

Abstract
Shiga toxins (Stxs) induce apoptosis via activation of the intrinsic and extrinsic pathways in many cell types. Toxin-mediated activation of the endoplasmic reticulum (ER) stress response was shown to be instrumental in initiating apoptosis in THP-1 myeloid leukemia cells. THP-1 cells responded to Shiga toxin type 1 (Stx1) in a cell maturation-dependent manner, undergoing rapid apoptosis in the undifferentiated state but reduced and delayed apoptosis in differentiated cells. The onset of apoptosis was associated with calpain activation and changes in expression of C/EBP homologous protein (CHOP), Bcl-2 family members, and death receptor 5 (DR5). Ligation of DR5 by tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) activates the extrinsic pathway of apoptosis. We show here that expression of TRAIL and DR5 is increased by Stx1 treatment. Addition of exogenous TRAIL enhances, and anti-TRAIL antibodies inhibit, Stx1-induced apoptosis of THP-1 cells. Silencing of CHOP or DR5 expression selectively prevented caspase activation, loss of mitochondrial membrane potential, and Stx1-induced apoptosis of macrophage-like THP-1 cells. In contrast, the rapid kinetics of apoptosis induction in monocytic THP-1 cells correlated with rates of calpain cleavage. The results suggest that CHOP-DR5 signaling and calpain activation differentially contribute to cell maturation-dependent Stx1-induced apoptosis. Inhibition of these signaling pathways may protect cells from Stx cytotoxicity.
Shiga toxins (Stxs) are major virulence factors expressed by the enteric pathogens Shigella dysenteriae serotype 1 and certain Escherichia coli serotypes referred to as Shiga toxin-producing E. coli (STEC). Infections with Stx-producing bacteria are associated with watery diarrhea that may progress to bloody diarrhea, acute renal failure, and central nervous system complications such as lethargy, seizures, and paralysis (60). STEC is a particular public health concern in developed nations, with approximately 73,000 cases annually of hemorrhagic colitis caused by E. coli O157:H7 and 37,000 annual cases caused by STEC non-O157 serotypes in the United States (42). The histopathological hallmark of disease caused by Stxs is damage to endothelial cells lining colonic capillaries, renal glomeruli and arterioles, and central nervous system (CNS) blood vessels (46). The essential role of Stxs in pathogenesis has been confirmed using animal models in which the infusion of the toxins causes extensive microvascular thromboses in the kidney and CNS and, in some cases, ataxia and limb paralysis (43, 61). S. dysenteriae serotype 1 produces Shiga toxin, while STEC may express one or more toxin variants categorized as Shiga toxin type 1 (Stx1) or Shiga toxin type 2 (Stx2) based on their antigenic similarity to Shiga toxin (56). All Stxs possess an AB5 structure composed of a monomeric A subunit in noncovalent association with a pentamer of B subunits (17). The B subunits mediate toxin binding by interaction with the membrane neutral glycolipid globotriaosylceramide (Gb3) (38). The toxins are then internalized and undergo a complex series of intracellular routing events, collectively termed retrograde transport, which ultimately deliver the toxins to the endoplasmic reticulum (ER) lumen (50). In the ER, the A subunit is proteolytically processed, and a fragment of the A subunit retrotranslocates into the cytosol. The N-glycosidase activity associated with the processed A subunit catalyzes the inactivation of eukaryotic ribosomes and inhibits protein synthesis (12, 51).
In addition to the capacity to inhibit protein synthesis, Stxs have been shown to induce apoptosis, or programmed cell death, in many cell types (5). The toxins appear to activate apoptotic signaling through an extrinsic (death receptor-mediated signaling) or an intrinsic (mitochondrion-mediated signaling) pathway. For example, the toxins have been shown to be capable of directly activating initiator and executioner caspase cascades but also to generate truncated BID (tBID) which translocates to mitochondrial membranes, leading to increased mitochondrial membrane permeability, release of cytochrome c, and formation of the apoptosome (6, 18, 34). As a result of signaling through the intrinsic or extrinsic pathway, intoxicated cells display characteristics of apoptosis such as DNA fragmentation, cell shrinkage, membrane blebbing, and chromatin condensation.
We previously showed that Stx1 induced apoptosis in the human myelogenous leukemia cell line THP-1 in a cell maturation-dependent manner. Undifferentiated, nonadherent monocytic THP-1 cells underwent rapid apoptosis when treated with Stx1, while differentiation to the adherent, macrophage-like state was associated with increased resistance to the cytotoxic action of the toxins, with only approximately 30% of cells undergoing delayed apoptosis (22). The induction of apoptosis by Stx1 involved the activation of the ER stress response in both monocytic and macrophage-like THP-1 cells (33, 36). Stx1 induced the expression of the ER stress effectors C/EBP homologous protein (CHOP), tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), and death receptor 5 (DR5) in monocytic THP-1 cells. Delivery of functional Stx1 into the cytosol of monocytic THP-1 cells led to downregulated expression of the prosurvival factor Bcl-2, while the delayed-apoptosis phenotype in macrophage-like cells was associated with increased Bcl-2 expression, phosphorylation, and mitochondrial translocation.
Increased expression of the apoptosis-inducing factor TRAIL and its death-inducing receptor, DR5, enhances cell death signals triggered during a prolonged ER stress response (23, 68). TRAIL may be membrane associated or may be cleaved from the cell surface by proteases to generate a soluble ligand (26, 40). Engagement of TRAIL with its cognate receptor DR5 activates the extrinsic pathway of apoptosis through DR5 aggregation, the recruitment of the Fas-associated death domain (FADD), and the formation of the death-inducing signaling complex (DISC) (31, 53). The observation that expression of TRAIL and DR5 was upregulated by Stx1 treatment of monocytic THP-1 cells suggested that this receptor-ligand pair may contribute to rapid apoptosis induced by the toxin in these cells. However, we also showed that calpains were rapidly activated by Stx1 in monocytic THP-1 cells, and calpains may directly cleave caspase-3 (36). The studies reported here were designed to characterize the roles of TRAIL/DR5 and calpains in the rapid apoptosis response of monocytic cells and in delayed apoptosis in macrophage-like cells. We show that Stx1-induced apoptotic signaling is amplified by the addition of soluble TRAIL (sTRAIL) and inhibited by exposure of cells to neutralizing anti-TRAIL antibodies prior to intoxication. A reduction in CHOP or DR5 expression using RNA interference (RNAi) techniques markedly protected cells from apoptosis induced by Stx1, linking activation of the ER stress response with apoptosis in this system. Signaling through CHOP and DR5 led to activation of the initiator caspase, caspase-8, and the executioner caspase, caspase-3, in macrophage-like THP-1 cells, but the effect of CHOP and DR5 knockdown on caspase activation and apoptosis of monocytic cells was minimal. In contrast, the rate of calpain activation (cleavage) was directly correlated with the rapid onset of apoptosis in monocytic THP-1 cells.
MATERIALS AND METHODS
Antibodies.
Anti-human calpain-1/2 antibody was purchased from Calbiochem, San Diego, CA. Antibodies directed against human DR5, caspase-8, caspase-3, and pan-actin were obtained from Cell Signaling Technologies, Beverly, MA. Rabbit polyclonal purified human anti-CHOP was purchased from Biolegend, San Diego, CA.
Cell culture.
The human myelogenous leukemia cell line THP-1 (American Type Culture Collection, Manassas, VA) was cultured in RPMI 1640 medium (Gibco-BRL, Grand Island, NY) containing 10% fetal bovine serum (HyClone Laboratories, Logan, UT), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in 5% CO2 in a humidified incubator. Cells maintained under these conditions were considered undifferentiated, monocytic cells. Monocytic cells (1 × 106 cells/ml) were differentiated to the adherent macrophage-like state with phorbol 12-myristate 13-acetate (PMA) (Sigma Chemical Co., St. Louis, MO) at a concentration of 50 ng/ml for 48 h. Plastic-adherent cells were washed three times with cold, sterile Dulbecco's phosphate-buffered saline (PBS) (Sigma) and then incubated with fresh medium lacking PMA but containing 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml). The medium was changed every 24 h for the next 3 days. Experiments were performed on the fourth day after PMA removal.
Toxin purification.
Stx1 purification was described previously (36). Briefly, Stx1 was expressed from Escherichia coli DH5α(pCKS112), a recombinant strain harboring a plasmid carrying the stx1 operon under the control of a thermoinducible promoter (62). Cells were lysed, and periplasmic extracts were subjected to sequential ion-exchange and chromatofocusing chromatographies. The purity of toxins was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis using anti-Stx1 specific antibodies. Prior to use, Stx1 preparations were shown to contain <0.1 ng of endotoxin/ml by use of the Limulus amoebocyte lysate assay (Associates of Cape Cod, East Falmouth, MA). Purified Stx1A−, a holotoxin with two point mutations (E167Q and R170L), was a kind gift from Shinji Yamasaki, Osaka Prefecture University, Osaka, Japan. The site-directed mutations in the Stx1 A subunit reduce toxin N-glycosidase activity by >5 log units (44). Purified Stx1 B subunits were a kind gift from Cheleste Thorpe, Tufts University School of Medicine, Boston, MA.
MTS-based cell cytotoxicity assay.
