 5 and 11), and pyopMYPTB (lanes 6 and 12) occurred as expected.
The complemented yopM mutants, as well as parental control Y. pseudotuberculosis strains, were then used to infect C57BL/6J mice by the orogastric route. As shown in Fig. 6B, expression of either the Y. pestis or the Y. pseudotuberculosis full-length YopM protein restored virulence to wild-type levels, with 9/13 mice infected with 32777ΔyopM + pyopMYP and 7/9 mice infected with 32777ΔyopM + pyopMYPTB succumbing to infection with a median survival time of 8 days, compared to 11/12 of 32777-infected mice with a median survival time of 7 days. In contrast, none of the mice infected with 32777ΔyopM were killed by the infection, 1/10 mice infected with 32777ΔyopM + pyopMΔ6-15 died, and 2/10 mice infected with 32777ΔyopM + pyopMΔ12-C died by the end of the monitoring period. There was no statistically significant difference among the survival curves of mice infected with the 32777ΔyopM or 32777ΔyopM strain complemented with pyopMΔ6-15 or pyopMΔ12-C, as determined by the log-rank test. Similarly, there was no difference among the survival curves of mice infected with 32777 or 32777ΔyopM complemented with pyopMYP or pyopMYPTB. These results suggested that the ability of the YopM protein to interact with both PRK2 and RSK1 is critical for the ability of the bacteria to cause virulence in the murine orogastric model of infection. In addition, a full-length allele of Y. pestis YopM in which the C terminus was modified by the presence of an eight-amino-acid FLAG epitope was also unable to complement the 32777ΔyopM mutant, despite being secreted normally (data not shown), supporting the hypothesis that the C terminus of the YopM protein is critical for virulence.
Both the RSK1 and PRK2 interaction domains of YopM are required for production of IL-10.
Due to the slight but significant defect in the ability of the 32777ΔyopM strain to colonize the spleen at 4 days postinfection (Fig. 3A), we wanted to determine whether or not the loss of IL-10 production in the 32777ΔyopM-infected mice was due to the lower organ burden at this time point or to some signaling defect due to the loss of yopM. We reasoned that while the strains containing the yopMΔ6-15 and yopMΔ12-C alleles are clearly defective for virulence, these alleles may still complement this early colonization defect. We infected mice via the intravenous route (1,000 CFU) with the strains shown in Fig. 7 and then looked at splenic colonization and serum IL-10 production in these animals at day 4 postchallenge.
FIG. 7.

IL-10 production is dependent on the RSK1 and PRK2 interaction domains of YopM. One thousand CFU of Y. pseudotuberculosis 32777, 32777ΔyopM, 32777ΔyopM + pyopMYP, pyopMΔ6-15, pyopMΔ12-C, or pyopMYPTB were delivered intravenously to C57BL/6J mice. (A) After 4 days, the mice were sacrificed and the level of splenic colonization was assessed. (B) Serum from the infected mice was assessed for IL-10 production by ELISA. Each filled circle represents the CFU in spleen (A) or the concentration of cytokine present in the serum (B) from a single mouse. Data shown are from 2 independent experiments with 2 to 5 mice per group. The horizontal line is the mean value from all mice.
As shown in Fig. 7A, the mice infected with 32777ΔyopM + pyopMΔ6-15 or 32777ΔyopM + pyopMΔ12-C had levels of splenic colonization similar to those of the 32777-infected mice, a level that is statistically significantly higher than that of 32777ΔyopM-infected mice. This observation suggests that the loss of the RSK1 and/or PRK2 interaction domains does not completely abrogate all YopM-dependent functions. In spite of this clear ability to restore wild-type levels of colonization, the mice infected with either 32777ΔyopM + pyopMΔ6-15 or 32777ΔyopM + pyopMΔ12-C had no induction of IL-10 in their serum (Fig. 7B), strongly suggesting that this altered cytokine level within the host was due to the loss of some YopM function and not to the lowered antigen load seen in the 32777ΔyopM-infected mice.
