

Abstract
The lectin actinohivin (AH) is a monomeric carbohydrate-binding agent (CBA) with three carbohydrate-binding sites. AH strongly interacts with gp120 derived from different X4 and R5 human immunodeficiency virus (HIV) strains, simian immunodeficiency virus (SIV) gp130, and HIV type 1 (HIV-1) gp41 with affinity constants (KD) in the lower nM range. The gp120 and gp41 binding of AH is selectively reversed by (α1,2-mannose)3 oligosaccharide but not by α1,3/α1,6-mannose- or GlcNAc-based oligosaccharides. AH binding to gp120 prevents binding of α1,2-mannose-specific monoclonal antibody 2G12, and AH covers a broader epitope on gp120 than 2G12. Prolonged exposure of HIV-1-infected CEM T-cell cultures with escalating AH concentrations selects for mutant virus strains containing N-glycosylation site deletions (predominantly affecting high-mannose-type glycans) in gp120. In contrast to 2G12, AH has a high genetic barrier, since several concomitant N-glycosylation site deletions in gp120 are required to afford significant phenotypic drug resistance. AH is endowed with broadly neutralizing activity against laboratory-adapted HIV strains and a variety of X4 and/or R5 HIV-1 clinical clade isolates and blocks viral entry within a narrow concentration window of variation (∼5-fold). In contrast, the neutralizing activity of 2G12 varied up to 1,000-fold, depending on the virus strain. Since AH efficiently prevents syncytium formation in cocultures of persistently HIV-1-infected HuT-78 cells and uninfected CD4+ T lymphocytes, inhibits dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin-mediated capture of HIV-1 and subsequent virus transmission to CD4+ T lymphocytes, does not upregulate cellular activation markers, lacks mitogenic activity, and does not induce cytokines/chemokines in peripheral blood mononuclear cell cultures, it should be considered a potential candidate drug for microbicidal use.
The envelope glycoproteins of human immunodeficiency virus (HIV) mediate attachment and viral entry into susceptible target cells. The initial steps in HIV infection include binding of the trimeric gp120 with three CD4 receptor molecules, a number of conformational changes in gp120, and interactions with chemokine receptor CCR5 or CXCR4, followed by the unfolding of gp41, which anchors the virion to the target membrane and which brings the viral and cellular membranes in close contact for further virus-cell fusion (17).
The HIV type 1 (HIV-1) gp120 envelope glycoprotein is highly glycosylated. Approximately half of its molecular weight is contributed by its carbohydrate content (22, 36). The recombinant HIV-1(IIIB) gp120 expressed in Chinese hamster ovary (CHO) cells is occupied by 11 high-mannose- or hybrid-type glycans and 13 complex-type glycans (29).
A variety of carbohydrate-binding agents (CBAs), such as the prokaryotic agent cyanovirin-N (CV-N) (7-10), the plant lectins Hippeastrum hybrid agglutinin (HHA) (1) and Galanthus nivalis agglutinin (GNA) (1), and the antibiotics pradimicin A and S (PRM-A and PRM-S, respectively) (4, 7), have been described to inhibit viral entry, presumably by their interaction with the glycans on HIV gp120. It has indeed been demonstrated that CBAs block virus entry by inhibiting the fusion of cell-free HIV virions with their target cells. They prevent the capture of virus particles by the dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN) and the macrophage mannose receptor, which are present on cells of the innate immune system. The subsequent transmission of the virus to CD4+ T cells is also efficiently hampered by CBAs, pointing to their potential as HIV microbicide drug candidates (6). By the lack of an efficient HIV vaccine, microbicides can evolve to be important tools for the prevention of HIV transmission and infection (23, 37, 38).
One CBA, namely, the prokaryotic agent CV-N, has been tested in animal vaginal and rectal virus transmission models (43, 44) and shown to successfully inhibit viral infections in the animals, suggesting that CV-N has the potential to act as an anti-HIV microbicide. However, recently, it was described that CV-N induced the production of a variety of cytokines and cellular activation markers in peripheral blood mononuclear cells (PBMCs), and in addition, the pronounced mitogenic activity of CV-N was also observed (2, 26). These potential side effects can compromise the application of CV-N as an efficient microbicide.
Actinohivin (AH) is an anti-HIV protein that has been isolated from a prokaryotic microorganism (the actinomycete Longispora albida K97-0003T) (14, 27). AH consists of 114 amino acids (12,524.3 Da) and exhibits a unique highly conserved internal sequence triplication (60% identity and ∼70% homology on each segment). It was shown that the three repeats of AH are necessary to block syncytium formation in recombinant cell cultures (HeLa /T env tat and HeLa CD4 lacZ cells) (40, 41). Recently, the crystal structure of AH was solved, and it was revealed that AH has three sugar-binding pockets (41). It was suggested before that AH-gp120 interactions are accomplished by N-linked high-mannose-type carbohydrates that are abundantly present on gp120 (15, 41).
We have now performed a detailed investigation of AH regarding its anti-HIV activity spectrum, potential side effects, kinetic interaction with the HIV-1 envelope proteins gp120 and gp41, and resistance spectrum and also added data on other CBAs that are related to AH in terms of their carbohydrate specificities (i.e., HHA and monoclonal antibody [MAb] 2G12) for comparative reasons. Based on its safety profile and unique kinetic/antiviral properties our data revealed that AH qualifies as a potential drug lead for further preclinical investigations.
MATERIALS AND METHODS
Test compounds.
AH was prepared and purified from a cultured broth of L. albida K97-0003T as described previously (14). The mannose-specific plant lectins HHA and Urtica dioica agglutinin (UDA) were derived and purified as described previously (39, 46) and were kindly provided by E. J. M. Van Damme (Ghent University, Belgium). Manα1,3manα1,6man (ß1,4-GlcNAc)3, (α1,2-man)2, and Man9GluNAc2 were obtained from Dextra Laboratories (Reading, United Kingdom). (α1,2-Man)3 and mannose were purchased from Carbohydrate Synthesis (Oxford, United Kingdom).
Cells.
Human CD4+ T-lymphocytic CEM, C8166, CEMX174, HuT-78, and Sup-T1 cells were obtained from the American Type Culture Collection (Manassas, VA). Persistently HIV-infected HuT-78 (HuT-78/HIV) cells were obtained upon cultivation for 3 to 4 weeks of HuT-78 cell cultures exposed to HIV-1(IIIB) or HIV-2(ROD). DC-SIGN-expressing Raji (Raji/DC-SIGN) cells were constructed by Geijtenbeek et al. (20, 21) and were kindly provided by L. Burleigh (Institut Pasteur, Paris, France). All cell lines were cultivated in RPMI 1640 medium (Invitrogen, Merelbeke, Belgium) supplemented with 10% fetal bovine serum (FBS; BioWhittaker Europe, Verviers, Belgium), 2 mM l-glutamine, 75 mM NaHCO3, and 20 μg/ml gentamicin (Invitrogen). The MT-4 cell line was a kind gift of L. Montagnier (who at that time was at the Pasteur Institute, Paris, France) and maintained in RPMI 1640 medium supplemented with 10% FBS and 2 mM l-glutamine. U87 CD4+ CCR5+ CXCR4+ cells were established as described previously (32) and cultivated in Dulbecco modified Eagle medium (Invitrogen) containing 10% FBS supplemented with 200 μg/ml Geneticin (Invitrogen), 20 μg/ml gentamicin, and 1 μg/ml puromycin (Sigma, St. Louis, MO). Buffy coat preparations for isolation of PBMCs from healthy humans were obtained from the Blood Transfusion Center in Leuven, Belgium.
Viruses.
HIV-1(IIIB) was a kind gift from R. C. Gallo (Institute of Human Virology, University of Maryland, Baltimore, MD), and HIV-2(ROD) was provided by L. Montagnier (Pasteur Institute). Simian immunodeficiency virus (SIV) mac251 [SIV(mac251)] was obtained from H. Egberink (Utrecht, Netherlands) and from R. Le Grand (Paris, France). Simian-human immunodeficiency virus strain sm239 [SHIV(sm239)] was kindly provided by K. Überla (Universität Erlangen, Nürnberg, Germany) and consists of SIV(mac239) in which the reverse transcriptase (RT) gene had been replaced by the HIV-1 RT gene (45).
Antiretrovirus assays.
CEM, MT-4, and CEMX174 cells (5 × 105 cells per ml) were suspended in fresh culture medium and infected with HIV-1 and HIV-2 (CEM, MT-4) or SHIV(sm239) (CEMX174) at 100 times the 50% cell culture infective dose per ml of cell suspension. One hundred microliters of the infected cell suspension was mixed with 100 μl of the appropriate dilutions of the test compounds, and the mixture was further incubated at 37°C. After 4 to 5 days, syncytium formation was recorded microscopically in the cell cultures, and the number of giant cells in the drug-treated cultures was estimated as a percentage of the number of giant cells present in the nontreated virus-infected cell cultures. SIV(mac251)-induced cytopathogenicity was also recorded in U87 CD4+ CCR5+ CXCR4+ cell cultures by microscopic inspection. Syncytium formation readings were performed for the drug-treated virus-infected cell cultures and consistently scored by different operators. The 50% effective concentration (EC50) corresponds to the compound concentrations required to prevent syncytium formation by 50% in the virus-infected CEM cell cultures.
Activities of test compounds against clinical HIV-1 clade isolates in PBMC cultures.
All the primary clinical isolates representing different HIV-1 clades (see Table 2) were kindly provided by L. Lathey (who was then at BBI Biotech Research Laboratories, Inc., Gaithersburg, MD), and their coreceptor use was determined in the U87 CD4 CXCR4 and U87 CD4 CCR5 cell lines (32). Testing of the activities of the CBAs against these isolates in PBMCs was performed as previously described in detail (2).
TABLE 2.
Inhibitory activities of AH, HHA, and MAb 2G12 against clinical HIV-1 clade isolates and laboratory-adapted virus strains and SIV(mac251) in PBMC cultures
| CBA | EC50a (μM) for the following strains (coreceptor use): |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clade A |
Clade B |
Clade C |
Clade D UG270 (X4) | Clade A/E ID12 (R5) | IIIB (X4) | NL4.3 (X4) | BaL (R5) | SIV(mac251) (R5) | ||||
| UG275 (R5) | UG273 (R5) | BZ167 (X4) | US2 (R5) | DJ 259 (R5) | ETH2220 (R5) | |||||||
| Actinohivin | 0.10 ± 0.03 | 0.15 ± 0.16 | 0.64 ± 0.85 | 0.49 ± 0.31 | 0.11 ± 0.10 | 0.41 ± 0.07 | 0.12 ± 0.12 | 0.54 ± 0.44 | >4 | |||
| HHAb | 0.58 | 0.11 | 0.88 | 0.10 | 0.02 | 0.01 | 0.12 | 0.12 | ||||
| 2G12b,c | 0.018 | 0.04 | >20 | >20 | >20 | 0.18 | 0.08 | 0.09 | >4 | |||
EC50, the compound concentration required to inhibit viral p24 production by 50%.