Monocytic and macrophage-like THP-1 cells (5 × 104 cells per well) were seeded in 96-well microtiter plates prior to treatment with Stx1 (400 ng/ml) or Stx1A− (400 ng/ml). In some experiments, neutralizing anti-tumor necrosis factor-related apoptosis-inducing ligand (anti-TRAIL) antibodies (10 to 50 ng/ml; R&D Systems, Minneapolis, MN) were added to the cells for 40 min prior to stimulation with Stx1 and incubation at 37°C in humidified 5% CO2. In some experiments, cells were treated with Stx1 or Stx1A− for 24 h in RPMI 1640 medium containing 0.5% fetal bovine serum (FBS) in the presence or absence of recombinant soluble TRAIL (sTRAIL) (10 to 50 ng/ml; R&D Systems, Minneapolis, MN). Control experiments with monocytic and macrophage-like THP-1 cells treated with sTRAIL or anti-TRAIL antibodies alone were included with each experiment. Cytotoxicity was determined by a colorimetric assay (8) using the novel tetrazolium compound 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inert salt (MTS) (Promega, Madison, WI). MTS (50 μl/5000 cells) was added to each well and incubation continued for 2 h at 37°C in 5% CO2. The optical density at 490 nm (OD490) was recorded with an automated microtiter plate reader (Dynatech MR5000; Molecular Dynamics, Chantilly, VA). The percentage of cell death was determined using the following equation: percentage of cell death = [(average OD490 of treated cells − average OD490 of control cells)/average OD490 of control cells] × 100. The background absorbance at 630 nm measured with untreated cells was subtracted from each sample reading. The reference wavelength of 630 nm was used to subtract background contributed by excess cell debris and other nonspecific absorbance.
Human TRAIL immunoassay.
Quantitation of human TRAIL/TNFSF10 protein was carried out using Quantikine colorimetric enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN). Briefly, differentiated THP-1 cells (5 × 106 cells/well) were stimulated with Stx1 (0 to 1,200 ng/ml) for 24 h. Cell-free supernatants and cell lysates were prepared, and kit-specified volumes of the samples were added in triplicate to 96-well polystyrene microtiter plates coated with a mouse monoclonal antibody against TRAIL. Human TRAIL immunoassay was performed according to the manufacturer's instructions. Amounts of human TRAIL in cell-free supernatants or cell lysates were estimated based on standard curves generated by serial dilutions of recombinant human TRAIL.
Analysis of apoptosis by annexin V and propidium iodide staining.
Monocytic THP-1 cells were incubated with soluble TRAIL (50 ng/ml) or neutralizing anti-TRAIL antibodies (50 ng/ml) for 30 min before treatment with Stx1 (400 ng/ml), Stx1A− (400 ng/ml), or cycloheximide (CHX) (100 μM) for various times. Following treatment, cells were centrifuged at 200 × g for 5 min, washed in ice-cold sterile PBS, and stained using the annexin V-Fluos staining kit (Roche Diagnostics, Indianapolis, IN). Cells were incubated in the provided incubation buffer containing annexin V (AV) and propidium iodide (PI) for 20 min at room temperature. After washing with PBS, apoptosis was measured by flow cytometry (Becton-Dickinson, Palo Alto, CA). Fluorescence parameters were gated using unstained and single-stained, untreated cells. At least 104 events were measured for each sample. Total percent apoptosis was expressed as follows: percentage of AV-positive (AV+) cells + percentage of AV+ PI+ cells − background fluorescence.
Measurement of ΔΨm.
Loss of mitochondrial membrane potential (ΔΨm) was measured as described previously (35). Briefly, differentiated THP-1 cells (5 × 106 cells/well) were transfected with DR5 small interfering RNA (siDR5) or CHOP siRNA (siCHOP) for 72 h, followed by treatment with Stx1 for 24 h. After stimulation, cells were detached by treatment with Accutase (Innovative Cell Technologies Inc., San Diego, CA) for 10 min. After centrifugation at 260 × g for 5 min, supernatants were removed, and cells were washed in ice-cold PBS and resuspended in 0.5 ml JC-1 assay buffer containing the reagent 5,5,6,6-tetrachloro-1,1,3,3-tetraethylbenz-imidazolocarbocyanine iodine (JC-1). Cells were incubated for 15 min at 37°C in the presence of 5% CO2, centrifuged at 400 × g for 5 min, and washed twice in JC-1 assay buffer. JC-1 fluorescence associated with mitochondrial membranes was detected using flow cytometry.
Preparation of cellular lysates and Western blotting.
Eighteen hours prior to stimulation, differentiated THP-1 cells (5 × 106 cells/well) were washed twice in cold Dulbecco's PBS and RPMI 1640 containing 0.5% FBS. Cells were stimulated with Stx1 (400 ng/ml), Stx1A− (400 ng/ml), Stx1 B subunit (800 ng/ml), or thapsigargin (Tg) (10 μm) for 0 to 24 h. Cells were harvested and lysed with modified radioimmunoprecipitation assay (RIPA) buffer as previously described (16). For detection of CHOP, cells were lysed with modified lysis buffer (137 mM NaCl, 15 mM EGTA, 0.1 mM sodium orthovanadate, 15 mM MgCl2, 0.1% Triton X-100, 25 mM morpholinepropanesulfonic acid [MOPS] [pH 7.2], 100 mM phenylmethylsulfonyl fluoride [PMSF], and 20 μM leupeptin). Extracts were collected and cleared by centrifugation at 15,000 × g for 10 min. Cell lysates from monocytic THP-1 cells were prepared as previously described (33). Protein concentrations in each extract were determined using the Micro bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL). Equal amounts of proteins (70 to 100 μg/lane) were separated by 8% or 12% Tris-glycine SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat milk prepared with TBST (20 mM Tris [pH 7.6], 137 mM NaCl, 0.1% Tween 20). Membranes were incubated with primary antibodies at 4°C for 24 h. After washing, the membranes were incubated with horseradish peroxidase-labeled secondary antibodies for 2 h at room temperature. Bands were visualized using the Western Lightning chemiluminescence system (NEN-Perkins-Elmer, Boston, MA). Data shown are from at least three independent experiments. Relative protein expression levels were measured using NIH image J software.
Analysis of apoptosis by TUNEL staining.
Apoptosis of adherent, macrophage-like THP-1 cells was determined using the in situ cell death detection kit (Roche). Briefly, THP-1 cells (2 × 105 cells/well) were differentiated on eight-well Lab-Tek chamber slides (Nalge-Nunc International, Naperville, IL), resulting in approximately 1 × 105 viable cells/well. Cells were treated with Stx1 in the presence or absence of sTRAIL or anti-TRAIL antibodies for 24 h. Cells were then fixed in freshly prepared 4% paraformaldehyde for 1 h, rinsed with PBS, and treated with permeabilization solution for 2 min on ice. Following washing, terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) reaction mix was added and left for 1 h at 37°C in the dark. Cells were washed with PBS, and fluorescent nuclei detected were quantified with a Bio-Tek (Richmond, VA) Synergy HT microplate reader. In order to count TUNEL-positive cells in the siRNA-transfected cells, the total number of TUNEL-positive cells was determined from 10 different fields (magnification, ×20) for each treatment from three independent experiments. In parallel, total numbers of cells were determined by counting 4′,6′-diamidino-2-phenylindole (DAPI) (Invitrogen, Eugene, OR)-stained nuclei for each treatment. Percentages of TUNEL-positive cells above untreated controls were calculated as follows: percent apoptosis = (number of TUNEL-positive cells/number of DAPI-positive total cells) × 100.
siRNA transfection.
Differentiated or undifferentiated THP-1 cells were transfected with 100 to 200 pmol ON-TARGETplus SMARTpool DR5 siRNA, CHOP siRNA, or nontargeting (NT) negative-control siRNA (Dharmacon, Inc., Chicago, IL) using transIT-TKO transfection reagent (Mirus Bio Corporation, Madison, WI) as per the manufacturer's instructions. Briefly, DR5, CHOP, or nontargeting siRNA was added to transfection reagent diluted in serum-free medium and incubated for 15 min to allow the formation of transfection complexes prior to transfection. The siRNA complexes were added to 1 × 106 to 2 × 106 cells/well grown in 6- or 12-well plates and incubated for 48 h, followed by the addition of fresh serum-containing medium to each well. Transfection efficiency was measured by observing Cy3-labeled fluorescence of the control siRNA (New England BioLabs, Ipswich, MA) with a fluorescence-activated cell sorter (FACSCalibur; Becton-Dickinson, Palo Alto, CA). Using this protocol, typical transfection efficiencies were 70 to 80%. After the transfected cells were incubated with Stx1 (400 ng/ml) for 0, 8, or 24 h, Western blotting was performed with specific antibodies against human DR5, CHOP, or caspase-3 and -8. In order to address the influence of silencing CHOP or DR5 on apoptosis, the TUNEL assay was carried out following exposure of cells to Stx1.
Statistics.
Data are reported as means ± standard errors of the mean (SEM) for at least three independent experiments. Statistical analyses of data were performed with EXCEL (Microsoft, Redmond, WA). The Student t test was used to measure differences in samples. Depending on the assay, a P value of <0.05, <0.01, or <0.001 was considered significant.
RESULTS
TRAIL contributes to Stx1-mediated apoptosis of monocytic THP-1 cells.
We have previously shown that Stx1 induces apoptosis of the myelogenous leukemia cell line THP-1 through activation of the ER stress response (36). Sensitivity to cell killing and the onset of apoptosis were cell maturation dependent; that is, replicating, nonadherent monocyte-like THP-1 cells were highly toxin sensitive and underwent rapid apoptosis, while differentiated, adherent macrophage-like THP-1 cells were relatively less toxin sensitive and the subset of cells that died in response to toxin treatment displayed a delayed onset of apoptosis (22, 35). Toxin activation of the ER stress response increased the expression of TRAIL and DR5 in toxin-sensitive monocytic cells (36), suggesting that TRAIL-DR5 ligation may contribute to the toxin-sensitive phenotype of undifferentiated cells. Annexin V (AV) binding to phosphatidylserine in the outer plasma membrane is a reliable early marker of apoptosis, while late-stage apoptosis includes the loss of plasma membrane integrity and uptake of propidium iodide (PI) (14). To explore the role of TRAIL-DR5 ligation in the rapid killing of monocytic THP-1 cells, we measured apoptosis by flow cytometry following exposure to Stx1 with or without the addition of exogenous soluble TRAIL (sTRAIL) or anti-TRAIL antibodies with subsequent staining with AV and PI over a 4- to 6-h time course.