DISCUSSION
The function of the YopM protein has remained elusive. Depending upon the strain examined, the protein contains between 13 and 21 LRRs. The crystal structure of the Y. pestis YopM protein was determined using X-ray crystallography, which showed that the protein has two N-terminal α helices that serve as a folding scaffold for the LRRs, which themselves take on a horseshoe-shaped structure. The C-terminal tail of the protein was unresolved in this structure, and it is therefore assumed to be disordered (17).
The YopM protein forms a complex with two host kinases, PRK2 and RSK1 (29). In vitro kinase reactions demonstrate that the formation of the YopM-PRK2-RSK1 complex results in increased phosphorylation of both PRK2 and RSK1 and therefore activates their kinase activity. This increased activation results in increased phosphorylation of a heterologous kinase substrate, myelin basic protein (MBP). Expression of a kinase-dead version of either PRK2 or RSK1 resulted in dramatically reduced phosphorylation of MBP. This produced a model whereby the YopM protein recruits PRK2 and RSK1 to a novel signaling complex that results in altered signaling that serves to promote bacterial virulence.
In order to determine the contribution of each of these protein interactions to virulence, we constructed a series of mutant YopM proteins that lack defined LRR regions and looked at whether or not these proteins were able to retain the ability to interact with PRK2 and/or RSK1. As shown in Fig. 1C, deletion of the entire structure from LRR1 to LRR15 (residues 74 to 379 of Y. pestis YopM) resulted in complete loss of GST-YopM binding to PRK2. A smaller deletion, removing the 10 LRRs from LRR6 to LRR15 also abrogated the binding of this protein. In spite of the loss of PRK2 interaction, these versions of YopM still exhibited robust binding to RSK1. In contrast, a truncation of the C terminus of the YopM protein through LRR12 eliminated the binding of the RSK1 protein to YopM while still maintaining near-normal levels of PRK2 binding (Fig. 1C). These results illustrate that different regions of the YopM protein are involved in the interaction with each of these signaling proteins.
The contribution of the YopM protein to virulence is fairly well established, although there are still many open questions as to its function. Loss of the yopM gene from Y. pestis has been reported to result in a 4-log-order increase in the LD50 for BALB/c and Swiss Webster mice (22, 27). Recently, Kerschen et al. also demonstrated a loss of virulence associated with the deletion of yopM in C57BL/6J mice as well (25). There does, however, appear to be some variation in the degree of attenuation of virulence in the Y. pestis ΔyopM mutant strains, as another publication reported seeing no attenuation of virulence in this strain (33). In accord with the latter result, we did not observe a defect in the virulence of a Y. pestis KIM5ΔyopM mutant following intravenous infection (Fig. 2A). However, significant attenuation of virulence was observed when Y. pseudotuberculosis 32777ΔyopM mutants were used to infect C57BL/6J mice via an intravenous model of infection (Fig. 2B) or via an orogastric model of infection (Fig. 6B). We then used the orogastric model to determine whether or not RSK1 and PRK2 binding is required for virulence.
The YopM protein exhibits a significant amount of sequence heterogeneity (8). Sequence analysis has demonstrated that, depending on the strain examined, Y. pestis expresses YopM proteins with either 13 or 15 LRRs, whereas the enteropathogens Y. enterocolitica and Y. pseudotuberculosis exhibit even more diversity, expressing versions with up to 21 LRRs. The consequence of this diversity is unknown; however, we demonstrate here that the Y. pestis 15-LRR YopM protein is able to restore wild-type levels of virulence to a Y. pseudotuberculosis ΔyopM mutant strain (Fig. 6B), suggesting that, at least under the conditions examined here, there is no difference between the abilities of these alleles to contribute to disease progression. A previous publication suggested that at least some of the internal LRRs are necessary for YopM to promote Y. pestis virulence. In a study of YopM proteins lacking LRR4 to LRR7 or LRR7 to LRR10, Hines et al. demonstrated that these mutant versions are incapable of complementing the virulence defect of Y. pestis ΔyopM; however, the reason for the virulence defect associated with these yopM alleles is unclear (22). Consistent with this result, we demonstrate that the loss of the PRK2 interaction domain in LRR6 to LRR15 also results in complete attenuation of virulence. In addition to this, the deletion of the RSK1 interaction domain in the C terminus of the Y. pestis YopM protein results in a complete attenuation of virulence when that allele is introduced into Y. pseudotuberculosis.