Data taken from Huskens et al. (25).
Data are expressed in μg/ml.
Cocultivation assay between uninfected Sup-T1 and persistently HIV-1-infected HuT-78 cells.
Persistently HIV-1(IIIB)-infected HuT-78 cells were washed to remove free virus from the culture medium, and 5 × 104 cells (50 μl) were transferred to 96-well microtiter plates. Next, a similar amount of Sup-T1 cells (50 μl) and appropriate concentrations of test compound (100 μl) were added to each well. After 2 days of coculturing at 37°C, the EC50s were quantified on the basis of the appearance of giant cells by microscopic inspection.
Capture of HIV-1(IIIB) by Raji/DC-SIGN cells and subsequent cocultivation with C8166 cells.
The capture of HIV-1(IIIB) by Raji/DC-SIGN cells and subsequent cocultivation with C8166 cells were performed as described previously (6). Briefly, exponentially growing B-lymphocyte Raji wild-type (WT) (Raji/0) and Raji/DC-SIGN cells were suspended in cell culture medium at 2 × 106 cells/ml. Next, 100 μl of HIV-1(IIIB) (∼250,000 pg p24) was added in the presence of serial dilutions of the test compounds (400 μl). After 60 min of incubation, the cells were carefully washed to remove unbound virions and resuspended in 1 ml of cell culture medium. The captured HIV-1(IIIB) was quantified by a p24 antigen (Ag) enzyme-linked immunosorbent assay (ELISA). From the Raji/DC-SIGN cell suspension, 200 μl was mixed in a 48-well microplate with 800 μl uninfected C8166 cells (2.5 × 105 cells/ml). These cocultures were further incubated at 37°C, and syncytium formation was evaluated microscopically after ∼18 to 42 h. Viral p24 Ag determination in the supernatants was also performed.
Selection and isolation of AH-resistant HIV-1 strains.
CEM cells were infected with HIV-1(IIIB) and seeded in 48-well plates in the presence of AH at a concentration equal to 1- to 2-fold its EC50. Three independent series of subcultivations were performed. When a full cytopathogenic effect was visible, the AH concentration was increased stepwise (∼1.5-fold higher). Subcultivations occurred after every 4 to 5 days by transferring 100 μl cell suspension or 200 μl supernatants of the AH-exposed HIV-infected CEM cell cultures to 900 and 800 μl uninfected cell suspensions, respectively.
Genotyping of HIV-1 env region.
Proviral DNA was extracted from cell pellets using DNeasy tissue kits (Qiagen, Hilden, Germany). The genotypes of both the gp120 and the gp41 genes were determined in this assay, as described previously (47).
Bio-Plex cytokine assay.
PBMCs were cultured in the presence of 1.6 μM AH, and the culture supernatant was collected after 72 h. The cytokine production profile was determined by a Bio-Plex 200 system (Bio-Plex human cytokine 27-plex assay; Bio-Rad, Hercules, CA), according to the manufacturer's instructions, and the list of cytokines tested has previously been described in detail (26). PBMCs from different donors were used for each individual experiment. The donors were screened to determine that they were cytomegalovirus and HIV negative.
Flow cytometric analysis.
MT-4 cells were infected with HIV-1(NL4.3) and analyzed when cytopathogenicity started to occur (3 to 4 days after infection). The HIV-1(NL4.3)-infected MT-4 cells were preincubated with or without AH at different concentrations for 30 min, washed, and then incubated with MAb 2G12 for 30 min at 4°C. Next, the cells were washed and incubated with rabbit anti-human (RaH) IgG-fluorescein isothiocyanate (FITC; DakoCytomation, Denmark) for 30 min at 4°C. As a control for background fluorescence, MT-4 cells were stained in parallel with RaH IgG-FITC only. Flow cytometric analysis was performed with a FACSCalibur instrument (BD Biosciences, San Jose, CA), as previously described in detail (25).
Surface plasmon resonance (SPR) analysis.
The following were covalently immobilized on the carboxymethylated dextran matrix of a CM5 sensor chip in 10 mM sodium acetate, pH 4.0, using standard amine coupling chemistry: recombinant gp120 proteins from the HIV-1(IIIB) strain (ImmunoDiagnostics Inc., Woburn, MA) [one batch produced by CHO cell cultures, referred to here as HIV-1(IIIB) (CHO), and another produced by the baculovirus system, referred to here as HIV-1(IIIB) (baculovirus)], from the HIV-1(MN) strain (ImmunoDiagnostics) (baculovirus), the HIV-1(LAV) strain (GenWay Biotech, San Diego, CA) (baculovirus), the HIV-1(ADA) strain (ImmunoDiagnostics) (baculovirus), the HIV-1(YU2) strain (ImmunoDiagnostics) (baculovirus), and the HIV-1(CM) strain (GenWay Biotech) (baculovirus); recombinant gp41 protein from HIV-1(HxB2) (Acris Antibodies GmbH, Herford, Germany) (produced by Pichia pastoris); soluble CD4 protein (GenWay Biotech) (baculovirus); recombinant gp105 protein from the HIV-2(ROD) strain (ImmunoDiagnostics) (baculovirus); recombinant gp130 protein from SIV(mac251) (USBiological, Swampscott, MA) (baculovirus); RNase B (Sigma); fetuin (Sigma); carbonic anhydrase (Sigma); and human serum albumin (Sigma). The exact chip densities are summarized in the Results section. A reference flow cell was used as a control for nonspecific binding and refractive index changes. All interaction studies were performed at 25°C on a Biacore T100 instrument (GE Healthcare, Uppsala, Sweden). The compound AH was serially diluted in 10 mM HEPES-150 mM NaCl-3 mM EDTA-0.05% surfactant P20, pH 7.4 (HBS-EP), over a wide concentration range by using 2-fold dilution steps. Samples were injected for 3 min at a flow rate of 45 μl/min, and the dissociation was followed for 5 min. One duplicate sample and several buffer blanks were used as a positive control and a double referencing, respectively. The CM5 sensor chip surface was regenerated with one injection of 50 mM NaOH. All interactions studied resulted in specific binding signals. The shape of the association and dissociation phases reveals that the curves do not have 1:1 Langmuir kinetics. The experimental data were fit using the 1:1 binding model (Biacore T100 Evaluation software, version 2.0.1) to determine the binding kinetics. These affinity and kinetic values are apparent values, as the injected concentrations of the evaluated compounds did result in biphasic binding signals.
To study in more detail the interaction between gp120 protein from the HIV-1(IIIB) strain and AH, the compound (instead of gp120) was covalently immobilized on a CM5 sensor chip, using conditions identical to those described above. Kinetic experiments were performed with IIIB gp120 produced by both CHO cells and insect (baculovirus) cells. Serial 2-fold analyte dilutions (concentration ranges, 1 to 32 nM for the IIIB gp120 [CHO] and 0.25 to 8 nM for the IIIB gp120 [baculovirus]) were injected over the AH-coated surface of the sensor chip (density, 142 resonance units [RU], or ∼11.4 fmol AH).
To determine the glycan specificity of AH, HIV-1(IIIB) gp120 and HIV-1(HxB2) gp41 were immobilized on a CM5 chip. An experimental setup similar to that described above was used, with the difference being that 20 nM AH was injected over the chip surface in the presence or absence of various mannose oligomers.
In the following set of experiments, an SPR-based experiment of the competition of different CBAs (HHA, UDA, and anti-gp120 MAb 2G12 [Polymun Scientific GmbH, Vienna, Austria]) with AH for binding to HIV-1(IIIB) gp120 (CHO origin) was performed.
To test whether AH keeps its full binding properties for HIV-1(IIIB) gp120 in an acidic (low-pH) environment, we diluted AH and HHA in HBS-EP, pH 4.0, and injected the drug at one concentration on the immobilized IIIB gp120 (CHO) with HBS-EP, pH 4.0, as the running buffer.
In the last set of experiments, the kinetic rate constants of the 2G12 MAb for IIIB gp120 (CHO), IIIB gp120 (baculovirus), and HxB2 gp41 (P. pastoris) were determined.
RESULTS
Antiviral activity of AH in HIV-infected cell cultures and in cocultures of persistently HIV-1-infected and uninfected CD4+ T-lymphocyte cells.
AH inhibited the HIV-1- and HIV-2-induced cytopathic effects in CEM cell cultures (Table 1). The EC50 values of AH for HIV-1(IIIB) and HIV-2(ROD) were 0.025 and 0.488 μM, respectively. AH at 4 μM was not inhibitory to SIV(mac251). AH was also found to be much less antivirally active in CEMX174 cell cultures (EC50, 0.68 μM) infected with the SHIV(sm239) strain, which contained the HIV-1 RT gene but that kept the SIV(mac239)-encoded envelope gene, against HIV-1. AH was not toxic to the cell cultures at the highest concentration tested (8 μM). The mannose-specific plant lectin HHA inhibited HIV-1 and HIV-2 with similar potencies (EC50 range, 0.004 to 0.006 μM) but proved less inhibitory to SIV(mac251) in U87 CD4+ CCR5+ CXCR4+ cells (EC50, 0.163 μM) and somewhat less inhibitory to SHIV(sm239) in CEMX174 cells (EC50, 0.01 μM) than to HIV-1. AH also efficiently prevented giant cell formation between HuT-78/HIV-1 cells and uninfected Sup-T1 cells, being ∼10-fold less active in this assay than HHA (Table 1).
TABLE 1.
Antiviral activities of AH and HHA in different cell lines
| CBA | EC50a (μM) |
||||
|---|---|---|---|---|---|
| HIV-1(IIIB) | HIV-2(ROD) | SIV(mac251) | SHIV(sm239) | HuT-78/HIV-1 + Sup-T1 | |
| Actinohivin | 0.025 ± 0.005 | 0.488 ± 0.238 | >4 | 0.68 ± 0.17 | 1.2 ± 0.1 |
| HHA | 0.006 ± 0.002 | 0.004 ± 0.002 | 0.163 ± 0.024 | 0.010 ± 0.001 | 0.110 ± 0.012 |
For HIV-1(IIIB), HIV-2(ROD), SIV(mac251), and SHIV(sm239), EC50s are the concentrations required to inhibit virus-induced cytopathogenicity in CEM (HIV) and U87 CD4+ CCR5+ CXCR4+ (SIV) cell cultures by 50%. For HuT-78/HIV-1 + Sup-T1, the EC50 is the concentration required to inhibit syncytium formation between HuT-78/HIV-1 and Sup-T1 cells by 50%. Data are the means of at least two to four independent experiments.