Characteristic fluorescence-activated cell sorter (FACS) scatter plots of monocytic cells treated with Stx1 with or without sTRAIL for 4 h are shown in Fig. 1 A. Additive percentages of cells staining AV+ (right lower quadrant) and AV+ PI+ cells (right upper quadrant) were calculated from three independent experiments to show the kinetics of apoptosis induction (Fig. 1B) using cells treated with Stx1, Stx1 plus sTRAIL, or Stx1 plus anti-TRAIL antibodies for 4 to 6 h. The addition of sTRAIL to Stx1-treated cells increased apoptosis. While significant protection was achieved at 4 h, the percentage of apoptosis at 5 to 6 h was lower in the presence of anti-TRAIL antibodies, although the differences at these times did not achieve statistical significance. Figure 1C shows the results of three independent FACS experiments to examine the role of anti-TRAIL antibodies in blocking Stx1-induced apoptosis. Untreated monocytic THP-1 cells displayed low levels (7.1%) of spontaneous apoptosis, and cells treated with sTRAIL alone (50 ng/ml) displayed modest increases (13.1%) in apoptosis which were eliminated by anti-TRAIL antibody treatment. Treatment of monocytic cells with Stx1 alone for 4 h resulted in approximately 37% of cells staining AV+ plus AV+ PI+. We noted a dose-dependent increase in apoptosis mediated by the addition of sTRAIL to Stx1-treated cells (10 to 50 ng/ml sTRAIL) (data not shown). The addition of sTRAIL (50 ng/ml) to Stx1-treated cells increased the percentage of apoptotic cells to 54%. Stx1-mediated monocytic cell apoptosis was significantly (P < 0.005) reduced to 24.6% by the addition of anti-TRAIL antibody. Interestingly, while anti-TRAIL antibody pretreatment protected cells from apoptosis induced by sTRAIL or Stx1 alone, antibody pretreatment failed to significantly protect cells from apoptosis induced by Stx1 plus sTRAIL.
FIG. 1.

Effects of Stx1, sTRAIL, and anti-TRAIL antibody on apoptosis of undifferentiated (monocytic) THP-1 cells. (A) Representative FACS scatter plots of untreated monocytic THP-1 cells and cells treated with soluble TRAIL, Stx1, or Stx1 plus TRAIL for 4 h, showing percentages of cells staining annexin V positive (lower right quadrants) or annexin V/propidium iodide (PI) double positive (upper right quadrants). (B) Monocytic THP-1 cells were either untreated or incubated with Stx1 with or without soluble TRAIL or neutralizing anti-TRAIL antibodies for 4 to 6 h. At each time point, cells were stained with annexin V and PI and analyzed by flow cytometry for apoptosis. The data shown are the means ± SEM of additive percentages of AV+ plus AV+ PI+ cells for three independent experiments. *, significant difference (P < 0.05) between cells treated with Stx1 versus Stx1 plus TRAIL for 4 h; **, significant difference (P < 0.01) between cells treated with Stx1 versus Stx1 plus anti-TRAIL antibodies for 4 h. (C) Monocytic THP-1 cells were incubated with Stx1 or Stx1A− for 4 h with or without treatment with soluble TRAIL (sTR) or neutralizing anti-sTRAIL antibodies (anti-sTR). Cells were stained with annexin V and PI, and the effects of treatment combinations on apoptosis were measured by flow cytometry. Results represent means ± SEM for three independent experiments. *, significant difference (P < 0.001) between treated cells and untreated cells; **, significant difference (P < 0.005) between cells treated with Stx1 plus sTR versus Stx1 plus anti-sTR; #, significant difference (P < 0.05) between cells treated with Stx1 versus Stx1 plus sTR; ##, significant difference (P < 0.01) between cells treated with Stx1 versus Stx1 plus anti-sTR. Cycloheximide (CHX) served as a negative control.
To assess the role of Stx1 enzymatic activity in apoptosis induction, monocytic THP-1 cells were treated with a purified Stx1 holotoxin molecule (Stx1A−) containing two point mutations which drastically reduce enzymatic activity (44). All of the treatment regimens using Stx1A− failed to induce apoptosis above spontaneous levels (Fig. 1C), confirming the necessity for toxin enzymatic activity in apoptosis induction. However, we also treated monocytic THP-1 cells with cycloheximide (CHX), which we have shown to be an effective inhibitor of protein synthesis in monocytic THP-1 cells (34), yet CHX failed to trigger apoptosis (Fig. 1C). We previously showed that Stx1 treatment of monocytic THP-1 cells robustly increased DR5 expression at transcriptional and translational levels (36). However, treatment of cells with CHX for 0 to 12 h did not activate DR5 expression (data not shown). Thus, protein synthesis inhibition per se does not appear to be required for apoptosis induction or activation of the ER stress response.
Stx1 induces the expression of TRAIL and DR5 by macrophage-like THP-1 cells.
The capacity of Stx1 to induce TRAIL and DR5 expression by macrophage-like THP-1 cells has not been characterized. We used ELISAs to measure the production of both soluble and cell-associated TRAIL by macrophage-like cells following treatment with increasing amounts of Stx1 for 24 h (Fig. 2 A). We noted a toxin dose-dependent increase in soluble and cell-associated TRAIL, consistent with earlier studies using Stx1-treated monocytic THP-1 cells. Using reverse transcription-PCR (RT-PCR) (Fig. 2B), we detected elevated levels of DR5 mRNA as early as 2 h after toxin treatment. Elevated levels of transcripts were maintained up to 4 h but then declined at 6 h after toxin treatment. Treatment of cells with Stx1A− for 4 h failed to induce DR5 mRNA expression. DR5 protein production was monitored by Western blotting of cell lysates prepared from macrophage-like cells treated with Stx1, Stx1A−, or purified Stx1 B subunits. As shown in Fig. 2C, elevated levels of DR5 protein were detected beginning at 4 h and peaking at 8 to 16 h after Stx1 treatment. The ER stress inducer thapsigargin (Tg) strongly stimulated DR5 protein expression. Neither Stx1A− nor Stx1 B subunits were capable of inducing DR5 synthesis above basal levels. Thus, the treatment of both monocytic (36) and macrophage-like THP-1 cells induces TRAIL and DR5 expression.
FIG. 2.

Stx1 induces TRAIL and DR5 expression by macrophage-like THP-1 cells. (A) Macrophage-like THP-1 cells were treated with the indicated amounts of Stx1 for 24 h. Cell-free supernatants and cellular lysates were collected and analyzed by ELISA specific for human TRAIL production. C, control (untreated) cells. The data shown are means ± SEM from at least three independent experiments. *, significant difference (P < 0.05) between Stx1-treated cells and control cells. (B) Analysis of DR5 mRNA expression by RT-PCR using total RNA extracted from macrophage-like THP-1 cells treated with Stx1 for 0 to 24 h (upper panel), or Stx1 or Stx1A− for 4 h (lower panel). Primers for amplification of β-actin were used as a control. The data shown are characteristic of three independent experiments. (C) Analysis of DR5 protein expression by Western blotting (WB) using cell lysates prepared from macrophage-like THP-1 cells treated with Stx1 or thapsigargin (Tg) for 0 to 16 h, with Stx1A− or Stx1 B subunits for 0 to 16 h, or with Tg for 4 h. Antibodies directed against β-actin were used as a loading control. The data shown are characteristic of three independent experiments.
Additive cytotoxic effect of TRAIL and protective effect of neutralizing anti-TRAIL antibody on Stx1-mediated cytotoxicity.
TRAIL binds to death domain-containing receptors DR4 and DR5, leading to recruitment of the death-inducing signaling complex and caspase-8-initiated apoptotic signaling (31, 55). We demonstrated using undifferentiated monocytic THP-1 cells that Stx1 rapidly activated the ER stress response, resulting in procaspase-8 cleavage and elevated levels of sTRAIL and DR5 expression (36). Given our finding that Stx1 treatment of macrophage-like THP-1 cells also induces TRAIL and DR5 expression, we determined if the addition of exogenous sTRAIL in the presence of Stx1 influenced cell death. Differentiated THP-1 cells either were treated with Stx1 for 24 h and posttreated 40 min later with sTRAIL or were pretreated with neutralizing anti-TRAIL antibodies for 40 min prior to Stx1 treatment to measure protection from Stx1-induced cell death using the indicator dye MTS. The percentage of cell death was normalized to control (untreated) cells. The addition of sTRAIL to Stx1-treated THP-1 cells showed a dose-dependent increase in percentages of cell death compared to that for cells treated with Stx1 alone (Fig. 3 A). Maximal cell death occurred when cells were treated with Stx1 plus 50 ng/ml sTRAIL. Anti-TRAIL antibody partially protected macrophage-like THP-1 cells from Stx1- and Stx1-plus-sTRAIL-induced apoptosis in a dose-dependent manner (Fig. 3A). We next performed comparative analyses to evaluate cytotoxicity in response to sTRAIL (50 ng/ml) or neutralizing anti-TRAIL (50 ng/ml) in Stx1- or Stx1A− -treated monocytic and macrophage-like THP-1 cells at 0, 6, and 24 h. In accordance with our earlier studies, monocytic THP-1 cells were more sensitive to Stx1 (Fig. 3B). By 24 h, monocytic cells treated with Stx1 plus sTRAIL showed >85% cytotoxicity, while macrophage-like cells treated with the same agents showed only 51% of cell death, a percentage of cell death that is only 4% increased compared to that at the 6-h time point (Fig. 3B). These data suggest not only that monocytic cells are more sensitive to cell death induced by Stx1 and TRAIL than are macrophage-like cells but that only a subset of differentiated cells are susceptible to apoptosis induction by Stx1 plus sTRAIL.
