

Abstract
Biofilm formation plays a multifaceted role in the life cycles of a wide variety of microorganisms. In the case of pathogenic Vibrio cholerae, biofilm formation in its native aquatic habitats is thought to aid in persistence during interepidemic seasons and to enhance infectivity upon oral ingestion. The structure of V. cholerae biofilms has been hypothesized to protect the bacteria during passage through the stomach. Here, we directly test the role of biofilm architecture in the infectivity of V. cholerae by comparing the abilities of intact biofilms, dispersed biofilms, and planktonic cells to colonize the mouse small intestine. Not only were V. cholerae biofilms better able to colonize than planktonic cells, but the structure of the biofilm was also found to be dispensable: intact and dispersed biofilms colonized equally, and both vastly out-colonized planktonic cells. The infectious dose for biofilm-derived V. cholerae was orders of magnitude lower than that of planktonic cells. This biofilm-induced hyperinfectivity may be due in part to a higher growth rate of biofilm-derived cells during infection. These results suggest that the infectious dose of naturally occurring biofilms of V. cholerae may be much lower than previously estimated using cells grown planktonically in vitro. Furthermore, this work implies the existence of factors specifically induced during growth in a biofilm that augment infection by V. cholerae.
Bacteria are often found in biofilms, surface-attached aggregates of microorganisms encased in an extracellular polysaccharide or protein matrix (14). Mature bacterial biofilms often assume a three-dimensional structure composed of pillars of bacteria separated by fluid-filled channels (15). Compared to their free-living, planktonic (PL) counterparts, biofilm-associated bacteria have been shown to be recalcitrant to a variety of stresses and antimicrobial agents, including chlorine, low pH, UV irradiation, antibiotics, host defenses, and more (4, 16, 21, 22, 39, 40, 44, 63, 72). The structure of the biofilm itself has been thought to physically protect the bacteria within. The decreased susceptibility of biofilms to antibiotics, for example, is understood to be due at least in part to decreased permeability of the biofilm to the antibiotic (20, 30, 60). However, growth rate and metabolic state have also been proposed to contribute to biofilm-related protection from certain antimicrobials and other stresses (5, 25, 59, 68). In addition, distinct genetic mechanisms of antibiotic resistance employed by biofilm microorganisms have been described (34, 41, 42).
The ability to form biofilms is a virulence determinant of many microorganisms. Classic examples of biofilm infections include chronic infections of the cystic fibrosis lung by Pseudomonas aeruginosa, Haemophilus influenzae and Streptococcus pneumoniae in chronic otitis media, uropathogenic Escherichia coli in recurrent urinary tract infections, and disease caused by microbial biofilms on a variety of indwelling medical devices (27, 35, 36, 52, 56). The reduced susceptibility of in vivo biofilms to antimicrobials has a huge impact on human health due to the difficulty involved in their eradication.
In other instances, biofilm formation contributes to a microorganism's survival in an environmental niche, such as in water system piping or on other solid surfaces, and consequently affects the likelihood of contact with a host (24, 58, 69). Vibrio cholerae is the causative agent of cholera and is a natural inhabitant of freshwater, marine, and estuarine environments. In the aquatic environment, V. cholerae has been observed to form biofilms on abiotic and biotic surfaces, including those of zooplankton, phytoplankton, algae, and crustaceans (23, 33, 62). This surface-attached state is thought to be the primary means of persistence of V. cholerae in the environment, providing protection from a variety of stresses and, when the bacteria are attached to chitin, a source of nutrients and a forum for acquiring new genetic material (43, 45, 46, 50, 67, 71). Moreover, biofilms are likely a form in which pathogenic (toxigenic) V. cholerae is consumed by humans, and they provide a means by which humans can obtain a concentrated infective dose (13, 28, 32). Because chitin- and biofilm-associated V. cholerae is better able to survive acid exposure (50, 72), it has been hypothesized that V. cholerae biofilms are protected during transit through the gastric acid barrier of the stomach, thus allowing more bacteria to reach the small intestine colonization site. Yet the role of stomach acid in susceptibility to cholera is unclear in humans (26), and the infant mouse, with neutral pH in the stomach, may be an irrelevant model of this phenomenon.
In this work, we test the hypothesis that the biofilm structure itself enhances colonization of the small intestine by V. cholerae. Instead, we found that V. cholerae bacteria dispersed from a biofilm are as infectious as those in an intact biofilm and that both are dramatically more infectious than free-living, planktonic cells in the infant mouse model. This work suggests that the physiological state of V. cholerae in biofilms, and not the biofilm structure, is the primary contributor to hyperinfectivity.
MATERIALS AND METHODS
Growth conditions.
V. cholerae was routinely grown in Luria-Bertani (LB) broth containing 100 μg/ml streptomycin (LB-Sm). Unless otherwise indicated, V. cholerae O1 El Tor strain C6709 was used. Where indicated, ampicillin (Ap) or kanamycin (Kn) was used at 50 μg/ml. For planktonic samples, V. cholerae was grown at 37°C with aeration for 16 h unless stated otherwise. Strains are described in Table 1.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or phenotype | Reference or source |
|---|---|---|
| V. cholerae strains | ||
| C6709 | O1 El Tor Ogawa; HapR+ | 55 |
| C6709 lacZ | lacZ::res-tet-res | 11 |
| N16961 | O1 El Tor Inaba; HapR− | 54 |
| N16961 lacZ | lacZ::res-tet-res | Lab strain |
| C6706str2 | O1 El Tor Inaba; HapR+ | 64 |
| C6706str2 lacZ | In-frame deletion of lacZ | This work |
| C6709(pACtsKan) | Temperature-sensitive plasmid in C6709 | This work |
| C6709 lacZ(pACtsKan) | Temperature-sensitive plasmid in C6709 lacZ | This work |
| C6709 lacZ(pVpsT) | pVpsT in C6709 lacZ; elevated VPS production | This work |
| C6709(pTrc99A) | pTrc99A in C6709 | This work |
| C6709 cheY-3 | Point mutation (D52N) in cheY-3; nonchemotactic | 38 |
| C6709 hapR | pGP704 plasmid insertion in hapR | This work |
| C6709 cdpA | pGP704 plasmid insertion in cdpA (VC0130) | 61 |
| C6709 pilTU | In-frame deletion of both pilT and pilU (adjacent open reading frames) | This work |
| C6709 cspA | In-frame deletion of VC0687 | This work |
| C6709 bug | In-frame deletion of VC1334 | This work |
| C6709 VC1516 | In-frame deletion of VC1516 | This work |
| C6709 VC2704 | In-frame deletion of VC2704 | This work |
| C6709 VC2705 | In-frame deletion of VC2705 | This work |
| C6709 napF | In-frame deletion of VCA0676 | This work |
| C6709 uhpA | In-frame deletion of VCA0682 | This work |
| E. coli strains | ||
| DH5α | λ− φ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | 29, 36 |
| DH5α λpir | F− Δ(lacZYA-argF)U169 recA1 endA1 hsdR17 supE44 thi-1 gvrA96 relA1 λpir | 29, 36 |
| SM10 λpir | thi recA thr leu tonA lacY supE RP4-2 Tc::Mu λpir | 48 |
| HB101 | pro leu thi lacY StrrendoI recA(r− m−) | 17 |
| Plasmids | ||
| pTrc99A | pBR322 derivative; Apr | 3 |
| pVpsT | vpsT cloned in pTrc99A; Apr | This work |
| pACtsKan | Temperature-sensitive origin; Knr | This work |
| pRK2013 | Helper plasmid, RK2 transfer genes, ColE1 ori; Knr | 17 |
| pCVD442 | oriR6K mobRP4 sacB; Apr | 18 |
| pCVD442::ΔlacZ | Allelic exchange vector for in-frame deletion of lacZ | This work |
| pGP704::hapR | Suicide vector for inactivating hapR | 70 |
| pCVD442::ΔpilTU | Allelic exchange vector for in-frame deletion of pilTU | This work |
| pCVD442::ΔVC0687 | Allelic exchange vector for in-frame deletion of VC0687 | This work |
| pCVD442::ΔVC1334 | Allelic exchange vector for in-frame deletion of VC1334 | This work |
| pCVD442::ΔVC1516 | Allelic exchange vector for in-frame deletion of VC1516 | This work |
| pCVD442::ΔVC2704 | Allelic exchange vector for in-frame deletion of VC2704 | This work |
| pCVD442::ΔVC2705 | Allelic exchange vector for in-frame deletion of VC2705 | This work |
| pCVD442::ΔVCA0676 | Allelic exchange vector for in-frame deletion of VCA0676 | This work |
| pCVD442::ΔVCA0682 | Allelic exchange vector for in-frame deletion of VCA0682 | This work |
Strain construction.