A number of publications have noted that the YopM protein traffics to the nuclei of both yeast and HeLa cells (4, 40, 41). This trafficking depends on the microtubule system of the cell, as the inhibitors colchicine and nocodazole both prevent YopM from accumulating in the nucleus, leading to a model whereby YopM either directly or indirectly interacts with a component of the endosomal pathway before entering the nucleus (40). This interaction appears to utilize multiple regions of the YopM protein, as either the N-terminal or the C-terminal half of the protein is capable of nuclear localization (41). Interestingly, although YopM is clearly found in the nucleus, there is a large proportion that is also located in the cytoplasmic fraction, a distribution pattern that also correlates with the activation state of RSK1 (11, 12).
Although virulence was clearly affected by the absence of the YopM protein in Y. pseudotuberculosis via either the orogastric or the intravenous route, there were small differences in the organ burdens at 4 days following intravenous infection with the 32777 or 32777ΔyopM strain. There were also significant differences in the recruitment of inflammatory monocytes to the spleens of 32777-infected mice compared to those of ΔyopM-infected mice (Fig. 4C). This led us to examine the cytokine levels in the serum of the infected mice, reasoning that any differences in cytokine levels would reflect alterations due to YopM activity rather than to altered colonization levels.
The IL-18 protein is involved in the upregulation of IFN-γ during bacterial infection, leading to the activation of macrophages and neutrophils and increased phagocytosis and antigen-presenting activity of these cell types. It has also been reported to increase NK cell activity (1). Yersinia-resistant strains of mice (C57BL/6J) express ∼4-fold higher levels of IL-18 mRNA than Yersinia-sensitive mice (BALB/c) (7), although there are clearly other potential factors involved in the differences in susceptibility between these two strains of mouse. Treatment of mice with anti-IL-18 antibodies led to 100- to 1,000-fold increases in splenic colonization following Y. enterocolitica infection of both mouse strains, indicating that IL-18 is an important part of the innate immune response to this pathogen (7). A previous report stated that the transcription of IL-18 and IFN-γ messages is increased in mice during infection with a yopM mutant strain of Y. pestis compared to infection with the wild-type strain (25), although actual serum cytokine levels were not determined in that study. In Y. enterocolitica-infected mice, antibody-mediated ablation of IFN-γ resulted in increased bacterial colonization of the spleen and increased death (3), further demonstrating the importance of this cytokine for host control of Yersinia infection.
Regulation of IL-18 production is subject to control at the transcriptional, translational, and posttranslational levels (42). Generally, during bacterial infection, the levels of IL-18 and IFN-γ form a positive feedback loop in which IL-12 and IL-18 produced by macrophages and dendritic cells are able to induce the production of IFN-γ by CD8+ T cells, NKT cells, and NK cells. In accord with this, we observed higher recruitment of inflammatory monocytes to the spleens of mice infected with the wild-type Y. pseudotuberculosis strain. This is also likely the reason for the elevated levels of IL-18 observed in wild-type-infected mice. Normally, increased IL-18 would lead to increased IFN-γ production, which in turn serves to activate antigen-presenting cells and increases their antibacterial effector function. Our results suggest that the YopM protein somehow disrupts this positive feedback loop by interfering with the ability of the host to increase IFN-γ production. We observed substantially higher levels of IFN-γ in the mice infected with Y. pseudotuberculosis 32777ΔyopM. This higher level of IFN-γ would be expected to lead to better control of the bacterial load and be manifested in better survival following infection with these strains, as was observed by us (Fig. 2B) and has been previously reported for yopM mutants of Y. pestis and Y. enterocolitica (27, 45). Previous work has suggested that neutrophils are critical for the control of Y. pestis KIM5ΔyopM in a model of septicemic plague (49), although we did not observe any difference in neutrophil recruitment to the spleens of the infected mice (Fig. 4B).