AH was next evaluated for its inhibitory activity against a broad variety of HIV-1 strains, such as laboratory-adapted strains HIV-1 IIIB (X4 coreceptor tropism), NL4.3 (X4), and BaL (R5) and representative clinical isolates belonging to clades A, B, C, D, and A/E, in PBMC cultures. AH proved markedly inhibitory to HIV-1 replication, independently of the virus clade and the coreceptor tropism (R5 or X4). The EC50s ranged from 0.10 to 0.64 μM (Table 2). This is in sharp contrast to the results for HHA, whose EC50s varied much more widely (up to 90-fold), depending on the nature of the virus clade, whereas the activity of mannose oligomer-specific MAb 2G12 varied even more, even exceeding >1,000-fold (Table 2). Both AH and MAb 2G12 were inactive against SIV(mac251) infection of PBMC cultures.
Inhibitory effect of AH on the capture of HIV-1 by Raji/DC-SIGN cells and subsequent transmission to uninfected CD4+ T-lymphocyte C8166 cells.
We also wanted to evaluate whether AH has the potential to prevent HIV-1(IIIB) capture by DC-SIGN-expressing Raji cells and to decrease the transmission of DC-SIGN-captured virions to uninfected CD4+ T cells. HIV-1 was exposed to different AH concentrations for 30 min before the virus was added to DC-SIGN-expressing Raji cells. One hour later, uncaptured virus particles and AH were carefully removed from the cell cultures by several washing (centrifugation) steps. p24 Ag ELISA analysis revealed that AH dose-dependently inhibited HIV-1(IIIB) capture by Raji/DC-SIGN cells, with the EC50 being 0.224 μM. In this assay, HHA was 5-fold more potent (Table 3). Next, the washed and AH-treated HIV-1-exposed Raji/DC-SIGN cells were cocultured with CD4+ T cells, and giant cell formation, determined microscopically within 24 to 48 h after cocultivation, was recorded. AH inhibited HIV-1 transmission at an EC50 of 0.032 μM, the potency of which was 10-fold less than that of HHA. Thus, AH markedly inhibited binding of HIV-1 to DC-SIGN and efficiently prevented subsequent transmission of the captured virions to uninfected CD4+ T-cell cultures.
TABLE 3.
Inhibitory effects of CBAs on HIV-1(IIIB) capture by Raji/DC-SIGN cells and subsequent transmission of the captured virus to cocultivated C8166 cells
| CBA | EC50a (μM) |
|
|---|---|---|
| Capture | Transmission | |
| Actinohivin | 0.224 ± 0.176 | 0.032 ± 0.024 |
| HHA | 0.045 ± 0.028 | 0.003 ± 0.002 |
EC50, compound concentration required to inhibit DC-SIGN-mediated HIV-1 capture by DC-SIGN and subsequent transmission to CD4+ T cells.
Binding of AH to the viral envelope and determination of its glycan-binding specificity.
AH binding to the virus envelope was subjected to a detailed kinetic characterization by means of SPR analysis. The properties of binding of AH to X4 HIV-1(IIIB) gp120 (expressed in either mammalian CHO cells and insect cells [baculovirus system]), X4 HIV-1(LAV) gp120, X4 HIV-1(MN) gp120, R5 HIV-1(ADA) gp120, R5 HIV-1(CM) gp120, R5 HIV-1(YU2) gp120, HIV-2(ROD) gp105, SIV(mac251) gp130, and HIV-1(HXB2) gp41 were evaluated (Table 4). All ligands were covalently immobilized on CM5 sensor chips. Twofold serial dilutions of AH (range, 2.5 to 80 nM) were applied to the immobilized ligands. The binding curves of the studied interactions could not be optimally fitted by 1:1 Langmuir kinetics due to the concomitant interactions of multiple carbohydrate-binding sites of AH with the glycosylated envelope molecules. Nevertheless, a 1:1 fit was applied to obtain the association rate constant (ka), the dissociation rate constant (kd), and the apparent kinetic rate constant (KD) (Table 4; Fig. 1A, C, and E). Since the kinetics were invariably characterized by an initial fast dissociation (designated the α phase) followed by a slower dissociation phase (designated the β phase), we also determined the off rate of the α dissociation phase (kdα; 0 to 30 s) and the off rate of the β dissociation phase (kdβ), which starts after the first 30 s of dissociation. Comparable apparent KD values (5 to 23 nM) were detected for the binding between AH and the gp120 envelope molecules, irrespective of the nature and origin of gp120 (derived from either X4 or R5 virus; expression in either mammalian CHO or insect cells; envelope origin from HIV-1, HIV-2, or SIV). Also, the kdβ off rates of the AH interactions with all the different gp120s fell within the same order of magnitude (2.71 × 10−3 to 8.97 × 10−3 s−1) (Table 4). Interestingly, the interaction of AH with HIV-1 gp41 was also comparable to the values found for HIV-1 gp120 with regard to the KD, kdα, and kdβ.
TABLE 4.
Kinetic data for the interaction of AH with gp120 and gp41 immobilized on the sensor chip
| Virus strain and glycoprotein (coreceptor) | KD (nM) | ka (1/Ms) | kd (1/s) | kdα (1/s) | kdβ (1/s) |
|---|---|---|---|---|---|
| IIIB gp120 (CHO) (X4) | 23.15 ± 0.96 | (1.40 ± 0.08)E+06 | (3.24 ± 0.05)E−02 | (3.57 ± 0.07)E−02 | (8.97 ± 0.27)E−03 |
| IIIB gp120 (baculovirus) (X4) | 19.53 ± 0.70 | (2.41 ± 0.34)E+06 | (4.72 ± 0.80)E−02 | (2.06 ± 0.22)E−02 | (4.12 ± 0.34)E−03 |
| MN gp120 (baculovirus) (X4) | 16.24 ± 1.35 | (6.15 ± 0.77)E+05 | (9.94 ± 0.42)E−03 | (1.01 ± 0.12)E−02 | (3.71 ± 0.06)E−03 |
| LAV gp120 (baculovirus) (X4) | 17.51 ± 3.28 | (7.51 ± 2.17)E+05 | (1.27 ± 0.17)E−02 | (9.37 ± 0.53)E−03 | (2.71 ± 0.12)E−03 |
| ADA gp120 (baculovirus) (R5) | 5.25 ± 1.84 | (1.29 ± 0.15)E+06 | (6.65 ± 1.96)E−03 | (3.26 ± 0.48)E−02 | (6.58 ± 2.05)E−03 |
| CM gp120 (baculovirus) (R5) | 8.04 ± 3.85 | (1.20 ± 0.68)E+06 | (7.99 ± 0.31)E−03 | (1.13 ± 0.14)E−02 | (4.74 ± 1.54)E−03 |
| YU2 gp120 (baculovirus) (R5) | 18.91 ± 2.83 | (7.05 ± 0.40)E+05 | (1.34 ± 0.27)E−02 | (9.81 ± 1.33)E−03 | (3.39 ± 0.22)E−03 |
| HIV-2(ROD) gp105 (baculovirus) | 14.79 ± 0.17 | (9.40 ± 2.10)E+05 | (1.39 ± 0.33)E−02 | (1.30 ± 0.08)E−02 | (6.56 ± 0.80)E−03 |
| SIV(mac251) gp130 (baculovirus) | 6.68 ± 1.10 | (1.32 ± 0.17)E+06 | (8.73 ± 0.31)E−03 | (1.28 ± 0.02)E−02 | (8.58 ± 0.97)E−03 |
| HxB2 gp41 (P. pastoris) | 13.53 ± 0.67 | (8.04 ± 0.16)E+05 | (1.09 ± 0.03)E−02 | (1.29 ± 0.13)E−02 | (2.87 ± 2.04)E−03 |
FIG. 1.

Kinetic analysis of AH interaction with IIIB gp120 isolated from CHO cell cultures (A) and from the baculovirus system (C) and with HxB2 gp41 isolated from P. pastoris (E) using the SPR technology. Serial 2-fold analyte dilutions (covering a concentration range of from 2.5 to 80 nM) were injected over the surface of the gp120-bound (A to D) or gp41-bound (E and F) sensor chip. The experimental data (colored curves) were fit using the 1:1 binding model (black lines) to determine the kinetic parameters. The data are representative examples of the results of three independent experiments. The biosensor chip densities were 822 RU (or 6.9 fmol) for gp120 from CHO (A and B), 725 RU (or 6.0 fmol) for gp120 from baculovirus (C and D), and 637 RU (or 15.5 fmol) for gp41 (E and F). For the titration experiments, AH at a fixed concentration (20 nM) was incubated for 30 min with increasing concentrations of (α1,2-man)3 (curve 1, 200 nM; curve 2, 2 μM; curve 3, 10 μM; curve 4, 20 μM; and curve 5, 200 μM) and injected over the three surfaces. The amplitudes at 3 min after injection were used to calculate the IC50s.
To verify the nature of the sugar specificity of AH for gp120/gp41 binding, AH at a fixed concentration (20 nM) was incubated for 30 min with increasing concentrations (from 200 nM to up to 200 μM) of (α1,2-man)3, manα1,3manα1,6man trimer, or (β1,4-GlcNAc)3. Binding of AH to gp120 and gp41 could be dose-dependently reversed by (α1,2-man)3 (50% inhibitory concentration [IC50], 5 to 11 μM) (Fig. 1B, D, and F) but not by the other oligosaccharide trimers (Table 5). When the binding of 20 nM AH to IIIB gp120 (baculovirus), IIIB gp120 (CHO), and HxB2 gp41 (P. pastoris) was evaluated in the presence of 200 μM mannose monosaccharide, no decrease in the binding amplitude was observed at all (data not shown). When similar SPR-based experiments were performed with (α1,2-man)2 and with the high-mannose-type glycan Man9GlcNAc2, the oligosaccharides inhibited the binding of AH to gp120 with IC50s as high as 455 and 38 μM, respectively (Table 5).
TABLE 5.
Inhibition of binding of AH to gp120 and gp41 immobilized on the sensor chip by oligosaccharide trimers, quantified by SPR technology
| Virus strain and glycoprotein | AH IC50a (μM) |
||||
|---|---|---|---|---|---|
| (α1,2-Man)3 | Manα1,3manα1,6 man | (β1,4-GlcNAc)3 | (α1,2-Man)2 | Man9GlcNAc2 | |
| IIIB gp120 (CHO) | 5.1 | >200 | >200 | 455 | 38 |
| IIIB gp120 (baculovirus) | 9.0 | >200 | >200 | NDb | ND |
| HxB2 gp41 (P. pastoris) | 11 | >200 | >200 | ND | ND |
IC50, oligosaccharide concentration required to inhibit binding of AH to HIV-1 gp120 or gp41 by 50%.
ND, not determined.
