

Abstract
Chagas' disease, a neglected tropical infection, affects about 18 million people, and 100 million are at risk. The only drug available, benznidazole, is effective in the acute form and in the early chronic form, but its efficacy and tolerance are inversely related to the age of the patients. Side effects are frequent in elderly patients. The search for new drugs is thus warranted. In the present study we evaluated the in vitro and in vivo effect of a cyclopalladated compound (7a) against Trypanosoma cruzi, the agent of Chagas' disease. The 7a compound inhibits trypomastigote cell invasion, decreases intracellular amastigote proliferation, and is very effective as a trypanocidal drug in vivo, even at very low dosages. It was 340-fold more cytotoxic to parasites than to mammalian cells and was more effective than benznidazole in all in vitro and in vivo experiments. The 7a cyclopalladate complex exerts an apoptosis-like death in T. cruzi trypomastigote forms and causes mitochondrion disruption seen by electron microscopy.
Chagas' disease, also known as American trypanosomiasis, is an infection caused by the parasite Trypanosoma cruzi. Humans are infected by contaminated feces and urine of the insect vector, transfusion with blood from infected patients, oral transmission and contaminated food, and placental or birth canal transmission (5). Inside humans, the parasite is present as a circulating nonreplicative trypomastigote form and inside cells as the replicative amastigote stage. The disease is endemic from Mexico to Argentina, where 10% to 30% of chronically infected persons develop cardiomyopathy and gastrointestinal problems (25). This neglected tropical disease affects about 18 million people, and another 100 million are at risk. Mortality ranges from 8% to 12% (31), with predicted deaths of 50,000 people per year (10).
Nowadays, chemotherapy treatment of Chagas' disease is limited, being successful in the acute form, less so in the chronic form, with severe side effects in elderly patients. No successful vaccine has been developed so far (15). Prevention is obtained by elimination of the insect vector and by increased control in blood banks and congenital transmission. Benznidazole, a nitroimidazole derivative, has been used for the treatment of acute and congenital T. cruzi infection. In a randomized, double-blind, placebo-controlled trial in a rural area in Brazil where Chagas' disease is endemic, a 60-day course of benznidazole treatment of early chronic T. cruzi infection in children was safe and 55.8% effective, with negative seroconversion of specific antibodies (6). The 6-year follow-up of adolescents enrolled in that trial showed successful chemotherapy in 84.7% by seronegativity using chemiluminescent enzyme-linked immunosorbent assay (ELISA) with a purified trypomastigote mucin antigen (2). Benznidazole, however, does not have all the conditions of an ideal drug, with its efficacy and tolerance being inversely related to the age of the patient (29). The other drug clinically available was Nifurtimox, but it has been discontinued from Bayer's production line. It is known that both drugs may increase the oxidative stress causing parasite death (14). Generally, it is accepted that new chemotherapeutic drugs should be developed for treatment of Chagas' disease together with improved methods to determine a parasitological cure based on negative serology (1).
In our laboratory we have studied cyclopalladated complexes with biological activity. They are much more stable and less toxic than other derivatives of palladium (II) (18) and were able to inhibit cathepsin B (3). One of these compounds, called complex 7a, showed specific in vitro and in vivo activities against tumor cells, with no significant toxicity in animals, even at high doses (20). The 7a compound causes anoikis and apoptosis of tumor cells (F. A. Serrano, D. C. Arruda, A. L. Matsuo, L. S. Silva, S. Smaili, A. C. F. Caires, L. R. Travassos, and E. G. Rodrigues, unpublished data), and its enantiomer is able to oxidize mitochondrial proteins leading to permeabilization and cytochrome c release (21).
Since the existing drugs against T. cruzi lead to oxidative stress, we considered testing the 7a compound against the parasite. T. cruzi, as well as other kinetoplastid protozoa, has a single and large mitochondrion containing multiple copies of DNA, forming a structure called a kinetoplast. The kinetoplastid mitochondrion also presents unique features regarding the electron transport chain. For example, they contain a cytochrome-independent alternative oxidase and have unique proteins for kinetoplast maintenance, as topoisomerases, and also an essential fatty acid pathway (22). Here we show that compound 7a targets the parasite mitochondrion, inhibits amastigote intracellular proliferation, and reduces experimental parasitemia and mortality with very low and nontoxic doses.
MATERIALS AND METHODS
Mice, cell line, parasites, and drugs.
BALB/c female mice (8 to 10 weeks old) were obtained from the CEDEME animal facility at UNIFESP. All animal experiments and procedures were approved by the institution's committee on the ethical handling of laboratory animals. Trypomastigote forms (Y strain) were obtained from the supernatant of infected LLCMK2 cells (ATCC CCL7) maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% of fetal bovine serum at 37°C in a humidified atmosphere with 5% CO2. For mouse infections, trypomastigotes were routinely maintained by weekly intraperitoneal (i.p.) infection in BALB/c mice with 500 parasites, as described previously (26). Benznidazole was a gift from Renato Mortara, Department of Parasitology, UNIFESP.
Cyclopalladated complex 7a.
The palladacycle compound 7a (Fig. 1) was obtained from N,N-dimethyl-1-phenethylamine (DMPA), complexed to 1,2-ethane-bis (diphenylphosphine) (DPPE) ligand. Binucleate complex 7a was fully characterized as described by Rodrigues et al. (20) and shown to have antitumor activity and no apparent toxicity in mice injected three times i.p. with doses of 240 nmol·kg−1 of body weight of complex 7a/animal/week.
FIG. 1.

