

Abstract
Generic versions of intravenous antibiotics are not required to demonstrate therapeutic equivalence with the innovator because therapeutic equivalence is assumed from pharmaceutical equivalence. To test such assumptions, we studied three generic versions of vancomycin in simultaneous experiments with the innovator and determined the concentration and potency of the active pharmaceutical ingredient by microbiological assay, single-dose pharmacokinetics in infected mice, antibacterial effect by broth microdilution and time-kill curves (TKC), and pharmacodynamics against two wild-type strains of Staphylococcus aureus by using the neutropenic mouse thigh infection model. The main outcome measure was the comparison of magnitudes and patterns of in vivo efficacy between generic products and the innovator. Except for one product exhibiting slightly greater concentration, vancomycin generics were undistinguishable from the innovator based on concentration and potency, protein binding, in vitro antibacterial effect determined by minimal inhibitory or bactericidal concentrations and TKC, and serum pharmacokinetics. Despite such similarities, all generic products failed in vivo to kill S. aureus, while the innovator displayed the expected bactericidal efficacy: maximum antibacterial effect (Emax) (95% confidence interval [CI]) was 2.04 (1.89 to 2.19), 2.59 (2.21 to 2.98), and 3.48 (2.92 to 4.04) versus 5.65 (5.52 to 5.78) log10 CFU/g for three generics and the innovator product, respectively (P < 0.0001, any comparison). Nonlinear regression analysis suggests that generic versions of vancomycin contain inhibitory and stimulatory principles within their formulations that cause agonistic-antagonistic actions responsible for in vivo failure. In conclusion, pharmaceutical equivalence does not imply therapeutic equivalence for vancomycin.
The World Health Organization (WHO) and all drug regulatory agencies (DRA) support commercialization of generic medicines because they control costs and are irreplaceable therapeutic options in countries lacking the innovator product (10, 41). WHO defines two products as therapeutically equivalent “if they are pharmaceutically equivalent and, after administration in the same molar dose, their effects with respect to both efficacy and safety are essentially the same, as determined from appropriate bioequivalence, pharmacodynamic, clinical, or in vitro studies” (41). Parenteral formulations, however, are not required to demonstrate therapeutic equivalence because it “may be considered self-evident” (41).
Such assumptions have never been challenged, but there are reasons to do so for parenteral antimicrobials. First, many antibacterials are secreted in nature by microorganisms, and industrial production of the active pharmaceutical ingredient (API) involves complex processes for biosynthesis, purification, and manufacture, hard to replicate even for the designer (22). Second, two molecules may look similar without being identical, displaying different biological effects (2). Third, makers of generic drugs do not necessarily know the nature, composition, and pharmacological interactions of excipients employed by the innovator to avoid polymorphs of the API (33). Fourth, while most medicines interact with the host only, antimicrobials also confront the invader organism, a dynamic triangle with numerous possibilities of biologic variation (3, 11, 17). Thus, mixing the exactitude of chemistry with the variability of biology could generate unpredictable effects in seriously sick patients, but differences between the generic and the innovator might pass unnoticed among the complexity of infectious diseases in which death is one of the expected outcomes.
Vancomycin (VAN) is a fermentation product of Amycolatopsis orientalis, an actinomycete discovered in 1955 in a dirt sample sent from Borneo to scientists at Eli Lilly (24, 27). Infusion reactions were common initially, but technology led the innovator to a safer product (8). Differences in composition are well known (36) and even advertised (Baxter promotional material; Baxter, Bogota, Colombia), but DRA worldwide support commercialization of vancomycin generics based on scant in vitro data claiming unaltered efficacy (9). After 50 years of unparalleled performance of vancomycin against Gram-positive pathogens, in vitro susceptibility has certainly decreased, and nowadays more than 20 clinical studies blame vancomycin for ineffectiveness and claim success for new, very expensive replacements (15). Without exception, all these studies fail to mention the manufacturer of the vancomycin products involved, despite the fact that most hospitals around the globe prefer generics. The present study was designed to fill the gap in evidence regarding in vivo efficacy of vancomycin generics compared with the innovator, spanned from November 2002 to November 2009, and allowed experimentation before and after Eli Lilly sold its brand name for the drug along with the secrets of manufacture (32). The null hypothesis was the assumption made by WHO and DRA, i.e., that pharmaceutical equivalence of vancomycin generics predicts their therapeutic equivalence with the innovator. Our data reject such a hypothesis.
MATERIALS AND METHODS
Antibacterial agents.
Antibiotics were bought from local drugstores and prepared following label instructions for clinical use (Table 1). Before 2004, four vancomycin (VAN) products were commercialized in Colombia: the innovator (Vancocin CP; Eli Lilly, Indianapolis, IN) (here called VAN-Lilly) and three generics manufactured by Abbott Laboratories (Chicago, IL), American Pharmaceutical Partners (APP; Los Angeles, CA), and Proclin Ltda. (Laboratorios Northia, Argentina) (here called VAN-Abbott US, VAN-APP, and VAN-Proclin, respectively). By November 2004, Eli Lilly terminated vancomycin production and sold its brand name to several manufacturers worldwide (32), a deal that generated these changes in the Colombian market: Baxter (Deerfield, IL) started commercialization of Vancocin CP (20) (here called VAN-Baxter), the vancomycin from Abbott in Chicago gave way to a product manufactured in France (here called VAN-Abbott France), VAN-APP was discontinued (APP, press release, 2003, and P. J. Vollmerhaus, ViroPharma, press release, 2004), and VAN-Proclin remained unchanged. By 2008, Abbott introduced additional changes, restarting manufacture in Chicago and commercialization under the brand name Hospira (here called VAN-Hospira). We kept enough provision of Vancocin CP from Eli Lilly for our experiments until 1 month before the expiration date (December 2005), when aliquots of the remaining stock were frozen at −70°C.
TABLE 1.
Characteristics of vancomycin products
| Vancomycin product | Form | Label | Batch/lot no. | Manufacturer | Importer |
|---|---|---|---|---|---|
| Lilly (innovator) | 0.5 g powder for i.v. injection | Vancocin CP | A050370, A048213, A014744 | Eli Lilly & Compañia de Mexico SA de CV | Eli Lilly Interamericana Inc., Bogota, Colombia |
| Vancocina CP | 5MJ42M, 5MT38P, 5MT66 M | Eli Lilly & Company, Indianapolis, IN | Eli Lilly Interamericana Inc., Bogota, Colombia | ||
| Abbott | 0.5 g powder for i.v. injection | Sterile vancomycin hydrochloride, USP | 18879Z7, 95826Z72 | Abbott Laboratories, North Chicago, IL | Abbott Laboratories de Colombia SA, Bogota, Colombia |
| Vancomicina IV | 19236TB21, 22826TB21, 83858Z7 | Abbott France, France | Abbott Laboratories de Colombia SA, Bogota, Colombia | ||
| Vancomicina IV | 85739Z7, 03703Z7, 09993Z7 | Abbott Laboratories, North Chicago, IL | Abbott Laboratories de Chile Ltda., Santiago, Chile | ||
| APP | 0.5 g and 1 g powder for i.v. injection | Vancomycin hydrochloride, USP | 121384, 120331, 120740 | American Pharmaceutical Partners Inc., Los Angeles, CA | Comedica Ltda., Bogota, Colombia |
| Proclin | 0.5 g powder for i.v. injection | Vancomicina 500 mg | 6679, 8872, 8690, 8441, 11471, 10049 | Laboratorios Northia S.A.C.I.F.I.A., Argentina | Proclin Pharma SA, Bogota, Colombia |
Bacterial strains.