The kinetics of the interaction between AH and gp120 (derived from mammalian CHO or insect cells) were also determined using AH (instead of gp120) immobilized on the sensor chip. Whereas the KD of AH was 1.19 nM for CHO-derived gp120, the affinity constant was as low as 0.056 nM when gp120 was derived from insect cells (Table 6). The markedly (20-fold) lower KD value for insect cell-derived gp120 was mainly due to a 10-fold higher ka. When other glycosylated glycoproteins, such as RNase B (containing one high-mannose-type glycan), carbonic anhydrase, and fetuin (containing a variety of complex-type glycans rich in GlcNAc), and also human serum albumin had been immobilized on the sensor chip, no interaction with AH could be detected by SPR analysis, pointing to the pronounced selectivity of this CBA.
TABLE 6.
Kinetic data for the interaction of gp120 with AH immobilized on the sensor chip
| HIV-1 strain | KD (nM) | ka (1/Ms) | kd (1/s) |
|---|---|---|---|
| IIIB (CHO) | 1.19 ± 0.05 | (7.19 ± 0.30)E+05 | (8.59 ± 0.50)E−04 |
| IIIB (baculovirus) | 0.056 ± 0.002 | (6.06 ± 0.18)E+06 | (3.53 ± 0.31)E−04 |
Binding of AH to HIV-1(IIIB) gp120 at pH 4.0.
Whereas all binding experiments with AH described above were performed at physiological pH, it was important in view of the microbicidal potential of AH to investigate whether AH keeps its HIV envelope-binding properties at low pH (i.e., pH 4.0). Therefore, AH (and HHA) were diluted in acidified running buffer (pH 4.0 and also, as a control, in running buffer at pH 7.4) and examined their binding to gp120. The data in Fig. 2 show that both CBAs were still able to substantially bind to gp120 at pH 4.0. The curve for HHA binding to gp120 showed an increased amplitude (∼60%) compared to the corresponding amplitude of gp120 binding at pH 7.4, whereas AH lost only ∼20% of its amplitude at pH 4.0 compared to the corresponding amplitude of gp120 binding at pH 7.4.
FIG. 2.

The CBAs AH and HHA (0.5 μM) were subjected for binding to IIIB gp120 (origin, CHO cells; chip density, 3,840 RU [∼32 fmol]) under both acidic (pH 4.0; red and blue) and neutral (pH 7.4; green and magenta) conditions. The association phase of AH and HHA was monitored for 3 min.
Interaction of MAb 2G12 with HIV-1 gp120 and gp41.
Anti-gp120 MAb 2G12 was also the subject of a kinetic SPR study of its binding to IIIB gp120 (CHO), IIIB gp120 (baculovirus), and HxB2 gp41 (P. pastoris) using the same sensor chips used for AH. Serial 2-fold dilutions of 2G12 (4 to 125 nM for gp120 and 4 nM to 4 μM for gp41) were exposed to the immobilized envelope proteins. Figure 3 demonstrates that 2G12 strongly binds to gp120 (KD, 5.6 to 16.1 nM), but it also binds to gp41, although at an affinity (KD) of 26 μM, which is 1,000- to 5,000-fold weaker than the binding kinetics observed for gp120 (Table 7).
FIG. 3.

Kinetic analysis of the MAb 2G12 interaction with the IIIB gp120 isolated from baculovirus (A) and CHO cell cultures (B) and with gp41 (HxB2) isolated from P. pastoris (C) using the SPR technology. Serial 2-fold analyte dilutions (covering concentration ranges of from 4 to 125 nM 2G12 for the gp120 interaction [A and B] and 4 nM to 4 μM for the gp41 interaction [C]) were injected over the surface. The experimental data (colored curves) were fit using the 1:1 binding model (black lines) to determine the kinetics. The same biosensor chip used for Fig. 1 was used in the experiment whose results are shown here.
TABLE 7.
Kinetics for interaction of MAb 2G12 with gp120 and gp41
| Virus strain and glycoprotein | KD | ka (1/Ms) | kd (1/s) |
|---|---|---|---|
| IIIB gp120 (CHO) | 16 nM | 5.57E+04 | 8.99E−04 |
| IIIB gp120 (baculovirus) | 5.6 nM | 7.25E+04 | 4.05E−04 |
| HxB2 gp41 (P. pastoris) | 26 μM | 1.34E+04 | 3.44E−01 |
Competition of CBAs with AH for binding to HIV-1 gp120.
To investigate whether it would still be possible for CBAs (i.e., HHA, UDA, and MAb 2G12) to bind to gp120 when gp120 is already bound to AH, the following experiment was performed (Fig. 4). AH (0.5 μM; ∼25-fold its KD value) was administered to gp120 immobilized on the sensor chip for 2 min (Fig. 4A, time point 1), and immediately at the end of this association phase AH was injected again, but in the presence of the other CBAs at concentrations that were at least >100-fold their KD values (Fig. 4A, time point 2). CBAs with specificities for glycans other than those to which AH is specific (i.e., α1,3/α1,6-mannose-specific HHA [Fig. 4, purple] and GlcNAc-specific UDA [Fig. 4, green]) can still independently bind to gp120 to which AH is bound. Instead, no further binding to gp120 could be detected for α1,2man-specific MAb 2G12 (Fig. 4, cyan) when gp120 was already covered by AH. In Fig. 4B, the interactions of the individual CBAs with gp120 (in the absence of AH) are shown.
FIG. 4.

(A) Competition between different CBAs (10 μM HHA [magenta], 10 μM UDA [green], 2 μM anti-gp120 G212 MAb [light blue]) with 0.5 μM AH (red) for binding to the IIIB gp120 (CHO origin; chip density, 360 RU [∼3 fmol]). AH (10 μM) was injected (time point 1), followed after 2 min by injection of 10 μM AH in the presence of another CBA (time point 2). (B) The test compounds were injected over the surface at a high concentration (>100-fold the KD).
Inhibition of MAb 2G12 binding to gp120 of HIV-1-infected MT-4 cells by AH.
Since binding of AH to SPR-immobilized gp120 inhibited the subsequent binding of MAb 2G12 to gp120 (Fig. 4), we also determined the IC50 of AH for inhibition of 2G12 binding to HIV-1-infected MT-4 cells by flow cytometric analysis. The data in Fig. 5 demonstrate that 1.6 μM AH afforded 91% inhibition of MAb 2G12 binding to gp120-expressing HIV-1-infected lymphocytes (Fig. 5A); 77% inhibition occurred at 0.32 μM AH (Fig. 5B) and 42% inhibition occurred at 0.064 μM AH (Fig. 5C), resulting in an IC50 of AH as low as 0.093 μM. Thus, AH clearly interferes with the MAb 2G12 binding epitope on HIV-1 gp120-expressing T cells.
FIG. 5.

Inhibition of the binding of anti-gp120 MAb 2G12 of HIV-1(NL4.3)-infected MT-4 cells in the presence of serial dilutions of AH: 1.6 μM (A), 0.32 μM (B), and 0.064 μM (C). Gray, blue, and red histograms represent the background fluorescence, MAb 2G12 binding when it was preincubated with AH, and MAb 2G12 binding, respectively. MFI, mean fluorescence intensity.
Selection of AH-resistant HIV-1(IIIB) strains.
To reveal the resistance pattern of AH against HIV-1, two independent series of AH selections were performed (Fig. 6). HIV-1(IIIB)-infected CEM T-cell cultures were initially exposed to AH at ∼0.02 μM. Subcultivations were performed every 4 to 5 days. Syncytium formation was recorded microscopically before each new subcultivation step, and the drug concentration was increased ∼1.5-fold once a full cytopathic effect was scored. Virus isolates (arrows in Fig. 6) were taken at 11 time points during the selection process and subjected to further genotypic and phenotypic analysis.
FIG. 6.

Schedule of selection of AH resistance development in HIV-1(IIIB)-infected CEM cell cultures exposed to increasing AH concentrations. Arrows indicate the time points when virus isolates were taken for further characterization. AH_1 and AH_2 represent two independent subcultivation schedules in which for each passage suspensions of AH-exposed CEM cell cultures were transferred. AH_3 represents the series of subcultivations that were started from the last passage of AH_2 and for which the supernatants of the AH-exposed CEM cell cultures were transferred at each passage. AH_4 was split from the AH_1 selection series after the 46th subcultivation and independently subcultured for an additional 12 passages.
Genotypic characterization of gp160 of AH-resistant HIV-1(IIIB) strains.
The envelope gene (encoding gp120 and gp41) of the virus isolates was sequenced and analyzed for the presence of mutations compared to the sequence of the envelope gene of WT HIV-1(IIIB) that was subcultivated in a parallel experiment but in the absence of AH pressure. Overall, deletions of several putative N-glycosylation motifs frequently appeared in gp120 of HIV-1 in the presence of AH. One new N-glycosylation motif appeared at amino acid position 29 in the gp120s of several virus isolates (Table 8; Fig. 7). Until passage 30 in AH subcultivation schedule 1 (AH_1/#30; ∼135 days), no N-glycan deletions could be detected in the viral envelopes of the virus isolates. However, virus isolates AH_1/#40 and AH_1/#43 each contained three N-glycosylation site deletions in gp120: T232T/M, S291S/F and N289N/S, T297T/I, and/or N339K/N. At a later passage, isolate AH_1/#58 regained the glycan at N339 but showed three additional N-glycan deletions at amino acid positions N186, N386, and N392. Interestingly, isolate AH_4/#58, which resulted from the AH_1 selection series after the cultures were separated and independently further subcultivated after passage 47, kept the N230 and N289 glycan deletions and also regained the N339 glycan (as was also found in AH_1/#58) as well as accumulated glycan deletions at positions N234 and N392 (Table 8). Whereas the virus isolates from passages AH_2/#25, AH_2/#30, and AH_2/#35 contained only mixtures of both mutant and wild-type nucleotide sequences at the particular N-glycosylation sites, the virus isolates at the end of the selection process (passages AH_3/#82 and AH_3/#100) contained up to six different deletions in N-glycosylation motifs, of which three were present as pure mutations. Also, in the latter set of virus isolates, a new potential glycosylation site is created by mutation of S29N/S, as was also observed in independently obtained isolate AH_1/#58 (Table 8). In the AH_2 and AH_3 selection series, the six N-glycosylation sites were deleted in all cases by mutation of the S or T in the corresponding glycosylation motifs. In the AH_1/#58 and AH_4/#58 HIV-1 isolates, a single N-glycosylation site deletion in gp41 was observed at amino acid position 811NAT/I813.
TABLE 8.