Plasmids used in this study are listed in Table 1. Sequences of primers used in this study are available upon request. In-frame deletions were made using standard allelic exchange methods as described previously (18, 60).
For ectopic expression of vpsT, the vpsT open reading frame was amplified by PCR using primers vpsTF and vpsTR and cloned into pTrc99A using the NcoI and SalI sites introduced with the primers. The desired construct was confirmed by PCR and named pVpsT. V. cholerae was transformed with pVpsT or pTrc99A by electroporation and confirmed to contain the plasmid by PCR.
Plasmid pACtsKan was constructed in this study and consists of the following sequences, in order: temperature-sensitive pSC101 repA101 (replication protein) and origin from pHSF422 (6), mobRP4 (mobilization sequence), E. coli lacI and lac promoter controlling expression of mar (Himar1 Mariner transposase), magellan6 mini-transposon (66), neo (neomycin and kanamycin resistance), and sacB (sucrose sensitivity). The complete sequence of pACtsKan is available from the authors upon request. Plasmid pACtsKan was introduced into C6709 and C6709 lacZ by triparental conjugation with E. coli DH5α λpir bearing pACtsKan and E. coli HB101(pRK2013::Tn9) (17).
Biofilms.
Biofilms of V. cholerae were grown at room temperature (21 to 23°C) under static conditions. In pilot studies using both intact and dispersed biofilms, biofilms were grown on glass slides propped in petri dishes containing 10 ml of LB-Sm broth. Robust biofilms were observed by 48 h. Slide biofilms were recovered by washing the slides four to five times in saline, and then the biofilm material was removed using a sterile razor blade and placed in saline. Parallel samples were either crudely broken up into uniform pieces (intact biofilms [iBF]) or dispersed (dBF) into individual cells by vortexing extensively with silica beads. Complete dispersion was confirmed by microscopy. Plating of serial dilutions showed consistent recovery of 6.3 × 107 ± 4.1 × 107 CFU per slide.
In subsequent studies comparing dBF to PL cells, biofilms were grown statically in 0.5 ml of LB-Sm medium in borosilicate glass tubes for 24 or 48 h. To obtain dBF samples, the supernatant was removed, and the remaining biofilm was washed with saline. The attached cells were resuspended in 1 ml of saline and dispersed by treatment in an ultrasonic water bath for 1 min (dBF). These biofilms were plated and determined to yield 1 × 107 to 3 × 107 CFU per tube. Ultrasonic disruption was confirmed by several means not to lyse cells or affect viability: by analyzing PL cells with and without ultrasonic treatment by plating for CFU, by direct counts by microscopy, and by live-dead staining.
Crystal violet staining of test tube biofilms was done as described previously, except 95% ethanol was used to solubilize the stain (37).
Competition experiments.
Competition experiments were done using 5-day-old CD-1 mice as well as in LB broth cultures with shaking as an in vitro control. Differentially marked samples were mixed at 1:1 ratios in saline prior to inoculation unless stated otherwise. The inocula were plated on LB agar plates containing 40 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) to differentiate wild-type and lacZ mutant colonies and to determine the input ratios and bacterial titers. The final inocula were ∼105 CFU per mouse. Mice were sacrificed at 24 h postinoculation, and the tissues indicated in the figure legends were harvested. Bacteria in these tissues were enumerated on LB-X-Gal plates. The competition index (CI) is calculated as the blue/white ratio of the output normalized to the blue/white ratio of the input. During initial studies, multiple independent dilutions of iBF/PL, dBF/PL, and dBF/iBF mixtures were plated to ensure consistent and accurate determination of the input ratio.
iBF and dBF samples were prepared as indicated above. In all cases, three independently grown biofilms were processed in parallel and pooled to obtain the iBF or dBF samples. Competition experiments used PL samples prepared by washing and diluting stationary-phase (16-h) bacteria in saline. The exceptions are the competitions using PL cells grown on LB agar plates or in the supernatant of a biofilm sample. For the former, colonies from a plate were suspended in saline to the desired concentration and combined 1:1 with differentially marked dBF cells. For the latter, differentially marked strains were both grown as biofilms. The PL cells were obtained from the broth phase of one strain, diluted in saline, and then combined 1:1 with the biofilm fraction (attached cells) of the second strain, dispersed as described.
To determine whether mutations that confer enhanced colonization arise during growth in a biofilm, V. cholerae biofilms grown in glass tubes were dispersed and passaged once in LB broth shaking culture to stationary phase; as a result of this process, these biofilm-derived cells were in planktonic form. The differentially marked strain was grown to stationary phase as usual and then combined 1:1 with passaged dBF cells for inoculation of mice.
For competitions between C6709 lacZ(pVpsT) and C6709(pTrc99A), both strains were grown in shaking culture for 16 h, with or without induction by 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). No agglutination was visible in any culture. All samples were washed and diluted in saline prior to being combined at 1:1 and inoculated into mice.
The study examining the replication of dBF and PL bacteria during infection was done as a competition between C6709 and C6709 lacZ, each carrying pACtsKan. The PL cultures were grown for 16 h at room temperature. An input mixture of 1 dBF to 10 PL cells was used to favor PL cells such that PL bacteria could be recovered from the small intestines in numbers comparable to those for dBF bacteria. At the times postinoculation indicated in Fig. 6, bacteria in the small intestine were enumerated on LB-Sm-X-Gal plates to determine the CI. The colonies were then replica plated on LB-Kn-X-Gal and LB-Sm-X-Gal plates, and the percentages of Kn-sensitive dBF and PL bacteria, i.e., those that had divided and lost the temperature-sensitive plasmid, were determined. As a control, the strains were grown in vitro as single strains and in competition. The CI and optical density at 600 nm (OD600) were determined.
FIG. 6.

Biofilm-derived bacteria replicate faster than planktonic bacteria during infection. The differentially marked dBF and PL bacteria, both containing pACtsKan, were mixed at 1 dBF to 10 PL bacteria and inoculated into mice. Growth in vivo was determined based on loss of Kn resistance conferred by the plasmid. (A) dBF bacteria show a higher rate of increase in Kn sensitivity than PL bacteria. At the indicated times postinoculation, the percentage increases in Kn-sensitive dBF and PL bacteria in the small intestine were determined. Each data point is the mean value for at least five mice. Error bars correspond to the standard deviation. (B) For the same set of mice, the CIs of dBF/PL were determined. Symbols are CI values from individual mice. Horizontal lines indicate the medians. *, P < 0.05 by Wilcoxon signed rank test, where the hypothetical value is 1. (C) dBF and PL bacteria containing pACtsKan grow at the same rate in vitro. In vitro competitions of dBF and PL bacteria, each initially containing pACtsKan, were done in triplicate.
Single-strain infections.
The 50% infectious dose (ID50) of V. cholerae biofilm bacteria was determined by inoculating mice with dBF serially diluted in saline. The number of CFU present in the small intestines at 24 h postinoculation was determined and plotted against the input dose. The distribution of dBF, PL, and nonchemotactic (cheY-3 PL cells) V. cholerae in the small intestine was investigated as described previously (38).
Determination of cell viability.
Biofilm and planktonic cell samples were assessed for viability by plating CFU, by direct counting by microscopy, and by live-dead staining. For the latter, a LIVE/DEAD BacLight Bacterial Viability and Counting Kit (Molecular Probes) was used according to the manufacturer's protocol.
RESULTS
V. cholerae cells grown in a biofilm, independent of biofilm architecture, have enhanced colonization compared to planktonic cells.
