

Abstract
Rifapentine and its primary metabolite, 25-desacetyl rifapentine, are active against mycobacterium tuberculosis. The objectives of this study were to describe the population pharmacokinetics of rifapentine and 25-desacetyl rifapentine in fasting and fed states. Thirty-five male healthy volunteers were enrolled in an open-label, randomized, sequential, five-way crossover study. Participants received a single 900-mg dose of rifapentine after meals with high fat (meal A), bulk and low fat (meal B), bulk and high fat (meal C), high fluid and low fat (meal D), or 200 ml of water (meal E). Venous blood samples were collected over 72 h after each rifapentine dose, and plasma was analyzed for rifapentine and 25-desacetyl rifapentine using high-performance liquid chromatography. Pharmacokinetic data were analyzed by nonlinear mixed-effect modeling using NONMEM. Compared with the fasting state, meal A had the greatest effect on rifapentine oral bioavailability, increasing it by 86%. Meals B, C, and D resulted in 33%, 46%, and 49% increases in rifapentine oral bioavailability, respectively. Similar trends were observed for 25-desacetyl rifapentine. As meal behavior has a substantial impact on rifapentine exposure, it should be considered in the evaluation of optimal dosing approaches.
Rifapentine (RFP), a cyclopentyl rifamycin, is an orally administered drug registered by the Food and Drug Administration (FDA) for the treatment of pulmonary tuberculosis (TB). It exerts its antibacterial activity through inhibition of DNA-dependent RNA polymerase in susceptible strains of Mycobacterium tuberculosis (32). RFP has a microbiologically active metabolite, 25-desacetyl rifapentine (25-DRFP) (10). RFP has a long half-life (5, 17) and superior in vitro potency against M. tuberculosis in comparison with rifampin and rifabutin (10), making it an attractive candidate for shortening and simplifying antitubercular therapy.
Currently, RFP is dosed at 600 mg either once weekly or twice weekly in human immunodeficiency virus (HIV)-negative patients with noncavitary TB (2). There is concern that the development of acquired rifamycin monoresistance (ARR), treatment failure, and relapse may be associated with intermittent dosing (4, 20), insufficient companion drug exposure (33), or low rifamycin concentrations (9, 21). RFP's sterilizing effect has been shown to be dose dependent in murine studies which suggest that daily doses of RFP may reduce treatment duration to just 3 months (25, 26, 37), and higher doses of RFP are associated with improved early bactericidal activity in humans (29). Clinical trials are currently evaluating new antituberculosis regimens containing higher doses of RFP used intermittently or daily doses of RFP. Concomitant food has a marked effect on RFP absorption (6). The effect of food on systemic RFP exposure may therefore impact treatment activity and safety. The aim of this study was to investigate the effect of meals differing in fat and bulk content on the rate and extent of RFP absorption and 25-DRFP disposition. The study was designed to include meals comprising largely maize, a staple cereal in many parts of Africa and South America, and a light meal (i.e., a reconstituted powdered chicken soup). The study was conducted in 1999 shortly after the release of results from studies evaluating intermittent 600-mg doses of rifapentine, which displayed unacceptably high relapse rates in patients with lung cavities or immune suppression. It was therefore anticipated that future studies would evaluate higher doses of the drug.
MATERIALS AND METHODS
Participant enrollment.
Adult male healthy volunteers (n = 35) were enrolled in an open-label, randomized, sequential, five-way, crossover design study at Groote Schuur Hospital, Cape Town, South Africa. Volunteers were eligible if they weighed at least 50 kg, had normal physical examination and baseline laboratory evaluation, and were nonsmokers. Exclusion criteria included a history of TB, active allergies, excessive coffee or alcohol consumption, recent blood donation of more than 500 ml, and clinically relevant cardiovascular, hepatic, neurologic, endocrine, or other major systemic disease. In order to limit the risks of unintended fetal exposure to the unregistered drug, women were excluded. Likewise HIV- or hepatitis B virus (HBV)-infected volunteers were excluded due to the risks of undiagnosed tuberculosis and adverse effects, respectively. Each participant provided written informed consent before being enrolled into the study. The study protocol (M000473/1LO1) was reviewed and approved by the Research Ethics Committee of the University of Cape Town and the Medicines Control Council of South Africa.