IFN-γ is released by a number of cell types, including NK cells, NKT cells, and CD8+ T cells following exposure to IL-12 and IL-18 (10). Previous reports have suggested that both CD8+ T cells and NK1.1+ cells are differentially localized to the spleens of mice infected with Y. pestis ΔyopM mutant strains (25), although we did not observe increased recruitment of NK1.1+ splenocytes to the spleens of mice infected with the 32777ΔyopM strain of Y. pseudotuberculosis (Fig. 4A). Although these cells may be a source of IFN-γ, they are likely not the only source, as depletion of NK1.1+ cells from mice did not restore the ability of the 32777ΔyopM strain to disseminate to the spleens of infected mice (data not shown). This result is consistent with previous work whereby depletion of NK1.1+ cells did not restore the virulence of the Y. pestis ΔyopM mutant strain used (49).
An attractive explanation for this YopM-mediated suppression of IFN-γ production is the increased levels of IL-10 observed in the Y. pseudotuberculosis 32777-infected mice. IL-10 is an antiinflammatory cytokine that is involved in dampening the production of both TNF-α and IFN-γ (16, 44). In previous studies, the induction of IL-10 has been associated with the presence of the Yersinia T3SS-secreted protein LcrV (9, 14). LcrV-mediated IL-10 production also caused suppression of TNF-α production (39). Changes in IL-10 production have also been associated with Y. pseudotuberculosis infection in BALB/c mice (43). A recent publication has demonstrated that IL-10 production is a host response to systemic infection with Y. pestis (35). This is a somewhat controversial area of research, as an independent study found no evidence for increased IL-10 levels in mice infected with Y. pseudotuberculosis (2). The dramatically increased IL-10 production we observed during Y. pseudotuberculosis 32777 infection (Fig. 5C) could lead to damping of the IFN-γ release by NK1.1+ cells and T cells in infected animals. This loss of IL-10 production observed in the absence of YopM would thus explain the increased IFN-γ levels observed in the serum of 32777ΔyopM-infected mice (Fig. 5A).
YopM-induced IL-10 production could explain the finding that YopM is not required for virulence in a pneumonic plague infection model (49). Numerous studies have demonstrated that the lung is an immunosuppressive environment, with high levels of IL-10 being produced by both alveolar macrophages and the alveolar epithelium (24). This constitutive IL-10 production would mitigate the necessity of Yersinia-induced immunosuppressive IL-10 production. Although it is clear that IL-10 does have an inhibitory effect on the release of proinflammatory cytokines, several studies have suggested that the proinflammatory cytokine IL-12 is necessary for IL-10 production by NK cells or NKT cells (20, 35). This demonstrates that a complex network of positive and negative feedback loops is induced in the host to control the pathogen load but also to limit damage by the inflammatory response.
The data presented here suggest that YopM functions by recruiting the RSK1 and PRK2 kinases to a signaling complex involved in modulating the innate immune response to Y. pseudotuberculosis. Previous in vitro work demonstrated that both RSK1 and PRK2 contribute to the ability of the signaling complex to phosphorylate a heterologous substrate, but as we show here, deletion of domains of the YopM protein that mediate interaction with RSK1 (C terminus) or PRK2 (LRR6 to LRR15) completely abrogates virulence in the orogastric infection model (Fig. 6B). This demonstrates for the first time that the ability to interact with these kinases involves different regions of the YopM protein and that both of these activities are critical for virulence. Further work is required to determine the nature of the signaling pathway affected, as well as the relative contribution of each kinase to this signaling pathway.
ADDENDUM
Following the initial submission of our manuscript, McCoy et al. (28) demonstrated that the C-terminal six amino acids are required for interaction with RSK1 and that this interaction is required for virulence. This is in concordance with the results presented here.
Acknowledgments
We thank Ali Ashkar at McMaster University for the generous gift of the anti-NK1.1 monoclonal antibody-producing hybridoma PK181 and for helpful discussions about NK1.1 depletion protocols. We also thank Sarit Lilo for constructing Y. pseudotuberculosis 32777ΔyopM. Yue Zhang is thanked for assistance with cell-sorting protocols and experiments, as well as helpful discussions.
This work was supported by grants from the National Institute of Allergy and Infectious Diseases (R01-AI043389, P01-AI055621) and the Northeast Biodefense Center (U54-AI057158-Lipkin) awarded to J.B.B.
The content of this report is solely our responsibility and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 1 June 2010.