Because of limitations of the infant mouse model of V. cholerae colonization, we could not test whether biofilm-associated V. cholerae is protected from gastric acid pH. However, we hypothesized that V. cholerae biofilms may provide other or additional benefits that aid in colonization. To test this, we performed competition experiments using differentially marked strains of V. cholerae O1 El Tor C6709 to compare the ability of intact biofilms (iBF) and stationary-phase planktonic (PL) cells to colonize the infant mouse small intestine. The iBF inocula were prepared from biofilms grown on glass slides in LB broth at room temperature under static conditions. Competitions of iBF/PL cells showed that intact biofilm bacteria out-competed PL bacteria for colonization by 5.8-fold (Fig. 1). In contrast, in vitro competitions in LB broth shaking cultures showed comparable growth of iBF and PL cells. Equal growth was observed in single-strain growth experiments as well (data not shown). Therefore, the in vivo advantage of the biofilm bacteria was not due to an overall growth benefit.
FIG. 1.
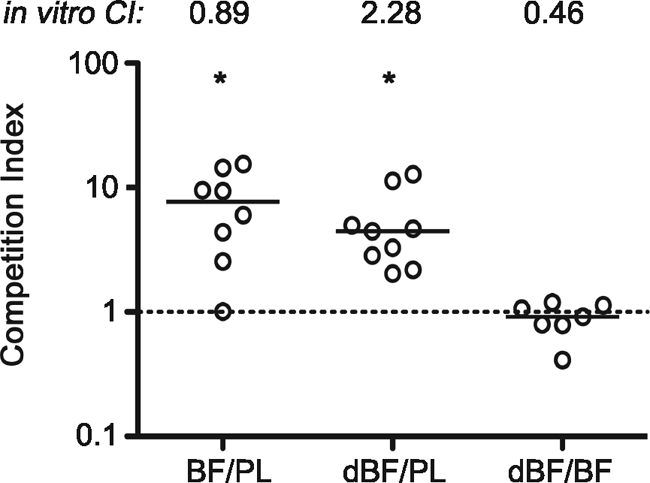
Biofilm-derived V. cholerae cells out-compete the planktonic counterparts in the small intestines of mice, independent of the biofilm architecture. V. cholerae iBF, dBF, and PL samples were paired as indicated on the x axis; all three combinations were C6709/C6709 lacZ. Each symbol represents the CI for an individual mouse. Horizontal lines indicate medians for each group. The same pairings of samples were simultaneously grown in LB broth shaking culture to determine the in vitro CI, shown at the top. Data from two independent experiments are shown. Both iBF (BF) and dBF out-compete PL bacteria for colonization (*, P < 0.01 by Wilcoxon signed rank test, where the hypothetical value is 1, i.e., no difference in colonization). The CIs for iBF/PL and dBF/PL do not differ from each other (P = 0.4), but both are significantly higher than that of dBF/iBF (P = 0.0014 by Kruskal-Wallis test).
We further postulated that the colonization benefits conferred to V. cholerae by the biofilm are not limited to physical protection by the biofilm structure but that the physiology of biofilm-derived V. cholerae enhances colonization. We tested this by comparing the colonization achieved by iBF and fully dispersed (dBF) V. cholerae biofilms. Like the iBF/PL bacteria competition, the dBF/PL bacteria comparison showed that dispersed biofilm out-competed planktonic bacteria for colonization but not in vitro (Fig. 1). Importantly, direct competition of differentially marked dBF/iBF samples in mice demonstrated that intact and dispersed biofilm bacteria colonize the small intestine equally, indicating that the architecture of the biofilm plays little or no role in the increased colonization observed.
Having observed that dBF and iBF bacteria taken from glass slides colonized mice equally well, we performed subsequent competition experiments using bacteria dispersed from biofilms grown in glass culture tubes, which facilitated preparation of inocula. The dBF samples derived from culture tube biofilms were confirmed to out-compete PL bacteria in the mouse but not in LB broth culture (Fig. 2A). By 24 h, the biofilm culture supernatants were turbid with planktonic cells, and biofilms could be detected at the air-liquid interface; both biofilm biomass and planktonic CFU counts increased by 48 h (data not shown). However, dBF from 24-h and 48-h biofilms showed levels of colonization comparable to those of stationary-phase PL cells (Fig. 2A). This result suggests that increased colonization ability is induced early during biofilm formation and does not require the robust biofilm present at 48 h.
FIG. 2.

Characterization of biofilm-induced infectivity. (A) Differentially marked dBF (24 or 48 h) and PL samples were paired for coinfection of mice. Bacteria were recovered from the small intestines at 24 h postinoculation and CIs were determined. In vitro competitions were done in parallel. Out-competition of PL bacteria by dBF was apparent in 24-h biofilms and maintained in 48-h biofilms. (B) Biofilm-induced infectivity is not strain specific. V. cholerae strains N16961 (hapR mutant) and C6706 (hapR+) were tested for biofilm-induced infectivity. Coinfections of mice and in vitro control competitions were done using differentially marked 24-h dBF cells and stationary-phase PL cells. (C) Out-competition of PL by dBF bacteria is not dependent on the method of culturing the PL samples. As an alternative to stationary-phase preparations, PL samples were grown either on LB agar plates or in the planktonic phase of a biofilm culture under static conditions. (D) Increased infectivity of biofilm-derived bacteria is not due to a mutation(s) that arises during growth in biofilm. Biofilm-derived bacteria were passaged in shaking culture, and then combined with stationary-phase PL bacteria for coinfection of mice; both biofilm-derived and PL samples were planktonic. In vitro competitions were done in parallel. For all panels, each symbol represents the CI in an individual mouse; horizontal bars mark the medians. *, P < 0.05 by the Wilcoxon signed rank test, where the hypothetical value is 1.
Other wild-type V. cholerae O1 El Tor strains, namely, N16961 (hapR mutant) and C6706 (hapR+), were tested for enhanced colonization after growth as a biofilm. The dBF and stationary-phase PL bacteria were tested in competition experiments in mice and in LB broth culture. For both strains, dBF outcompeted PL bacteria in the mouse small intestine but not in vitro (Fig. 2B). Therefore, this phenomenon is not strain specific but is common to several O1 El Tor strains isolated in different years from different continents.
Since the above experiments used stationary-phase PL bacteria in competition with the biofilms, it was possible that nutrient deprivation, the level of aeration, or other conditions in shaking stationary-phase cultures might attenuate the PL sample, thus leading to a reduction in virulence. To address this, dBF bacteria were competed against PL bacteria grown under alternative conditions. The dBF bacteria still showed increased colonization compared to PL bacteria grown on an LB agar plate (and thus having cells with a variety of growth phases); these samples grew equally well in vitro (Fig. 2C, dBF/plate). Additionally, dBF bacteria were tested in competition with PL bacteria obtained directly from the broth phase of biofilm samples of the differentially marked strain. Thus, bacteria grown under identical conditions, but with dBF obtained from the attached fraction of cells and PL bacteria obtained from the unattached fraction, were directly assessed for colonization of the mouse. Once again, the dBF cells out-competed PL cells by 10-fold for colonization (Fig. 2C, dBF/sup). The dBF bacteria did not out-compete PL bacteria for growth in vitro.
Finally, because increased mutation rates have been observed for P. aeruginosa during growth in a biofilm (7, 19), it remained a formal possibility that mutations could arise in the biofilm population of V. cholerae during static growth and that these mutants might have an increased ability to colonize the mouse. To test this, V. cholerae was grown under biofilm conditions in culture tubes. The attached bacteria were then removed and passaged once in LB broth shaking culture (to become planktonic) to stationary phase prior to being tested in competition with stationary-phase PL bacteria. There was no difference in colonization between the LB-passaged biofilm bacteria and PL bacteria, indicating that the enhanced colonization observed in biofilm-derived V. cholerae is a transient phenotype induced during growth in a biofilm (Fig. 2D).
VPS does not protect V. cholerae during infection of the mouse.