Dosage schedules and sample handling.
After an overnight fast, participants received a single 900-mg dose of RFP (200 ml of water with six 150-mg Priftin tablets; Hoechst Marion Roussel, Italy) 30 min after the meal (Table 1). Individuals were randomized to receive the RFP dose in the fasted state (meal E) on one occasion. Other scheduled meals included a standardized lunch and dinner at 6 or 12 h postdose. At each visit a 20-ml blood sample was collected prior to drug administration, and 10-ml samples were collected 2, 3, 4, 5, 6, 7, 8, 10, 12, 14, 24, 36, 48, and 72 h after the dose to obtain plasma for quantification of RFP and 25-DRFP. Each dose was separated by a 14-day washout period. Seven participants were randomized to each of five different meal sequences. Due to the large number of potential meal sequences, the design was not fully balanced. The blood samples were collected by venipuncture into lithium-heparin-coated glass tubes and centrifuged at 2,500 rpm within 1 h of collection. The supernatants were transferred into two dry polypropylene tubes and stored at −80°C away from light until analysis.
TABLE 1.
Description of different meals ingested 30 min before administration of a single 900 mg single dose of RFP
| Meal | Description | Content (g) of: |
kJ | Wt (g) | ||
|---|---|---|---|---|---|---|
| Protein | Fat | Carbohydrates | ||||
| A (high-fat breakfast) | 2 rashers of bacon (20 g), 1 fried egg (50 g), 1 slice white toast (30 g) with butter (7 g) and marmalade (10 g), 2 cups decaffeinated coffee (400 ml) with full-cream milk (100 ml) and 2 teaspoons of sugar (10 g) (English breakfast) | 18.9 | 27 | 38 | 1,966 | 627 |
| B (low-fat and bulky breakfast) | 1 1/2 cups soft maize meal porridge (375 g cooked) with 3 teaspoons of sugar (15 g), 1 cup of decaffeinated coffee (200 ml) with full-cream milk (50 ml) and 1 teaspoon of sugar (5 g) (maize meal porridge) | 6 | 3 | 66 | 1,285 | 645 |
| C (high-fat and bulky breakfast) | 1 1/2 cups soft maize meal porridge (375 g cooked) with 3 teaspoons of sugar (15 g) and 5 teaspoons of lard (25 g), 1 cup of decaffeinated coffee (200 ml) with full-cream milk (50 ml) and 1 teaspoon of sugar (5 g) (maize meal porridge with lard) | 6 | 28 | 66 | 2,229 | 670 |
| D (low-fat and high-fluid breakfast) | 2 cups of reconstituted (powder) chicken noodle soup (400 ml), 1 cup of decaffeinated coffee (200 ml) with skim milk (50 ml) and 1 teaspoon of sugar (5 g) | 9 | 4 | 28 | 774 | 660 |
Drug determination.
Plasma concentrations of RFP and 25-DRFP were determined using a validated high-performance tandem liquid chromatography (HPLC) method developed at the Division of Clinical Pharmacology, Cape Town, South Africa (19). The assay was validated over the concentration range of 0.5 to 30 μg/ml. Linearity values of the calibration curve (r2) were 0.9975 and 0.9946 for RPT and 25-DRFP, respectively.
Population PK analysis.