FIG. 3.

Comparative cytotoxicities of undifferentiated and differentiated THP-1 cells treated with Stx1 or Stx1A− with or without sTRAIL or anti-TRAIL antibodies. (A) Macrophage-like THP-1 cells were treated with Stx1 alone or with various amounts of soluble TRAIL (TR) (10 to 50 ng/ml) for 40 min before Stx1 treatment or were treated with neutralizing anti-TRAIL antibodies (anti-TR) (10 to 50 ng/ml) for 40 min after Stx1 treatment. Cells were then incubated for 24 h, followed by measurement of cytotoxicity. The data shown are means ± SEM from at least three independent experiments. *, significant difference (P < 0.05) between Stx1-treated cells and Stx1-plus-TR- or anti-TR-treated cells. (B) Undifferentiated (monocytic) and differentiated (macrophage-like) THP-1 cells were seeded in 96-well microtiter plates. In some wells, cells were treated with Stx1 or Stx1A− for 6 or 24 h in the presence or absence of sTRAIL (TR) (50 ng/ml). In some wells, cells were treated with anti-TRAIL antibodies (antiTR) (50 ng/ml) for 40 min prior to stimulation with Stx1 or Stx1A−. Controls included untreated cells and cells treated with sTRAIL or neutralizing anti-TRAIL antibodies only. Cytotoxicity was determined by colorimetric assay using MTS as described in Materials and Methods. The percentage cell death was normalized to control (untreated) cells. Data shown are the means ± SEM for at least three independent experiments.
TRAIL and DR5 contribute to apoptosis induction in Stx1-treated macrophage-like THP-1 cells.
We used TUNEL staining to examine the sensitivity of adherent, macrophage-like THP-1 cells to apoptosis induced by Stx1 with or without sTRAIL or anti-TRAIL antibodies. Characteristic fluorescent micrographs are shown in Fig. 4 A, while the statistical analyses of measurements of TUNEL fluorescence intensities from three independent experiments are shown in Fig. 4B. Untreated cells and cells treated with sTRAIL, anti-TRAIL antibodies, or sTRAIL plus anti-TRAIL antibodies for 24 h showed few fluorescent nuclei characteristic of apoptotic cells. Macrophage-like cells treated with Stx1 displayed significantly (P < 0.001) increased fluorescence intensity, which was further increased by the addition of sTRAIL (50 ng/ml). Treatment of cells with Stx1 plus anti-TRAIL significantly (P < 0.01) reduced TUNEL fluorescence. Use of the Stx1A− holotoxin showed that toxin enzymatic activity was essential for apoptosis induction. The ER stress inducer thapsigargin significantly increased TUNEL fluorescence. The TUNEL assay is likely to dramatically underestimate the degree of cell death induced by Stx1 and Stx1 plus sTRAIL, since it detects fluorescence of adherent cells. We showed that approximately 60% of macrophage-like THP-1 cells detach from the wells following treatment with Stx1 or Stx1 plus sTRAIL for 24 h (Fig. 4C). However, pretreatment of toxin-treated cells with anti-TRAIL antibodies reduced cell detachment by approximately 20%. Taken together, these data suggest that, just as was the case with monocytic THP-1 cells, Stx1-induced expression of TRAIL may contribute, in part, to apoptosis of macrophage-like THP-1 cells, albeit with different kinetics.
FIG. 4.

Effects of Stx1 and TRAIL on apoptosis in macrophage-like THP-1 cells. (A) Macrophage-like THP-1 cells were treated with Stx1, Stx1A−, sTRAIL (sTR) (50 ng/ml), or anti-TRAIL antibodies (antiTR) (50 ng/ml) for 24 h. Control, untreated cells. Cells were fixed, permeabilized, and incubated with TUNEL reaction solution. Representative TUNEL-positive cells visualized by fluorescence microscopy are shown. (B) The relative fluorescence intensity of each treatment was measured using a microplate reader. Results shown are the means ± SEM from at least three independent experiments. *, significant difference (P < 0.001) between treated cells and control cells; **, significant difference (P < 0.01) between Stx1 plus sTR and Stx1 plus anti-TR antibodies. Thapsigargin was used as a positive control for ER stress-induced apoptosis. (C) To determine the extent of cell detachment after various treatments, differentiated THP-1 cells were treated with Stx1, Stx1A−, or thapsigargin for 24 h. In some experiments, cells were pre- or posttreated with sTRAIL or anti-TRAIL antibodies. Culture supernatants were removed, and 100-μl suspensions of trypsinized THP-1 cells were transferred to microcentrifuge tubes. Trypan blue exclusion was used to distinguish viable and nonviable cells. Two hundred microliters of the trypan blue suspensions were transferred to hemocytometer chambers for counting. Viable cells were counted and expressed as a percentage of untreated control cell numbers.
Silencing of DR5 or CHOP protects macrophage-like, but not monocytic, THP-1 cells from Stx1-induced apoptosis.
Since activation of the ER stress response leading to increased TRAIL and DR5 expression appears to contribute to Stx1-induced apoptosis and since apoptotic cell death induced by Stx1 plus sTRAIL is greater than cell death induced by TRAIL or Stx1 alone, we hypothesized that silencing of DR5 expression may protect cells from intoxication. By use of RNA interference (RNAi), an elegant strategy to achieve specific gene silencing at the posttranscriptional level (25), DR5 expression was inhibited and its knockdown verified by Western blotting using DR5-specific antibodies with extracts prepared from Stx1-treated macrophage-like THP-1 cells (Fig. 5 A). Thapsi-gargin (Tg), a well characterized inducer of the ER stress response, was used as a positive control to evaluate DR5 RNAi knockdown efficacy. Quantitative analysis showed that transfection of Stx1- and Tg-treated cells with DR5 siRNA significantly reduced (P < 0.01; Stx1 versus siRNA DR5 plus Stx1) DR5 protein expression (Fig. 5A, bar graph). Earlier studies have shown that increased expression of the transcriptional activator C/EBP homologous protein (CHOP) is required for increased DR5 expression (57, 68), and we have shown that Stx1-mediated activation of the ER stress response upregulated CHOP expression (36). We reasoned, therefore, that RNAi knockdown of the regulator CHOP would inhibit Stx1-induced DR5 expression. CHOP expression was reduced by 62% in comparison to cells treated with Stx1 alone or Stx1 plus nontargeting (NT) siRNA (Fig. 5B). As predicted, CHOP knockdown negatively affected DR5 mRNA expression, manifesting a 60% reduction in DR5 expression, while CHOP transcript levels were not susceptible to DR5 knockdown in the Stx1-treated macrophage-like cells (Fig. 5B, bar graph). These data indicate that CHOP is an upstream regulator in the ER stress pathway involved in DR5 expression.
FIG. 5.

Silencing of DR5 and CHOP expression reduces CHOP-dependent expression of DR5- and Stx1-induced apoptosis in macrophage-like, but not monocytic, THP-1 cells. (A and B) Differentiated THP-1 cells were transfected with or without 200 pmol siRNA duplexes targeting DR5 (siDR5) or CHOP (siCHOP) or with nontargeting siRNA duplexes (NTsiRNA) as described in Materials and Methods. After 72 h, cells were exposed to Stx1 for 8 h, followed by Western blotting using anti-DR5 (A and B) and anti-CHOP (B) antibodies, which recognize 46-kDa (precursor form)/41-kDa (mature form) and 29-kDa proteins, respectively. β-Actin was used as an equal-protein-loading control. The bar graphs show the quantitative analysis (means ± SEM) of DR5 or CHOP protein expression from three independent experiments. *, significant difference in DR5 protein expression (P < 0.01; Stx1 versus siRNA DR5 plus Stx1 treatment). (C) Effect of DR5 or CHOP knockdown on apoptosis of macrophage-like THP-1 cells. Differentiated THP-1 cells were transfected with DR5 siRNA (siDR5), CHOP siRNA (siCHOP), or nontargeting siRNA (NTsiRNA) or without siRNA (mock) for 72 h, followed by Stx1 treatment for another 24 h. Cells were fixed, permeabilized, and incubated with TUNEL reaction solution or DAPI to stain nuclei. Cells were visualized by fluorescence microscopy, and 10 different fields (magnification, ×20) for each treatment were selected to count total numbers of TUNEL positive cells. Total numbers of cells per field (magnification, ×20) were also counted, and the percentage of apoptotic cells above that for untreated controls were calculated as described in Materials and Methods. Upper panels, representative fields showing TUNEL-positive cells from each treatment. Lower bar graph, quantitative data showing percentage of apoptosis (means ± SEM from at least four independent experiments) for each treatment. *, significant difference (P < 0.001) for cells treated with Stx1 alone versus siDR5- plus Stx1-treated cells. (D) Monocytic THP-1 cells transfected with siRNAs specific for DR5 (siDR5), CHOP (siCHOP), or nontargeting siRNA (NTsiRNA) or mock transfected were treated with Stx1 for 5 h. Expression of DR5 and CHOP was assessed by Western blotting as outlined above. The bar graph depicts quantitative analysis (means ± SEM) of DR5 or CHOP protein expression from three independent experiments. *, significant difference in DR5 and CHOP protein expression (P < 0.01). (E) Quantitative data showing percentages of apoptosis for each treatment from the FACS scatter plots of untreated monocytic THP-1 cells, cells treated with Stx1 alone, or cells treated with siRNAs plus Stx1. Results are the means ± SEM from at least four independent experiments.