Amino acid mutations that appeared in HIV-1 strains under escalating AH concentrations
| Glycosylation motif in HIV-1(IIIB) gp120 | Type of N-glycan | Amino acid mutationa |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AH_1/#40 | AH_1/#43 | AH_1/#58 | AH_4/#58 | AH_2/#25 | AH_2/#30 | AH_2/#35 | AH_3/#82 | AH_3/#100 | ||
| T19T/I | T19T/I | |||||||||
| S29N/Sb | S29N/S | S29N/S | S29N/S | S29N/S | ||||||
| V42G/V | ||||||||||
| V84I/V | ||||||||||
| 88NVT90 | Complex | |||||||||
| 136NDT138 | Complex | T138T/I | T138T/I | |||||||
| 141NSS143 | Complex | |||||||||
| E150K/E | ||||||||||
| 156NCS158 | Complex | |||||||||
| 160NIS162 | Complex | |||||||||
| D185N/D | D185D/G | |||||||||
| 186NDT188 | Complex | T188N/T | ||||||||
| T192T/R | T192T/M | |||||||||
| 197NTS199 | Complex | |||||||||
| A204T/A | ||||||||||
| P214P/L | ||||||||||
| 230NKT232 | High mannose | T232T/M | T232T/M | T232K/T | T232K | T232 M | T232 M | |||
| 234NGT236 | High mannose | N234K/N | ||||||||
| 241NVS243 | High mannose | |||||||||
| T257T/P | T257T/P | |||||||||
| 262NGS264 | High mannose | |||||||||
| A266E/A | ||||||||||
| E269K/E | ||||||||||
| V270I/V | ||||||||||
| V271I/V | ||||||||||
| 276NFT278 | Complex | N276N/I | ||||||||
| A281T/A | ||||||||||
| T283T/I | ||||||||||
| 289NQS291 | High mannose | N289N/S | N289N/Y | N289Y | ||||||
| S291S/F | S291S/F | S291S/F | S291S/F | S291V/F | S291F | S291F | ||||
| V292G/V | ||||||||||
| 295NCT297 | High mannose | T297T/I | T297T/A | T297T/A | T297A | T297A | ||||
| 301NNT303 | Complex | T303T/I | T303T/I | T303T/I | ||||||
| R304K/R | R304K/R | R304K/R | ||||||||
| 332NIS334 | High mannose | |||||||||
| A336T/A | ||||||||||
| 339NNT341 | High mannose | N339K/N | N339N/D | |||||||
| K343K/N | ||||||||||
| 356NKT358 | Complex | |||||||||
| V360I/V | V360I/V | |||||||||
| T363T/I/M | ||||||||||
| S364P/S | S364P/S | |||||||||
| D368D/E | ||||||||||
| E381/K/N/E/D | ||||||||||
| 386NST388 | High mannose | N386K/N | ||||||||
| 392NST394 | High mannose | T394T/I | T394I | T394T/I | T394T/I | |||||
| 397NST399 | Complex | |||||||||
| 401NNT403 | Complex | T403T/I | T403T/I | |||||||
| E424K/E | ||||||||||
| S435S/I | ||||||||||
| 448NIT450 | High mannose | |||||||||
| 463NGS465 | Complex | |||||||||
| R471K/R | ||||||||||
| P493P/L | ||||||||||
The amino acid sequence numbering is according to Kwong et al. (28), and assignment of high-mannose- or complex-type glycans is according to Leonard et al. (29). Mutated amino acids in boldface result in the deletion of a glycosylation motif. Virus isolates AH_1#15 and AH_1#30 contain no amino acid mutations. Virus isolates AH_1#58 and AH_4#58 also contain an N-glycan deletion in gp41, being 811NAT813 to 811NAI813, as a mixture with the wild type.
This amino acid change results in the creation of a new putative N-glycosylation site (italics).
FIG. 7.
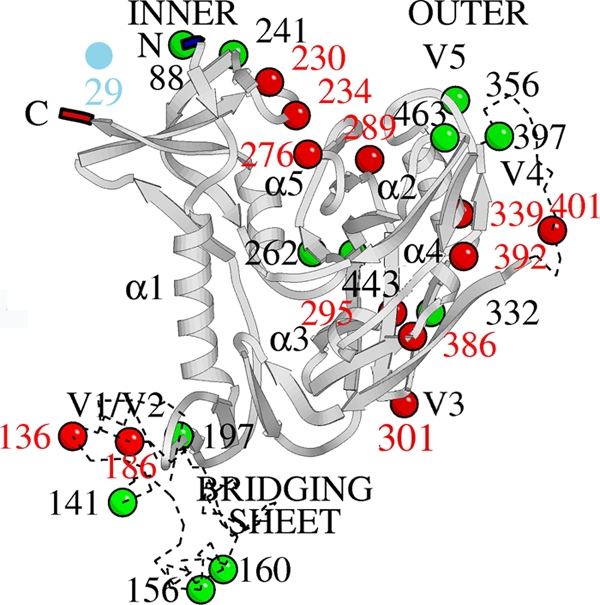
Mapping of the mutated glycosylation sites (deleted [red balls], unchanged [green balls], or created [blue ball]) in gp120 of HIV-1(IIIB) strains isolated upon AH drug pressure in CEM cell cultures.
It was shown before that HIV-1 gp120 expressed in CHO cells contains 24 putative N-glycosylation sites, of which approximately 11 were defined to carry a high-mannose-type structure (29). Seven out of the 12 deleted N-glycans found to occur in the AH selection experiments were previously determined to be high-mannose type. This observation points to the fact that AH may selectively bind to α1,2-mannose glycans and therefore preferentially selects for the deletion of high-mannose-type glycans in HIV-1 gp120. The mutated glycosylation sites were mapped on the three-dimensional structure of the HIV-1 envelope gp120 (Fig. 7). In addition to the glycosylation site mutations, several other amino acid mutations (not belonging to a glycosylation motif) occurred in several virus isolates, usually appearing as mixtures with the wild-type amino acids. When the wild-type virus envelope was analyzed for genotypic changes after ∼40 passages in CEM cell cultures in the absence of AH, no glycosylation site mutations had occurred.
Phenotypic characterization of AH-resistant HIV-1(IIIB) strains.
Four different virus isolates (AH_1/#43, AH_1/#58, AH_3/#82, and AH_3/#100) recovered in the presence of different concentrations of AH during the drug resistance selection process were evaluated for their sensitivities to a variety of CBAs (including AH, PRM-A, HHA, UDA, and MAb 2G12) (Table 9) that were carefully selected according to their carbohydrate specificities. The three N-glycan deletions observed in virus isolate AH_1/#43 seems to make the virus only 2-fold phenotypically resistant to AH. Two more N-glycan deletions (as in AH_1/#58) make the virus 19-fold phenotypically resistant to AH. CBAs such as HHA (with α1,3/α1,6-mannose specificity) and PRM-A (with α1,2-mannose specificity) kept sensitivities to all AH-resistant virus strains equal to that of WT HIV-1(IIIB). The AH-resistant isolates also showed no cross-resistance to the N-acetylglucosamine-specific compound UDA. In contrast, 2G12, which recognizes high-mannose-type N-glycans in HIV-1 gp120 (i.e., at positions N295, N332, and N392) (34, 35, 42) markedly lost (>25-fold) its potential for activity against the AH_1/#58, AH_3/#82, and AH_3/#100 HIV-1 isolates (Table 9).
TABLE 9.
Phenotypic sensitivity of wild-type HIV-1 and mutant HIV-1 strains to CBAs
| CBA | EC50a (μM) |
|||||
|---|---|---|---|---|---|---|
| WT | AH_1/#43 | AH_1/#58 | AH_4/#58 | AH_3/#82 | AH_3/#100 | |
| AH | 0.025 ± 0.005 | 0.065 ± 0.012 | 0.480 ± 0.089 | 0.229 ± 0.055 | 0.360 ± 0.339 | 0.309 ± 0.047 |
| 2G12 | 0.009 ± 0.003 | 0.036 ± 0.020 | >0.250 | 0.081 ± 0.044 | >0.250 | >0.250 |
| HHA | 0.006 ± 0.003 | 0.004 ± 0.001 | 0.008 ± 0.002 | 0.004 ± 0.001 | 0.004 ± 0.001 | 0.005 ± 0.001 |
| UDA | 0.14 ± 0.04 | 0.09 ± 0.01 | 0.19 ± 0.06 | 0.13 ± 0.02 | 0.15 ± 0.08 | 0.15 ± 0.08 |
| PRM-A | 5.2 ± 4.2 | 4.5 ± 0.7 | 5.1 ± 0.3 | 4.7 ± 0.1 | 7.8 ± 0.2 | 6.5 ± 0.5 |
EC50, compound concentration required to inhibit virus-induced cytopathogenicity by 50%.
Lack of cytokine and chemokine induction in AH-exposed PBMC cultures.
The Bio-Plex system is a powerful tool that enables the simultaneous quantification of a wide variety of cytokines in a single sample (26). By means of the Bio-Plex system, the potential of AH to induce chemokines and cytokines in PBMCs was investigated for a total of 27 different cytokines/chemokines. PBMCs were cultured in the presence of 1.6 μM AH, and phytohemagglutinin (PHA; as a mitogenic lectin and cytokine-inducing control) was tested at a 100-fold lower concentration (0.016 μM). The drug concentration chosen for AH equals more than 50-fold the EC50 against HIV-1 replication in cell culture. After 72 h, PBMC supernatant was collected from up to four different donors and was subjected to Bio-Plex analysis. As was also found for the plant lectin HHA, no cytokines/chemokines were markedly stimulated in the presence of 1.6 μM AH, whereas 0.016 μM PHA or 0.18 μM CV-N clearly induced a variety of cytokines/chemokines (Table 10). It should be noticed that the data obtained for CV-N and AH were derived from different experimental assays using PBMCs from different donors in each experiment. Therefore, an appropriate statistical analysis could not be performed. However, it is clear from the data that in the majority of cases, the stimulatory action of CV-N is so much higher than that of AH (>100- to 500-fold and <2-fold or undetectable, respectively), so we may reasonably conclude that AH has a much less pronounced (or nonexisting) stimulatory effect, in contrast to the stimulatory effect of CV-N (or PHA). Indeed, repeat experiments using PBMCs from different donors give much lower interindividual variation for a single compound than the values seen between AH and CV-N or PHA in the independent experiments. AH also had no mitogenic activity in PBMCs and did not induce cellular CD25, CD69, or HLA-DR activation markers (data not shown). This is in sharp contrast to the results for PHA (data not shown) and also to the anti-HIV α1,2-man-specific lectin CV-N (26).
TABLE 10.