RFP and 25-DRFP plasma concentration versus time data were modeled using nonlinear mixed-effects modeling in NONMEM (version VI, double precision, level 2.0) (1). The analysis was done in two steps. First the RFP model was developed. Thereafter, the fixed and random effects estimates of oral clearance (CL/F), volume of distribution (V/F), first-order absorption rate constant (ka), mean transit time (MTT), oral bioavailability (F), and number of hypothetical transit compartments (NN) were fixed, and the 25-DRFP model was developed using all data. Xpose version 4 (13) and Census (35) were used during model building for graphical analysis and data tracking, respectively. For the RFP data, various pharmacokinetic (PK) models, including one or two compartments with first-order absorption and first- or zero-order elimination (12), incorporating either lag times (to describe the delay in the appearance of drug in plasma) or transit absorption compartments (27, 28), time-varying clearance (18), and enterohepatic recirculation (24), were fitted to the data during model development. Time-varying clearance was introduced into the model as CLi = {TV(CL1/F)·[1 − mpast(1)] + TV(CL2/F)·mpast(1)}· exp(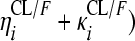 , where CLi is the oral clearance for the ith individual, TV(CL1/F) is the typical oral clearance which later changes to TV(CL2/F) at model event time (MTIME), mpast(1) remains zero until MTIME when it changes to 1,
, where CLi is the oral clearance for the ith individual, TV(CL1/F) is the typical oral clearance which later changes to TV(CL2/F) at model event time (MTIME), mpast(1) remains zero until MTIME when it changes to 1, 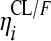 represents the interindividual variability (IIV), and
represents the interindividual variability (IIV), and 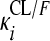 represents the interoccasional variability (IOV). The above-mentioned models were also evaluated for the 25-DRFP data. In addition, models accounting for presystemic formation of the metabolite (16), loss of the parent drug to other metabolites (30), and saturable clearance of the metabolite (23) and a two-compartment model with time-varying CL/F were tested when developing the 25-DRFP model.
represents the interoccasional variability (IOV). The above-mentioned models were also evaluated for the 25-DRFP data. In addition, models accounting for presystemic formation of the metabolite (16), loss of the parent drug to other metabolites (30), and saturable clearance of the metabolite (23) and a two-compartment model with time-varying CL/F were tested when developing the 25-DRFP model.
Covariates (age and body weight) were evaluated with respect to their impact on IIV in F and CL/F parameters. The covariate analysis was done by a forward inclusion procedure (α = 0.01, change in objective function value [ΔOFV] ≥ 6.63) followed by a backwards deletion step (α = 0.001, ΔOFV ≥ 10.83). Clinical relevance was assumed when the typical value of CL/F or F changed by at least 10% in order to prevent the detection of an irrelevant statistically significant relationship. The effect of different meals was investigated simultaneously on the typical value of F, MTT, and ka.
The following equation was used to quantify meal effects: TVF = θF·(1 + RXF). TVF is the typical bioavailability, RXF is the fractional change (all five visits) in F due to a given meal relative to the value for the fasted state, and θF is the value of F under fasting conditions (fixed to 1). A similar approach was used to evaluate meal effects on the fixed effects for MTT and ka. The delay in absorption was modeled using a transit absorption model where drug absorption is described as drug movement through a series of hypothetical presystemic compartments, as suggested by Savic et al. (28). Carryover effects on clearance and MTIME values at each treatment period were evaluated relative to the first treatment period using the equations TV(CL/F) = {TV(CL1/F)·[1 − mpast(1)] + TV(CL2/F)·mpast(1)}·(1 + θclocc) and TVMT = θMTIME·(1 − θmtocc), where θclocc is the fractional change in the typical value of oral clearance at a given treatment occasion relative to the first occasion, θMTIME is the typical value of MTIME (TVMT), and θmtocc is the fractional change in MTIME at a given occasion relative to the first occasion.
Estimation of typical population PK parameters and random IIV and IOV associated with observed and predicted plasma concentrations was done using a first-order conditional estimation method with ɛ-η interaction (FOCE INTER) in NONMEM. IIV and IOV were described using an exponential model (15). Model discrimination was based on graphical assessment of conditional weighted residuals (CWRES) versus time, basic goodness of fit (GOF) plots, and changes in the NONMEM OFV during model development. OFV is equal to approximately −2 × log likelihood, and ΔOFV is assumed to be chi squared distributed. Statistical significance was set at 5% (ΔOFV > 3.84) for a single degree of freedom (i.e., addition of one model parameter) and at 1% significance level (ΔOFV > 6.63) for deletion of one parameter (31). Assessment of IIV estimates and their corresponding standard errors (SE) was done to check for η shrinkage since η and ɛ shrinkage values above 30% result in a model with a low power to detect model and residual error misspecification, which may hide true relationships (14). Correlations between parameters' variability components were identified, and covariance between random effects was explored. The correlation coefficients were calculated in Census using estimates from the NONMEM covariance step. Final model qualification included simulation-based diagnostics, i.e., visual predictive checks (VPC) (11). The residual variability and its corresponding standard error were estimated. Additive error, power function, constant coefficient of variation, and additive plus proportional error models were evaluated (1).