Antibacterial efficacy was tested in vitro and in vivo against Staphylococcus aureus GRP-0057, a wild-type clinical isolate from a patient with community-acquired bacteremia. S. aureus ATCC 29213 was the quality control organism for susceptibility tests, as recommended by the Clinical and Laboratory Standards Institute (CLSI) (7). Bacillus subtilis ATCC 6633 was the seeding strain in microbiological assays.
Microbiological assays.
Difco antibiotic medium no. 8 was seeded with B. subtilis, with plating of (10-μl) quintuplet series of 8 2-fold concentrations attainable in human serum (1 to 128 mg/liter), starting 1 dilution above detection limit. Vancomycin concentrations plotted against the diameters of their respective inhibition zones produced standard curves to compute concentration and potency of API. Protein binding was determined simultaneously for generics and the innovator by ultrafiltration of two concentrations (16 and 64 mg/liter) with a 50-kDa membrane (Amicon Ultra-15; Millipore Corp., Billerica, MA) (14). To minimize interassay variation, we made a special apparatus that allowed simultaneous runs of all assays needed for each antibiotic (4). Within-day and overall between-day coefficients of variation were less than 11%. We validated recently the application of this method to establish pharmaceutical equivalence of generic antibiotics (42), and others demonstrated that it is comparable to high-pressure liquid chromatography (HPLC) and even superior to polarization fluoroimmunoassay (Abbott TDX) in accuracy and precision in determining concentrations of vancomycin (40).
Single-dose serum pharmacokinetics (PK) in the mouse.
To study each vancomycin product, 2 groups of 3 neutropenic mice infected with S. aureus GRP-0057 received 2 h after infection subcutaneous vancomycin at 50 mg/kg of body weight dissolved in 200 μl (6 mice per product). Each group was bled (100 μl by retro-orbital puncture) four times, one at 20, 45, 75, and 120 min and the other at 30, 60, 90, and 150 min postdose, spanning 5 vancomycin murine half-lives. Each PK experiment included the simultaneous comparison of one generic with the innovator. Serum was obtained by blood centrifugation at 10,000 × g for 10 min and plated immediately (10 μl) in duplicate for microbiological assays. After 20 min at room temperature, plates were placed within the incubator under an air atmosphere at 37°C for 18 h and then inhibition zones were measured with an electronic caliper (Mitutoyo Corp., Kawasaki, Japan). Inhibition zones were interpolated by linear regression against the standard curve of the innovator and the respective generic (Prism 4; GraphPad Software Inc., San Diego, CA), and the time-concentration data were analyzed by population PK (Kinetica; Thermo Scientific, Waltham, MA). For dose levels for which no kinetics were determined, PK parameters were extrapolated from the values obtained with 50 mg/kg.
Susceptibility testing.
Broth microdilution followed CLSI guidelines (7). Duplicates of MICs and minimal bactericidal concentrations (MBCs) of all products against S. aureus GRP-0057 were read simultaneously after 18 to 21 h of incubation and repeated two to three times. Results for quality control strain S. aureus ATCC 29213 remained within the accepted range.
Time-kill curves (TKC).
S. aureus GRP-0057 inoculated (105 to 106 CFU/ml) into 30 ml cation-adjusted Mueller-Hinton broth (MHB) was incubated with shaking in a water bath (37°C) for 48 h. Culture samples (0.1 ml) taken at 0, 6, 12, 18, 24, 36, and 48 h were serially diluted before being plated on Trypticase soy agar. All experiments included untreated and treated cultures with 1, 2, 4, 10, 20, and 40 times the MIC (1.0 mg/liter), with simultaneous testing of the innovator (VAN-Lilly) and generics (VAN Abbott US, VAN-APP, and VAN-Proclin).
In vivo pharmacodynamics (PD): the animal model.
We used the neutropenic mouse thigh infection model to determine PD of each product. Mice were bred and housed in a murine pathogen-free barrier facility (Micro-Isolator system; Lab Products, Seaford, DE), with free access to sterile water plus vitamin K (Sigma-Aldrich, St. Louis, MO) and sterile mouse chow (Zeigler Bros., Gardners, PA). The University of Antioquia Animal Experimentation Ethics Committee approved each experimental procedure involving mice. For the model, 6-week-old Udea:ICR(CD-1) females weighing 23 to 27 g were rendered neutropenic by 2 intraperitoneal injections of cyclophosphamide (Cytoxan; BMS, New York, NY), 4 days (150 mg/kg) and 1 day (100 mg/kg) before infection; ≤10 neutrophils/μl were confirmed from infection point to day 4 afterwards (43). Infection was induced by intramuscular inoculation of 100 μl log-phase bacteria in each thigh; treatment started 2 h later and lasted 24 h. At least five 24-h total doses (24hTD), spanning from no effect to maximum effect, were studied per product. Each 24hTD was given to groups of 2 mice (10 mice to test five 24hTD) and administered by 200-μl subcutaneous injections given every 1 h (q1h). Although the area under the concentration-time curve over 24 h in the steady state divided by the MIC (AUC/MIC ratio) is the pharmacodynamic index that predicts its efficacy, vancomycin is actually a time-dependent antibiotic with prolonged persistent effects. For instance, to maintain maximum kill rates, serum levels should constantly exceed the MIC (12). Since its half-life in the mouse (∼30 min) is approximately 12 times shorter than that in humans (∼360 min), the q1h dosing schedule was selected to constantly exceed the MIC, as happens with q12h dosing in adult humans (12). Untreated controls were sacrificed in groups of 2 mice right after inoculation (hour −2, to confirm inoculum size) and at the time of starting (hour 0) and ending therapy (hour 24). Treated animals were sacrificed at hour 24, and their thighs were dissected aseptically, homogenized, serially diluted, plated in duplicate, and incubated at 37°C for 18 h. Detection of antibiotic carryover was part of the protocol in every experiment, but it was found only with doses of ≥2,400 mg/kg per day (used once, thighs were washed three times to eliminate vancomycin from tissues; no trace of vancomycin was detected after this procedure, and no signs of carryover effect were seen on plates). After colonies were counted for each thigh, data were stored in an Excel database (Microsoft, Seattle, WA). Each data point in the figures represents the mean of both thighs from one mouse, unless indicated otherwise. The limit of detection was 100 CFU per thigh, and each thigh in this model weighs 1 g; therefore, any thigh with zero colonies was entered in the database as 100 CFU/g.
Statistical analysis.