Structure of the 7a compound [Pd2 (S(−)C2,N-DMPA)2 (m-DPPE)]Cl2.
Parasite and mammalian cell drug cytotoxicity.
Parasite killing levels for the tested drugs were determined as described by Muelas-Serrano et al. (17). Briefly, parasites recovered from the supernatants of disrupted infected cells were counted in a Neubauer chamber and distributed in 96-well flat-bottom plates (5 × 106 parasites per well). Drug 7a or benznidazole at 50 pmol·100 μl−1·well−1 (500 nM) or 12.5 nmol·100 μl−1·well−1 (125 μM), respectively, was added, and the time viability of parasites was measured by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay in a period up to 24 h.
Alternatively, for 50% inhibitory concentration (IC50) determination, wells containing 2 × 104 parasites or LLCMK2 cells were incubated for 24 h with different concentrations of 7a ranging from 5 pmol·100 μl−1·well−1 (50 nM) to 100 fmol·100 μl−1·well−1 (1 nM) or 1 nmol·100 μl−1·well−1 (10 μM) to 50 pmol·100 μl−1·well−1 (500 nM), respectively. Viable parasites were counted in a Neubauer chamber, and viable cells were measured by the MTT assay.
Cell invasion experiments.
LLCMK2 cells (4 × 104) were plated onto 13-mm glass coverslips inside a 24-well plate. After 24 h, cells were washed twice with phosphate-buffered saline (PBS) and incubated with 5 × 106 trypomastigotes in the presence of 7a ranging from 500 pmol·ml−1·well−1 (500 nM) to 500 fmol·ml−1·well−1 (500 pM) or 100 nmol·ml−1·well−1 (100 μM) of benznidazole in DMEM supplemented with 10% fetal bovine serum (FBS) for 2 h at 37°C with 5% CO2. Cells were then washed five times with PBS to remove extracellular trypomastigotes and further incubated in DMEM/10% FBS at 37°C for two additional hours. Infected cells were examined for the presence of internalized parasites by optical microscopy using standard Giemsa staining (9).
Drug effect on the proliferation of intracellular amastigotes.
After invasion, as described above, noninternalized parasites were washed away with phosphate-buffered saline, and cells were incubated in the presence of 500 pmol·ml−1·well−1 (500 nM) of 7a or 50 nmol·ml−1·well−1 (50 μM) of benznidazole in DMEM containing 10% FBS. Fresh medium containing each compound was replaced every 24 h. After 48 h, cells were fixed and stained with Giemsa, and intracellular parasites counted by observation using an optical microscope.
DNA degradation.
Trypomastigotes released from LLCMK2 cultures (1 × 108) were treated with 10 nmol·ml−1 (10 μM) of 7a in DMEM supplemented with FBS at 37°C for 6 h. Parasites were centrifuged, resuspended in 500 μl of 62.5 mM EDTA, 4% Triton X-100, 2.5 M LiCl2, and 50 mM Tris-HCl, pH 8.0 (TELT), and gently homogenized for 5 min. The same volume of chloroform-phenol (1: 1) equilibrated in 0.1 M Tris-HCl, pH 8.0, was added, and phases were separated by centrifugation. The aqueous phase was recovered, and the DNA was precipitated with 2 volumes of ethanol and 0.1 M sodium acetate. The DNA was recovered by centrifugation, washed with 70% ethanol, resuspended in Milli-Q water with 20 μg·ml−1 of RNase, and incubated at 37°C for 1 h. DNA integrity was observed by electrophoresis using 0.8% agarose gel in standard Tris-acetate EDTA buffer.
Indirect immunofluorescence.
Trypomastigotes (5 × 106 per ml) were treated with 500 pmol·ml−1 (500 nM) of 7a in DMEM supplemented with FBS at 37°C for 30 min, then attached onto a glass slide for 5 min and subsequently fixed with 4% p-formaldehyde in PBS for 20 min. The slides were washed three times with PBS, treated with Triton X-100 0.1% in PBS for 5 min, and then incubated with 1% bovine serum albumin in PBS for 30 min. The slides were washed three times with PBS, incubated with anti-dihydrolipoamide dehydrogenase (a mitochondrial matrix protein) antiserum (27) (kindly provided by Kevin Taylor, University of East Anglia, United Kingdom) diluted in PBS for 1 h. Labeling was detected with anti-rabbit IgG-Alexa Fluor 594 (Invitrogen) in the presence of 0.01 mM 4,6-diamidino-2-phenylindole (DAPI). Vectashield (Vector Laboratories) was used for mounting, and the slides were visualized with a 100× oil immersion objective (1.41 numerical aperture) in a Olympus BX61 microscope. The images were acquired using cellM̂ software and processed by Blind 3D deconvolution using the software AutoQuant X2 and Adobe Photoshop CS4.
Propidium iodide and annexin V staining.
Trypomastigotes (1 × 107) were treated with 250 pmol·ml−1 (250 nM) of 7a in DMEM supplemented with FBS at 37°C for 6 h and labeled for propidium iodide (PI) and annexin V using the annexin V-FITC apoptosis detection kit (Sigma, MO) according to the manufacturer's instructions. Data acquisition was performed using a FACSCalibur flow cytometer (Becton Dickinson, CA), and data were analyzed with Cell Quest software (Joseph Trotter, CA).
Transmission electron microscopy (TEM).