RESULTS
RFP and 25-DRFP plasma concentrations in 2,272 samples from 34 participants were available. Their means (±standard deviations) for age, weight, and height were 23.9 ± 4.82 years, 74.4 ± 12.3 kg, and 177.2 ± 7.33 cm, respectively. Data for 1 participant, who withdrew after a single visit, were not available. Less than 1% of the concentration-time data were below the limit of quantification and therefore excluded in the PK analysis. A one-compartment model with first-order absorption and time-varying clearance best described the RFP data (Fig. 1). The delay in absorption was described using a transit absorption model. The residual error model, selected by goodness-of-fit (GOF) plots, individual weighted residuals (IWRES) versus time, and decrease in OFV had both additive (θADD) and proportional (θPROP) error terms. The model described the data well, as shown in Fig. 2. The population PK parameter estimates for RFP and meal effects on RFP are tabulated in Tables 2 and 3, respectively. All the meals we investigated increased the bioavailability of RFP relative to fasting conditions. No clinically significant demographic covariates were supported by the data. Accounting for the correlation between F and MTT (correlation coefficient = 0.65) and between CL/F and MTT (correlation coefficient = −0.56) in the RFP model significantly improved the model. The η shrinkage values for RFP CL/F, MTT, and F were 6%, 40%, and 13%, respectively. 25-DRFP PK data were best described by a two-compartment model with time-varying clearance (Fig. 1). Final PK parameter estimates for 25-DRFP are summarized in Table 4.
FIG. 1.

Illustration of the parent metabolite model. All rifapentine (RFP) is assumed to be converted to the major metabolite (25-DRFP). N1 represents the first hypothetical transit compartment up to Nn compartment. ktr is the transit rate constant. ka is the absorption rate constant from the hypothetical drug depot compartment to plasma. k (calculated as CL/V) is the elimination rate constant of rifapentine. CLm is the time-varying metabolite clearance. Vm represents volume of distribution of the metabolite. k34 is the first-order rate constant of the metabolite from plasma to the peripheral compartment, and k43 is the first-order rate constant of the metabolite from the peripheral compartment back to plasma.
FIG. 2.

Visual predictive check for the final (left) and metabolite (right) models. The lower, middle, and upper solid lines are the 5th, 50th, and 95th percentiles of the observed data, respectively. The dotted and dashed-dotted (50th percentile) lines around each percentile show the 95% confidence interval from the model prediction. The circles are the observed concentration-time data points.
TABLE 2.
Final parameter estimates for RFP
| Parameter | Estimate (RSEa [%]) | IIVb (RSE [%]) | IOVc (RSE [%]) |
|---|---|---|---|
| CL1/F (liters/h)d | 2.14 (13.6) | 19.2 (24.3) | 12.2 (9.1) |
| CL2/F (liters/h)e | 3.22 (11.9) | 19.2 (24.3) | 12.2 (9.1) |
| MTT (h) | 1.45 (10.8) | 9.5 (87.0) | 24.2 (13.4) |
| F | 1 (fixed) | 21.4 (35.3) | 29 (16.6) |
| MTIME (h)f | 43 (2.6) | ||
| V/F (liters) | 60.6 (9.2) | ||
| NN | 10.9 (9.6) | ||
| ka (h−1) | 1.66 (13.1) | ||
| Residual variability | |||
| Additive error (mg/liter) | 0.206 (12.3) | ||
| Proportional error (%) | 10.6 (1.81) |
RSE, relative standard error expressed as percentage of estimate.
IIV, interindividual variability expressed as coefficient of variation (CV).
IOV, interoccasional variability expressed as CV.
Oral clearance of RFP before MTIME.
Oral clearance of RFP after MTIME.
MTIME is the estimated time when oral clearance of RFP changes.
TABLE 3.