Fold change in level of cytokine and chemokine production in PBMCs incubated with CBAs at 72 h postadministrationa
| Cytokine/chemokine | Fold change |
|||
|---|---|---|---|---|
| AH (1.6 μM) | HHA (0.4 μM) | CV-Nb (0.18 μM) | PHAb (0.016 μM) | |
| IL-1beta | ND | 1.6 ± 0.4 | 100-500 | 100-500 |
| IL-1ra | 2.4 ± 0.03 | 1.6 ± 0.2 | 10-100 | 10-100 |
| IL-2 | 1.4 ± 0.9 | 0.58 ± 0.08 | 3-10 | 1-3 |
| IL-4 | ND | 1.0 ± 0.2 | 10-100 | 3-10 |
| IL-5 | ND | 0.59 ± 0.04 | >500 | 10-100 |
| IL-6 | 2.52 | 1.1 ± 0.1 | >500 | >500 |
| IL-7 | ND | 1.92 | 3-10 | 1-3 |
| IL-8 | 1.5 ± 0.02 | 1.06 | 100-500 | 100-500 |
| IL-9 | 0.9 ± 0.3 | 1.1 ± 0.2 | 10-100 | 10-100 |
| IL-10 | 1.4 ± 0.0 | 1.6 ± 0.3 | 100-500 | 10-100 |
| IL-12 | ND | 4.9 | 3-10 | 3-10 |
| IL-13 | ND | 0.73 ± 0.46 | >500 | 100-500 |
| IL-15 | 0.38 | 1.5 ± 0.3 | 10-100 | 10-100 |
| IL-17 | ND | 1.8 ± 0.02 | 100-500 | 10-100 |
| Eotaxin | ND | 1.1 ± 0.1 | 10-100 | 3-10 |
| FGF | ND | >7.9 | 3-10 | 1-3 |
| G-CSF | ND | 1.1 ± 0.2 | 10-100 | 10-100 |
| GM-CSF | ND | 1.1 ± 0.2 | 10-100 | 3-10 |
| λ-IFN | ND | 1.1 ± 0.3 | >500 | 10-100 |
| IP-10 | 4.3 ± 1.2 | 0.59 ± 0.15 | 3-10 | 1-3 |
| MCP-1 | 2.0 ± 1.3 | 1.0 | 100-500 | 100-500 |
| MIP-1alpha | 1.50 | 1.1 ± 0.3 | >500 | 10-100 |
| MIP-1beta | 1.1 ± 0.3 | 1.1 ± 0.1 | 10-100 | 10-100 |
| PDGF-BB | 1.2 ± 0.1 | 0.95 ± 0.07 | 1-3 | 1-3 |
| RANTES | 0.9 ± 0.01 | 0.85 ± 0.15 | 10-100 | 1-3 |
| TNF-α | ND | 1.2 ± 0.5 | >500 | 10-100 |
| VEGF | 0.7 ± 0.4 | 1.9 ± 0.8 | 1-3 | 1-3 |
PBMCs from two to four different donors were used for these studies. Abbreviations: ND, not detectable (the cytokine levels in the presence or absence of AH were under the detection limit of the assay [<2 pg/ml]); IL, interleukin; FGF, fibroblast growth factor; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; γ-IFN, gamma interferon; IP-10, interferon-inducible protein 10; MCP-1, monocyte chemoattractant protein 1; MIP-1α and MIP-1β macrophage inflammatory proteins 1α and 1β, respectively; PDGF-BB, platelet-derived growth factor BB; TNF-α, tumor necrosis factor alpha; VEGF, vascular endothelial growth factor.
Fold change values represent the range of cytokine/chemokine stimulation afforded in at least 50% of the CBA-exposed PBMC donors. Data taken from Huskens et al. (26).
DISCUSSION
AH, a 12.5-kDa protein consisting of 114 amino acids (14), showed similarities to CV-N (11) for binding to high-mannose-type glycans. Whereas AH exhibits a sequence that consists of a highly conserved triple tandem repeat that displays three similar carbohydrate-binding sites (41), CV-N can form a domain-swapped dimer containing two sites with a high affinity for α1,2-man carbohydrates and two sites with a lower affinity for α1,2-man carbohydrates on opposite ends of the dimer (8-10, 49). Our antiviral data and previous observations (15, 16) point to AH as a potential candidate microbicide drug. AH efficiently inhibits HIV-1 in various cell cultures, blocks syncytium formation between persistently HIV-1(IIIB)-infected T cells and uninfected CD4+ T-lymphocytes, prevents DC-SIGN-directed HIV-1 capture, and blocks the subsequent transmission of captured virus to uninfected CD4+ T cells (Tables 1 and 3). Interestingly, AH has broad neutralizing activity against a variety of laboratory-adapted and clinical HIV isolates and does not discriminate between X4 and R5 viruses (Table 2). Moreover, its broad neutralizing activity is striking, in the sense that AH displays minimal variation (up to 5-fold) in antiviral activity, irrespective of the nature of the viral strain. Such a narrow variation in the potency of AH (in contrast to that of HHA) represents an interesting property with respect to the potential use of AH as a microbicide drug lead. So-called broadly neutralizing MAb 2G12, which has the same α1,2-mannose oligosaccharide specificity as AH, displays up to >1,000-fold variation in its antiviral neutralization capabilities (Table 2). Thus, AH neutralizes a much broader range of HIV strains and clades than MAb 2G12 and should therefore be considered a prime candidate microbicidal drug for further studies. Although the reasons for the consistent suppression of the virus variants by AH compared to 2G12 (and HHA) are not entirely clear, these observations can be related to the fact that AH has preferential specificity for α1,2-mannose oligomers that are abundantly present on the envelopes of all HIV strains. Although monoclonal antibody 2G12 is also specific for α1,2-mannose oligomers, it solely recognizes a specific configuration of three glycans (at positions N392, N339, and N386) (34, 35, 42), and therefore, virus strains that have different glycan configurations at these amino acid positions (or that lack a glycan at these amino acid positions) should show diminished recognition by (affinity for) this monoclonal antibody. HHA has preferential α1,3/α1,6-mannose oligomer specificity (46), and such specificity may result in a higher degree of variation among different HIV-1 strains than among those with α1,2-mannose oligomer specificity. The more consistent suppression of a broad variety of virus clade members has also been observed for the α1,2-specific agent cyanovirin-N and pradimicin A and S derivatives (2, 4, 7) but not for the α1,3-mannose-specific agent GNA (3).
In the drug resistance selection experiments, a total of 12 N-glycan deletions were observed when the sequences of all mutant virus isolates were taken into account (Fig. 7). Seven of the above-mentioned glycosylation sites were high-mannose-type N-glycans, and only five mutations reflected a complex-type N-glycan deletion. Such predominant selectivity for the deletion of high-mannose-type glycans under AH pressure has also been observed for the cyanobacterial lectin CV-N (2, 43, 48) and the α1,2-mannose-specific antibiotics pradimicin A and S (4, 7). This points to the pronounced preference of AH to bind to high-mannose-type glycans. Such a preference of AH for α1,2-mannose oligomers on high-mannose-type glycans of gp120 was very recently demonstrated by Tanaka et al. (41) by ELISA and by glycosidase-treated envelope gp120 and is also further confirmed by our SPR results showing that (α1,2-man)3, but not the manα1,3manα1,6man trimer or (ß1,4-GlcNAc)3 was able to efficiently inhibit the interaction between AH and gp120. The α1,2-mannose oligomer preference of AH also became evident from the much lower (∼20-fold) KD value of AH binding to insect cell-derived gp120 (containing a high density of high-mannose-like glycan structures) than to CHO cell-derived gp120 (Table 6) when binding experiments were performed with AH that was immobilized onto the sensor chip. The high-mannose-type glycans of gp120 involved in the selection of resistance to AH were also involved in the selection of resistance to other α1,2-man-specific CBAs. Indeed, six corresponding high-mannose-type glycan deletions were also observed in pradimicin-exposed HIV-1-infected cell cultures (4, 7); deletions at glycan positions N230, N339, N386, and N392 were also selected under CV-N pressure (2, 48); and deletions at glycan positions N295, N339, and N392 emerged with MAb 2G12 pressure (25). However, it is interesting to notice that a single N-glycan deletion in HIV-1 gp120 (i.e., N295) is sufficient to render a virus with such a mutation fully insensitive to the neutralizing activity of MAb 2G12 (25), whereas three concomitant N-glycan deletions in HIV-1 gp120 (i.e., at positions N230, N289, and N339) resulted in only a 2- to 3-fold decreased antiviral activity of AH. Up to five N-glycan deletions were required to render the mutant virus at least 20-fold phenotypically resistant to AH. Thus, AH has a much higher genetic barrier than 2G12, which is in agreement with its broad neutralizing potential against a variety of HIV strains. Our SPR findings of the binding affinity of AH showing similar affinities of binding to gp120s of different origins (including X4- and R5-derived envelopes) are in agreement with these conclusions. In this respect, however, it was surprising to notice that AH proved inactive or poorly active against SIV(mac251) and SHIV(sm239) when it was evaluated in three different cellular assays [SIV(mac251) infection of U87 CD4 CCR5 CXCR4 and PBMCs and SHIV(sm239) infection of CEMX174 cell cultures], although it efficiently binds to SIV(mac251) gp130, as revealed by the SPR analyses. However, the recombinant SIV gp130 used in our SPR binding studies was produced in a baculovirus system. Proteins secreted from insect cells generally have a higher degree of mannose-rich (rather than complex) oligosaccharides than envelope proteins expressed in mammalian cell lines. Therefore, the possibility that AH may show a higher affinity for the baculovirus-expressed SIV envelope proteins than for SIV gp130 expressed by mammalian (CHO) cells cannot be excluded. However, given that AH binding to baculovirus- or mammalian cell-expressed HIV-1 gp120 is not substantially different, it might be unlikely that the lack of significant anti-SIV activity of AH in our cell cultures and the good affinity of AH for SIV gp130 (baculovirus expressed) is just due to such potential differences in insect and mammalian cell glycosylation. On the other hand, although SIV gp130 is as heavily glycosylated as HIV gp120 (18) and the number of high-mannose-type glycans on HIV-1 gp120 has been well determined (∼40% of total glycans) (29), the degree of mannosylated glycans (with end-standing α-1,2-mannose oligomers) on SIV gp130 is far more unclear from the literature (12, 13). It is therefore well possible that SIV gp130 has a much lower level of high-mannose-type glycans than HIV gp120. The points of interaction of AH with SIV gp130 glycans may also be different from the points of AH interaction with HIV gp120 glycans. Therefore, in an attempt to explain the markedly lower levels of activity of AH against SIV(mac251) and SHIV(sm239) as well as against HIV-2(ROD), an alignment of the sequences was made for the gp120s of laboratory strains HIV-1(IIIB), HIV-1(NL4.3), and HIV-1(BaL); clinical HIV-1 isolates UG275, BZ167, DJ259, and UG270; HIV-2(ROD); SIV(mac251); and SIV (sm239) (Fig. 8). The glycosylation motifs are highlighted in yellow. The gp120 glycosylation sites that were found to be deleted in the mutant HIV-1(IIIB) isolates under AH pressure are marked in red in the HIV-1(IIIB) gp120 sequence. As can be seen, six to seven of these glycosylation sites that were found to be deleted under AH pressure in the HIV-1 strains have no corresponding glycosylation site in HIV-2(ROD) gp120 and SIV(mac251) or SIV(sm239) gp130 (i.e., at positions N136, N276, N295, N339, N386, N392, and N401). It may therefore well be possible that the weak anti-HIV-2/SIV activity of AH observed in cell culture can also be explained by the lack of the envelope glycans at these locations in the envelope. In fact, MAb 2G12 is known to specifically interact with HIV-1 gp120 at positions N295, N332, and N392; and its binding to gp120 is further influenced by the presence of the glycans at positions N339 and N386 (34, 35, 42). Interestingly, 2G12 is not inhibitory to HIV-2(ROD) or SIV(mac251) (25); and the 2G12-specific glycosylation sites at positions N295, N332, N392, and N339 have no corresponding counterpart in the HIV-2/SIV envelopes. Thus, the different distribution of the N-glycans on the HIV-2/SIV envelopes compared with that on HIV-1 gp120 may be responsible for the observed differential antiviral activities of 2G12 and AH. To further explain why AH strongly binds to the HIV-2 and SIV envelopes but displays poor, if any, activity against HIV-2 and SIV, it should be kept in mind that the binding of an agent (i.e., monoclonal antibodies) to the viral envelope does not necessarily result in virus neutralization. Indeed, a variety of monoclonal antibodies that bind to HIV gp120 but that lack significant antiviral activity exist. For example, Luallen et al. (31) recently reported on the strong binding activities of immunoglobulins against manα1,2manα1,2man trisaccharides and HIV-1 gp120 but a lack of neutralizing activity against HIV-1 in cell culture. Also, Forsman et al. (19) could discriminate between neutralizing and nonneutralizing epitopes on the HIV-1 envelope by the use of llama antibodies that were generated in these animals upon immunization with HIV-1 gp120. Therefore, we currently hypothesize that AH may predominantly bind to nonneutralizing glycan-containing epitopes on HIV-2(ROD) gp120 and SIV gp130 and cannot interact with one or more specific glycan epitopes that are present on the HIV-1 envelope but not on the HIV-2 and SIV envelopes, resulting in the eventual lack of substantial activity against the HIV-2 and SIV strains.