All experiments included the innovator (gold standard) and at least one generic product; tests to assess the magnitude and significance of the differences between groups varied according to the parameters involved. Comparisons of concentration and potency of the API as well as protein binding of each product required curve fitting analysis (CFA) with Prism. Primary population PK parameters volume of distribution and clearance were computed under a two-compartment model (Kinetica) and used to calculate secondary PK parameters half-life and area under the concentration-time curve (AUC), and then the AUCs of generics and innovator were compared by analysis of variance (ANOVA) followed by Bonferroni's post hoc test (Prism); our design had 86.9% power to detect a 25% difference (α = 0.05) in AUC between any of 3 generics and the innovator, expecting 11% maximum variance in standard deviation. To compare MICs, MBCs, or MBC/MIC ratios, we used the Kruskal-Wallis test with StatXact-5 (Cytel Software, Cambridge, MA); for TKC, we determined the intensity of the effect (IE) of each VAN product by subtracting the AUC from 0 to 48 h (AUC0-48) of treated cultures from that of untreated cultures (19) and then compared IE values by one-way ANOVA with Prism. To facilitate extrapolation to the clinical setting, we used in some graphs the AUC/MIC ratio for the free, unbound fraction of vancomycin (fAUC/MIC) instead of 24-h total dose as the independent variable.
The data process for the thigh model starts by subtracting CFU/g of untreated control mice (hour 24) from CFU/g remaining in mice treated for 24 h. This value represents the antibacterial effect (E) for each mouse (dependent variable) and is designed to span from no effect to maximum effect; E is a negative number because that implies effective bacterial killing, except under ineffective doses that eventually allow growth beyond that of untreated mice. Least-squares nonlinear regression (NLR) produces three primary pharmacodynamic parameters (PDP) that describe the sigmoid dose-effect relationship typical of bactericidal antibiotics fitting the Hill model: Emax or maximum effect, 50% effective dose (ED50) or the dose required for attaining half the Emax, and N or Hill's slope:
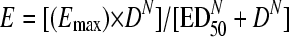 |
(1) |
where E is the antibacterial effect caused by D, the 24hTD. Each primary PDP has its own biological meaning under the Hill model: Emax quantifies the efficacy of the antibiotic, ED50 is an approximate measure of potency, and N describes the affinity of the drug's API for binding its molecular target in the bacterial cell. Secondary PDP represent the exact potency of the antibiotic (28, 38): the doses (mg/kg per day) required in vivo to reach a net bacteriostatic effect (BD) or to kill the first log (1LKD). To calculate secondary PDP, the net bacterial growth in the absence of therapy (G = CFU/gh24 − CFU/gh0) replaces E in equation 1 when the antibiotic action prevents bacterial growth (BD) or when it kills the first log of organisms (1LKD):
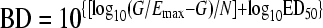 |
(2) |
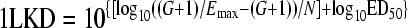 |
(3) |
Due to the aberrant pharmacodynamic behavior of generic products (unfitted by the classical sigmoid Hill model), it was necessary to fit their data to a U-shaped curve described by the Gaussian model:
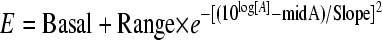 |
(4) |
where
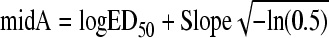 |
(5) |
Basal represents response in the absence of antibiotic, Range is the maximal inhibitory response value lying within the Basal and the deepest point of the Gaussian curve (Emax − Basal), and Slope is a fitting constant that describes the particular form of the bell-shaped curve (not to be confused with Hill's slope) (6). LogED50 is the logarithm of the effective dose needed to reach 50% of the Emax. The expression 10log[A] in equation 4 corresponds to the logarithmic form in which the dose is introduced in all dose-response relationships: [A] is the independent variable, represented here by the 24hTD. Since Basal is zero (CFU/gcontrols − CFU/gtreated = 0 without treatment), Range equals Emax in our Gaussian model:
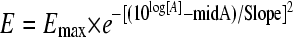 |
(6) |
If any generic and the innovator fit different PD models, their effects are not “essentially the same”; therefore, there is not therapeutic equivalence. To establish which model appropriately described the dose-effect relationship of each product, the individual probability of the Hill and Gaussian model being correct was computed by Akaike's information criterion (AICc) with Prism. Besides, we ran all products simultaneously under the Hill model (multiple NLR [M-NLR]), fixing the Emax to the innovator's value, a strategy that permits calculation of hypothetical ED50 and N values for generic products assuming that the null hypothesis is correct (generic = innovator). The experimental design (inclusion of the innovator product in every experiment to guarantee simultaneous comparisons with all generics) allows this approach, giving significant PDP for all products without violating NLR assumptions, an absolute requirement for valid comparison of PDP by CFA (23). Simple NLR permits independent analysis of each product to determine its PD profile without the influence of the others, and M-NLR allows comparison of several generics against the innovator, assuming that all have identical proportions of the same chemical entities (the null hypothesis).
Accepting a 5% chance for a type I error under CFA (a specialized ANOVA), the treatment of 10 animals per product to compare 3 generics with the innovator (one experiment with 40 treated and 6 untreated mice) confers 96.6% power to reject the null hypothesis if the magnitude of the difference in antibacterial efficacy between generics and innovator is ≥1 log10 CFU/g. Such difference represents in this model a net bactericidal effect of ≥0.1 million bacterial cells, a threshold value several orders of magnitude greater than what would be considered important in clinical medicine.
RESULTS
Microbiological assays.
The concentration and potency of VAN-APP and VAN-Proclin were indistinguishable from those of the innovator. VAN-Abbott US displayed equivalent potency (parallel slopes, P = 0.9434) but contained a greater concentration of API (124.7%, different intercepts, P = 0.0085). Vancomycin protein binding in mouse serum ranged from 22.7 to 27.2% (64 mg/liter) and from 24.2 to 36.4% (16 mg/liter) for all products, without difference between generics (VAN-Hospira and VAN-Proclin) and the innovator available at the time of this assay (VAN-Baxter) or between the concentrations tested (mean protein binding, 28.4%).
Single-dose serum PK in infected mice.
Table 2 contains primary and secondary population PK parameters for VAN-Lilly, VAN-Abbott US, VAN-APP, and VAN-Proclin after one subcutaneous injection of 50 mg/kg. Prediction curves for population PK parameters were highly correlated with observed data for all products (r2 = 0.979 for VAN-Lilly and >0.999 for generics). As expected from its pharmaceutical nonequivalence, VAN-Abbott US exceeded significantly serum AUC (123%), while pharmaceutically equivalent generics VAN-APP (99%) and VAN-Proclin (103%) remained indistinguishable from VAN-Lilly.
TABLE 2.