Different meal effects, relative to fasted state, estimated from the parent (RFP) model
Meals are as defined in Table 1. The model was parameterized for meal effects on oral bioavailability (F), first-order absorption rate constant (ka), and mean transit time (MTT). Meal effects on ka and MTT were dropped in the final model since they became insignificant.
RSE, relative standard error.
TABLE 4.
Final parameter estimates for 25-DRFP
| Parameter | Estimate (RSEa [%]) | IIVb (RSE [%]) | IOVc (RSE [%]) |
|---|---|---|---|
| CLM1/F (liters/h)d | 1.81 (8.7) | 23.9 (23.9) | 30.2 (10.6) |
| CLM2/F (liters/h)e | 4.63 (8.1) | 23.9 (23.9) | 30.2 (10.6) |
| MTIME (h)f | 46.8 (0.3) | ||
| VMC/F (liters)g | 6.36 (5.8) | ||
| VMP/F (liters)h | 22.1 (4.8) | ||
| QM (liters/h)i | 4.4 (6.9) | ||
| Residual variability | |||
| Additive error (mg/liter) | 0.211 (9.7) | ||
| Proportional error (%) | 19.1 (0.7) |
RSE, relative standard error expressed as percentage of esimate.
IIV, interindividual variability expressed as coefficient of variation (CV).
IOV, interoccasional variability expressed as CV.
Oral clearance of 25-DRFP before MTIME.
Oral clearance of 25-DRFP after MTIME.
MTIME is the estimated time when oral clearance of 25-DRFP changes.
VMC/F, apparent volume of distribution of 25-DRFP in the central compartment.
VMP/F, apparent volume of distribution of 25-DRFP in the peripheral compartment.
QM, intercompartmental clearance of 25-DRFP.
Clinical adverse events were mild. Three volunteers were withdrawn due to elevated alanine transaminase (ALT) levels. One volunteer withdrew after the first RPE dose and had an ALT level 2 times the upper normal limit (UNL). Two volunteers were withdrawn after the second dose; they had ALT levels 2.2 and 6.9 times the ULN. ALT levels in these 3 participants returned to the normal range after withdrawal, and they remained clinically well and asymptomatic throughout. There was no evidence of a relationship between toxicity and drug concentrations or meal type.
DISCUSSION
The bioavailability of RFP and consequently 25-DRFP exposure were increased when single 900-mg RFP oral doses were administrated immediately after food. The high-fat meal (an English breakfast which included one fried egg) had the greatest effect (Table 3), increasing the oral bioavailability by 85.7%. This finding is consistent with previous studies (6, 18). A study conducted in Hong Kong found that 2 eggs with toast increased the bioavailability of RFP to almost the same extent as a high-fat English breakfast (6). This finding forms the basis for the concomitant meal used in the ongoing Rifaquin study, in which patients have 2 boiled eggs and bread immediately before each weekly 1,200-mg dose or twice-weekly 900-mg dose of RFP. Our finding that a high-fat breakfast without eggs (meal C) increased bioavailability of RFP by only 45.7% supports the notion proposed by Chan et al. (6) that eggs may be effective in promoting RFP absorption, although the mechanism is unclear. Meal effects on tablet dissolution, gastric emptying time (hence duration of absorption), and pH (which may affect solubility or absorption) may, in part, account for the pharmacokinetic differences observed with the different meals. Surprisingly, the low-fat chicken noodle soup (meal D) increased RFP bioavailability to an extent comparable to that of the maize meal porridge with lard (meal C). Monosodium glutamate (MSG) was one of the ingredients in the chicken noodle soup. MSG accelerates gastric emptying time in a high-energy, high-protein liquid diet (36), and we suspect that it could have played a role in meal D's effect. Even though in our final model we could not find statistically significant effects of all meals on ka and MTT, meal D gave the highest increase in ka (100% against the fasting state), showing its influence on RFP pharmacokinetics.