REFERENCES
- 1.Akira, S. 2000. The role of IL-18 in innate immunity. Curr. Opin. Immunol. 12:59-63. [DOI] [PubMed] [Google Scholar]
- 2.Auerbuch, V., and R. R. Isberg. 2007. Growth of Yersinia pseudotuberculosis in mice occurs independently of Toll-like receptor 2 expression and induction of interleukin-10. Infect. Immun. 75:3561-3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Autenrieth, I. B., and J. Heesemann. 1992. In vivo neutralization of tumor necrosis factor-alpha and interferon-gamma abrogates resistance to Yersinia enterocolitica infection in mice. Med. Microbiol. Immunol. 181:333-338. [DOI] [PubMed] [Google Scholar]
- 4.Benabdillah, R., L. J. Mota, S. Lutzelschwab, E. Demoinet, and G. R. Cornelis. 2004. Identification of a nuclear targeting signal in YopM from Yersinia spp. Microb. Pathog. 36:247-261. [DOI] [PubMed] [Google Scholar]
- 5.Black, D. S., and J. B. Bliska. 1997. Identification of p130Cas as a substrate of Yersinia YopH (Yop51), a bacterial protein tyrosine phosphatase that translocates into mammalian cells and targets focal adhesions. EMBO J. 16:2730-2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black, D. S., and J. B. Bliska. 2000. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol. Microbiol. 37:515-527. [DOI] [PubMed] [Google Scholar]
- 7.Bohn, E., A. Sing, R. Zumbihl, C. Bielfeldt, H. Okamura, M. Kurimoto, J. Heesemann, and I. B. Autenrieth. 1998. IL-18 (IFN-gamma-inducing factor) regulates early cytokine production in, and promotes resolution of, bacterial infection in mice. J. Immunol. 160:299-307. [PubMed] [Google Scholar]
- 8.Boland, A., S. Havaux, and G. R. Cornelis. 1998. Heterogeneity of the Yersinia YopM protein. Microb. Pathog. 25:343-348. [DOI] [PubMed] [Google Scholar]
- 9.Brubaker, R. R. 2003. Interleukin-10 and inhibition of innate immunity to yersiniae: roles of Yops and LcrV (V antigen). Infect. Immun. 71:3673-3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaix, J., M. S. Tessmer, K. Hoebe, N. Fuseri, B. Ryffel, M. Dalod, L. Alexopoulou, B. Beutler, L. Brossay, E. Vivier, and T. Walzer. 2008. Cutting edge: priming of NK cells by IL-18. J. Immunol. 181:1627-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaturvedi, D., M. S. Cohen, J. Taunton, and T. B. Patel. 2009. The PKARIalpha subunit of protein kinase A modulates the activation of p90RSK1 and its function. J. Biol. Chem. 284:23670-23681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Lorenzo, V., M. Herrero, U. Jakubzik, and K. N. Timmis. 1990. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J. Bacteriol. 172:6568-6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Depaolo, R. W., R. Tang, I. Kim, M. Han, N. Levin, N. Ciletti, A. Lin, D. Anderson, O. Schneewind, and B. Jabri. 2008. Toll-like receptor 6 drives differentiation of tolerogenic dendritic cells and contributes to LcrV-mediated plague pathogenesis. Cell Host Microbe 4:350-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Derbise, A., B. Lesic, D. Dacheux, J. M. Ghigo, and E. Carniel. 2003. A rapid and simple method for inactivating chromosomal genes in Yersinia. FEMS Immunol. Med. Microbiol. 38:113-116. [DOI] [PubMed] [Google Scholar]
- 16.Dirix, V., V. Verscheure, T. Goetghebuer, M. Hainaut, A. S. Debrie, C. Locht, and F. Mascart. 2009. Monocyte-derived interleukin-10 depresses the Bordetella pertussis-specific gamma interferon response in vaccinated infants. Clin. Vaccine Immunol. 16:1816-1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evdokimov, A. G., D. E. Anderson, K. M. Routzahn, and D. S. Waugh. 2001. Unusual molecular architecture of the Yersinia pestis cytotoxin YopM: a leucine-rich repeat protein with the shortest repeating unit. J. Mol. Biol. 312:807-821. [DOI] [PubMed] [Google Scholar]