To investigate the functional significance of RNAi silencing of DR5 or CHOP expression, we explored effects on apoptosis induction using TUNEL staining with Stx1-treated macrophage-like THP-1 cells transfected or not with DR5 or CHOP siRNA (Fig. 5C). Examination of characteristic fluorescence micrographs (Fig. 5C, upper panels) shows that fluorescence intensities were reduced by DR5 and CHOP knockdown. Statistical analysis of four independent experiments using DR5 or CHOP siRNA-transfected cells reveals that fluorescent signal was significant reduced (P < 0.001) compared to that in cells treated with Stx1 alone (Fig. 5C, bar graph). In contrast, neither the nontargeting (NT) siRNA plus Stx1 nor mock transfection treatment plus Stx1 significantly altered the percentage of apoptotic cells compared to treatment with Stx1 alone. To determine whether targeting of DR5 and CHOP is involved in reduction of apoptosis in undifferentiated monocytic cells, cells were transfected with DR5 or CHOP siRNA and then incubated with Stx1 for 5 h, followed by annexin-PI apoptosis analysis. Although siRNAs effectively reduced DR5 and CHOP expression in monocytic THP-1 cells (Fig. 5D), unlike in macrophage-like cells, silencing of DR5 and CHOP had modest effects on cytotoxicity mediated by Stx1 treatment in the monocytic THP-1 cells (Fig. 5E). Treatment of cells with DR5 or CHOP siRNA alone did not cause a cytotoxic effect above spontaneous control levels (data not shown). We conclude that the ablation of DR5 or CHOP expression protects macrophage-like cells from Stx1-induced apoptosis. However, the data also support the concept that signaling through DR5 may play a relatively less important role in apoptosis induction in the highly toxin-sensitive monocytic THP-1 cells.
Signaling through CHOP-DR5 is involved in caspase-3 and -8 activation in Stx1-treated macrophage-like cells but not monocytic cells.
The upregulation of DR5 and subsequent binding of TRAIL may result in the formation of the death-inducing signaling complex (DISC) involved in the activation of the extrinsic and intrinsic pathways of apoptosis induction (21, 39). Initiator caspases which associate with the DISC are cleaved and, in turn, activate executioner caspases which then activate proximal mediators of apoptosis or inhibit prosurvival mediators, e.g., cleavage and activation of proapoptotic Bcl-2 family members, and cleavage and inactivation of DNA repair enzymes (30, 32, 37, 63). In response to Stx1 treatment, caspase-3 and -8 were the major caspases activated in both undifferentiated and differentiated THP-1 cells, showing maximal activation at 8 h after toxin treatment (34, 35). To further understand the requirements necessary to activate caspase-3 and -8, differentiated THP-1 cells were treated with Stx1, Stx1A− holotoxin, and Stx1 B subunits for 8 h. As shown in Fig. 6 A, Stx1 treatment increased caspase-3 and -8 activation (cleavage), while the activity was not detectable in Stx1A− and Stx1 B subunit-treated cells. Thus, as was the case with apoptosis induction, active Stx1 appeared to be necessary for caspase activation. We reasoned that if Stx1-induced upregulation of CHOP, DR5, and sTRAIL activates death receptor-mediated apoptotic pathways, then DR5 DISC-associated caspases should be activated. To evaluate this mechanism, both differentiated and undifferentiated THP-1 cells were transfected with siRNA duplexes to specifically silence DR5 and CHOP expression. After intoxication, Western blotting was carried out to measure caspase-3 and -8 activation. DR5 and CHOP knockdown reduced levels of caspase-3 and -8 activation in differentiated THP-1 cells (Fig. 6B), although some caspase activation was evident in siRNA-transfected lysates (Fig. 6B, bar graph). In contrast, caspase activation was not reduced following DR5 or CHOP knockdown in monocytic THP-1 cells (Fig. 6C).
FIG. 6.

Caspase activation following Stx1 treatment of DR5 or CHOP siRNA-transfected macrophage-like and monocytic THP-1 cells. (A) Differentiated THP-1 cells were treated with Stx1, Stx1A−, or Stx1 B subunits for 8 h. Cells were lysed, followed by Western blotting with anti-caspase-3 and -8 antibodies. Membranes were then stripped and reprobed with antiactin antibody for an equal-protein-loading control. (B) Differentiated THP-1 cells (D-THP1) were transfected with siRNAs specific for DR5 (siDR5), CHOP (siCHOP), or nontargeting siRNA (NTsiRNA) or were mock transfected (Mock) with reagent only for 72 h. After washing, cells were incubated with or without Stx1 for 8 h. Cell lysates were analyzed for caspase-3 and -8 cleavage as described above. A representative Western blot is shown in the left panel; the bar graph (right panel) depicts results from at least three independent experiments with caspase activation expressed as fold increase compared to control (untreated) cells. (C) Undifferentiated THP-1 cells (UD-THP1) were transfected with siRNAs and treated with Stx1 as outlined above. A representative Western blot showing caspase-3 and -8 cleavage is shown.
Signaling through the CHOP-DR5 pathway is involved in activating the mitochondrion-mediated pathway of apoptosis.
We have shown that Stx1 induced THP-1 cell apoptosis by the intrinsic (mitochondrial-mediated) pathway, involving BiD cleavage, cytochrome c release and disruption of mitochondrial membrane potential (34, 35). To understand cross talk between mitochondrion- and death receptor-mediated cytotoxic pathways activated by Stx1, CHOP or DR5 expression in differentiated THP-1 cells was silenced, and after siRNA transfection, the relative loss of mitochondrial membrane potential (ΔΨm) following Stx1 treatment for 24 h was detected using the JC-1 dye, which accumulates in the mitochondria and is released into the cytoplasm with the loss of mitochondrial membrane potential. As shown in Fig. 7, a partial reduction in ΔΨm following intoxication was observed in CHOP and DR5 knockdown cells (14% ± 0.01% and 16% ± 4.6%, respectively) compared to treatment with Stx1 alone (37.3% ± 4.7%). ΔΨm was not affected by Stx1A− holotoxin or Stx1 B-subunit treatment.
FIG. 7.
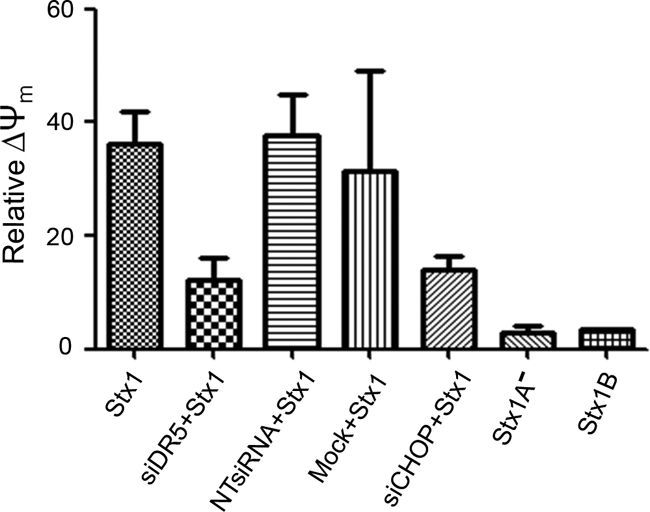
CHOP and DR5 signaling activates the mitochondrion-mediated pathway of apoptosis in Stx1-treated macrophage-like THP-1 cells. Differentiated THP-1 cells were transfected with siRNAs specific for DR5 (siDR5), CHOP (siCHOP), or nontargeting siRNA (NTsiRNA) or were mock transfected (Mock) with reagent only for 72 h and then treated without or with Stx1, Stx1A−, or Stx1 B subunits for 24 h. After incubation, JC-1 was used to measure ΔΨm (loss of mitochondrial membrane potential) by flow cytometry. Control cells were maintained in medium for 24 h. The percent ΔΨm was compared to that for control (untreated) cells. The data are the means ± SEM from three independent experiments.
Differential rates of Stx1-induced calpain cleavage correlate with different rates of Stx1-induced apoptosis in monocytic versus macrophage-like THP-1 cells.
It is tempting to speculate that siRNA-mediated DR5 and CHOP gene silencing reveals apoptosis-inducing pathways selectively operative in undifferentiated THP-1 cells, which would account for the failure of DR5 and CHOP knockdown to reduce caspase-3 and -8 activation (Fig. 6C). Furthermore, our finding that apoptosis is rapidly induced in monocytic THP-1 cells suggests that Stx1 may activate apoptotic mechanisms prior to upregulated TRAIL and DR5 expression. Calcium-dependent death signaling via proteolysis of calpain has been well characterized as an alternative pathway to activate caspases (2, 65). We reported that the ER stress response induced by Stx1 in monocytic THP-1 cells triggered the rapid release of Ca2+ from intracellular stores and the rapid activation of calpain, with calpain cleavage products evident within 1 h of Stx1 treatment (36). To examine whether a similar phenomenon occurs in macrophage-like THP-1 cells, cells were left untreated or treated with Stx1 for various times, cellular lysates were prepared, and calpain cleavage was detected by immunoblotting. Stx1 activated calpain cleavage in macrophage-like THP-1 cells, but with kinetics very different from those in monocytic cells. Calpain cleavage was first detected beginning 4 h after toxin treatment and progressively increased throughout the duration of the experiment (Fig. 8 A, left panel). It is noteworthy that we failed to detect calpain proteolysis in macrophage-like cells treated with Stx1A− for 12 h (Fig. 8A, right panel). In terms of kinetics and band intensities, Stx1-induced calpain cleavage was markedly faster and higher in undifferentiated monocytic cells than in toxin-treated differentiated macrophage-like cells (Fig. 8B). Thus, the early activation of calpain and caspases may contribute to the rapid-apoptosis phenotype of Stx1-treated monocytic THP-1 cells.