Trypomastigotes (5 × 107) submitted to treatment with 1 nmol·ml−1 (1 μM) of 7a in DMEM supplemented with FBS at 37°C for 1 h were fixed for 1 h at room temperature with 2% formaldehyde and 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer at pH 7.2. The fixed parasites were then decanted overnight on aclar film precoated with 0.01% poly-l-lysine. The aclar film was washed three times with 0.1 M cacodylate, pH 7.2 (10 min each), incubated with 1% osmium tetroxide in the same buffer for 30 min at room temperature, washed with cacodylate buffer, and washed three times with water. Subsequently the aclar was treated for 30 min with 0.4% uranyl acetate for membrane contrast enhancement, gradually dehydrated in a series of ethanol solutions, and embedded in Epon resin. Ultrathin sections were stained with uranyl acetate and lead citrate, mounted on Formvar grids, and observed in a JEOL 1200 EX II transmission electron microscope at 80 kV.
Animal infection.
Blood parasitemia in the infected animal was quantified by counting motile parasites in 10 μl of fresh blood sample drawn from lateral tail veins (26). Each mouse received a single dose i.p. of 200 nmol·kg−1 of body weight of the testing compound, or PBS as a control, 30 min before the inoculation of 500 blood trypomastigotes. The parasitemia and animal survival were recorded daily. In the therapeutic approach, infected mice were treated with each compound for 13 consecutive days with up to 3 doses per day of 40 nmol·kg−1 of body weight, with intervals of 4 h, starting 1 day after parasite inoculation. Parasitemia was measured as described above, and survival was followed every day until the 60th day postinfection. For histopathological analysis, animals were sacrificed on the 13th day postinfection. Experiments were performed with 10 animals per group.
Preparation of tissues for histopathological analysis.
The hematoxylin and eosin (H&E) staining of paraffin sections was performed with heart, lung, bladder, spleen, intestine, and muscle tissues. The sections obtained from the experimental and control groups were fixed in neutral buffered formalin (10% formalin in PBS), dehydrated, and embedded in paraffin by a routine technique, and semiserial sections of 5 μm were stained with H&E. The results are expressed as the mean number of amastigote nests per microscope field. The data presented for each group are the mean ± standard deviation (SD) of results for three animals; 20 fields in 4 sections were counted in each tissue.
Statistical analyses.
The Student t test was used throughout, except for with the parasitemia and the survival curves, for which one-way analysis of variance (ANOVA) and the Mantel-Cox test were employed, respectively.
RESULTS
Cyclopalladated 7a is highly toxic for T. cruzi trypomastigotes compared to mammalian cells in culture.
To evaluate the relative toxicity of the cyclopalladated compound 7a, we measured the IC50 for mammalian cells and trypomastigote forms of the parasite. Cell culture-derived trypomastigotes (2 × 104) were incubated for 24 h with different concentrations of 7a, and the number of moving, refringent parasites was counted in a Neubauer chamber. An IC50 of 15 nM was obtained (data not shown). It is known that some metallic drugs have a nephrotoxic effect. For this reason we used the mammalian kidney cell lineage LLCMK2 for mammalian cell cytotoxic evaluation. About 2 × 104 cells were distributed into a 96-well plate and incubated with different concentrations of 7a. After 24 h, 7a showed an IC50 of 5.1 μM, measured by the MTT method (data not shown). Thus, 7a drug affects preferentially the parasite, being effective at concentrations 340 times less than the cytotoxic concentration for mammalian cells.
A trypanocide effect of drug 7a was observed at a much lower concentration than that of benznidazole.
Benznidazole is the drug of choice for treatment of Chagas' disease. To compare the efficacies of drug 7a and benznidazole, cell-derived trypomastigotes (5 × 106) were treated for 0, 2, 6, and 24 h with 7a or benznidazole at 37°C in DMEM supplemented with 10% FBS. As shown in Fig. 2, drug 7a at 50 pmol·100 μl−1·well−1 (500 nM) causes a rapid decrease in parasite viability compared with benznidazole (P < 0.05). About 50% of the parasites died in the second hour of incubation. In contrast, a much higher concentration of benznidazole, 12.5 nmol·100 μl−1·well−1 (125 μM), was required to obtain the same killing after 24 h of incubation.
FIG. 2.

Time-dependent viability (MTT method) of 5 × 106 trypomastigotes treated with compound 7a (50 pmol·100 μl−1·well−1; 500 nM) or benznidazole (12.5 nmol·100 μl−1·well−1; 125 μM) during 24 h. *, P < 0.05.
Inhibition of T. cruzi cell invasion by 7a.
Cell invasion by trypomastigotes is a key step for establishment of T. cruzi infection in the mammalian host. Therefore, we evaluated whether 7a could prevent mammalian cell invasion in culture by trypomastigotes. As shown in Fig. 3, drug 7a inhibited cell invasion (P < 0.05) at concentration as low as 5 pmol·ml−1·well−1 (5 nM) with 90% inhibition at 500 pmol·ml−1·well−1 (500 nM). This inhibitory effect cannot be attributed to a direct parasite killing, since about 50% of parasites kept motility at 50 pmol·100 μl−1·well−1 (500 nM) of 7a after 2 h of incubation. In contrast, benznidazole had no effect on trypomastigote invasion, even at high concentrations, probably due to its slow action.
FIG. 3.

Drug 7a and benznidazole effects on parasite invasion of mammalian cells. Parasites were plated on LLC-MK2 cells with compound 7a or benznidazole for 2 h. Noninvading parasites were washed away, and after 4 h, cells were stained with Giemsa. The number of infected cells was determined by direct analysis under a light microscope. *, P < 0.05.
Compound 7a prevents intracellular growth of amastigotes.