The pharmacokinetics of RFP was best described by a one-compartment model with transit absorption compartments, first-order absorption, and first-order elimination with time-varying clearance. Time-varying clearance is suggestive of autoinduction (8, 38), and this finding is consistent with a recent study by Dooley et al. (7) in which thrice-weekly doses of RFP were administered. In our study, the autoinduction effect was detected with a 2-week washout between single 900-mg doses of RFP. The baseline CL/F tended to increase slightly in a cumulative manner with subsequent dosing occasions, and there was a tendency for the time of change (i.e., MTIME) to decrease slightly after each subsequent dose. Given that RFP and 25-DRFP were still identifiable in plasma up to 72 h postdose and that RFP is a potent enzyme inducer, it is not surprising that the duration of its effect on clearance should be similar to that of rifampin (22).
As RFP concentrations had a double peak, an enterohepatic recirculation model was investigated. However, based on ΔOFV and other diagnostic procedures the data did not support an enterohepatic recirculation model. Other possible explanations include analytical interference due to food ingested 6 h after the dose, absorption windows, or progressive solubilization along the gastrointestinal tract and variable gastric emptying. Our final PK models for both RFP and 25-DRFP differ from a previously published model from our group (18). The previous model was developed to describe patient data where the participants were preinduced by rifampin and were on other antituberculosis drugs. Furthermore, wider variability is expected among the patients described in the previous model than in healthy volunteers. We identified a positive correlation between IIV of oral bioavailability and MTT (r = 0.65), which was expected since increased MTT allows more time for drug absorption, while negative correlations between η values for CL/F and MTT (r = −0.56) were due to the association between IIVs for CL/F and F.
The prominent food effect has important implications for the interpretation of studies evaluating antituberculosis regimens. Assuming linear pharmacokinetics of RFP over different dose ranges (34), our results indicate that drug exposure following a 600-mg dose of RFP after a high fat meal is 24% higher than exposure after a 900-mg dose under fasting conditions. While higher RFP exposure may be beneficial in enhancing the efficacy of regimens, there is concern that the risk of adverse events is increased (3). Thus, rigorous trial of a new dosing regimen would include proof of efficacy and safety when RFP is taken with and without food, as is likely to occur under operational conditions.
In conclusion, as RFP has dose-related activity, concomitant food should be considered when evaluating optimal RFP doses in RFP-based regimens. The effects of RFP should be evaluated under the meal conditions that can feasibly be provided by tuberculosis control programs in high-burden countries; in many settings provision of meals may be an unrealistic expectation.
Acknowledgments
At the time of the study, P. B. Fourie was attached to the Tuberculosis Research Program of South Africa MRC, which financially supported the work through a grant from Hoechst Marion Roussel, France. S. P. Zvada is supported by the Wellcome Trust (grant number WT081199/Z/06/Z).
We thank P. Denti in our department for his assistance with modeling.
Footnotes
Published ahead of print on 1 June 2010.
REFERENCES
- 1.Beal, S. L., L. B. Sheiner, and A. Boeckmann. 1996. NONMEM users' guides. University of California, San Fransisco, San Francisco, CA.
- 2.Blumberg, H. M., W. J. Burman, R. E. Chaisson, C. L. Daley, S. C. Etkind, L. N. Friedman, P. Fujiwara, M. Grzemska, P. C. Hopewell, M. D. Iseman, R. M. Jasmer, V. Koppaka, R. I. Menzies, R. J. O'Brien, R. R. Reves, L. B. Reichman, P. M. Simone, J. R. Starke, and A. A. Vernon. 2003. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am. J. Respir. Crit. Care Med. 167:603-662. [DOI] [PubMed] [Google Scholar]