FIG. 8.

Stx1 treatment of macrophage-like THP-1 cells triggers the delayed activation of calpain, which requires toxin enzymatic activity. (A) Differentiated macrophage-like THP-1 cells were incubated without or with Stx1 for 0 to 24 h (left panel), or with Stx1 or Stx1A− for 12 h (right panel). Cell lysates were prepared and subjected to SDS-PAGE. Calpain cleavage was detected by Western blotting with calpain-specific antibodies. (B) Undifferentiated (U) and differentiated (D) THP-1 cells were incubated with Stx1 for various times. At the indicated time points, cell lysates were prepared and subjected to Western blotting to assess calpain cleavage as described for panel A. C, control (untreated) cells. Representative gels from at least three independent experiments are shown. The bar graph (below in panel B) depicts results from at least three independent experiments with calpain activation (cleavage) as fold increase compared to control values (untreated cells).
DISCUSSION
Apoptosis is programmed cell death which is initiated by intracellular signaling pathways. Apoptosis is essential for normal development, tissue/organ homeostasis, and removal of damaged cells. It has become clear that a number of bacterial pathogens have evolved mechanisms to manipulate the host apoptosis response, presumably to the benefit of the pathogen (10, 15). Apoptotic signaling can be categorized into two pathways, the intrinsic and extrinsic pathways. The intrinsic pathway is initiated in response to cellular stress, such as DNA damage, hypoxia, or the presence of misfolded proteins within the ER. The intrinsic pathway is activated through changes in mitochondria that affect membrane permeability and potential. The extrinsic pathway is activated through engagement of death domain-containing receptors of the TNF receptor superfamily, TNFR1, Fas, and the TRAIL receptors, with their cognate apoptosis-inducing ligands, TNF-α, FasL, and TRAIL, respectively (11, 27). We have previously shown that the treatment of monocytic and macrophage-like THP-1 cells with Stx1 activates signaling cascades which may contribute to initiation of both intrinsic and extrinsic pathways of apoptosis induction, including signaling through the ER stress response, the rapid activation of caspase-8 and BID, the release of intracellular Ca2+ from ER, stores and activation of calpains (22, 35, 36).
We show here that Stx1 treatment of monocytic and macrophage-like THP-1 cells resulted in increased expression of TRAIL and DR5. TRAIL is a type II transmembrane protein originally cloned based on its sequence homology to TNF-α and FasL (45, 66). Like other members of the TNF superfamily, TRAIL may be cleaved by metalloproteases to form bioactive soluble TRAIL (41). Similar to other members of the TNF superfamily, TRAIL forms homotrimers and therefore induces the trimerization of its membrane receptors. Five TRAIL receptors have been characterized, i.e., DR4, DR5, DcR1, DcR2, and osteoprotegerin (OPG). In humans, only DR4 and DR5 possess intact death domains, while DcR1 and DcR2 are considered “decoy” receptors lacking functional death domains, and TRAIL binds OPG with lower affinity than it does DR4 and DR5. When DR4 or DR5 binds TRAIL, trimerization recruits the adaptor protein FADD, as well as procaspase-8 or procaspase-10, to the receptor-ligand complex. Following autoproteolysis to generate functional caspase-8, caspase-3, -6, and -7 may be directly activated, or BID may be cleaved to activate apoptotic signaling cascades through mitochondrial membrane perturbation.
The RT-PCR analyses presented here suggest that DR5 expression is positively regulated by Stx1, at least in part, at the transcriptional level. Apoptotic signaling through TRAIL-DR5 ligation is functional in monocytic and macrophage-like THP-1 cells, as evidenced by the increased percentages of apoptotic cells detected when exogenous sTRAIL was added to intoxicated cells. Anti-TRAIL antibody treatment protected cells from cytotoxicity mediated by Stx1. Toxin enzymatic activity was required for increased TRAIL and DR5 expression. However, the protein synthesis inhibitor cycloheximide, which does not undergo retrograde transport to the ER or undergo retrotranslocation across the ER membrane, failed to induce apoptosis or increased DR5 expression. TRAIL binding to the death domain-containing receptor DR5 results in receptor trimerization and recruitment of the DISC, which contains procaspase-8. Within this macromolecular scaffold, procaspase-8 is thought to be activated by proximity-dependent autoproteolysis (19). Caspase-8 may then directly cleave procaspase-3 or, alternatively, may cleave the BH3-only Bcl2 family member BID. In its truncated form, tBID intercalates within the mitochondrial membrane, where it may facilitate pore formation by the proapoptotic Bcl2 family members Bax and Bak, leading to increased mitochondrial membrane permeability, release of cytochrome c, formation of the apoptosome, activation of caspase-9 and -3, and apoptosis (11, 27). We have previously shown that these downstream events follow treatment of monocytic and macrophage-like THP-1 cells with Stx1 (22, 35, 36), suggesting that TRAIL-DR5 ligation occurs and facilitates apoptosis induction in these cells. Furthermore, TRAIL and DR5 seem to be uniquely involved in Stx1-induced apoptosis in THP-1 cells, as we have shown that other members of the tumor necrosis factor and tumor necrosis factor receptor superfamilies, TNF/TNFR1 and FasL (Apo)/Fas, are not involved in apoptotic signaling in THP-1 cells (34, 35).
The precise mechanism by which Stx1 regulates TRAIL and DR5 expression remains to be characterized, although the signaling proteins TRAF-2 and RIP are known to associate with the TRAIL-DR5 DISC. Signaling through TRAF-2 and RIP activates the transcriptional factor NF-κB (52). We have previously shown that Stx1 activates NF-κB signaling in macrophage-like THP-1 cells, and TRAIL and DR5 gene expression is regulated by NF-κB (20, 48, 54, 64). DR5 expression is also regulated by CHOP (68), and Stx1 treatment of monocytic and macrophage-like THP-1 cells increases CHOP expression (33, 36). Furthermore, we show here that RNAi knockdown of CHOP and DR5 expression partially protects macrophage-like cells from the cytotoxic action of Stx1. Taken together, these findings suggest that apoptosis induction by Stx1 may occur in multiple stages involving both the intrinsic and extrinsic pathways. Functional toxins must be routed to the ER, processed, and translocated into the cytoplasm. Retrograde transport and toxin enzymatic activity then initiate the ER stress response, leading to rapid caspase-8 activation and activation of NF-κB and CHOP signaling pathways. This initial response to Stx1 leads to increased TRAIL and DR5 expression, which, acting in an autocrine or paracrine manner, may augment apoptotic signaling initiated by the toxin. However, TRAIL-DR5 signaling may be relatively more important in the delayed-apoptosis phenotype of differentiated, macrophage-like THP-1 cells, as inhibition of DR5 or CHOP expression with siRNAs did not fully attenuate caspase activation or apoptosis in undifferentiated, monocytic THP-1 cells. These data suggest that other proapoptotic signaling cascades may contribute to rapid apoptosis induction. In this regard, the differential kinetics of calpain activation leading to direct caspase activation may contribute to the cell maturation-dependent differences in apoptosis induction that we have observed using THP-1 cells (Fig. 6). As shown in Fig. 8, calpain is rapidly cleaved in Stx1-treated monocytic cells, while calpain activation is delayed in macrophage-like cells.
While we have focused on the elucidation of Stx1-induced apoptotic signaling mechanisms, it is important to note that only a fraction of differentiated THP-1 cells die following prolonged (72-h) exposure to Stx1 (22). The mechanism of cell survival in the face of intoxication is not clear, but it is important to note that following binding to DR4 or DR5, TRAIL has been reported to activate both apoptotic and prosurvival signaling in some cell types. Prosurvival signaling is mediated through activation of NF-κB, Akt (or protein kinase B), and the mitogen-activated protein kinases (MAPKs) extracellular signal-regulated kinase (ERK), Jun N-terminal protein kinase (JNK), and p38 (7). We have shown that NF-κB, Akt, and MAPK pathways are activated by Stxs in macrophage-like THP-1 cells (4, 16, 48), suggesting that survival of toxin challenge by some differentiated THP-1 cells, and the delayed onset of apoptosis seen with other cells, may be related to the activation of “balanced” survival versus apoptosis signaling pathways in the macrophage-like cells, while signaling in the monocytic cells primarily triggers apoptosis.