Vibrio exopolysaccharide (VPS) is a common extracellular matrix of V. cholerae biofilms and is required for biofilm formation on glass surfaces (71). While the dispersal methods we used were confirmed by microscopy to break up the biofilm completely into individual cells, the possibility remained that VPS might not have been entirely removed from cells despite washing. This polysaccharide coating, if present, might have a protective effect during infection of the mouse or could conceivably have adhesive properties leading to increased colonization. The dBF cells were thus examined to determine whether VPS was present on the surface. Staining of V. cholerae biofilm-derived cells using wheat germ agglutinin and concanavalin A lectins failed to differentiate between iBF, dBF, and PL cells (data not shown). Therefore, the effect of VPS on colonization was assessed by testing V. cholerae with increased VPS production for altered colonization of mice. For this, vpsT, encoding a direct activator of transcription of VPS biosynthesis genes (12), was cloned into an expression vector, yielding pVpsT, and ectopically expressed. When grown in static culture, this strain showed dramatically increased biofilm formation compared to V. cholerae carrying an empty vector and to a non-biofilm-forming vpsR mutant, indicating elevated VPS production (Fig. 3A). Induction of VpsT expression with IPTG was not required to achieve elevated biofilm formation although addition of IPTG did augment biofilm production further. Though we did not measure it, we presumed that this strain will have elevated VPS production during planktonic growth when VpsT is induced. V. cholerae carrying pVpsT was grown planktonically in shaking culture, with and without IPTG, and tested in competition with differentially marked V. cholerae carrying an empty vector for colonization of mice and growth in vitro. Expression of VpsT did not affect colonization as the pVpsT strain competed equally with the control strain (Fig. 3C). Even after induction of high levels of VpsT, no change in colonization occurred. These data and the data above indicate that the presence of VPS on the bacterial surface is not sufficient to confer the increased colonization rates observed in biofilm-derived bacteria.
FIG. 3.

The role of VPS in biofilm-induced infectivity. (A) Ectopic expression of VpsT leads to dramatically increased biofilm formation. Differentially marked V. cholerae bacteria containing pVpsT or empty vector (WT), as well as a vpsR negative control, were assayed for biofilm formation under static conditions by crystal violet staining. Both in the presence and in the absence of 0.1 mM IPTG, the pVpsT strain produced significantly more biofilm than the vector control strain, indicating increased VPS production (*, P < 0.05 by Student's t test). Bars indicate the mean A570; error bars show standard deviations. (B) Ectopic expression of VpsT does not result in increased infectivity. Differentially marked strains containing pVpsT or vector control were grown in shaking culture, with or without 0.1 mM IPTG. The strains were assessed for colonization of the mouse small intestine using a competition assay. In vitro competitions were done in parallel. Symbols represent data for individual mice or in vitro control samples; horizontal lines indicate the median CI.
VBNC cells do not contribute to the observed increase in competitiveness of biofilm-derived V. cholerae.
V. cholerae has been shown to enter a “viable but nonculturable” (VBNC) state in which they can be detected by direct fluorescence antibodies but cannot be cultured using routine methods (2, 8). During growth in a biofilm, it is possible that V. cholerae might enter a VBNC but still infectious state. If so, the input ratio used to calculate the competitive index (CI) would underestimate the true number of infectious bacteria in the biofilm sample. This phenomenon would result in an artificially high CI. To address this possibility, the dBF and PL samples were subjected to live-dead staining and direct counting of bacteria. The dBF and PL samples did not show significant differences in the percentages of live (SYTO-9 positive) cells, with 65.6% ± 14.2% and 47.9% ± 7.3% live cells, respectively. In addition, the direct counting of bacteria in dBF samples by microscopy correlated well with the numbers of bacteria estimated by determination of CFU counts. The dBF samples had 2.7 × 107 ± 0.3 × 107 bacteria by direct counting and 2.5 × 107 ± 1.1 × 107 CFU. Therefore, VBNC V. cholerae did not contribute substantially to the observed high CI values obtained for dBF/PL bacteria competition experiments.
V. cholerae cells derived from a biofilm are more infectious than planktonic cells.
The observed out-competition by biofilm-derived cells over PL cells in the mouse small intestine could be due to heightened infectivity or possibly to other reasons such as an increased growth rate at later stages of infection. We tested the former possibility by measuring the 50% infectious dose (ID50) of dBF and PL bacteria. Serial dilutions of dBF and PL samples were used to inoculate mice, and then the bacteria present in the small intestines after 24 h were enumerated. The ID50 for V. cholerae derived from a biofilm was dramatically lower, <7 CFU, the smallest input dose tested; the ID50 for stationary-phase planktonic cells was approximately 3 × 104. Interestingly, the bacterial burden decreased with inoculum size but only to a point, since input doses of 200 CFU or less resulted in a baseline number of bacteria being recovered from the small intestines (Fig. 4). It is not certain but it can be expected that a longer infection would have resulted in increased bacterial burden in these mice. The ID50 studies indicate that biofilm-derived V. cholerae cells are hyperinfectious relative to their planktonic counterparts.
FIG. 4.
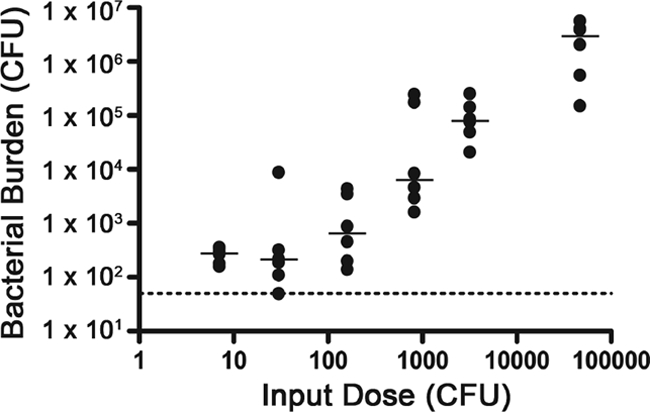
Bacterial burden decreases with inoculum size of biofilm-derived V. cholerae. Samples of dBF were serially diluted and then inoculated into mice. At 24 h postinoculation, the bacteria present in the small intestines were enumerated. The dotted line indicates the limit of detection. Shown are the numbers of CFU per small intestine at different input doses, where each circle represents the value from an individual mouse. The horizontal lines show the median. The bacterial burdens from the highest two input doses are each significantly higher than those from the three lowest doses (P < 0.05 for the indicated comparisons by the Kruskal-Wallis test).
The dynamics of an infection with biofilm-derived V. cholerae differ from those of planktonic cells.
The ID50 studies suggested that biofilm-derived bacteria must have an altered dynamic of colonization of the small intestine. To investigate the nature of this alteration, the temporal and spatial differences in colonization between PL and biofilm-derived bacteria were analyzed. First, dBF and PL bacteria were tested in competition in mice, and the CIs in the small intestine and large bowel were determined at different time points. At 1 h postinoculation, no differences in colonization between the dBF and PL cells were observed in the small or large intestines (Fig. 5A A). However, by 5 h the dBF cells clearly out-competed PL cells in the small intestine; in the large intestine, the CI remained at ∼1, suggesting that the advantage of dBF cells is specific to the environment of the small intestine. At 24 h, when colonization by V. cholerae is well-established, dBF bacteria greatly outnumbered PL bacteria in both small and large intestines. The increased CI in the large intestine at 24 h may be representative of the bacteria that detached from the small intestine. Analysis of the bacterial burden in the different sections of the intestine showed that while the number of PL cells dropped by 2 orders of magnitude, the number dBF cells remained relatively constant (Fig. 5B). This suggests that the dBF cells are either more resistant to a killing mechanism in the gut or that they grow faster than PL cells.
FIG. 5.