Single-dose mouse serum population pharmacokinetics of three generics and the innovator product of vancomycin after injection of 50 mg/kg subcutaneouslya
| VAN product | EHL (min) | CV (%) | Cmax (mg/liter) | CV (%) | V (liters) | CV (%) | Clearance (liters/min) | CV (%) | AUC (min·mg/liter) | CV (%) | PAUC (ANOVA)b |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lilly | 33.0 | 12.3 | 51.2 | 23.9 | 0.01332 | 1.38 | 0.00028 | 11.3 | 4,460 | 11.0 | Reference |
| Abbott | 21.6 | 3.00 | 73.3 | 33.2 | 0.00712 | 3.18 | 0.00023 | 0.69 | 5,479 | 0.69 | <0.001 |
| APP | 36.1 | 3.00 | 49.5 | 12.6 | 0.01482 | 1.16 | 0.00028 | 2.13 | 4,397 | 2.12 | 1.000 |
| Proclin | 34.1 | 0.93 | 55.1 | 9.59 | 0.01280 | 0.19 | 0.00027 | 0.99 | 4,617 | 0.55 | 0.878 |
Abbreviations: EHL, effective half-life; CV, coefficient of variation; V, volume of distribution.
Bonferroni's post hoc test.
In vitro susceptibility testing.
Vancomycin products did not differ in MIC, MBC, or MBC/MIC ratio against S. aureus GRP-0057 or ATCC 29213. Geometric means of MIC and MBC against the first strain ranged from 1.19 to 1.41 and from 1.68 to 2.38 mg/liter, respectively (Table 3).
TABLE 3.
Determination of MICs and MBCs of vancomycin products against two strains of S. aureus by broth microdilution (all products were tested simultaneously)
| VAN product | Geometric mean (range)a |
|||||
|---|---|---|---|---|---|---|
|
S. aureus GRP-0057 |
S. aureus ATCC 29213 |
|||||
| MIC | MBC | MBC/MIC | MIC | MBC | MBC/MIC | |
| Lilly | 1.19 (1-2) | 2.00 (2-2) | 1.68 (1-2) | 1.00 (1-1) | 2.00 (1-4) | 2.00 (1-4) |
| Abbott | 1.41 (1-2) | 2.38 (2-4) | 1.68 (1-4) | 1.19 (1-2) | 1.41 (1-2) | 1.19 (0.5-2) |
| APP | 1.41 (1-2) | 1.68 (1-4) | 1.19 (0.5-2) | 1.19 (0.5-1) | 1.41 (1-2) | 1.19 (1-4) |
| Proclin | 1.19 (1-2) | 1.68 (1-2) | 1.41 (1-2) | 1.19 (1-2) | 2.00 (2-2) | 1.68 (1-2) |
| P (Kruskal-Wallis) | 0.81 | 0.35 | 0.35 | 0.35 | 0.49 | 0.80 |
Obtained after two or three duplicate assays.
Time-kill curves (TKC).
Before addition of vancomycin, MHB cultures had (S. aureus GRP-0057) 105.56 to 106.19 CFU/ml; untreated controls grew up to 108.68 to 109.13 CFU/ml by 24 to 48 h (growth, 2.49 to 3.57 log10 CFU/ml). Bacteriostatic and bactericidal concentrations acted as expected, but no product or concentration achieved culture sterilization. IE comparisons showed no differences between generics and the innovator (P ≥ 0.22 for all vancomycin concentrations).
In vivo pharmacodynamics.
We obtained identical results from three independent experiments in the neutropenic mouse thigh infection model designed to compare the dose-effect curves of VAN-Abbott US, VAN-APP, and VAN-Proclin against those of VAN-Lilly. Untreated control mice from these three experiments had on average (±standard error of the mean [SEM]) 104.30 ± 0.16 and 107.82 ± 0.11 CFU per thigh at hours 0 and 24, respectively (24-h growth range, 3.39 to 3.65; weighted growth mean, 3.47 ± 0.08 log10 CFU/g). As expected, there was no difference among the three dose-effect curves of the innovator (P = 0.2594), allowing the combination of the data in a single NLR. Equation 1 (Hill model) described VAN-Lilly's dose-effect relationship with an excellent fit, producing multicollinearity-free, very significant PDP and a sound NLR fulfilling normality, constant variance, and independence assumptions (Table 4). Emax was 5.65 ± 0.07 log10 CFU/g, and ED50 was 62.7 ± 1.61 mg/kg per day. The steep N (5.6 ± 0.70) suggests that vancomycin-receptor interaction is an all-or-none phenomenon, exquisitely dose dependent. 1LKD (79.6 ± 1.54) was only 16.7% greater than BD (68.2 ± 1.26 mg/kg/day), as expected from highly bactericidal antibiotics (low MBC/MIC ratios).
TABLE 4.
In vivo efficacies of three generics and the innovator product of vancomycina
| Model PDP (unit) or statistical test | Vancomycin product PDP magnitude ± SE |
P value by CFA (IP vs GP) | |||
|---|---|---|---|---|---|
| Lilly | Abbott | APP | Proclin | ||
| Hill's Emax (log10 CFU/g)c | 5.65 ± 0.07b | 2.04 ± 0.07b | 2.59 ± 0.18b | 3.48 ± 0.27b | <0.0001 (all GP) |
| Gaussian Emax (log10 CFU/g)c | 6.70 ± 0.29b | 2.30 ± 0.17b | 3.28 ± 0.13b | 5.07 ± 0.39b | <0.0001 (all) |
| Hill's ED50 (mg/kg/day) | 62.7 ± 1.61b | NS | 58.6 ± 7.17b | NS | 0.57 (APP) |
| Gaussian logED50 | 1.90 ± 0.04b | 1.75 ± 0.09b | 1.82 ± 0.02b | 1.80 ± 0.04b | 0.14 (all) |
| Hill's slope (N) | 5.60 ± 0.70b | NS | 6.32 ± 2.78 | NS | 0.72 (APP) |
| Gaussian slope | 0.92 ± 0.06b | 0.91 ± 0.11b | 0.43 ± 0.02b | 0.45 ± 0.04b | <0.001 (APP, Proclin) |
| Hill's BD (mg/kg/day) | 68.2 ± 1.26b | NC | No bacteriostatic effect | NC | NA |
| Hill's 1LKD (mg/kg/day) | 79.6 ± 1.54b | NC | No bactericidal effect | NC | NA |
| Adjusted R2 | 0.99 | 0.94 | 0.96 | 0.85 | NA |
| Sy|x | 0.24 | 0.25 | 0.25 | 0.74 | NA |
| Model probability of being correct | Hill, >0.9999 | Hill, >0.9999 | Gaussian, >0.9999 | Gaussian, 0.9793 | NA |
Simple nonlinear regression analysis of each product based on the pharmacodynamic equation (Hill or Gaussian) best fitting its dose-effect relationship (all equations passed normality and constant variance tests). Abbreviations: GP, generic product; IP, innovator product; NA, nonapplicable; NC, not computable; NS, nonsignificant PDP (the PDP value was not significantly different from zero). Data in bold refer exclusively to the model best fitting each vancomycin product.
P < 0.0001 (other PDP had P values between 0.0001 and 0.050).
Emax values are negative because they represent a decrease in the number of microorganisms. The minus sign is eliminated to facilitate reading.