- 3.Bock, N. N., T. R. Sterling, C. D. Hamilton, C. Pachucki, Y. C. Wang, D. S. Conwell, A. Mosher, M. Samuels, and A. Vernon. 2002. A prospective, randomized, double-blind study of the tolerability of rifapentine 600, 900, and 1,200 mg plus isoniazid in the continuation phase of tuberculosis treatment. Am. J. Respir. Crit. Care Med. 165:1526-1530. [DOI] [PubMed] [Google Scholar]
- 4.Burman, W., D. Benator, A. Vernon, A. Khan, B. Jones, C. Silva, C. Lahart, S. Weis, B. King, B. Mangura, M. Weiner, and W. El-Sadr. 2006. Acquired rifamycin resistance with twice-weekly treatment of HIV-related tuberculosis. Am. J. Respir. Crit. Care Med. 173:350-356. [DOI] [PubMed] [Google Scholar]
- 5.Burman, W. J., K. Gallicano, and C. Peloquin. 2001. Comparative pharmacokinetics and pharmacodynamics of the rifamycin antibacterials. Clin. Pharmacokinet. 40:327-341. [DOI] [PubMed] [Google Scholar]
- 6.Chan, S. L., W. W. Yew, J. H. Porter, K. P. McAdam, B. W. Allen, J. M. Dickinson, G. A. Ellard, and D. A. Mitchison. 1994. Comparison of Chinese and Western rifapentines and improvement of bioavailability by prior taking of various meals. Int. J. Antimicrob. Agents 3:267-274. [DOI] [PubMed] [Google Scholar]
- 7.Dooley, K., C. Flexner, J. Hackman, C. A. Peloquin, E. Nuermberger, R. E. Chaisson, and S. E. Dorman. 2008. Repeated administration of high-dose intermittent rifapentine reduces rifapentine and moxifloxacin plasma concentrations. Antimicrob. Agents Chemother. 52:4037-4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordi, T., R. Xie, N. V. Huong, D. X. Huong, M. O. Karlsson, and M. Ashton. 2005. A semiphysiological pharmacokinetic model for artemisinin in healthy subjects incorporating autoinduction of metabolism and saturable first-pass hepatic extraction. Br. J. Clin. Pharmacol. 59:189-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gumbo, T., A. Louie, M. R. Deziel, W. Liu, L. M. Parsons, M. Salfinger, and G. L. Drusano. 2007. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob. Agents Chemother. 51:3781-3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heifets, L. B., P. J. Lindholm-Levy, and M. A. Flory. 1990. Bactericidal activity in vitro of various rifamycins against Mycobacterium avium and Mycobacterium tuberculosis. Am. Rev. Respir. Dis. 141:626-630. [DOI] [PubMed] [Google Scholar]
- 11.Holford, N. 2005. The visual predictive check-superiority to standard diagnostic (Rorschach) plots, abstr. 738. Abstr. 14th PAGE Meeting.
- 12.Holford, N. H., R. J. Ambros, and K. Stoeckel. 1992. Models for describing absorption rate and estimating extent of bioavailability: application to cefetamet pivoxil. J. Pharmacokinet. Biopharm. 20:421-442. [DOI] [PubMed] [Google Scholar]
- 13.Jonsson, E. N., and M. O. Karlsson. 1999. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58:51-64. [DOI] [PubMed] [Google Scholar]
- 14.Karlsson, M. O., and R. M. Savic. 2007. Diagnosing model diagnostics. Clin. Pharmacol. Ther. 82:17-20. [DOI] [PubMed] [Google Scholar]
- 15.Karlsson, M. O., and L. B. Sheiner. 1993. The importance of modeling interoccasion variability in population pharmacokinetic analyses. J. Pharmacokinet. Biopharm. 21:735-750. [DOI] [PubMed] [Google Scholar]
- 16.Kerbusch, T., U. Wahlby, P. A. Milligan, and M. O. Karlsson. 2003. Population pharmacokinetic modelling of darifenacin and its hydroxylated metabolite using pooled data, incorporating saturable first-pass metabolism, CYP2D6 genotype and formulation-dependent bioavailability. Br. J. Clin. Pharmacol. 56:639-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keung, A., M. G. Eller, K. A. McKenzie, and S. J. Weir. 1999. Single and multiple dose pharmacokinetics of rifapentine in man: part II. Int. J. Tuberc. Lung Dis. 3:437-444. [PubMed] [Google Scholar]