After cell invasion, trypomastigote forms transform into amastigotes, which represent the intracellular replicative form of T. cruzi. To verify whether 7a could have any effect against amastigotes, preinfected cells were incubated with pmol·ml−1·well−1 (500 nM) of 7a or 50 nmol·ml−1·well−1 (50 μM) of benznidazole for 48 h. After treatment, cells were stained with Giemsa and observed by optical microscopy in order to evaluate the relative number of infected cells and amastigotes per infected cell. As shown in Fig. 4, both drugs at much different concentrations significantly reduced (P < 0.05) the relative number of infected cells (Fig. 4A) with ∼50% parasites per infected cell (Fig. 4B).
FIG. 4.
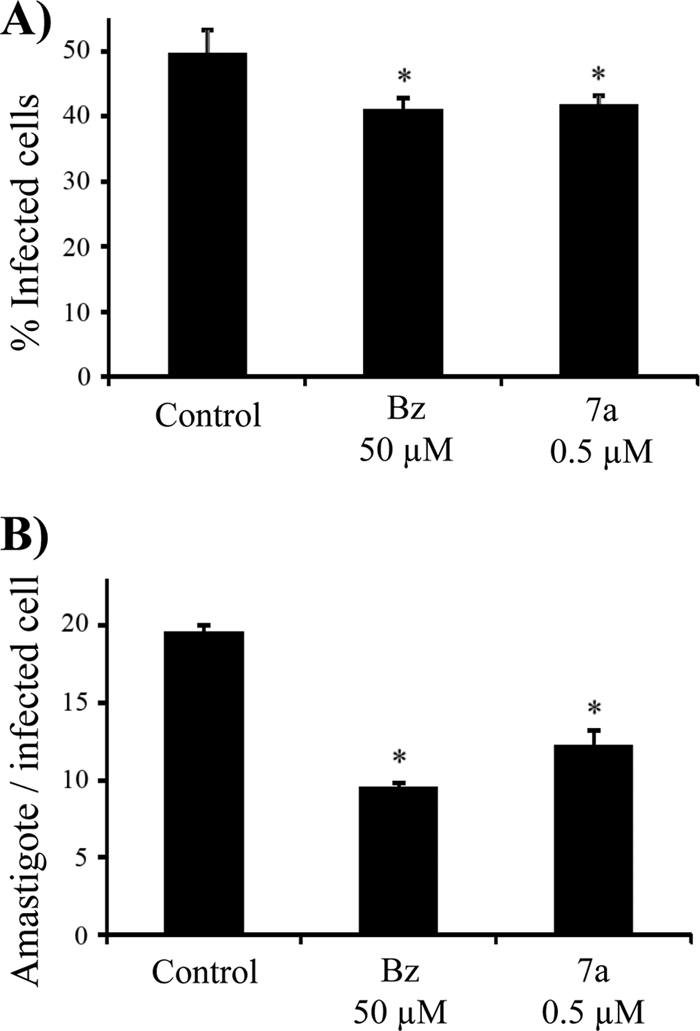
Inhibition of amastigote growth. After 48 h treatment with 7a (500 pmol·ml−1·well−1 concentration at 500 nM) or benznidazole (50 nmol·ml−1·well−1; 50 μM), infected cells were stained with Giemsa and analyzed. (A) Percentage of infected cells after treatment; (B) relative number of amastigotes per infected cell. *, P < 0.05.
Compound 7a induces an apoptosis-like parasite death.
To understand the mechanism by which compound 7a promotes parasite death we compared the observed killing at 37°C with the effect at 4°C (Fig. 5). We found that at 4°C much less parasite lysis was observed, suggesting that metabolically active parasites are required and that the effect of 7a does not occur as a consequence of simple membrane destruction. Previous work from our group suggests that compound 7a interacts with mitochondria, causing a collapse in the cell extrusion of protons (20). Therefore, the parasites were stained by immunofluorescence with a mitochondrial matrix marker after incubation with 7a. As shown in Fig. 5A to C, the mitochondrial structure remains intact in control cells or parasites treated at 4°C, whereas parasites incubated with compound 7a at 37°C for 5 min showed morphological alterations in the mitochondrial structure (Fig. 5D). On prolonged incubation (>30 min), only those parasites incubated at 37°C died.
FIG. 5.

Effects of compound 7a on parasite mitochondrion at 4°C or 37°C. DNA staining was performed with DAPI (blue) and the mitochondrion matrix marker with Alexa fluor 594 (red). (A) Nontreated trypomastigotes at 4°C; (B) 7a-treated trypomastigotes at 4°C; (C) nontreated trypomastigotes at 37°C; (D) 7a-treated trypomastigotes at 37°C.
As death involving mitochondria is found in apoptosis, we looked for other features of apoptotic death in the parasite. DNA was extracted from 7a-treated and -untreated parasites, and its integrity was analyzed by agarose gel electrophoresis. Differently from the control, treated parasites showed DNA degradation in a step pattern, suggestive of apoptosis (Fig. 6). We then labeled parasites treated with 7a for a short period, to avoid lysis, with annexin V to stain eventual phosphatidylserine flipping to the membrane surface, which is another common biochemical alteration observed in the apoptotic process. As shown in Fig. 7, there is a clear increase in annexin V labeling but no change in propidium iodide uptake in treated parasites compared to control parasites as detected by fluorescence-activated cell sorter (FACS), suggesting that labeling occurs owing to the surface expression of the phospholipid.
FIG. 6.