Generics' PD were completely different from those of the innovator. Emax was statistically different from zero under equation 1 for all three generics, but their magnitudes were much lower, killing ∼445,000 fewer microorganisms per gram of tissue than did VAN-Lilly (P < 0.0001). VAN-Abbott US could not reach bacteriostasis (Emax = 2.04 ± 0.07 log10 CFU/g), allowing bacterial growth even at maximal dosing (1,200 mg/kg per day; AUC/MIC ratio, 1,068 h) and therefore preventing computation of ED50 and N. VAN-APP had significant PDP but marginal antibacterial efficacy (Emax = 2.59 ± 0.18 log10 CFU/g), violating the constant variance assumption under the Hill model. VAN-Proclin performed best among generics (Emax = 3.48 ± 0.27 log10 CFU/g), but the data did not fit the Hill model either, violating the constant variance assumption and giving nonsignificant values for ED50 and N (Table 4).
Analysis of dose-effect relationships explains generics' unfitness to the Hill model (Fig. 1). VAN-Abbott US was completely ineffective; it is shown in comparison with the innovator in a separate graph because it was not pharmaceutically equivalent to VAN-Lilly; thus, the two products have different AUC/MIC ratios despite their identical dosing regimens (Fig. 1A). VAN-APP and VAN-Proclin achieved bacteriostasis or killed 1 log at 75 to 150 mg/kg (fAUC/MIC, 66.8 to 133.5 h), but greater doses caused paradoxical bacterial growth in a U-shaped, Gaussian pattern (Eagle effect) (Fig. 1B). AICc model comparison confirmed that while the dose-effect relationships of VAN-Lilly and VAN-Abbott US fitted equation 1 (the Hill model) with a probability of correctness of >0.9999, those of VAN-APP and VAN-Proclin fitted equation 6 (the Gaussian model) with probabilities of correctness of >0.9999 and 0.9793, respectively (Table 4). Comparison of all generic products with the innovator under Hill's model (M-NLR) demonstrated that 2.1 (VAN-Proclin), 4.3 (VAN-APP), and infinite (VAN-Abbott US) dose increments would be required to reach the innovator's efficacy (Table 5).
FIG. 1.

In vivo efficacy against S. aureus GRP-0057 (years 2002 and 2003) at a low inoculum (4.30 ± 0.05 log10 CFU per thigh when subcutaneous treatment q1h started). Vancomycin generic products are compared with the innovator (VAN-Lilly) in dose-effect experiments (2.34 to 1,200 mg/kg per day) using the neutropenic mouse thigh infection model (each data point represents the mean CFU/g of both thighs from a single mouse). (A) Pharmacodynamic patterns of VAN-Abbott US and VAN-Lilly fitted to the Hill model. Despite containing a significantly greater concentration of API (125%), VAN-Abbott US was completely ineffective in vivo. VAN-Abbott US is shown in a separate graph because of its greater AUC/MIC ratio than that of VAN-Lilly (123%; their dosing regimens were identical). (B) VAN-APP and VAN-Proclin were both pharmaceutically equivalent to VAN-Lilly, but neither was therapeutically equivalent due to their marked Eagle effect. The curve for VAN-APP ends at 300 mg/kg (fAUC/MIC, 267 h) because this product was discontinued and the remaining amount was insufficient for the highest doses.
TABLE 5.
| PDP (unit) | PDP magnitude ± SE for each vancomycin product: |
P value by CFA (IP vs GP) | |||
|---|---|---|---|---|---|
| Lilly | Abbott | APP | Proclin | ||
| Emax (log10 CFU/g) | −5.65 ± 0.25 (<0.0001) | −5.65 ± 0.25 (<0.0001) | −5.65 ± 0.25 (<0.0001) | −5.65 ± 0.25 (<0.0001) | NA |
| ED50 (mg/kg/day) | 62.7 ± 6.21b | 1,877 ± 1,684d | 270.7 ± 112.6c | 129.5 ± 33.0c | <0.0001 (all) |
| N (Hill slope) | 5.55 ± 2.67c | 0.41 ± 0.14c | 0.74 ± 0.25c | 0.77 ± 0.16b | <0.0001 (all) |
| BD (mg/kg/day) | 68.2 ± 1.26b | No bacteriostatic effect | No bacteriostatic effect | No bacteriostatic effect | NA |
| 1LKD (mg/kg/day) | 78.6 ± 1.57b | No bactericidal effect | No bactericidal effect | No bactericidal effect | NA |
Abbreviations: GP, generic product; IP, innovator product; NA, nonapplicable.
P < 0.0001.
P between 0.0001 and 0.050.
P = 0.268.
All data shown so far were obtained before November 2004; results from experiments carried out after 1 December 2004 are shown below (this is relevant to understanding why some products did and others did not change their PD profile during the execution of this study). Despite the fact that serum PK demonstrated comparable absorptions from subcutaneous space for all products, we ruled out any impact of the injection site or the inoculum size on the results, adapting the thigh model for intravenous (i.v.) treatment and increasing the number of microorganisms per thigh at hour 0. After infection with S. aureus GRP-0057, groups of 15 mice received 2 h later VAN-Abbott US or VAN-Lilly q8h i.v., in 5 doses ranging from 75 to 1,200 mg/kg per day (3 mice per dose), starting when mice had 6.74 ± 0.12 log10 CFU per thigh (24-h growth, 1.52 ± 0.21 log10 CFU/g). Confronting this higher inoculum by the i.v. route, both products became less potent (VAN-Lilly, 3.5-fold; VAN-Abbott, 3.8-fold) and showed the Eagle effect, but it was more conspicuous in VAN-Abbott US (Fig. 2), which displayed minimal efficacy compared to the innovator (Emax = 3.82 ± 0.40 versus 5.35 ± 0.15, respectively; P < 0.0001). This lot of VAN-Abbott US was manufactured and imported directly from Chicago by the maker (labeled in English), while all lots employed in previous experiments came from Chile (labeled in Spanish but also manufactured in Chicago). We also determined if this new lot of VAN-Abbott US was equivalent to VAN-Lilly by other routes (intraperitoneal and subcutaneous) or under different dosing regimens (q1h, q3h, q6h, and q12h) at 1,200 mg/kg per day: it had some efficacy independently of these variables but always less than that of the innovator. M-NLR analysis confirmed the significant inferiority of VAN-Abbott US (P < 0.001) related to the product itself (P = 0.003), not to the route (P > 0.05) or schedule (P > 0.05) of administration (not shown).
FIG. 2.

In vivo efficacy against S. aureus GRP-0057 (year 2004) at a high inoculum (6.74 log10 CFU per thigh when intravenous treatment q8h started). VAN-Abbott US was compared with the innovator (VAN-Lilly) after intravenous administration (75 to 1,200 mg/kg per day) but with 2.5-log increases in the inoculum size. The greater bacterial load required four times more vancomycin to reach maximum effect (600 mg/kg; fAUC/MIC, 534.1 h) and caused the Eagle effect in both products, but the efficacy of VAN-Abbott US was significantly inferior to that of VAN-Lilly (Emax, 3.82 ± 0.33 versus 5.35 ± 0.13, respectively; P < 0.0001). Note that despite the use of identical dosing regimens, the AUC/MIC ratio of VAN-Abbott US is 124% of that of VAN-Lilly due to pharmaceutical nonequivalence.