- 18.Fürste, J. P., W. Pansegrau, R. Frank, H. Blocker, P. Scholz, M. Bagdasarian, and E. Lanka. 1986. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48:119-131. [DOI] [PubMed] [Google Scholar]
- 19.Galyov, E. E., S. Hakansson, A. Forsberg, and H. Wolf-Watz. 1993. A secreted protein kinase of Yersinia pseudotuberculosis is an indispensable virulence determinant. Nature 361:730-732. [DOI] [PubMed] [Google Scholar]
- 20.Grant, L. R., Z. J. Yao, C. M. Hedrich, F. Wang, A. Moorthy, K. Wilson, D. Ranatunga, and J. H. Bream. 2008. Stat4-dependent, T-bet-independent regulation of IL-10 in NK cells. Genes Immun. 9:316-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamid, N., A. Gustavsson, K. Andersson, K. McGee, C. Persson, C. E. Rudd, and M. Fallman. 1999. YopH dephosphorylates Cas and Fyn-binding protein in macrophages. Microb. Pathog. 27:231-242. [DOI] [PubMed] [Google Scholar]
- 22.Hines, J., E. Skrzypek, A. V. Kajava, and S. C. Straley. 2001. Structure-function analysis of Yersinia pestis YopM's interaction with alpha-thrombin to rule on its significance in systemic plague and to model YopM's mechanism of binding host proteins. Microb. Pathog. 30:193-209. [DOI] [PubMed] [Google Scholar]
- 23.Hoang, T. T., R. R. Karkhoff-Schweizer, A. J. Kutchma, and H. P. Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77-86. [DOI] [PubMed] [Google Scholar]
- 24.Jose, P., M. G. Avdiushko, S. Akira, A. M. Kaplan, and D. A. Cohen. 2009. Inhibition of interleukin-10 signaling in lung dendritic cells by Toll-like receptor 4 ligands. Exp. Lung Res. 35:1-28. [DOI] [PubMed] [Google Scholar]
- 25.Kerschen, E. J., D. A. Cohen, A. M. Kaplan, and S. C. Straley. 2004. The plague virulence protein YopM targets the innate immune response by causing a global depletion of NK cells. Infect. Immun. 72:4589-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim, H. M., B. S. Park, J. I. Kim, S. E. Kim, J. Lee, S. C. Oh, P. Enkhbayar, N. Matsushima, H. Lee, O. J. Yoo, and J. O. Lee. 2007. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130:906-917. [DOI] [PubMed] [Google Scholar]
- 27.Leung, K. Y., B. S. Reisner, and S. C. Straley. 1990. YopM inhibits platelet aggregation and is necessary for virulence of Yersinia pestis in mice. Infect. Immun. 58:3262-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCoy, M. W., M. L. Marre, C. F. Lesser, and J. Mecsas. 2010. The C-terminal tail of Yersinia YopM is critical for interacting with RSK1 and for virulence. Infect. Immun. 78:2584-2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald, C., P. O. Vacratsis, J. B. Bliska, and J. E. Dixon. 2003. The Yersinia virulence factor YopM forms a novel protein complex with two cellular kinases. J. Biol. Chem. 278:18514-18523. [DOI] [PubMed] [Google Scholar]
- 30.Mittal, R., S. Y. Peak-Chew, and H. T. McMahon. 2006. Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc. Natl. Acad. Sci. U. S. A. 103:18574-18579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukherjee, S., G. Keitany, Y. Li, Y. Wang, H. L. Ball, E. J. Goldsmith, and K. Orth. 2006. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312:1211-1214. [DOI] [PubMed] [Google Scholar]
- 32.Navarro, L., A. Koller, R. Nordfelth, H. Wolf-Watz, S. Taylor, and J. E. Dixon. 2007. Identification of a molecular target for the Yersinia protein kinase A. Mol. Cell 26:465-477. [DOI] [PubMed] [Google Scholar]
- 33.Nemeth, J., and S. C. Straley. 1997. Effect of Yersinia pestis YopM on experimental plague. Infect. Immun. 65:924-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palmer, L. E., S. Hobbie, J. E. Galan, and J. B. Bliska. 1998. YopJ of Yersinia pseudotuberculosis is required for the inhibition of macrophage TNF-alpha production and downregulation of the MAP kinases p38 and JNK. Mol. Microbiol. 27:953-965. [DOI] [PubMed] [Google Scholar]