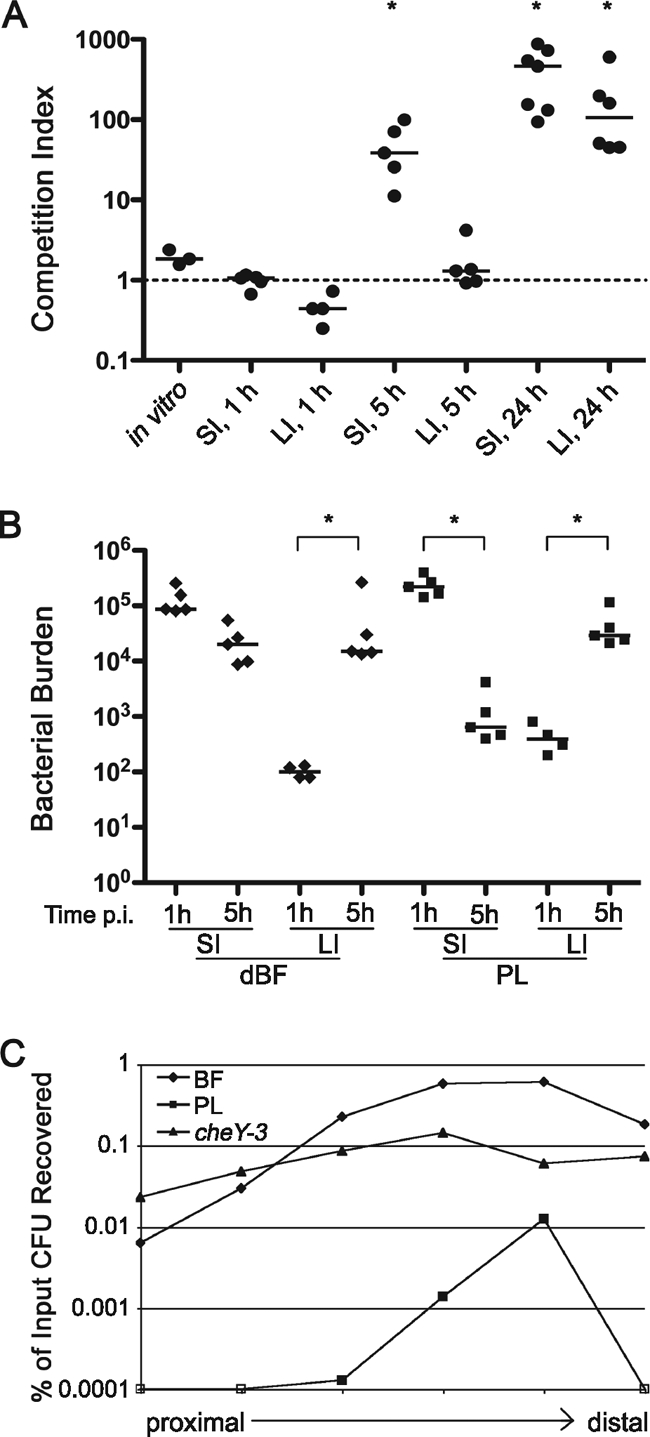
The dynamics of biofilm-induced infectivity. (A) Biofilm-induced hyperinfectivity is detectable early, but not immediately, during infection and is specific to the small intestine. Differentially marked dBF and PL bacteria were used to coinfect mice. At 1, 5, or 24 h postinoculation, the stomachs, small intestines (SI), and large intestines (LI) were harvested, and the bacteria were enumerated on LB agar containing X-Gal. The CIs were calculated for each tissue at each time point. Each symbol corresponds to the CI value of an individual mouse. Horizontal lines indicate the median. *, P < 0.05 by Wilcoxon signed rank test, where the hypothetical value is 1. (B) Bacterial burden (CFU) of dBF and PL bacteria in the SI and LI at 1 and 5 h postinoculation. *, P < 0.05 for the indicated comparisons by Kruskal-Wallis test. (C) Biofilm-derived bacteria assume a distribution in the small intestine similar to that of PL bacteria but achieve a higher level of colonization. Mice were inoculated with PL, dBF, or nonchemotactic (cheY mutant) bacteria. The number of CFU present in each of six equal-sized segments at 5 h postinoculation was determined and normalized to the input CFU count. Data points are the mean number of CFU present in at least five mice.
Previous studies have shown that V. cholerae that has been passaged in vivo, in both mice and humans, show hyperinfectivity (1, 10). This phenotype is associated with repression of chemotaxis; nonchemotactic V. cholerae has been shown to have a reduced ID50 compared to the wild type (9, 10). The nonchemotactic bacteria show an altered distribution along the small intestine. Whereas wild-type bacteria favor colonization of the distal half of the small intestine, nonchemotactic bacteria colonize relatively uniformly throughout the small intestine (9). Moreover, the nonchemotactic bacteria, presumably because they are less restricted for areas of colonization, reach higher overall numbers in the small intestine than the wild type.
We showed that, like nonchemotactic mutants, biofilm-derived V. cholerae has a lower ID50 and attains a higher burden in the small intestines of mice. To determine whether biofilm-derived bacteria also have a wider distribution in the small intestine, dBF and PL cells of the wild-type and nonchemotactic (cheY-3) strains were inoculated into mice, and small intestines were harvested at 5 h postinoculation. The intestines were divided into six equal segments, and the bacteria were enumerated. The dBF bacteria, like wild-type PL cells, were found to primarily colonize the distal portion of the small intestine, consistent with dBF having wild-type chemotaxis (Fig. 5B). However, dBF bacteria colonized the small intestine in similar numbers to the nonchemotactic mutant and so demonstrated hyperinfectivity. The dBF V. cholerae thus followed altered colonization dynamics and displayed an intermediate phenotype between PL and nonchemotactic bacteria.
Targeted mutagenesis of candidate biofilm-induced hyperinfectivity genes.
Several groups have used transcriptional profiling to compare V. cholerae planktonic bacteria with cells from biofilms at various stages of development as well as with mutants that produce altered biofilms (49, 57, 72). A wide variety of genes have been identified that are upregulated in a biofilm and thus could participate in biofilm-induced hyperinfectivity (42, 52, 61, 62). We selected 10 genes covering a variety of putative functions, as listed in Table 2, for mutagenesis and examined the mutants for loss of hyperinfectivity.
TABLE 2.
Colonization by dispersed biofilms of candidate biofilm-induced hyperinfectivity genes
| Locus | Annotation or predicted function | Biofilm formationa | Median in vivo CIb | Mean in vitro CIb |
|---|---|---|---|---|
| cdpA | c-di-GMP phosphodiesterase | 1.25 | 1.17 | |
| pilTU | Putative ATPases; pilus associated | 0.68 | 0.59 | 0.26 |
| VC0687 | CspA; carbon starvation protein A | 0.93 | 0.77 | 1.5 |
| VC1334 | Bordetella uptake gene (Bug) homologue | 1.08 | 2.19 | 2.78 |
| VC1516 | Formate dehyrogenase related | 0.81 | 0.63 | 1.7 |
| VC2704 | Hypothetical protein | 1.05 | 1.34 | 0.97 |
| VC2705 | Putative sodium/solute symporter | 1.1 | 1.54 | 1.17 |
| VCA0676 | NapF; nitrate acquisition | 0.63 | 1.08 | 1.20 |
| VCA0682 | UhpA; transcriptional regulator | 1.1 | 1.30 | 1.18 |
| hapR | Quorum-sensing regulator | 3.5 | 1.43 | 0.57 |
Measured by crystal violet staining and A570 readings and expressed relative to values in the isogenic C6709 parent strain.
Comparisons were between dispersed biofilms of differentially marked mutant and wild-type strains, normalized to the input ratio. For in vivo experiments, five to nine mice were used; for in vitro experiments, three replicates were done.
All mutants grew equivalently to the wild type in LB broth in vitro (data not shown). All strains formed wild-type levels of biofilm as measured by crystal violet staining except for the hapR mutant, which formed 3.5 times more biofilm than the wild type (Table 2). To test for loss of biofilm-induced hyperinfectivity, the mutants and differentially marked wild-type strain were grown separately as biofilms, dispersed into planktonic cells, combined at 1:1 ratios, and used to infect infant mice. All mutants colonized the small intestine comparably to the wild type, indicating that the selected genes do not play a role in this phenotype (Table 2). The pilTU mutant was slightly attenuated in vivo and also in vitro, so the in vivo attenuation was likely due to a small growth defect.
Biofilm-derived bacteria may replicate faster than planktonic cells initially during infection.