We found during in vivo TKC experiments with S. aureus GRP-0057 that VAN-Abbott France displayed essentially the same efficacy as did VAN-Lilly (not shown), in contrast to data from VAN-Abbott US. This was confirmed by repeating the thigh model with S. aureus ATCC 29213, a strain more susceptible in vivo to vancomycin than is S. aureus GRP-0057. VAN-APP was not tested because it became unavailable in Colombia in 2005; VAN-Baxter had bought the manufacturing secrets from Eli Lilly and introduced the same brand name as the innovator (Vancocin CP). Four groups of 10 animals received subcutaneous treatment q1h (18.75 to 300 mg/kg per day), starting when mice had 4.13 log10 CFU per thigh (24-h growth, 4.58 log10 CFU/g). There was no difference (P = 0.7681) in efficacy between VAN-Abbott France, VAN-Baxter, and VAN-Lilly, all three fitting Hill's model. VAN-Proclin, however, differed significantly from VAN-Lilly, displaying again the paradoxical U-shaped pattern, with 99.7% probability of better fit to the Gaussian model than to the Hill model by AICc (Fig. 3).
FIG. 3.

In vivo efficacy against S. aureus ATCC 29213 (year 2005) at a low inoculum (4.13 log10 CFU per thigh when subcutaneous treatment q1h started), after some makers of generics acquired manufacturing secrets from Eli Lilly. Vancomycin generic products were compared with the innovator (VAN-Lilly) in dose-effect experiments (18.75 to 300 mg/kg per day) using the neutropenic mouse thigh infection model (each data point represents the mean CFU/g of both thighs from a single mouse). VAN-Abbott France, VAN-Baxter, and VAN-Lilly fitted to the Hill model and were indistinguishable (P = 0.7681). VAN-Proclin, on the other hand, displayed again the Eagle effect, fitting the Gaussian instead of the Hill model, as happened before 2005.
To determine if extreme bacterial inocula could impact in vivo results, we repeated the thigh model with a very high inoculum of S. aureus GRP-0057 and tested all vancomycin products available in Colombia during 2008. VAN-Baxter (substituting for the innovator due to discontinuation of VAN-Lilly) was compared with VAN-Proclin and VAN-Hospira (new generic made by Abbott in Lake Forest, IL, commercialized under the brand name Hospira). Low inoculum (4.07 log10 CFU per thigh when treatment started) led to vigorous bacterial growth (4.17 log10 CFU/g in 24 h), failure of VAN-Proclin (Emax = 2.73 ± 0.10 log10 CFU/g; P < 0.0001 compared to VAN-Baxter), and therapeutic success of VAN-Hospira and VAN-Baxter (Emax = 5.43 ± 0.10 and 5.60 ± 0.15 log10 CFU/g, respectively, P = 0.3497) (Fig. 4 A). Very high inoculum (8.34 log10 CFU per thigh when treatment started) led to minimal bacterial growth (1.12 log10 CFU/g in 24 h) and decreased efficacy of both products, but VAN-Proclin still differed significantly from VAN-Baxter (P = 0.0021), particularly in terms of the exact potency of each vancomycin (BD = 274.2 ± 17.3 and 151.2 ± 23.1 mg/kg, respectively, P = 0.0006) (Fig. 4B). Of note, these lots of VAN-Proclin did not show an Eagle effect.
FIG. 4.

In vivo efficacy of vancomycin products available in Colombia during 2008 against S. aureus GRP-0057 at low (A) and very high (B) inocula (4.07 and 8.34 log10 CFU per thigh when subcutaneous treatment q1h started, respectively). After VAN-Lilly was discontinued, VAN-Baxter replaced it as the innovator product; both panels show its dose-effect relationship compared with those of the newest version of VAN-Abbott (commercialized under the brand name Hospira) and VAN-Proclin. At a low inoculum, VAN-Hospira was indistinguishable from VAN-Baxter while VAN-Proclin was again ineffective; the very high inoculum had a marked impact on vancomycin pharmacodynamics, but VAN-Proclin remained inferior despite losing its Eagle effect.
DISCUSSION
These data indicate that, before 2005, all generic versions of vancomycin commercialized in Colombia were ineffective in vivo, i.e., they lacked therapeutic equivalence with respect to the innovator. The findings were consistently reproduced under diverse conditions in neutropenic mice infected in the thighs with two wild-type clinical strains of S. aureus and occurred independently of the manufacturer's reputation. Unexpectedly, two products (VAN-APP and VAN-Proclin) were indistinguishable from the innovator in terms of concentration and potency of the API, protein binding, MIC, MBC, MBC/MIC ratios, standard TKC, and PK profiles, and the only product that differed (VAN-Abbott) had 125% of the API concentration and 123% of the AUC of VAN-Lilly, but none of it made these generics effective. One uncomfortable aspect uncovered by this study is that all these tests have been used for decades to guarantee therapeutic equivalence of generic drugs, except in vivo pharmacodynamics. On the positive side, we also found that some generic products evolved and reached therapeutic equivalence after 2005, and one maker was able to produce effective vancomycin (VAN-Baxter) right from the beginning after buying manufacturing secrets from the innovator.
Two potential limitations deserve consideration. Determination of pharmaceutical equivalence was based on microbiological assays, a nonchemical technique unsuitable for finding fermentation impurities or degradation products that probably explain therapeutic failure of generic vancomycin (see below). However, the microbiological assay was better suited to the exploratory nature of this study because it gives accurate estimates of potency (besides concentration), a specific requirement from DRA for pharmaceutical equivalence (21). Another limitation was the use of the maximum dose (50 mg/kg) as the only level at which PK parameters were obtained (we lost to technical errors the data from the other two dose levels, 12.5 and 3.125 mg/kg). One dose level is enough to establish bioequivalence (31), but additional dose levels would have provided a more accurate extrapolation of PK parameters to the lowest dose used in the animal model (0.78 mg/kg).
The animal model demonstrated that the innovator of vancomycin required an AUC/MIC ratio of 133.5 h for maximal efficacy. Generic products, in contrast, would fail in the clinical setting if such a target were attained, because VAN-Abbott US and VAN-APP would not even reach bacteriostatic effect, while VAN-Proclin would kill ∼445,000 fewer microorganisms per gram than would the innovator. Efficacy would never be obtained with VAN-Abbott US, independently of the dose prescribed by the physician (Fig. 1). If, on the other hand, VAN-APP and VAN-Proclin are prescribed to reach the commonly recognized target for maximal efficacy of vancomycin (400 h), an even less effective response would ensue due to the Eagle effect (Fig. 1). This paradoxical PD profile, reported in 1948 for S. aureus exposed to increasing concentrations of penicillin (18), is described by the Gaussian model, which is used to fit concentration-response curves with both inhibitory and stimulatory components (6).