The human leukemia cell line THP-1 expresses Gb3 (47) and contains a mutation in the gene encoding the tumor suppressor p53 (58). Gb3-expressing tumors are frequently aggressive and resistant to anticancer drugs, and p53-deficient cells frequently develop resistance to chemotherapeutic agents. Thus, novel treatment options for these malignant tumors are needed. The capacity of Stx1 to induce apoptosis in Gb3+ p53− leukemia cells highlights the potential for the toxins to be utilized as anticancer agents. A single intratumor injection of Stx1 into athymic nude mice bearing Gb3+ human malignant meningioma xenografts improved survival. Survival was associated with the marked increase of TUNEL+ cells within the graft (49). In a screening of 25 breast cancer tissues for sensitivity to Stx1-induced apoptosis, it was shown that sensitivity correlated with Gb3 expression (28). Using immunohistochemical techniques, only 17 of the 25 tissue samples expressed Gb3. However, arterioles serving the tumors were rich in Gb3, suggesting that Shiga toxins may be used to disrupt the vascular networks serving tumors. A major hurdle in the use of bacterial toxins in cancer therapy is targeting of the cytotoxic effect to transformed cells while sparing normal cells. Our observation that Stx1 increases TRAIL and DR5 expression in a myelogenous leukemia cell line is interesting in light of the fact that TRAIL is reported to be selectively involved in apoptosis induction in transformed cells, while most nontransformed cells are refractory to apoptosis induction by TRAIL (1, 27). Furthermore, downregulation of expression of the antiapoptotic Bcl-2 family member Bcl-2 (3, 59), the caspase-8 activation inhibitor c-FLIP (24, 29), or the apoptosis inhibitor XIAP (9) has been shown to sensitize transformed cells to the cytotoxic action of TRAIL. Shiga toxins have been shown to be capable of downregulating expression of Bcl-2 and IAPs (35, 36) in monocytic THP-1 cells and of inducing rapid c-FLIP degradation in endothelial cells (13). Thus, our observation that the addition of sTRAIL increases apoptosis in the presence of Stx1 may be of potential clinical relevance. Recombinant human TRAIL (rhTRAIL) is currently undergoing clinical trials for efficacy in treatment of tumors and hematological malignancies (67). A number of chemotherapeutic agents, such as rituximab or paclitaxel, carboplatin, and bevacizumab combination therapy, have been shown to increase the efficacy of rhTRAIL. The potential for Shiga toxins to be employed as specific cytotoxic agents for transformed cells will require a more thorough survey of multiple transformed cells types as well as screening of primary cells for TRAIL and DR5 expression and apoptosis induction following intoxication.
Acknowledgments
This work was supported by Public Health Service grant RO1 AI34530 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (to V.L.T.).
We thank Shinji Yamasaki and Cheleste Thorpe for gifts of reagents necessary to carry out the experiments.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 1 June 2010.
REFERENCES
- 1.Ashkenazi, A., and V. M. Dixit. 1998. Death receptors: signaling and modulation. Science 281:1305-1308. [DOI] [PubMed] [Google Scholar]
- 2.Blomgren, K., C. Zhu, X. Wang, J. O. Karlsson, A. L. Leverin, B. A. Bahr, C. Mallard, and H. Hagberg. 2001. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J. Biol. Chem. 276:10191-10198. [DOI] [PubMed] [Google Scholar]
- 3.Chawla-Sarkar, M., S. I. Bae, F. J. Reu, B. S. Jacobs, D. J. Lindner, and E. C. Borden. 2004. Downregulation of Bcl-2, FLIP or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 11:915-923. [DOI] [PubMed] [Google Scholar]
- 4.Cherla, R. P., S.-Y. Lee, R. A. Mulder, M.-S. Lee, and V. L. Tesh. 2009. Shiga toxin 1-induced proinflammatory cytokine production is regulated by the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling pathway. Infect. Immun. 77:3919-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cherla, R. P., S.-Y. Lee, and V. L. Tesh. 2003. Shiga toxins and apoptosis. FEMS Microbiol. Lett. 228:159-166. [DOI] [PubMed] [Google Scholar]
- 6.Ching, J. C., N. L. Jones, P. J. Ceponis, M. A. Karmali, and P. M. Sherman. 2002. Escherichia coli Shiga-like toxins induce apoptosis and cleavage of poly(ADP-ribose) polymerase via in vitro activation of caspases. Infect. Immun. 70:4669-4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corazza, N., D. Kassahn, S. Jakob, A. Badmann, and T. Brunner. 2009. TRAIL-induced apoptosis: between tumor therapy and immunopathology. Ann. N. Y. Acad. Sci. 1171:50-58. [DOI] [PubMed] [Google Scholar]
- 8.Cory, A. H., T. C. Owen, J. A. Barltrop, and J. G. Cory. 1991. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 3:207-212. [DOI] [PubMed] [Google Scholar]
- 9.Cummins, J. M., M. Kohli, C. Rago, K. W. Kinzler, B. Vogelstein, and F. Bunz. 2004. X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Res. 64:3006-3008. [DOI] [PubMed] [Google Scholar]
- 10.DeLeo, F. R. 2004. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis 9:399-413. [DOI] [PubMed] [Google Scholar]
- 11.Elmore, S. 2007. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35:495-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Endo, Y., K. Tsurugi, T. Yutsudo, Y. Takeda, T. Ogasawara, and K. Igarashi. 1988. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 171:45-50. [DOI] [PubMed] [Google Scholar]
- 13.Erwert, R. D., R. K. Winn, J. M. Harlan, and D. D. Bannerman. 2002. Shiga-like toxin inhibition of FLICE-like inhibitory protein expression sensitizes endothelial cells to bacterial lipopolysaccharide-induced apoptosis. J. Biol. Chem. 277:40567-40574. [DOI] [PubMed] [Google Scholar]
- 14.Fadok, V. A., D. R. Voelker, P. A. Campbell, J. J. Cohen, D. L. Bratton, and P. M. Henson. 1992. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148:2207-2216. [PubMed] [Google Scholar]
- 15.Faherty, C. S., and A. T. Maurelli. 2008. Staying alive: bacterial inhibition of apoptosis during infection. Trends Microbiol. 16:173-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster, G. H., and V. L. Tesh. 2002. Shiga toxin 1-induced activation of c-Jun NH(2)-terminal kinase and p38 in the human monocytic cell line THP-1: possible involvement in the production of TNF-alpha. J. Leukoc. Biol. 71:107-114. [PubMed] [Google Scholar]
- 17.Fraser, M. E., M. M. Chernaia, Y. V. Kozlov, and M. N. G. James. 1994. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 Å resolution. Nat. Struct. Biol. 1:59-64. [DOI] [PubMed] [Google Scholar]
- 18.Fujii, J., T. Matsui, D. P. Heatherly, K. H. Schlegel, P. I. Lobo, T. Yutsudo, G. M. Ciraolo, R. E. Morris, and T. Obrig. 2003. Rapid apoptosis induced by Shiga toxin in HeLa cells. Infect. Immun. 71:2724-2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fulda, S. 2009. Caspase-8 in cancer biology and therapy. Cancer Lett. 281:128-133. [DOI] [PubMed] [Google Scholar]
- 20.Gong, B., and A. Almasan. 2000. Apo2 ligand/TNF-related apoptosis-inducing ligand and death receptor 5 mediate the apoptotic signaling induced by ionizing radiation in leukemic cells. Cancer Res. 60:5754-5760. [PubMed] [Google Scholar]
- 21.Green, D. R. 2000. Apoptotic pathways: paper wraps stone blunts scissors. Cell 102:1-4. [DOI] [PubMed] [Google Scholar]
- 22.Harrison, L. M., R. P. Cherla, C. van den Hoogen, W. C. van Haaften, S.-Y. Lee, and V. L. Tesh. 2005. Comparative evaluation of apoptosis induced by Shiga toxin 1 and/or lipopolysaccharides in human monocytic and macrophage-like cells. Microb. Pathog. 38:63-76. [DOI] [PubMed] [Google Scholar]
- 23.He, Q., D. I. Lee, R. Rong, M. Yu, X. Luo, M. Klein, W. S. El-Deiry, Y. Huang, A. Hussain, and M. S. Sheikh. 2002. Endoplasmic reticulum calcium pool depletion-induced apoptosis is coupled with activation of the death receptor 5 pathway. Oncogene 21:2623-2633. [DOI] [PubMed] [Google Scholar]
- 24.Hietakangas, V., M. Poukkula, K. M. Heiskanen, J. T. Karvinen, L. Sistonen, and J. E. Eriksson. 2003. Erythroid differentiation sensitizes K562 leukemia cells to TRAIL-induced apoptosis by downregulation of c-FLIP. Mol. Cell. Biol. 23:1278-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huppi, K., S. E. Martin, and N. J. Caplen. 2005. Defining and assaying RNAi in mammalian cells. Mol. Cell 17:1-10. [DOI] [PubMed] [Google Scholar]
- 26.Hymowitz, S. G., M. P. O'Connell, M. H. Ultsch, A. Hurst, K. Totpal, A. Ashkenazi, A. M. de Vos, and R. F. Kelley. 2000. A unique zinc-binding site revealed by a high-resolution X-ray structure of homotrimeric Apo2L/TRAIL. Biochemistry 39:633-640. [DOI] [PubMed] [Google Scholar]
- 27.Jin, Z., and W. S. El-Deiry. 2005. Overview of cell death signaling pathways. Cancer Biol. Ther. 4:139-163. [DOI] [PubMed] [Google Scholar]
- 28.Johansson, D., E. Kosovac, J. Moharer, I. Ljuslinder, T. Brannstrom, A. Johansson, and P. Behnam-Motlagh. 2009. Expression of verotoxin-1 receptor Gb3 in breast cancer tissue and verotoxin-1 signal transduction to apoptosis. BMC Cancer 9:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim, Y., N. Suh, M. Sporn, and J. C. Reed. 2002. An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. J. Biol. Chem. 277:22320-22329. [DOI] [PubMed] [Google Scholar]