FIG. 8.

Alignment of the envelope protein sequences of a variety of laboratory HIV-1 strains, clinical HIV-1 isolates, HIV-2(ROD), SIV(mac251), and SIV(sm239). The sequences are derived from the NCBI database, and the alignment was performed using the MUSCLE program (http://www.ebi.ac.uk). The N-glycosylation motifs are indicated in yellow. The glycosylation sites that were found to be mutated in AH-exposed HIV-1(IIIB) isolates are indicated in red.
When competition experiments between AH and other CBAs for binding to gp120 were performed using SPR and flow cytometric analyses, AH binding of gp120 prevents the additional binding of 2G12 MAb on gp120 but not that of other CBAs, such as HHA, which has specificity for α1,3- and α1,6-linked mannosyl units, and UDA, which has specificity for GlcNAc oligomers. These data are in agreement with the findings of Tanaka et al. (41), who demonstrated by ELISA that 2G12 binding to gp120 was dose-dependently inhibited by AH, whereas AH binding to gp120 was not inhibited by MAb 2G12. In addition to the pronounced binding of AH to SIV gp130, MAb 2G12 also binds to SIV gp130, although at a lower affinity than HIV-1 gp120 (SPR analysis [data not shown]), and neither AH nor 2G12 efficiently neutralizes HIV-2 or SIV in cell cultures. These findings indicate that AH and MAb 2G12 must interact with a partially overlapping epitope on HIV-1 gp120 and that AH covers a broader epitope on gp120 than MAb 2G12, explaining its broader neutralizing potential against virus clades and higher genetic barrier, as outlined above. The AH resistance selection data are in agreement with this assumption.
Interestingly, although the efficient binding of AH to gp41 has also been observed (in the SPR studies, AH has affinity for gp41 at nanomolar levels), only one glycan deletion could be detected in the HIV-1 envelope gp41 of two virus isolates during the drug resistance selection experiments. All other N-glycosylation site mutations (n = 12) were detected in gp120. This can be interpreted to indicate that the selection pressure by AH acts predominantly on gp120 and not on the more hidden gp41 glycans. This phenomenon has so far been observed for all CBAs investigated. Thus, the binding of AH to gp41 does seem to be directly related to its antiviral activity.
Under AH pressure, a significant part of the HIV-1 gp120 N-glycans becomes deleted. This may affect the integrity of the N-glycan shield of the HIV envelope, resulting in the creation of holes in the protective glycan layer of the virus. This might, in turn, trigger the immune system of the host against potential (conservative) immunogenic epitopes that then become uncovered upon AH pressure. Hu et al. (24) have demonstrated that CV-N-resistant virus strains in which a few glycosylation sites were deleted in their gp120 envelope became highly susceptible to the neutralizing activities of antibodies directed against the V3 loop of the HIV-1 gp120, providing a proof of concept that targeting HIV-1 glycans may represent a novel therapeutic strategy. Moreover, Reitter and Desrosiers (33) had previously shown that sera from monkeys infected with mutant SIV strains that lacked dual combinations of two N-linked glycosylation sites in their envelope contained markedly increased levels of antibody binding to specific peptides from this envelope region and showed significantly increased neutralizing activity against SIV. These results demonstrate a role for N-linked glycosylation in limiting the neutralizing antibody response to SIV in monkeys and in shielding the virus from immune recognition (33). CBAs such as AH may therefore belong to an interesting class of drugs that allow the immune system to get involved in concerted but complementary action with chemotherapeutics to suppress HIV infection (5).
In conclusion, AH prevents cell-free virus infection, cell-cell virus transfer, virus capture, and the subsequent transmission of HIV by DC-SIGN-expressing cells and may therefore qualify as a potential microbicide candidate drug. Its pronounced and consistent neutralizing activity against X4 and R5 HIV-1 strains is invariably in the nanomolar range. Although CV-N is considered the prototype of a microbicidal lectin, CV-N dramatically stimulates the production of a wide variety of cytokines and chemokines upon exposure in PBMC cultures, whereas almost all levels of cytokines/chemokines in AH-exposed PBMC cultures were beneath the detection level. AH recognizes a broader N-glycans (high-mannose type)-containing epitope on gp120 than MAb 2G12. It is inhibitory to a wider variety of HIV-1 clades and has a higher genetic barrier than MAb 2G12.
Acknowledgments
This work was supported by the Concerted Actions of the K. U. Leuven (grant GOA 05/19), the Center of Excellence (grant EF/05/15), the Foundation of Scientific Research (FWO no. G-485-08), the CHAARM project of the European Commission, and the Foundation Dormeur.
We are grateful to Leen Ingels, Becky Provinciael, Yoeri Schrooten, Sandra Claes, and Eric Fonteyn for excellent technical assistance.
Footnotes
Published ahead of print on 24 May 2010.
REFERENCES
- 1.Balzarini, J., S. Hatse, K. Vermeire, K. Princen, S. Aquaro, C. F. Perno, E. De Clercq, H. Egberink, G. Vanden Mooter, W. Peumans, E. Van Damme, and D. Schols. 2004. Mannose-specific plant lectins from the Amaryllidaceae family qualify as efficient microbicides for prevention of human immunodeficiency virus infection. Antimicrob. Agents Chemother. 48:3858-3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balzarini, J., K. Van Laethem, W. J. Peumans, E. J. Van Damme, A. Bolmstedt, F. Gago, and D. Schols. 2006. Mutational pathways, resistance profile, and side effects of cyanovirin relative to human immunodeficiency virus type 1 strains with N-glycan deletions in their gp120 envelopes. J. Virol. 80:8411-8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balzarini, J., K. Van Laethem, S. Hatse, K. Vermeire, E. De Clercq, W. Peumans, E. Van Damme, A.-M. Vandamme, A. Bolmstedt, and D. Schols. 2004. Profile of resistance of human immunodeficiency virus to mannose-specific plant lectins. J. Virol. 78:10617-10627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balzarini, J., K. Van Laethem, D. Daelemans, S. Hatse, A. Bugatti, M. Rusnati, Y. Igarashi, T. Oki, and D. Schols. 2007. Pradimicin A, a carbohydrate-binding nonpeptidic lead compound for treatment of infections with viruses with highly glycosylated envelopes, such as human immunodeficiency virus. J. Virol. 81:362-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balzarini, J. 2007. Targeting the glycans of glycoproteins: a novel paradigm for antiviral therapy. Nat. Rev. Microbiol. 5:583-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balzarini, J., Y. Van Herrewege, K. Vermeire, G. Vanham, and D. Schols. 2007. Carbohydrate-binding agents efficiently prevent dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN)-directed HIV-1 transmission to T-lymphocytes. Mol. Pharmacol. 71:3-11. [DOI] [PubMed] [Google Scholar]
- 7.Balzarini, J., K. François, K. Van Laethem, B. Hoorelbeke, M. Renders, J. Auwerx, S. Liekens, T. Oki, Y. Igarashi, and D. Schols. 2010. Pradimicin S, a highly-soluble non-peptidic small-size carbohydrate-binding antibiotic, is an anti-HIV drug lead for both microbicidal and systemic use. Antimicrob. Agents Chemother. 54:1425-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bewley, C. A., K. R. Gustafson, M. R. Boyd, D. G. Covell, A. Bax, G. M. Clore, and A. M. Gronenborn. 1998. Solution structure of cyanovirin-N, a potent HIV-inactivating protein. Nat. Struct. Biol. 5:571-578. [DOI] [PubMed] [Google Scholar]
- 9.Bewley, C. A. 2001. Solution structure of a cyanovirin-N:Man alpha 1-2Man alpha complex: structural basis for high-affinity carbohydrate-mediated binding to gp120. Structure 9:931-940. [DOI] [PubMed] [Google Scholar]
- 10.Botos, I., B. R. O'Keefe, S. R. Shenoy, L. K. Cartner, D. M. Ratner, P. H. Seeberger, M. R. Boyd, and A. Wlodawer. 2002. Structures of the complexes of a potent anti-HIV protein cyanovirin-N and high mannose oligosaccharides. J. Biol. Chem. 277:34336-34342. [DOI] [PubMed] [Google Scholar]
- 11.Boyd, M. R., K. R. Gustafson, J. B. McMahon, R. H. Shoemaker, B. R. O'Keefe, T. Mori, R. J. Gulakowski, L. Wu, M. I. Rivera, C. M. Laurencot, M. J. Currens, J. H. Cardellina II, R. W. Buckheit, Jr., P. L. Nara, L. K. Pannell, R. C. Sowder II, and L. E. Henderson. 1997. Discovery of cyanovirin-N, a novel human immunodeficiency virus-inactivating protein that binds viral surface envelope glycoprotein gp120: potential applications to microbicide development. Antimicrob. Agents Chemother. 41:521-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, B., E. M. Vogan, H. Gong, J. J. Skehel, D. C. Wiley, and S. C. Harrison. 2005. Determining the structure of an unliganded and fully glycosylated SIV gp120 envelope glycoprotein. Structure 13:197-211. [DOI] [PubMed] [Google Scholar]