Biofilm bacteria could achieve higher numbers in the mouse small intestine by being more adherent, by increased resistance to a host defense mechanism, and/or by an in vivo-specific growth advantage. We cannot differentiate between these mechanisms simply by measuring the number of CFU during infection. We tested the hypothesis that biofilm V. cholerae bacteria out-compete planktonic cells because they replicate faster during infection. To test this we introduced a temperature-sensitive plasmid, pACtsKan, into differentially marked (lacZ+ and lacZ mutant) V. cholerae. The strains were used in a mouse competition experiment, and the loss of this plasmid, based on loss of Kn resistance, was monitored over time of infection. During infection, the percentage of Kn-sensitive CFU increased faster for the dBF cells than for PL bacteria, indicating that the dBF bacteria lost the plasmid at a higher rate due to a higher rate of replication (Fig. 6A). The increase in Kn sensitivity in dBF cells corresponded with increased colonization, beginning between 2 and 4 h postinoculation (Fig. 6A and 6). In vitro, the dBF and PL cells grew equally both in single culture and coculture (Fig. 6C), and the plasmid was lost at comparable rates in both biofilm-derived and planktonic cells (data not shown). These data indicate that biofilm-derived V. cholerae bacteria grow faster than planktonic cells during infection; however, the results do not preclude a contribution by other mechanisms such as increased stress resistance or adherence.
DISCUSSION
Biofilm formation by the facultative human pathogen V. cholerae in its natural habitat is predicted to be a survival strategy and, importantly, a primary source for human infections (13, 32). Previous studies have shown that V. cholerae biofilms offer protection from acid pH, bile acids, chlorine, and predation (31, 43, 71, 72). Biofilm resistance to acid pH was hypothesized to protect the bacteria during passage through the human stomach due to protection by the biofilm structure (50, 72) though this cannot be tested using the infant mouse model. In this work, we tested the hypotheses that biofilm formation by V. cholerae enhances colonization and that the architecture of the biofilm is critical in that process. Intact and fully dispersed biofilms were assayed for the ability to colonize the mouse small intestine compared to planktonic V. cholerae. The dispersed biofilms were found to colonize the infant mouse as well as intact biofilms of V. cholerae; both dispersed and intact biofilms vastly out-competed planktonic bacteria. This effect could not be attributed to VBNC cells, to biofilm-induced mutations, or to the growth phase of the planktonic cells used. Nor was the effect found to be strain specific. Together, these data indicate that the physiological state of the biofilm-grown bacteria enhances virulence and that the three-dimensional structure of the biofilm per se is not required for enhanced virulence.
We considered the possibility that despite the process of dispersion of the biofilms, VPS remained on the surfaces of individual bacteria. This VPS coating might protect the bacteria from a host killing mechanism or enhance adhesion within the host small intestine. Multiple lines of evidence suggest that even if VPS indeed remains on the bacterial surface, it does not cause the observed hyperinfectivity. First, ectopic overexpression of VpsT, the direct activator of the vps biosynthetic operons (12), did not affect the ability of planktonic cells to colonize the small intestine. One caveat to this is that it was not possible to confirm maintained production of VPS by the VpsT-expressing strain during infection. Second, previous work has shown that a rugose isolate, which has elevated VPS biosynthesis and biofilm production, is attenuated in the mouse model (53). Third, a hapR mutant, which produces rugose colonies and overproduces biofilm, behaved like the wild type in vivo. Finally, we have previously shown that V. cholerae with elevated cyclic di-GMP (c-di-GMP), a bacterial second messenger that enhances VPS production and biofilm formation, is attenuated in vivo (61). Importantly, though these bacteria are “programmed” for high VPS expression, inactivation of vps in this strain does not affect colonization (61). Taken together, these data suggest that if VPS remains present on the surface of biofilm-derived V. cholerae, it alone does not enhance colonization.
Several possible mechanisms could result in the out-competition of planktonic cells by biofilm-derived V. cholerae. Analysis of the bacterial load in the small intestine showed that while the number of biofilm-derived bacteria remained at around 104 to 105 per small intestine, the number of planktonic cells dropped by almost 3 orders of magnitude. These results are consistent with better adaptation of biofilm-derived V. cholerae for survival and growth in the host small intestine. To address this possibility, the presence of a temperature-sensitive plasmid was monitored over the course of infection, allowing the in vivo growth rate of biofilm and planktonic bacteria to be assessed. The higher rate of plasmid loss by biofilm-derived bacteria during infection, with no difference in in vitro growth rate, suggests that they indeed grow at a higher rate than planktonic cells in the mouse small intestine. Thus, growth in a biofilm may prime V. cholerae for growth upon entry into the human host and contributes to the hyperinfectious phenotype observed in the mouse model of intestinal infection. This result does not exclude the possibility that other biofilm-induced factors contribute to hyperinfectivity as any mechanism that enhances bacterial survival during infection may factor into increased growth. The possible roles of increased resistance to a host killing mechanism or increased adherence to host tissues by biofilm V. cholerae has not yet been determined.
Host passage of V. cholerae has been shown to cause hyperinfectivity in both humans and the mouse model (1, 47). These host-passaged bacteria showed a much lower infectious dose and achieved a higher load in the mouse small intestine. This phenotype persists even after incubation in pond water and may play a critical role in the ability of V. cholerae to cause epidemic disease by facilitating person-to-person spread of the organism (29, 47, 51). The infectious dose for biofilm-derived V. cholerae was similarly substantially lower than that for planktonic cells. Yet unlike host passage, which results in colonization along the entire length of the small intestine, likely due to altered chemotaxis, growth in a biofilm still favors the colonization of the distal portion of the small intestine. Thus, the increased infectivity and bacterial burden observed for biofilm-derived V. cholerae occur through a distinct process(es).
The structure of a biofilm is the most overt contributor to the reduced susceptibility of microbial biofilms to myriad stresses and chemical assaults. A few studies have directly tested the role of biofilm structure in conferring resistance to a variety of antimicrobials in vitro, with mixed results. Limited work has been done to understand how growth as a biofilm in the environment affects the virulence of facultative pathogenic bacteria. In vitro grown biofilms of Listeria monocytogenes were less virulent than their planktonic counterparts (73). Biofilms of Salmonella enterica serovar Typhimurium appeared to confer a transient advantage in the spleens of mice after intraperitoneal inoculation, but ultimately biofilm and planktonic cells showed comparable virulence levels (65). These results are in contrast to our findings using V. cholerae biofilms and may reflect the pathogenesis of the organisms. While L. monocytogenes and S. Typhimurium invade host tissues, V. cholerae remains in the intestinal lumen and proliferates before the host develops an effective immune response.
Several transcriptional profiling studies have been done comparing biofilm and planktonic V. cholerae cells (49, 57, 72), identifying a wide variety of genes that are differentially transcribed and that could participate in biofilm-induced hyperinfectivity. We investigated the roles of 10 candidate genes covering a variety of putative functions, but these did not contribute to biofilm-induced hyperinfectivity. Interestingly, a hapR mutant that is defective for quorum sensing and “locked” in a low cell density state was unaffected in biofilm-induced hyperinfectivity. Previous work by Zhu and Mekalanos showed that, when intact biofilms are used, a hapR mutant is attenuated for colonization of the mouse compared to wild-type cells (72). Their results, in combination with our data, support their hypothesis that hapR mediates detachment from a biofilm; in this study, biofilms are dispersed prior to infection, so hapR is dispensable.
The work described herein suggests that microbes within a biofilm are physiologically distinct from planktonic cells and can thus affect other phenotypes including pathogenesis. Thus, V. cholerae biofilms, potentially including those present in the aquatic environment, may have a lower infectious dose in humans than previously determined, which has interesting implications for the natural acquisition of V. cholerae. While our targeted mutagenesis approach did not uncover the mechanism(s) responsible for the observed phenotype, we expect that many biofilm-induced factors contribute to the hyperinfectivity of V. cholerae and represent an important group of virulence factors that remain to be identified.
Acknowledgments
We thank members of the Camilli lab for helpful suggestions, particularly Anne Bishop for advice on statistical methods.
This work was supported by National Institutes of Health grants NIAID R01 AI055058 to A.C. and NIDDK T32 DK007542 to R.T. A.C. is a Howard Hughes Medical Institute investigator.
R.T. and A.C. designed experiments; R.T. and B.P. performed research; R.T. analyzed data and wrote the report.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 1 June 2010.