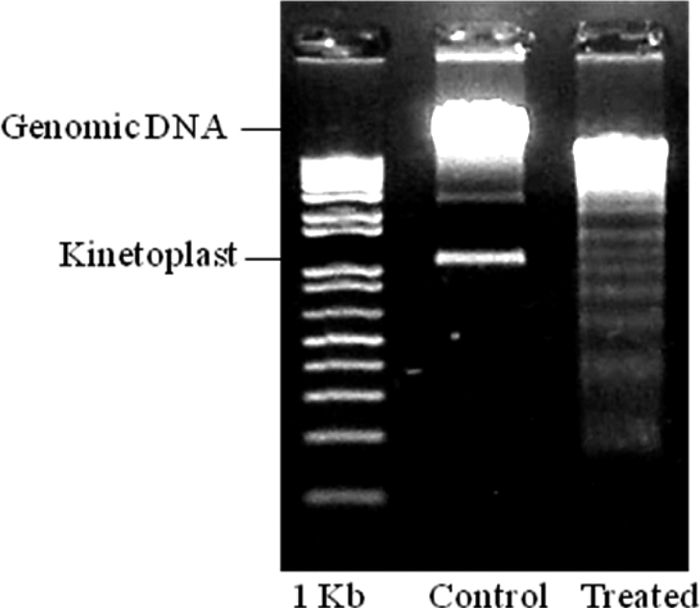
DNA degradation after trypomastigote treatment with compound 7a for 6 h. A clear ladder pattern of degradation as found in apoptosis was observed with treated parasites.
FIG. 7.

Analysis of phosphatidylserine surface exposure by FACS, in T. cruzi trypomastigotes treated with compound 7a for 6 h. The ordinate represents PI-marked parasites, and abscissa corresponds to the annexin V-positive parasites. (A) Nontreated parasites; (B) treated parasites.
Ultrastructural changes in the 7a-treated parasites.
We determined the ultrastructural disturbances in trypomastigote forms caused by compound 7a using electron microscopy. Figure 8 A shows a typical mitochondrion (M), nucleus (N), kinetoplast (K), and flagellum (F). In contrast, as shown in Fig. 8B and C, treated parasites showed mitochondrion swelling (asterisk), formation of abnormal membrane structures inside the organelle (black arrowhead), and atypical vacuoles (triangle). This result corroborates the fluorescence microscopy in which the morphological alterations of mitochondria are shown.
FIG. 8.

Ultrastructural changes in 7a-treated trypomastigotes. (A) Control parasite: normal mitochondrion (M), nucleus (N), kinetoplast (K), and flagellum (F); (B and C) treated parasites, showing mitochondrion swelling (asterisk), formation of abnormal membrane structures inside the organelle (black arrows), and atypical vacuoles (triangle).
Preinjection of compound 7a protects mice from acute infection.
To verify the potential use of 7a compound for Chagas' disease therapy, a single dose i.p. of 200 nmol of 7a per kg of body weight was intraperitoneally administered 30 min before mouse infection by highly virulent bloodstream forms of the T. cruzi Y strain. As shown in Fig. 9, we observed a significant delay in the mortality of mice pretreated with compound 7a (P < 0.01). Specimens of heart, skeletal muscle, spleen, intestine, lungs, bladder, and kidney collected on day 13 postinfection were examined for the presence of parasite nests by regular H&E histopathology. The numbers of parasite nests decreased for all observed tissues of treated animals, but only for the bladder and heart were the differences statistically significant (P < 0.05). No parasites were seen in the spleen (Table 1).
FIG. 9.

Survival of pretreated mice. To evaluate a possible in vivo drug effect, we treated female BALB/c mice with a single dose of 240 nmol·kg−1 body weight of compound 7a or benznidazole 30 min before infection with 500 trypomastigote forms. Percent survival is shown; n = 10. *, P < 0.01.
TABLE 1.
Number of parasite nests in tissues from infected mice pre-treated with 7aa
| Tissue | Amastigote nests/20 fieldsb |
|
|---|---|---|
| Control (n = 3) | 7a (n = 3) | |
| Heart | 28 (± 8.5) | 4.8 (± 9)* |
| Bladder | 56 (± 9.1) | 8 (± 10)* |
| Lung | 3 (± 4.4) | 0.2 (± 0.45) |
| Intestine | 1.7 (± 2.1) | 0.2 (0.45) |
| Spleen | 0 | 0 |
| Muscle | 0.67 (± 1.2) | 0.2 (± 0.45) |
The sections of hematoxylin/eosin-stained tissues from mice injected i.p. with 7a at 200 nmol·kg−1 body weight 30 min before infection with 500 bloodstream trypomastigotes (Y strain) were scored for determination of the number of parasite nests on the indicated tissues. *, P < 0.05.
Values are means ± standard deviations from 20 fields; magnification, ×400.
Compound 7a is effective in the treatment of acute Chagas' disease.
As compound 7a proved to be effective against T. cruzi infection when injected in mice prior to infection, we tested whether it could eliminate the parasite from infected animals. Three i.p. daily doses of 40 nmol of compound 7a or benznidazole per kg of body weight within 4-h intervals were administered in mice 1 day after mouse infection with 500 trypomastigote forms, until the 14th day. Maximal parasitemia and animal mortality were significantly reduced (P < 0.01) in 7a-treated animals (Fig. 10 A and B). Histopathological analysis of tissues collected on day 13 postinfection still revealed the presence of amastigote nests in 7a-treated animals but at significantly (P < 0.05) lower levels than those for the saline control, in the bladder, intestine, and heart, whereas benznidazole significantly (P < 0.05) decreased nests in the intestine (Table 2). A higher dose, up to 200 nmol kg−1, that was not toxic when administered three times a week was not well tolerated when injected daily for 10 days and showed no improvement in animal survival (data not shown).