- 30.Kischkel, F. C., D. A. Lawrence, A. Chuntharapai, P. Schow, K. J. Kim, and A. Ashkenazi. 2000. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 12:611-620. [DOI] [PubMed] [Google Scholar]
- 31.Kuang, A. A., G. E. Diehl, J. Zhang, and A. Winoto. 2000. FADD is required for DR4- and DR5-mediated apoptosis: lack of TRAIL-induced apoptosis in FADD-deficient mouse embryonic fibroblasts. J. Biol. Chem. 275:25065-25068. [DOI] [PubMed] [Google Scholar]
- 32.Lee, J. H., J. Y. Jung, Y. J. Jeong, J. H. Park, K. H. Yang, N. K. Choi, S. H. Kim, and W. J. Kim. 2008. Involvement of both mitochondrial- and death receptor-dependent apoptotic pathways regulated by Bcl-2 family in sodium fluoride-induced apoptosis of the human gingival fibroblasts. Toxicology 243:340-347. [DOI] [PubMed] [Google Scholar]
- 33.Lee, M.-S., R. P. Cherla, D. Leyva-Illades, and V. L. Tesh. 2009. Bcl-2 regulates the onset of Shiga toxin 1-induced apoptosis in THP-1 cells. Infect. Immun. 77:5233-5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee, S.-Y., R. P. Cherla, I. Caliskan, and V. L. Tesh. 2005. Shiga toxin 1 induces apoptosis in the human myelogenous leukemia cell line THP-1 by a caspase-8-dependent, tumor necrosis factor receptor-independent mechanism. Infect. Immun. 73:5115-5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee, S.-Y., R. P. Cherla, and V. L. Tesh. 2007. Simultaneous induction of apoptotic and survival signaling pathways in macrophage-like THP-1 cells by Shiga toxin 1. Infect. Immun. 75:1291-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee, S.-Y., M.-S. Lee, R. P. Cherla, and V. L. Tesh. 2008. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 10:770-780. [DOI] [PubMed] [Google Scholar]
- 37.Li, H., H. Zhu, C. J. Xu, and J. Yuan. 1998. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491-501. [DOI] [PubMed] [Google Scholar]
- 38.Lingwood, C. A. 2003. Shiga toxin receptor glycolipid binding. Pathology and utility. Methods Mol. Med. 73:165-186. [DOI] [PubMed] [Google Scholar]
- 39.MacFarlane, M., M. Ahmad, S. M. Srinivasula, T. Fernandes-Alnemri, G. M. Cohen, and E. S. Alnemri. 1997. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J. Biol. Chem. 272:25417-25420. [DOI] [PubMed] [Google Scholar]
- 40.Malhi, H., and G. J. Gores. 2006. TRAIL resistance results in cancer progression: a TRAIL to perdition? Oncogene 25:7333-7335. [DOI] [PubMed] [Google Scholar]
- 41.Mariani, S. M., and P. H. Krammer. 1998. Differential regulation of TRAIL and CD95 ligand in transformed cells of the T and B lymphocyte lineage. Eur. J. Immunol. 28:973-982. [DOI] [PubMed] [Google Scholar]
- 42.Mead, P. S., L. Slutsker, V. Dietz, L. F. McCaig, J. S. Bresee, C. Shapiro, P. M. Griffin, and R. V. Tauxe. 1999. Food-related illness and death in the United States. Emerg. Infect. Dis. 5:607-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moxley, R. A., and D. H. Francis. 1998. Overview of animal models, p. 249-260. In J. B. Kaper, A. D. O'Brien, (ed.), Escherichia coli O157:H7 and other Shiga toxin-producing E. coli strains. ASM Press, Washington, DC.
- 44.Ohmura, M., S. Yamasaki, H. Kurazono, K. Kashiwagi, K. Igarashi, and Y. Takeda. 1993. Characterization of non-toxic mutant toxins of Vero toxin 1 that were constructed by replacing amino acids in the A subunit. Microb. Pathog. 15:169-176. [DOI] [PubMed] [Google Scholar]
- 45.Pitti, R. M., S. A. Marsters, S. Ruppert, C. J. Donahue, A. Moore, and A. Ashkenazi. 1996. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 271:12687-12690. [DOI] [PubMed] [Google Scholar]
- 46.Proulx, F., and V. L. Tesh. 2007. Renal diseases in the pediatric intensive care unit: thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura, p. 1189-1204 In H. R. Wong, D. S. Wheeler, and T. P. Shanley (ed.), Pediatric critical care medicine: basic science and clinical evidence. Springer Verlag, London, United Kingdom.
- 47.Ramegowda, B., and V. L. Tesh. 1996. Differentiation-associated toxin receptor modulation, cytokine production, and sensitivity to Shiga-like toxins in human monocytes and monocytic cell lines. Infect. Immun. 64:1173-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakiri, R., B. Ramegowda, and V. L. Tesh. 1998. Shiga toxin type 1 activates tumor necrosis factor-alpha gene transcription and nuclear translocation of the transcriptional activators nuclear factor-kappaB and activator protein-1. Blood 92:558-566. [PubMed] [Google Scholar]
- 49.Salhia, B., J. T. Rutka, C. Lingwood, A. Nutikka, and W. R. Van Furth. 2002. The treatment of malignant meningioma with verotoxin. Neoplasia 4:304-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sandvig, K. 2001. Shiga toxins. Toxicon 39:1629-1635. [DOI] [PubMed] [Google Scholar]
- 51.Saxena, S. K., A. D. O'Brien, and E. J. Ackerman. 1989. Shiga toxin, Shiga-like toxin II variant, and ricin are all single-site RNA N-glycosidases of 28 S RNA when microinjected into Xenopus oocytes. J. Biol. Chem. 264:596-601. [PubMed] [Google Scholar]
- 52.Schneider, P., M. Thome, K. Burns, J. L. Bodmer, K. Hofmann, T. Kataoka, N. Holler, and J. Tschopp. 1997. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 7:831-836. [DOI] [PubMed] [Google Scholar]
- 53.Sheikh, M. S., and Y. Huang. 2004. Death receptors as targets of cancer therapeutics. Curr. Cancer Drug Targets 4:97-104. [DOI] [PubMed] [Google Scholar]
- 54.Shetty, S., J. B. Gladden, E. S. Henson, X. Hu, J. Villanueva, N. Haney, and S. B. Gibson. 2002. Tumor necrosis factor-related apoptosis inducing ligand (TRAIL) up-regulates death receptor 5 (DR5) mediated by NFkappaB activation in epithelial derived cell lines. Apoptosis 7:413-420. [DOI] [PubMed] [Google Scholar]
- 55.Strasser, A. 2005. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 5:189-200. [DOI] [PubMed] [Google Scholar]
- 56.Strockbine, N. A., L. R. M. Marques, J. W. Newland, H. Willams-Smith, R. K. Holmes, and A. D. O'Brien. 1986. Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect. Immun. 53:135-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Su, R. Y., K. H. Chi, D. Y. Huang, M. H. Tai, and W. W. Lin. 2008. 15-Deoxy-Delta12,14-prostaglandin J2 up-regulates death receptor 5 gene expression in HCT116 cells: involvement of reactive oxygen species and C/EBP homologous transcription factor gene transcription. Mol. Cancer Ther. 7:3429-3440. [DOI] [PubMed] [Google Scholar]
- 58.Sugimoto, K., H. Toyoshima, R. Sakai, K. Miyagawa, K. Hagiwara, F. Ishikawa, F. Takaku, Y. Yazaki, and H. Hirai. 1992. Frequent mutations in the p53 gene in human myeloid leukemia cell lines. Blood 79:2378-2383. [PubMed] [Google Scholar]
- 59.Sun, S. Y., P. Yue, J. Y. Zhou, Y. Wang, H. R. Choi Kim, R. Lotan, and G. S. Wu. 2001. Overexpression of BCL2 blocks TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in human lung cancer cells. Biochem. Biophys. Res. Commun. 280:788-797. [DOI] [PubMed] [Google Scholar]
- 60.Tarr, P. I., C. A. Gordon, and W. L. Chandler. 2005. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365:1073-1086. [DOI] [PubMed] [Google Scholar]
- 61.Taylor, F. B., Jr., V. L. Tesh, L. DeBault, A. Li, A. C. K. Chang, S. D. Kosanke, T. J. Pysher, and R. L. Siegler. 1999. Characterization of the baboon responses to Shiga-like toxin: descriptive study of a new primate model of toxic responses to Stx-1. Am. J. Pathol. 154:1285-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tesh, V. L., J. A. Burris, J. W. Owens, V. M. Gordon, E. A. Wadolkowski, A. D. O'Brien, and J. E. Samuel. 1993. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 61:3392-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thorburn, A. 2007. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) pathway signaling. J. Thorac. Oncol. 2:461-465. [DOI] [PubMed] [Google Scholar]
- 64.Wang, Q., Y. Ji, X. Wang, and B. M. Evers. 2000. Isolation and molecular characterization of the 5′-upstream region of the human TRAIL gene. Biochem. Biophys. Res. Commun. 276:466-471. [DOI] [PubMed] [Google Scholar]
- 65.Waterhouse, N. J., D. M. Finucane, D. R. Green, J. S. Elce, S. Kumar, E. S. Alnemri, G. Litwack, K. Khanna, M. F. Lavin, and D. J. Watters. 1998. Calpain activation is upstream of caspases in radiation-induced apoptosis. Cell Death Differ. 5:1051-1061. [DOI] [PubMed] [Google Scholar]
- 66.Wiley, S. R., K. Schooley, P. J. Smolak, W. S. Din, C. P. Huang, J. K. Nicholl, G. R. Sutherland, T. D. Smith, C. Rauch, C. A. Smith, and R. G. Goodwin. 1995. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3:673-682. [DOI] [PubMed] [Google Scholar]
- 67.Wu, G. S. 2009. TRAIL as a target in anti-cancer therapy. Cancer Lett. 285:1-5. [DOI] [PubMed] [Google Scholar]
- 68.Yamaguchi, H., and H. G. Wang. 2004. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 279:45495-45502. [DOI] [PubMed] [Google Scholar]