REFERENCES
- 1.Alam, A., R. C. Larocque, J. B. Harris, C. Vanderspurt, E. T. Ryan, F. Qadri, and S. B. Calderwood. 2005. Hyperinfectivity of human-passaged Vibrio cholerae can be modeled by growth in the infant mouse. Infect. Immun. 73:6674-6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alam, M., M. Sultana, G. B. Nair, A. K. Siddique, N. A. Hasan, R. B. Sack, D. A. Sack, K. U. Ahmed, A. Sadique, H. Watanabe, C. J. Grim, A. Huq, and R. R. Colwell. 2007. Viable but nonculturable Vibrio cholerae O1 in biofilms in the aquatic environment and their role in cholera transmission. Proc. Natl. Acad. Sci. U. S. A. 104:17801-17806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amann, E., and J. Brosius. 1985. “ATG vectors” for regulated high-level expression of cloned genes in Escherichia coli. Gene 40:183-190. [DOI] [PubMed] [Google Scholar]
- 4.Anderl, J. N., M. J. Franklin, and P. S. Stewart. 2000. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemother. 44:1818-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderl, J. N., J. Zahller, F. Roe, and P. S. Stewart. 2003. Role of nutrient limitation and stationary-phase existence in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemother. 47:1251-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benjamin, W. H., Jr., P. Hall, S. J. Roberts, and D. E. Briles. 1990. The primary effect of the Ity locus is on the rate of growth of Salmonella typhimurium that are relatively protected from killing. J. Immunol. 144:3143-3151. [PubMed] [Google Scholar]
- 7.Boles, B. R., M. Thoendel, and P. K. Singh. 2004. Self-generated diversity produces “insurance effects” in biofilm communities. Proc. Natl. Acad. Sci. U. S. A. 101:16630-16635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brayton, P. R., M. L. Tamplin, A. Huq, and R. R. Colwell. 1987. Enumeration of Vibrio cholerae O1 in Bangladesh waters by fluorescent-antibody direct viable count. Appl. Environ. Microbiol. 53:2862-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butler, S. M., and A. Camilli. 2004. Both chemotaxis and net motility greatly influence the infectivity of Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 101:5018-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butler, S. M., E. J. Nelson, N. Chowdhury, S. M. Faruque, S. B. Calderwood, and A. Camilli. 2006. Cholera stool bacteria repress chemotaxis to increase infectivity. Mol. Microbiol. 60:417-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camilli, A., and J. J. Mekalanos. 1995. Use of recombinase gene fusions to identify Vibrio cholerae genes induced during infection. Mol. Microbiol. 18:671-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casper-Lindley, C., and F. H. Yildiz. 2004. VpsT is a transcriptional regulator required for expression of vps biosynthesis genes and the development of rugose colonial morphology in Vibrio cholerae O1 El Tor. J. Bacteriol. 186:1574-1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colwell, R. R., A. Huq, M. S. Islam, K. M. Aziz, M. Yunus, N. H. Khan, A. Mahmud, R. B. Sack, G. B. Nair, J. Chakraborty, D. A. Sack, and E. Russek-Cohen. 2003. Reduction of cholera in Bangladeshi villages by simple filtration. Proc. Natl. Acad. Sci. U. S. A. 100:1051-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costerton, J. W., Z. Lewandowski, D. E. Caldwell, D. R. Korber, and H. M. Lappin-Scott. 1995. Microbial biofilms. Annu. Rev. Microbiol. 49:711-745. [DOI] [PubMed] [Google Scholar]
- 15.Costerton, J. W., Z. Lewandowski, D. DeBeer, D. Caldwell, D. Korber, and G. James. 1994. Biofilms, the customized microniche. J. Bacteriol. 176:2137-2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Beer, D., R. Srinivasan, and P. S. Stewart. 1994. Direct measurement of chlorine penetration into biofilms during disinfection. Appl. Environ. Microbiol. 60:4339-4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ditta, G., S. Stanfield, D. Corbin, and D. R. Helinski. 1980. Broad host range DNA cloning system for gram-negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc. Natl. Acad. Sci. U. S. A. 77:7347-7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donnenberg, M. S., and J. B. Kaper. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59:4310-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Driffield, K., K. Miller, J. M. Bostock, A. J. O'Neill, and I. Chopra. 2008. Increased mutability of Pseudomonas aeruginosa in biofilms. J. Antimicrob. Chemother. 61:1053-1056. [DOI] [PubMed] [Google Scholar]
- 20.Dunne, W. M., Jr., E. O. Mason, Jr., and S. L. Kaplan. 1993. Diffusion of rifampin and vancomycin through a Staphylococcus epidermidis biofilm. Antimicrob. Agents Chemother. 37:2522-2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elasri, M. O., and R. V. Miller. 1999. Study of the response of a biofilm bacterial community to UV radiation. Appl. Environ. Microbiol. 65:2025-2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Espeland, E. M., and R. G. Wetzel. 2001. Complexation, stabilization, and UV photolysis of extracellular and surface-bound glucosidase and alkaline phosphatase: implications for biofilm microbiota. Microb. Ecol. 42:572-585. [DOI] [PubMed] [Google Scholar]
- 23.Faruque, S. M., M. J. Albert, and J. J. Mekalanos. 1998. Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 62:1301-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feazel, L. M., L. K. Baumgartner, K. L. Peterson, D. N. Frank, J. K. Harris, and N. R. Pace. 2009. Opportunistic pathogens enriched in showerhead biofilms. Proc. Natl. Acad. Sci. U. S. A. 106:16393-16399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilbert, P., P. J. Collier, and M. R. Brown. 1990. Influence of growth rate on susceptibility to antimicrobial agents: biofilms, cell cycle, dormancy, and stringent response. Antimicrob. Agents Chemother. 34:1865-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gitelson, S. 1971. Gastrectomy, achlorhydria and cholera. Isr. J. Med. Sci. 7:663-667. [PubMed] [Google Scholar]
- 27.Hall-Stoodley, L., F. Z. Hu, A. Gieseke, L. Nistico, D. Nguyen, J. Hayes, M. Forbes, D. P. Greenberg, B. Dice, A. Burrows, P. A. Wackym, P. Stoodley, J. C. Post, G. D. Ehrlich, and J. E. Kerschner. 2006. Direct detection of bacterial biofilms on the middle-ear mucosa of children with chronic otitis media. JAMA 296:202-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hall-Stoodley, L., and P. Stoodley. 2005. Biofilm formation and dispersal and the transmission of human pathogens. Trends Microbiol. 13:7-10. [DOI] [PubMed] [Google Scholar]
- 29.Hartley, D. M., J. G. Morris, Jr., and D. L. Smith. 2006. Hyperinfectivity: a critical element in the ability of V. cholerae to cause epidemics? PLoS Med. 3:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoyle, B. D., J. Alcantara, and J. W. Costerton. 1992. Pseudomonas aeruginosa biofilm as a diffusion barrier to piperacillin. Antimicrob. Agents Chemother. 36:2054-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hung, D. T., J. Zhu, D. Sturtevant, and J. J. Mekalanos. 2006. Bile acids stimulate biofilm formation in Vibrio cholerae. Mol. Microbiol. 59:193-201. [DOI] [PubMed] [Google Scholar]
- 32.Huo, A., B. Xu, M. A. Chowdhury, M. S. Islam, R. Montilla, and R. R. Colwell. 1996. A simple filtration method to remove plankton-associated Vibrio cholerae in raw water supplies in developing countries. Appl. Environ. Microbiol. 62:2508-2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huq, A., E. B. Small, P. A. West, M. I. Huq, R. Rahman, and R. R. Colwell. 1983. Ecological relationships between Vibrio cholerae and planktonic crustacean copepods. Appl. Environ. Microbiol. 45:275-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khot, P. D., P. A. Suci, R. L. Miller, R. D. Nelson, and B. J. Tyler. 2006. A small subpopulation of blastospores in Candida albicans biofilms exhibit resistance to amphotericin B associated with differential regulation of ergosterol and β-1,6-glucan pathway genes. Antimicrob. Agents Chemother. 50:3708-3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumamoto, C. A. 2002. Candida biofilms. Curr. Opin. Microbiol. 5:608-611. [DOI] [PubMed] [Google Scholar]