FIG. 10.

Survival of acutely infected mice. One day after infection, animals were treated with three doses of 40 nmol·kg−1 body weight of each drug per day until the 14th day. (A) Measurement of peak parasitemia; (B) percent mouse survival in the control, benznidazole, and 7a groups (n = 10). *, P < 0.05.
TABLE 2.
Histopathological analysis of tissuesa
| Tissue | Amastigote nests/20 fieldsb |
||
|---|---|---|---|
| Control (n = 5) | Bz (n = 5) | 7a (n = 4) | |
| Heart | 15.7 (± 4.6) | 12.3 (± 5.0) | 5.3 (± 4.6)* |
| Bladder | 54.7 (± 17.9) | 36.7 (± 5.5) | 4.7 (± 4.5)* |
| Lung | 1.3 (±1.5) | 0 | 0.3 (± 0.58) |
| Intestine | 21.5 (± 0.7) | 0.67 (± 1.2)* | 0* |
| Spleen | 4.7 (± 3.2) | 2.3 (± 3.2) | 0.67 (± 1.2) |
| Muscle | 0 | 0 | 0 |
The sections of hematoxylin/eosin-stained tissues from mice treated i.p. with three doses per day of 7a at 40 nmol·kg−1 body weight during 13 days after infection with 500 bloodstream trypomastigotes (Y strain) were scored for the number of parasite nests on the indicated tissues. *, P < 0.05.
Values are means ± standard deviations from 20 fields; magnification, ×400. Bz, benznidazole.
DISCUSSION
In the present work we demonstrate the potential of the cyclopalladated compound 7a against T. cruzi infection. This compound was previously shown to be effective against syngeneic murine melanoma in vivo (20). It seems to target mitochondria of eukaryotic cells (F. A. Serrano et al., unpublished data). Our experiments provide clear evidence that T. cruzi invasive and intracellular proliferative forms are about 300 times more sensitive to compound 7a than mammalian cells. Compound 7a destroys trypomastigotes probably by targeting the mitochondrion, followed by DNA fragmentation and phosphatidylserine surface exposure, in a process that resembles apoptosis of mammalian cells. It is well known that mitochondrial damage can initiate a series of events that lead cells to apoptosis characterized by nuclear condensation and fragmentation, caspase activation, and phosphatidyl serine cell surface exposure, in an active process (28). The reason for this apoptosis-like process in a unicellular organism is still controversial. In parasites like Leishmania, it has been proposed that a few dead cells in the population stimulate infection (30). It has also been proposed that T. cruzi insect forms are killed by complement through extensive free radical production by the mitochondrion and induction of a similar apoptotic-like mechanism (12). Reports using venom from Bothrops jararaca (7) and aromatic diamidines (8) also promoted a similar apoptosis-like death of T. cruzi.
The distinctive mitochondrion of the parasite could explain its increased susceptibility, with an IC50 340-fold less than that in mammalian cells. Moreover, compound 7a acts very quickly on trypomastigotes, unlike benznidazole, which, even at high concentrations, may take more than 24 h to kill the parasite. The inefficiency of benznidazole with the Y strain infection may be explained by the partial resistance of this strain (16) and was confirmed here, as even a single dose of 400 nmol·kg−1 body weight of benznidazole had minimal effect. As it has been proposed that benznidazole toxicity for T. cruzi is due to oxidative stress generated preferentially in the parasite (19), compound 7a could easily circumvent the resistance mechanism.
Recently, NO-liberating drugs have been tested against T. cruzi with successful in vivo results, leading to 60 to 80% survival and even 100% survival of infected mice in combination with benznidazole (11, 23, 24). In vitro, all these drugs were active, with IC50s of >52 μM. In comparison, compound 7a was even more effective with a very low IC50. Probably, using other treatment protocols or drug combinations, the effectiveness of compound 7a could be further improved to achieve 100% survival of infected animals.
The inhibition of cell invasion by 7a does not appear to occur as a consequence of trypomastigote death. The most probable explanation for this effect is that cyclopalladated compounds may directly inhibit cysteine proteases by direct oxidation of the catalytic domain (4). One of these enzymes is T. cruzi cruzain, which is important for parasite invasion (13).
Previous studies have shown that compound 7a was used in cancer therapy and was not toxic in mice, even at 240 nmol·kg−1 body weight, injected three times a week. We performed preliminary analysis of acute administration of a 4,000-nmol·kg−1 single dose, i.p., and found that it was not lethal to mice (data not shown). Presently, the therapeutic in vivo test showed that compound 7a was able to significantly reduce the parasitemia and the number of parasite nests in the bladder, heart, and intestine (P < 0.05), with 80% animal survival after 60 days of infection (P < 0.01). It is important to note that the treatment terminated on day 14 and that the first 7a-treated mouse died after 34 days of infection, while both PBS- and benznidazole-treated groups of mice were all dead on day 22. We report here on a promising chemotherapeutic approach against T. cruzi that might possibly be extended to all kinetoplastidae because the cyclopalladated compound rapidly disrupts the mitochondrion. This compound was demonstrated to have a fast antiparasite effect, decreasing its viability and invasion capacity and leading to an apoptosis-like death. In preclinical assays, it was very effective in protecting 80% of treated animals. Due to its high efficacy in vivo, it could be an alternative treatment for Chagas' disease. Further studies on the treatment protocols may help us to improve the compound efficacy in the treatment of experimental trypanosomiasis.