Vancomycin had so many fermentation impurities that it was nicknamed “Mississippi Mud” 50 years ago (30). After several attempts, Eli Lilly developed a chromatographic purification method that led to a product with at least 92% factor B and less than 4% impurities (Vancocin CP). Such impurities, known as crystalline degradation products or CDP-1 (minor and major fractions) (25), explained the greater frequency of adverse reactions reported for generics elsewhere (36) and, we propose, the Eagle effect found here. Antibacterial efficacy depends entirely on factor B (5, 29, 39), but CDP-1 binds d-Ala-d-Ala (36) with less affinity (>1,000×) and efficacy (7 to 14×) (5, 29, 39). Generics have less factor B (84% at most) and two to three times more CDP-1 than does the innovator (9, 36). A vancomycin agonistic-antagonistic pharmacodynamic pattern is also evident with the innovator, but only at the greatest dose and by the intravenous route, without impact on efficacy compared with generic products (Fig. 2). It is not surprising if we consider that a 25-g mouse will pass through its body ∼1 × 1018 molecules of vancomycin after 100 mg/kg per day and that one S. aureus cell has 106 false (cell wall) and 103 to 104 vital (periplasmic space) d-Ala-d-Ala targets. If bacterial growth reaches 109 CFU/g, the number of false targets will be 1015. That would leave only 103 molecules of vancomycin in the mouse to confront 1012 to 1013 vital d-Ala-d-Ala targets per gram of tissue (26). Under this adverse balance, every molecule of vancomycin counts, and so protein binding, renal clearance, and, of course, concentrations of factor B and its antagonist CDP-1 become critical for in vivo efficacy. It explains why vancomycin is so susceptible to inoculum size of S. aureus in vivo (Fig. 4), as was demonstrated by Craig et al. (13). Based on this interpretation of the data, we postulate that the lower efficacy of generics in vivo is due to a relative absence of free factor B molecules, wasted in binding more d-Ala-d-Ala to counteract the antagonistic competence of CDP-1. This hypothesis would explain the PD profile of generics displaying the Eagle effect, not that of VAN-Abbott US (before November 2004) and VAN-Proclin (from 2008), devoid of in vivo efficacy altogether. For these products, one potential explanation would be faster degradation of factor B in vivo, but we have no data to substantiate such a claim.
The dogma proclaiming that pharmaceutical equivalence predicts therapeutic equivalence is not true for vancomycin. In vivo failure of generics was consistent, independently of their route of administration or the dosing schedule. Although the poor quality of generic products has always been a matter of concern worldwide (37), we describe here an entirely different situation: good quality, as currently defined (41), does not imply efficacy in vivo, and this has clinical implications (34). “Similar” standards seem as insufficient to guarantee therapeutic equivalence as do pharmaceutical equivalence and so-called “bioequivalence” (1, 16, 35). Given their medical importance and the variety of epidemiological consequences emerging from their improper use, all antimicrobials deserve the same scrutiny as that presented here for generic products of vancomycin.
Acknowledgments
This work was supported by the Research Committee of the University of Antioquia and Colciencias grant 1115-04-731-98.
Conflicts of interest are as follows: Omar Vesga has received honoraria for lectures and financial support to participate in international meetings from GlaxoSmithKline, Bristol-Myers Squibb, AstraZeneca, Merck Sharp & Dohme, and Wyeth and has participated in advisory boards for Wyeth and Johnson & Johnson; Maria Agudelo received financial support from AstraZeneca and Wyeth to participate in international meetings; Beatriz E. Salazar received financial support from Bristol-Myers Squibb to participate in one international meeting; Carlos A. Rodriguez received financial support from AstraZeneca and Wyeth to participate in international meetings; and Andres F. Zuluaga received honoraria for lectures from Pfizer and Roche and financial support from Merck Sharp & Dohme to participate in one international meeting.
Footnotes
Published ahead of print on 14 June 2010.
REFERENCES
- 1.Adelman, C. C., and J. Norris. 2001. Usefulness of foreign aid for health care in less-developed countries. Lancet 358:2174. [DOI] [PubMed] [Google Scholar]
- 2.Bauer, J., S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter, and J. Morris. 2001. Ritonavir: an extraordinary example of conformational polymorphism. Pharm. Res. 18:859-866. [DOI] [PubMed] [Google Scholar]
- 3.Beam, T. R., Jr., D. N. Gilbert, and C. M. Kunin. 1992. General guidelines for the clinical evaluation of anti-infective drug products. Infectious Diseases Society of America and the Food and Drug Administration. Clin. Infect. Dis. 15(Suppl. 1):S5-S32. [DOI] [PubMed] [Google Scholar]
- 4.Bennett, J. V., J. L. Brodie, E. J. Benner, and W. M. Kirby. 1966. Simplified, accurate method for antibiotic assay of clinical specimens. Appl. Microbiol. 14:170-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Best, G. K., N. H. Best, and N. N. Durham. 1968. Chromatographic separation of the vancomycin complex. Antimicrob. Agents Chemother. 8:115-119. [PubMed] [Google Scholar]
- 6.Christopoulos, A., M. K. Grant, N. Ayoubzadeh, O. N. Kim, P. Sauerberg, L. Jeppesen, and E. E. El-Fakahany. 2001. Synthesis and pharmacological evaluation of dimeric muscarinic acetylcholine receptor agonists. J. Pharmacol. Exp. Ther. 298:1260-1268. [PubMed] [Google Scholar]
- 7.Clinical and Laboratory Standards Institute. 2009. Performance standards for antimicrobial susceptibility testing, approved standard M100-S19. Clinical and Laboratory Standards Institute, Wayne, PA.
- 8.Conley, N. S., R. S. Weiner, and J. W. Hiemenz. 1991. Rigors with vancomycin. Ann. Intern. Med. 115:330. [DOI] [PubMed] [Google Scholar]
- 9.Conte, J. E., Jr. 1987. Comparative antibacterial activity of Vancocin and generic vancomycin. Antimicrob. Agents Chemother. 31:333-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook, A., J. P. Acton, and E. Schwartz. 1998. How increased competition from generic drugs has affected prices and returns in the pharmaceutical industry. Congressional Budget Office, Washington, DC. http://www.cbo.gov/ftpdocs/6xx/doc655/pharm.pdf1-75.
- 11.Craig, W. A. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 26:1-10. [DOI] [PubMed] [Google Scholar]
- 12.Craig, W. A. 2003. Basic pharmacodynamics of antibacterials with clinical applications to the use of beta-lactams, glycopeptides, and linezolid. Infect. Dis. Clin. North Am. 17:479-501. [DOI] [PubMed] [Google Scholar]
- 13.Craig, W. A., D. Lee, S. Kethireddy, and D. R. Andes. 2008. Comparison of in vitro and in vivo activity of vancomycin against MRSA at 105 and 107 inocula, abstr. A-986. Abstr. 48th Intersci. Conf. Antimicrob. Agents Chemother. (ICAAC)-Infect. Dis. Soc. Am. (IDSA) 46th Annu. Meet. American Society for Microbiology and Infectious Diseases Society of America, Washington, DC.