- 36.Lam, J., R. Chan, K. Lam, and J. W. Costerton. 1980. Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect. Immun. 28:546-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lauriano, C. M., C. Ghosh, N. E. Correa, and K. E. Klose. 2004. The sodium-driven flagellar motor controls exopolysaccharide expression in Vibrio cholerae. J. Bacteriol. 186:4864-4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee, S. H., S. M. Butler, and A. Camilli. 2001. Selection for in vivo regulators of bacterial virulence. Proc. Natl. Acad. Sci. U. S. A. 98:6889-6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leid, J. G., M. E. Shirtliff, J. W. Costerton, and A. P. Stoodley. 2002. Human leukocytes adhere to, penetrate, and respond to Staphylococcus aureus biofilms. Infect. Immun. 70:6339-6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mah, T. F., and G. A. O'Toole. 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 9:34-39. [DOI] [PubMed] [Google Scholar]
- 41.Mah, T. F., B. Pitts, B. Pellock, G. C. Walker, P. S. Stewart, and G. A. O'Toole. 2003. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426:306-310. [DOI] [PubMed] [Google Scholar]
- 42.Mateus, C., S. A. Crow, Jr., and D. G. Ahearn. 2004. Adherence of Candida albicans to silicone induces immediate enhanced tolerance to fluconazole. Antimicrob. Agents Chemother. 48:3358-3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matz, C., D. McDougald, A. M. Moreno, P. Y. Yung, F. H. Yildiz, and S. Kjelleberg. 2005. Biofilm formation and phenotypic variation enhance predation-driven persistence of Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 102:16819-16824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McNeill, K., and I. R. Hamilton. 2003. Acid tolerance response of biofilm cells of Streptococcus mutans. FEMS Microbiol. Lett. 221:25-30. [DOI] [PubMed] [Google Scholar]
- 45.Meibom, K. L., M. Blokesch, N. A. Dolganov, C. Y. Wu, and G. K. Schoolnik. 2005. Chitin induces natural competence in Vibrio cholerae. Science 310:1824-1827. [DOI] [PubMed] [Google Scholar]
- 46.Meibom, K. L., X. B. Li, A. T. Nielsen, C. Y. Wu, S. Roseman, and G. K. Schoolnik. 2004. The Vibrio cholerae chitin utilization program. Proc. Natl. Acad. Sci. U. S. A. 101:2524-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Merrell, D. S., S. M. Butler, F. Qadri, N. A. Dolganov, A. Alam, M. B. Cohen, S. B. Calderwood, G. K. Schoolnik, and A. Camilli. 2002. Host-induced epidemic spread of the cholera bacterium. Nature 417:642-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller, V. L., and J. J. Mekalanos. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moorthy, S., and P. I. Watnick. 2004. Genetic evidence that the Vibrio cholerae monolayer is a distinct stage in biofilm development. Mol. Microbiol. 52:573-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nalin, D. R., V. Daya, A. Reid, M. M. Levine, and L. Cisneros. 1979. Adsorption and growth of Vibrio cholerae on chitin. Infect. Immun. 25:768-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson, E. J., A. Chowdhury, J. B. Harris, Y. A. Begum, F. Chowdhury, A. I. Khan, R. C. Larocque, A. L. Bishop, E. T. Ryan, A. Camilli, F. Qadri, and S. B. Calderwood. 2007. Complexity of rice-water stool from patients with Vibrio cholerae plays a role in the transmission of infectious diarrhea. Proc. Natl. Acad. Sci. U. S. A. 104:19091-19096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Otto, M. 2008. Staphylococcal biofilms. Curr. Top. Microbiol. Immunol. 322:207-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rashid, M. H., C. Rajanna, D. Zhang, V. Pasquale, L. S. Magder, A. Ali, S. Dumontet, and D. K. Karaolis. 2004. Role of exopolysaccharide, the rugose phenotype and VpsR in the pathogenesis of epidemic Vibrio cholerae. FEMS Microbiol. Lett. 230:105-113. [DOI] [PubMed] [Google Scholar]
- 54.Richardson, K., and C. D. Parker. 1985. Identification and occurrence of Vibrio cholerae flagellar core proteins in isolated outer membrane. Infect. Immun. 47:674-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roberts, A., G. D. Pearson, and J. J. Mekalanos. 1992. Cholera vaccine strains derived from a 1991 Peruvian isolate of Vibrio cholerae and other El Tor strains. Proceedings of the 28th Joint Conference, U.S.-Japan Cooperative Medical Science Program on Cholera and Related Diarrheal Diseases. National Institutes of Health, Bethesda, MD.
- 56.Rosen, D. A., T. M. Hooton, W. E. Stamm, P. A. Humphrey, and S. J. Hultgren. 2007. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 4:e329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schoolnik, G. K., M. I. Voskuil, D. Schnappinger, F. H. Yildiz, K. Meibom, N. A. Dolganov, M. A. Wilson, and K. H. Chong. 2001. Whole genome DNA microarray expression analysis of biofilm development by Vibrio cholerae O1 E1 Tor. Methods Enzymol. 336:3-18. [DOI] [PubMed] [Google Scholar]
- 58.September, S. M., F. A. Els, S. N. Venter, and V. S. Brozel. 2007. Prevalence of bacterial pathogens in biofilms of drinking water distribution systems. J. Water Health 5:219-227. [PubMed] [Google Scholar]
- 59.Spoering, A. L., and K. Lewis. 2001. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J. Bacteriol. 183:6746-6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stewart, P. S. 1996. Theoretical aspects of antibiotic diffusion into microbial biofilms. Antimicrob. Agents Chemother. 40:2517-2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tamayo, R., S. Schild, J. T. Pratt, and A. Camilli. 2008. Role of cyclic di-GMP during el tor biotype Vibrio cholerae infection: characterization of the in vivo-induced cyclic di-GMP phosphodiesterase CdpA. Infect. Immun. 76:1617-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tamplin, M. L., A. L. Gauzens, A. Huq, D. A. Sack, and R. R. Colwell. 1990. Attachment of Vibrio cholerae serogroup O1 to zooplankton and phytoplankton of Bangladesh waters. Appl. Environ. Microbiol. 56:1977-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Teitzel, G. M., and M. R. Parsek. 2003. Heavy metal resistance of biofilm and planktonic Pseudomonas aeruginosa. Appl. Environ. Microbiol. 69:2313-2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thelin, K. H., and R. K. Taylor. 1996. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect. Immun. 64:2853-2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turnock, L. L., E. B. CSomers, N. G. Faith, C. C. J., and A. C. Lee Wong. 2002. The effects of prior growth as a biofilm on the virulence of Salmonella typhimurium for mice. Comp. Immunol. Microbiol. Infect. Dis. 25:43-48. [DOI] [PubMed] [Google Scholar]
- 66.van Opijnen, T., K. L. Bodi, and A. Camilli. 2009. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6:767-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wai, S. N., Y. Mizunoe, A. Takade, S. I. Kawabata, and S. I. Yoshida. 1998. Vibrio cholerae O1 strain TSI-4 produces the exopolysaccharide materials that determine colony morphology, stress resistance, and biofilm formation. Appl. Environ. Microbiol. 64:3648-3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walters, M. C., III, F. Roe, A. Bugnicourt, M. J. Franklin, and P. S. Stewart. 2003. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 47:317-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong, A. C. 1998. Biofilms in food processing environments. J. Dairy Sci. 81:2765-2770. [DOI] [PubMed] [Google Scholar]
- 70.Yildiz, F. H., N. A. Dolganov, and G. K. Schoolnik. 2001. VpsR, a member of the response regulators of the two-component regulatory systems, is required for expression of vps biosynthesis genes and EPSETr-associated phenotypes in Vibrio cholerae O1 El Tor. J. Bacteriol. 183:1716-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yildiz, F. H., and G. K. Schoolnik. 1999. Vibrio cholerae O1 El Tor: identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. Proc. Natl. Acad. Sci. U. S. A. 96:4028-4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu, J., and J. J. Mekalanos. 2003. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev. Cell 5:647-656. [DOI] [PubMed] [Google Scholar]
- 73.Zundel, E., Y. Gallois, E. Constanty, S. M. Roche, F. Mompart, T. Meylheuc, and P. Pardon. 2007. Virulence levels of biofilm-grown Listeria monocytogenes LO28 are lower than those of planktonic cells in an oral inoculation test on mice. J. Food Saf. 27:30-42. [Google Scholar]