Acknowledgments
The present work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and the Centro de Microscopia Eletronica (CEME-UNIFESP).
Footnotes
Published ahead of print on 17 May 2010.
REFERENCES
- 1.Almeida, I. C., D. T. Covas, L. M. Soussumi, and L. R. Travassos. 1997. A highly sensitive and specific chemiluminescent enzyme-linked immunosorbent assay for diagnosis of active Trypanosoma cruzi infection. Transfusion 37:850-857. [DOI] [PubMed] [Google Scholar]
- 2.Andrade, A. L., C. M. Martelli, R. M. Oliveira, S. A. Silva, A. I. Aires, L. M. Soussumi, D. T. Covas, L. S. Silva, J. G. Andrade, L. R. Travassos, and I. C. Almeida. 2004. Short report: benznidazole efficacy among Trypanosoma cruzi-infected adolescents after a six-year follow-up. Am. J. Trop. Med. Hyg. 71:594-597. [PubMed] [Google Scholar]
- 3.Bincoletto, C., I. L. Tersariol, C. R. Oliveira, S. Dreher, D. M. Fausto, M. A. Soufen, F. D. Nascimento, and A. C. Caires. 2005. Chiral cyclopalladated complexes derived from N,N-dimethyl-1-phenethylamine with bridging bis(diphenylphosphine)ferrocene ligand as inhibitors of the cathepsin B activity and as antitumoral agents. Bioorg. Med. Chem. 13:3047-3055. [DOI] [PubMed] [Google Scholar]
- 4.Caires, A. C. 2007. Recent advances involving palladium (II) complexes for the cancer therapy. Anticancer Agents Med. Chem. 7:484-491. [DOI] [PubMed] [Google Scholar]
- 5.Coura, J. R., and J. C. Dias. 2009. Epidemiology, control and surveillance of Chagas disease: 100 years after its discovery. Mem. Inst. Oswaldo Cruz 104(Suppl. 1):31-40. [DOI] [PubMed] [Google Scholar]
- 6.de Andrade, A. L., F. Zicker, R. M. de Oliveira, S. S. Almeida, A. Luquetti, L. R. Travassos, I. C. Almeida, S. S. de Andrade, J. G. de Andrade, and C. M. Martelli. 1996. Randomised trial of efficacy of benznidazole in treatment of early Trypanosoma cruzi infection. Lancet 348:1407-1413. [DOI] [PubMed] [Google Scholar]
- 7.Deolindo, P., A. S. Teixeira-Ferreira, E. J. Melo, A. C. Arnholdt, W. Souza, E. W. Alves, and R. A. DaMatta. 2005. Programmed cell death in Trypanosoma cruzi induced by Bothrops jararaca venom. Mem. Inst. Oswaldo Cruz 100:33-38. [DOI] [PubMed] [Google Scholar]
- 8.De Souza, E. M., R. Menna-Barreto, T. C. Araujo-Jorge, A. Kumar, Q. Hu, D. W. Boykin, and M. N. Soeiro. 2006. Antiparasitic activity of aromatic diamidines is related to apoptosis-like death in Trypanosoma cruzi. Parasitology 133:75-79. [DOI] [PubMed] [Google Scholar]
- 9.Fichera, L. E., M. C. Albareda, S. A. Laucella, and M. Postan. 2004. Intracellular growth of Trypanosoma cruzi in cardiac myocytes is inhibited by cytokine-induced nitric oxide release. Infect. Immun. 72:359-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gelb, M. H., and W. G. Hol. 2002. Parasitology. Drugs to combat tropical protozoan parasites. Science 297:343-344. [DOI] [PubMed] [Google Scholar]
- 11.Guedes, P. M., F. S. Oliveira, F. R. Gutierrez, G. K. da Silva, G. J. Rodrigues, L. M. Bendhack, D. W. Franco, M. A. Do Valle Matta, D. S. Zamboni, R. S. da Silva, and J. S. Silva. 2010. Nitric oxide donor trans-[RuCl([15]aneN)NO] as a possible therapeutic approach for Chagas' disease. Br. J. Pharmacol. 160:270-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irigoin, F., N. M. Inada, M. P. Fernandes, L. Piacenza, F. R. Gadelha, A. E. Vercesi, and R. Radi. 2009. Mitochondrial calcium overload triggers complement-dependent superoxide-mediated programmed cell death in Trypanosoma cruzi. Biochem. J. 418:595-604. [DOI] [PubMed] [Google Scholar]
- 13.Kerr, I. D., J. H. Lee, C. J. Farady, R. Marion, M. Rickert, M. Sajid, K. C. Pandey, C. R. Caffrey, J. Legac, E. Hansell, J. H. McKerrow, C. S. Craik, P. J. Rosenthal, and L. S. Brinen. 2009. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem. 284:25697-25703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maya, J. D., S. Bollo, L. J. Nunez-Vergara, J. A. Squella, Y. Repetto, A. Morello, J. Perie, and G. Chauviere. 2003. Trypanosoma cruzi: effect and mode of action of nitroimidazole and nitrofuran derivatives. Biochem. Pharmacol. 65:999-1006. [DOI] [PubMed] [Google Scholar]
- 15.Maya, J. D., B. K. Cassels, P. Iturriaga-Vasquez, J. Ferreira, M. Faundez, N. Galanti, A. Ferreira, and A. Morello. 2007. Mode of action of natural and synthetic drugs against Trypanosoma cruzi and their interaction with the mammalian host. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 146:601-620. [DOI] [PubMed] [Google Scholar]