- 35.Perona-Wright, G., K. Mohrs, F. M. Szaba, L. W. Kummer, R. Madan, C. L. Karp, L. L. Johnson, S. T. Smiley, and M. Mohrs. 2009. Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe 6:503-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prehna, G., M. I. Ivanov, J. B. Bliska, and C. E. Stebbins. 2006. Yersinia virulence depends on mimicry of host Rho-family nucleotide dissociation inhibitors. Cell 126:869-880. [DOI] [PubMed] [Google Scholar]
- 37.Shao, F., and J. E. Dixon. 2003. YopT is a cysteine protease cleaving Rho family GTPases. Adv. Exp. Med. Biol. 529:79-84. [DOI] [PubMed] [Google Scholar]
- 38.Simonet, M., and S. Falkow. 1992. Invasin expression in Yersinia pseudotuberculosis. Infect. Immun. 60:4414-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sing, A., A. Roggenkamp, A. M. Geiger, and J. Heesemann. 2002. Yersinia enterocolitica evasion of the host innate immune response by V antigen-induced IL-10 production of macrophages is abrogated in IL-10-deficient mice. J. Immunol. 168:1315-1321. [DOI] [PubMed] [Google Scholar]
- 40.Skrzypek, E., C. Cowan, and S. C. Straley. 1998. Targeting of the Yersinia pestis YopM protein into HeLa cells and intracellular trafficking to the nucleus. Mol. Microbiol. 30:1051-1065. [DOI] [PubMed] [Google Scholar]
- 41.Skrzypek, E., T. Myers-Morales, S. W. Whiteheart, and S. C. Straley. 2003. Application of a Saccharomyces cerevisiae model to study requirements for trafficking of Yersinia pestis YopM in eucaryotic cells. Infect. Immun. 71:937-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugawara, I. 2000. Interleukin-18 (IL-18) and infectious diseases, with special emphasis on diseases induced by intracellular pathogens. Microbes Infect. 2:1257-1263. [DOI] [PubMed] [Google Scholar]
- 43.Tansini, A., and B. M. de Medeiros. 2009. Susceptibility to Yersinia pseudotuberculosis infection is linked to the pattern of macrophage activation. Scand. J. Immunol. 69:310-318. [DOI] [PubMed] [Google Scholar]
- 44.Tripp, C. S., K. P. Beckerman, and E. R. Unanue. 1995. Immune complexes inhibit antimicrobial responses through interleukin-10 production. Effects in severe combined immunodeficient mice during Listeria infection. J. Clin. Invest. 95:1628-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trülzsch, K., T. Sporleder, E. I. Igwe, H. Russmann, and J. Heesemann. 2004. Contribution of the major secreted Yops of Yersinia enterocolitica O:8 to pathogenicity in the mouse infection model. Infect. Immun. 72:5227-5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uff, S., J. M. Clemetson, T. Harrison, K. J. Clemetson, and J. Emsley. 2002. Crystal structure of the platelet glycoprotein Ib(alpha) N-terminal domain reveals an unmasking mechanism for receptor activation. J. Biol. Chem. 277:35657-35663. [DOI] [PubMed] [Google Scholar]
- 47.Une, T., and R. R. Brubaker. 1984. In vivo comparison of avirulent Vwa− and Pgm− or Pstr phenotypes of yersiniae. Infect. Immun. 43:895-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Viboud, G. I., and J. B. Bliska. 2005. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59:69-89. [DOI] [PubMed] [Google Scholar]
- 49.Ye, Z., E. J. Kerschen, D. A. Cohen, A. M. Kaplan, N. van Rooijen, and S. C. Straley. 2009. Gr1+ cells control growth of YopM-negative Yersinia pestis during systemic plague. Infect. Immun. 77:3791-3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang, Y., A. T. Ting, K. B. Marcu, and J. B. Bliska. 2005. Inhibition of MAPK and NF-kappa B pathways is necessary for rapid apoptosis in macrophages infected with Yersinia. J. Immunol. 174:7939-7949. [DOI] [PubMed] [Google Scholar]