- 14.Crandon, J. L., M. A. Banevicius, and D. P. Nicolau. 2009. Pharmacodynamics of tigecycline against phenotypically diverse Staphylococcus aureus isolates in a murine thigh model. Antimicrob. Agents Chemother. 53:1165-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deresinski, S. 2007. Vancomycin: does it still have a role as an antistaphylococcal agent? Expert Rev. Anti Infect. Ther. 5:393-401. [DOI] [PubMed] [Google Scholar]
- 16.Dettelbach, H. R. 1986. A time to speak out on bioequivalence and therapeutic equivalence. J. Clin. Pharmacol. 26:307-308. [DOI] [PubMed] [Google Scholar]
- 17.Drusano, G. L. 2004. Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug.’ Nat. Rev. Microbiol. 2:289-300. [DOI] [PubMed] [Google Scholar]
- 18.Eagle, H., and A. D. Musselman. 1948. The rate of bactericidal action of penicillin in vitro as a function of its concentration, and its paradoxically reduced activity at high concentrations against certain organisms. J. Exp. Med. 88:99-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Firsov, A. A., S. N. Vostrov, A. A. Shevchenko, and G. Cornaglia. 1997. Parameters of bacterial killing and regrowth kinetics and antimicrobial effect examined in terms of area under the concentration-time curve relationships: action of ciprofloxacin against Escherichia coli in an in vitro dynamic model. Antimicrob. Agents Chemother. 41:1281-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Food and Drug Administration. 2006. New and generic drug approvals. Center for Drug Evaluation and Research, Food and Drug Administration, Silver Spring, MD.
- 21.Food and Drug Administration. 2001. Guidance for industry: bioanalytical method validation, no. 22. Center for Drug Evaluation and Research, Food and Drug Administration, U.S. Department of Health and Human Services, Rockville, MD.
- 22.Gardner, C. R., C. T. Walsh, and O. Almarsson. 2004. Drugs as materials: valuing physical form in drug discovery. Nat. Rev. Drug Discov. 3:926-934. [DOI] [PubMed] [Google Scholar]
- 23.Glantz, S. A. 2002. How to test for trends, p. 230-297. In S. A. Glantz (ed.), Primer of biostatistics, 5th ed. McGraw-Hill Companies, Inc., New York, NY.
- 24.Griffith, R. S. 1981. Introduction to vancomycin. Rev. Infect. Dis. 3(Suppl.):S200-S204. [PubMed] [Google Scholar]
- 25.Harris, C. M., H. Kopecka, and T. M. Harris. 1983. Vancomycin: structure and transformation to CDP-I. J. Am. Chem. Soc. 105:6915-6922. [Google Scholar]
- 26.Hiramatsu, K. 1998. Vancomycin resistance in staphylococci. Drug Resist. Updat. 1:135-150. [DOI] [PubMed] [Google Scholar]
- 27.Kim, S. J., L. Cegelski, M. Preobrazhenskaya, and J. Schaefer. 2006. Structures of Staphylococcus aureus cell-wall complexes with vancomycin, eremomycin, and chloroeremomycin derivatives by 13C{19F} and 15N{19F} rotational-echo double resonance. Biochemistry 45:5235-5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lees, P., F. M. Cunningham, and J. Elliott. 2004. Principles of pharmacodynamics and their applications in veterinary pharmacology. J. Vet. Pharmacol. Ther. 27:397-414. [DOI] [PubMed] [Google Scholar]
- 29.Marshall, F. J. 1965. Structure studies on vancomycin. J. Med. Chem. 8:18-22. [DOI] [PubMed] [Google Scholar]
- 30.Moellering, R. C., Jr. 2006. Vancomycin: a 50-year reassessment. Clin. Infect. Dis. 42(Suppl. 1):S3-S4. [DOI] [PubMed] [Google Scholar]
- 31.Nation, R. L., and L. N. Sansom. 1994. Bioequivalence requirements for generic products. Pharmacol. Ther. 62:41-55. [DOI] [PubMed] [Google Scholar]
- 32.Pollack, A. 9 November 2005. Crucial antibiotic rescues biotech maker's finances. N. Y. Times, p. C1-C4.
- 33.Remenar, J. F., J. M. MacPhee, B. K. Larson, V. A. Tyagi, J. H. Ho, D. A. McIlroy, M. B. Hickey, P. B. Shaw, and O. Almarsson. 2003. Salt selection and simultaneous polymorphism assessment via high-throughput crystallization: the case of sertraline. Org. Process Res. Dev. 7:990-996. [Google Scholar]
- 34.Rodriguez, C. A., M. Agudelo, J. C. Cataño, A. F. Zuluaga, and O. Vesga. 2009. Potential therapeutic failure of generic vancomycin in a liver transplant patient with MRSA peritonitis and bacteremia. J. Infect. 59:277-280. [DOI] [PubMed] [Google Scholar]
- 35.Senn, S. 1998. In the blood: proposed new requirements for registering generic drugs. Lancet 352:85-86. [DOI] [PubMed] [Google Scholar]
- 36.Somerville, A. L., D. H. Wright, and J. C. Rotschafer. 1999. Implications of vancomycin degradation products on therapeutic drug monitoring in patients with end-stage renal disease. Pharmacotherapy 19:702-707. [DOI] [PubMed] [Google Scholar]
- 37.Taylor, R. B., O. Shakoor, R. H. Behrens, M. Everard, A. S. Low, J. Wangboonskul, R. G. Reid, and J. A. Kolawole. 2001. Pharmacopoeial quality of drugs supplied by Nigerian pharmacies. Lancet 357:1933-1936. [DOI] [PubMed] [Google Scholar]
- 38.Toutain, P. L. 2002. Pharmacokinetic/pharmacodynamic integration in drug development and dosage-regimen optimization for veterinary medicine. AAPS Pharm. Sci. 4:E38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vollmerhaus, P. J., E. Breukink, and A. J. Heck. 2003. Getting closer to the real bacterial cell wall target: biomolecular interactions of water-soluble lipid II with glycopeptide antibiotics. Chemistry 9:1556-1565. [DOI] [PubMed] [Google Scholar]
- 40.White, L. O., R. Edwards, H. A. Holt, A. M. Lovering, R. G. Finch, and D. S. Reeves. 1988. The in-vitro degradation at 37 degrees C of vancomycin in serum, CAPD fluid and phosphate-buffered saline. J. Antimicrob. Chemother. 22:739-745. [DOI] [PubMed] [Google Scholar]
- 41.WHO/DMP/RGS. 1998. Marketing authorization of pharmaceutical products with special reference to multisource (generic) products: a manual for a drug regulatory authority. World Health Organization, Geneva, Switzerland. http://apps.who.int/prequal/info_general/documents/WHO_DMP_RGS_98_5_R.pdf.
- 42.Zuluaga, A. F., M. Agudelo, C. A. Rodriguez, and O. Vesga. 2009. Application of microbiological assay to determine pharmaceutical equivalence of generic intravenous antibiotics. BMC Clin. Pharmacol. 9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuluaga, A. F., B. E. Salazar, C. A. Rodriguez, A. X. Zapata, M. Agudelo, and O. Vesga. 2006. Neutropenia induced in outbred mice by a simplified low-dose cyclophosphamide regimen: characterization and applicability to diverse experimental models of infectious diseases. BMC Infect. Dis. 6:55. [DOI] [PMC free article] [PubMed] [Google Scholar]