- 16.Michailowsky, V., S. M. Murta, L. Carvalho-Oliveira, M. E. Pereira, L. R. Ferreira, Z. Brener, A. J. Romanha, and R. T. Gazzinelli. 1998. Interleukin-12 enhances in vivo parasiticidal effect of benznidazole during acute experimental infection with a naturally drug-resistant strain of Trypanosoma cruzi. Antimicrob. Agents Chemother. 42:2549-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muelas-Serrano, S., J. J. Nogal-Ruiz, and A. Gomez-Barrio. 2000. Setting of a colorimetric method to determine the viability of Trypanosoma cruzi epimastigotes. Parasitol. Res. 86:999-1002. [DOI] [PubMed] [Google Scholar]
- 18.Navarro-Ranninger, C., I. Lopez-Solera, J. M. Perez, J. Rodriguez, J. L. Garcia-Ruano, P. R. Raithby, J. R. Masaguer, and C. Alonso. 1993. Analysis of two cycloplatinated compounds derived from N-(4-methoxyphenyl)-alpha-benzoylbenzylidenamine. Comparison of the activity of these compounds with other isostructural cyclopalladated compounds. J. Med. Chem. 36:3795-3801. [DOI] [PubMed] [Google Scholar]
- 19.Pedrosa, R. C., A. F. De Bem, C. Locatelli, R. C. Pedrosa, R. Geremias, and F. D. Wilhelm. 2001. Time-dependent oxidative stress caused by benznidazole. Redox Rep. 6:265-270. [DOI] [PubMed] [Google Scholar]
- 20.Rodrigues, E. G., L. S. Silva, D. M. Fausto, M. S. Hayashi, S. Dreher, E. L. Santos, J. B. Pesquero, L. R. Travassos, and A. C. Caires. 2003. Cyclopalladated compounds as chemotherapeutic agents: antitumor activity against a murine melanoma cell line. Int. J. Cancer 107:498-504. [DOI] [PubMed] [Google Scholar]
- 21.Santana, D. P., P. A. Faria, E. J. Paredes-Gamero, A. C. Caires, I. L. Nantes, and T. Rodrigues. 2009. Palladacycles catalyse the oxidation of critical thiols of the mitochondrial membrane proteins and lead to mitochondrial permeabilization and cytochrome c release associated with apoptosis. Biochem. J. 417:247-256. [DOI] [PubMed] [Google Scholar]
- 22.Sen, N., and H. K. Majumder. 2008. Mitochondrion of protozoan parasite emerges as potent therapeutic target: exciting drugs are on the horizon. Curr. Pharm. Des. 14:839-846. [DOI] [PubMed] [Google Scholar]
- 23.Silva, J. J., A. L. Osakabe, W. R. Pavanelli, J. S. Silva, and D. W. Franco. 2007. In vitro and in vivo antiproliferative and trypanocidal activities of ruthenium NO donors. Br. J. Pharmacol. 152:112-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silva, J. J., P. M. Guedes, A. Zottis, T. L. Balliano, F. O. Nascimento Silva, L. G. Franca Lopes, J. Ellena, G. Oliva, A. D. Andricopulo, D. W. Franco, and J. S. Silva. 2010. Novel ruthenium complexes as potential drugs for Chagas's disease: enzyme inhibition and in vitro/in vivo trypanocidal activity. Br. J. Pharmacol. 160:260-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanowitz, H. B., F. S. Machado, L. A. Jelicks, J. Shirani, A. C. de Carvalho, D. C. Spray, S. M. Factor, L. V. Kirchhoff, and L. M. Weiss. 2009. Perspectives on Trypanosoma cruzi-induced heart disease (Chagas disease). Prog. Cardiovasc. Dis. 51:524-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trocoli Torrecilhas, A. C., R. R. Tonelli, W. R. Pavanelli, J. S. da Silva, R. I. Schumacher, W. de Souza, N. Cunha e Silva, I. de Almeida Abrahamsohn, W. Colli, and M. J. Manso Alves. 2009. Trypanosoma cruzi: parasite shed vesicles increase heart parasitism and generate an intense inflammatory response. Microbes Infect. 11:29-39. [DOI] [PubMed] [Google Scholar]
- 27.Tyler, K. M., K. R. Matthews, and K. Gull. 1997. The bloodstream differentiation-division of Trypanosoma brucei studied using mitochondrial markers. Proc. Biol. Sci. 264:1481-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaux, D. L., and A. Strasser. 1996. The molecular biology of apoptosis. Proc. Natl. Acad. Sci. U. S. A. 93:2239-2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viotti, R., C. Vigliano, B. Lococo, M. G. Alvarez, M. Petti, G. Bertocchi, and A. Armenti. 2009. Side effects of benznidazole as treatment in chronic Chagas disease: fears and realities. Expert Rev. Anti Infect. Ther. 7:157-163. [DOI] [PubMed] [Google Scholar]
- 30.Wanderley, J. L., A. Benjamin, F. Real, A. Bonomo, M. E. Moreira, and M. A. Barcinski. 2005. Apoptotic mimicry: an altruistic behavior in host/Leishmania interplay. Braz. J. Med. Biol. Res. 38:807-812. [DOI] [PubMed] [Google Scholar]
- 31.WHO. 2002. World Health Organization (WHO), 2002. Control of Chagas' disease. Second report of the WHO Expert Committee. World Health Organ. Tech. Rep. Ser. 905:1-109. [PubMed] [Google Scholar]