- 13.Chen, X., M. Lu, B. K. Poon, Q. Wang, and J. Ma. 2009. Structural improvement of unliganded simian immunodeficiency virus gp120 core by normal-mode-based X-ray crystallographic refinement. Acta Crystallogr. D Biol. Crystallogr. 65:339-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiba, H., J. Inokoshi, M. Okamoto, S. Asanuma, K. Matsuzaki, M. Iwama, K. Mizumoto, H. Tanaka, M. Oheda, K. Fujita, H. Nakashima, M. Shinose, Y. Takahashi, and S. Omura. 2001. Actinohivin, a novel anti-HIV protein from an actinomycete that inhibits syncytium formation: isolation, characterization, and biological activities. Biochem. Biophys. Res. Commun. 282:595-601. [DOI] [PubMed] [Google Scholar]
- 15.Chiba, H., J. Inokoshi, H. Nakashima, S. Omura, and H. Tanaka. 2004. Actinohivin, a novel anti-human immunodeficiency virus protein from an actinomycete, inhibits viral entry to cells by binding high-mannose type sugar chains of gp120. Biochem. Biophys. Res. Commun. 316:203-213. [DOI] [PubMed] [Google Scholar]
- 16.Chiba, H., S. Asanuma, M. Okamoto, J. Inokoshi, H. Tanaka, K. Fujita, and S. Omura. 2001. A simple screening system for anti-HIV drugs: syncytium formation assay using T-cell line tropic and macrophage tropic HIV env expressing cell lines—establishment and validation. J. Antibiot. 54:818-826. [DOI] [PubMed] [Google Scholar]
- 17.Doms, R. W. 2000. Beyond receptor expression: the influence of receptor conformation, density, and affinity in HIV-1 infection. Virology 276:229-237. [DOI] [PubMed] [Google Scholar]
- 18.Douglas, N. W., G. H. Munro, and R. S. Daniels. 1997. HIV/SIV glycoproteins: structure-function relationships. J. Mol. Biol. 273:122-149. [DOI] [PubMed] [Google Scholar]
- 19.Forsman, A., E. Beirnaert, M. M. Aasa-Chapman, B. Hoorelbeke, K. Hijazi, W. Koh, V. Tack, A. Szynol, C. Kelly, A. McKnight, T. Verrips, H. de Haard, and R. A. Weiss. 2008. Llama antibody fragments with cross-subtype human immunodeficiency virus type 1 (HIV-1)-neutralizing properties and high affinity for HIV-1 gp120. J. Virol. 82:12069-12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geijtenbeek, T. B., R. Torensma, S. J. van Vliet, G. C. van Duijnhoven, G. J. Adema, Y. van Kooyk, and C. G. Figdor. 2000. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell 100:575-585. [DOI] [PubMed] [Google Scholar]
- 21.Geijtenbeek, T. B., D. S. Kwon, R. Torensma, S. J. van Vliet, G. C. van Duijnhoven, J. Middel, I. L. Cornelissen, H. S. Nottet, V. N. Kewal Ramani, D. R. Littman, C. G. Figdor, and Y. van Kooyk. 2000. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100:587-597. [DOI] [PubMed] [Google Scholar]
- 22.Geyer, H., C. Holschbach, G. Hunsmann, and J. Schneider. 1988. Carbohydrates of human immunodeficiency virus. Structures of oligosaccharides linked to the envelope glycoprotein 120. J. Biol. Chem. 263:11760-11767. [PubMed] [Google Scholar]
- 23.Grant, R. M., D. Hamer, T. Hope, R. Johnston, J. Lange, M. M. Lederman, J. Lieberman, C. J. Miller, J. P. Moore, D. E. Mosier, D. D. Richman, R.T. Schooley, M. S. Springer, R. S. Veazey, and M. A. Wainberg. 2008. Whither or wither microbicides? Science 321:532-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu, Q., N. Mahmood, and R. J. Shattock. 2007. High-mannose-specific deglycosylation of HIV-1 gp120 induced by resistance to cyanovirin-N and the impact on antibody neutralization. Virology 368:145-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huskens, D., K. Van Laethem, K. Vermeire, J. Balzarini, and D. Schols. 2007. Resistance of HIV-1 to the broadly HIV-1-neutralizing, anti-carbohydrate antibody 2G12. Virology 360:294-304. [DOI] [PubMed] [Google Scholar]
- 26.Huskens, D., K. Vermeire, E. Vandemeulebroucke, J. Balzarini, and D. Schols. 2008. Safety concerns for the potential use of cyanovirin-N as a microbicidal anti-HIV agent. Int. J. Biochem. Cell Biol. 40:2802-2814. [DOI] [PubMed] [Google Scholar]
- 27.Inokoshi, J., H. Chiba, S. Asanuma, A. Takahashi, S. Omura, and H. Tanaka. 2001. Molecular cloning of actinohivin, a novel anti-HIV protein from an actinomycete, and its expression in Escherichia coli. Biochem. Biophys. Res. Commun. 281:1261-1265. [DOI] [PubMed] [Google Scholar]
- 28.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leonard, C. K., M. W. Spellman, L. Riddle, R. J. Harris, J. N. Thomas, and T. J. Gregory. 1990. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J. Biol. Chem. 265:10373-10382. [PubMed] [Google Scholar]
- 30.Reference deleted.
- 31.Luallen, R. J., C. Agrawal-Gamse, H. Fu, D. F. Smith, R. W. Doms, and Y. Geng. 2010. Antibodies against Manalpha1,2-Manalpha1,2-Man oligosaccharide structures recognize envelope glycoproteins from HIV-1 and SIV strains. Glycobiology 20:280-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Princen, K., S. Hatse, K. Vermeire, E. De Clercq, and D. Schols. 2004. Establishment of a novel CCR5 and CXCR4 expressing CD4+ cell line which is highly sensitive to HIV and suitable for high-throughput evaluation of CCR5 and CXCR4 antagonists. Retrovirology 1:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reitter, J. N., and R. C. Desrosiers. 1998. Identification of replication-competent strains of simian immunodeficiency virus lacking multiple attachment sites for N-linked carbohydrates in variable regions 1 and 2 of the surface envelope protein. J. Virol. 72:5399-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanders, R. W., M. Venturi, L. Schiffner, R. Kalyanaraman, H. Katinger, K. O. Lloyd, P. D. Kwong, and J. P. Moore. 2002. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J. Virol. 76:7293-7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scanlan, C. N., R. Pantophlet, M. R. Wormald, S. E. Ollmann, R. Stanfield, I. A. Wilson, H. Katinger, R. A. Dwek, P. M. Rudd, and D. R. Burton. 2002. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of α(1-2) mannose residues on the outer face of gp120. J. Virol. 76:7306-7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scanlan, C. N., J. Offer, N. Zitzmann, and R. A. Dwek. 2007. Exploiting the defensive sugars of HIV-1 for drug and vaccine design. Nature 446:1038-1045. [DOI] [PubMed] [Google Scholar]
- 37.Shattock, R. J., and R. W. Doms. 2002. AIDS models: microbicides could learn from vaccines. Nat. Med. 8:425. [DOI] [PubMed] [Google Scholar]
- 38.Shattock, R. J., and J. P. Moore. 2003. Inhibiting sexual transmission of HIV-1 infection. Nat. Rev. Microbiol. 1:25-34. [DOI] [PubMed] [Google Scholar]
- 39.Shibuya, N., I. J. Goldstein, J. A. Shafer, W. J. Peumans, and W. F. Broekaert. 1986. Carbohydrate binding properties of the stinging nettle (Urtica dioica) rhizome lectin. Arch. Biochem. Biophys. 249:215-224. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi, A., J. Inokoshi, H. Chiba, S. Omura, and H. Tanaka. 2005. Essential regions for antiviral activities of actinohivin, a sugar-binding anti-human immunodeficiency virus protein from an actinomycete. Arch. Biochem. Biophys. 437:233-240. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka, H., H. Chiba, J. Inokoshi, A. Kuno, T. Sugai, A. Takahashi, Y. Ito, M. Tsunoda, K. Suzuki, A. Takénaka, T. Sekiguchi, H. Umeyama, J. Hirabayashi, and S. Omura. 2009. Mechanism by which the lectin actinohivin blocks HIV infection of target cells. Proc. Natl. Acad. Sci. U. S. A. 106:15633-15638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trkola, A., M. Purtscher, T. Muster, C. Ballaun, A. Buchacher, N. Sullivan, K. Srinivasan, J. Sodroski, J. P. Moore, and H. Katinger. 1996. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 70:1100-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsai, C. C., P. Emau, Y. Jiang, B. Tian, W. R. Morton, K. R. Gustafson, and M. R. Boyd. 2003. Cyanovirin-N gel as a topical microbicide prevents rectal transmission of SHIV89.6P in macaques. AIDS Res. Hum. Retrovir. 19:535-541. [DOI] [PubMed] [Google Scholar]
- 44.Tsai, C. C., P. Emau, Y. Jiang, M. B. Agy, R. J. Shattock, A. Schmidt, W. R. Morton, K. R. Gustafson, and M. R. Boyd. 2004. Cyanovirin-N inhibits AIDS virus infections in vaginal transmission models. AIDS Res. Hum. Retrovir. 20:11-18. [DOI] [PubMed] [Google Scholar]
- 45.Überla, K., C. Stahl-Hennig, D. Böttiger, K. Mätz-Rensing, F. J. Kaup, J. Li, W. A. Haseltine, B. Fleckenstein, G. Hunsmann, B. Öberg, and J. Sodroski. 1995. Animal model for the therapy of acquired immunodeficiency syndrome with reverse transcriptase inhibitors. Proc. Natl. Acad. Sci. U. S. A. 92:8210-8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Damme, E. J. M., A. K. Allen, and W. J. Peumans. 1988. Related mannose-specific lectins from different species of the family Amaryllidaceae. Physiol. Plant 73:52-57. [Google Scholar]
- 47.Van Laethem, K., Y. Schrooten, P. Lemey, E. Van Wijngaerden, S. De Wit, M. Van Ranst, and A. M. A. Vandamme. 2005. Genotypic resistance assay for the detection of drug resistance in the human immunodeficiency virus type 1 envelope gene. J. Virol. Methods 123:25-34. [DOI] [PubMed] [Google Scholar]
- 48.Witvrouw, M., V. Fikkert, A. Hantson, C. Pannecouque, B. R. O'Keefe, J. McMahon, L. Stamatatos, E. De Clercq, and A. Bolmstedt. 2005. Resistance of human immunodeficiency virus type 1 to the high-mannose binding agents cyanovirin N and concanavalin A. J. Virol. 79:7777-7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang, F., C. A. Bewley, J. M. Louis, K. R. Gustafson, M. R. Boyd, A. M. Gronenborn, G. M. Clore, and A. Wlodawer. 1999. Crystal structure of cyanovirin-N, a potent HIV-inactivating protein, shows unexpected domain swapping. J. Mol. Biol. 288:403-412. [DOI] [PubMed] [Google Scholar]