- 18.Langdon, G., J. Wilkins, L. McFadyen, H. McIlleron, P. Smith, and U. S. Simonsson. 2005. Population pharmacokinetics of rifapentine and its primary desacetyl metabolite in South African tuberculosis patients. Antimicrob. Agents Chemother. 49:4429-4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langdon, G., J. J. Wilkins, P. J. Smith, and H. McIlleron. 2004. Consecutive-dose pharmacokinetics of rifapentine in patients diagnosed with pulmonary tuberculosis. Int. J. Tuberc. Lung Dis. 8:862-867. [PubMed] [Google Scholar]
- 20.Menzies, D., A. Benedetti, A. Paydar, I. Martin, S. Royce, M. Pai, A. Vernon, C. Lienhardt, and W. Burman. 2009. Effect of duration and intermittency of rifampin on tuberculosis treatment outcomes: a systematic review and meta-analysis. PLoS Med. 6:e1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitchison, D. A. 1998. Development of rifapentine: the way ahead. Int. J. Tuberc. Lung Dis. 2:612-615. [PubMed] [Google Scholar]
- 22.Niemi, M., J. T. Backman, M. F. Fromm, P. J. Neuvonen, and K. T. Kivisto. 2003. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin. Pharmacokinet. 42:819-850. [DOI] [PubMed] [Google Scholar]
- 23.Relling, M. V., R. R. Evans, S. Groom, W. R. Crom, and C. B. Pratt. 1993. Saturable elimination and saturable protein binding account for flavone acetic acid pharmacokinetics. J. Pharmacokinet. Biopharm. 21:639-651. [DOI] [PubMed] [Google Scholar]
- 24.Roberts, M. S., B. M. Magnusson, F. J. Burczynski, and M. Weiss. 2002. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin. Pharmacokinet. 41:751-790. [DOI] [PubMed] [Google Scholar]
- 25.Rosenthal, I. M., K. Williams, S. Tyagi, C. A. Peloquin, A. A. Vernon, W. R. Bishai, J. H. Grosset, and E. L. Nuermberger. 2006. Potent twice-weekly rifapentine-containing regimens in murine tuberculosis. Am. J. Respir. Crit. Care Med. 174:94-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenthal, I. M., M. Zhang, K. N. Williams, C. A. Peloquin, S. Tyagi, A. A. Vernon, W. R. Bishai, R. E. Chaisson, J. H. Grosset, and E. L. Nuermberger. 2007. Daily dosing of rifapentine cures tuberculosis in three months or less in the murine model. PLoS Med. 4:e344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rousseau, A., F. Leger, Y. Le Meur, F. Saint-Marcoux, G. Paintaud, M. Buchler, and P. Marquet. 2004. Population pharmacokinetic modeling of oral cyclosporin using NONMEM: comparison of absorption pharmacokinetic models and design of a Bayesian estimator. Ther. Drug Monit. 26:23-30. [DOI] [PubMed] [Google Scholar]
- 28.Savic, R. M., D. M. Jonker, T. Kerbusch, and M. O. Karlsson. 2007. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 34:711-726. [DOI] [PubMed] [Google Scholar]
- 29.Sirgel, F. A., P. B. Fourie, P. R. Donald, N. Padayatchi, R. Rustomjee, J. Levin, G. Roscigno, J. Norman, H. McIlleron, and D. A. Mitchison. 2005. The early bactericidal activities of rifampin and rifapentine in pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 172:128-135. [DOI] [PubMed] [Google Scholar]
- 30.Taft, D. R., G. R. Iyer, L. Behar, and R. V. DiGregorio. 1997. Application of a first-pass effect model to characterize the pharmacokinetic disposition of venlafaxine after oral administration to human subjects. Drug Metab. Dispos. 25:1215-1218. [PubMed] [Google Scholar]
- 31.Wahlby, U., E. N. Jonsson, and M. O. Karlsson. 2001. Assessment of actual significance levels for covariate effects in NONMEM. J. Pharmacokinet. Pharmacodyn. 28:231-252. [DOI] [PubMed] [Google Scholar]
- 32.Wehrli, W. 1983. Rifampin: mechanisms of action and resistance. Rev. Infect. Dis. 5(Suppl. 3):S407-S411. [DOI] [PubMed] [Google Scholar]
- 33.Weiner, M., D. Benator, W. Burman, C. A. Peloquin, A. Khan, A. Vernon, B. Jones, C. Silva-Trigo, Z. Zhao, and T. Hodge. 2005. Association between acquired rifamycin resistance and the pharmacokinetics of rifabutin and isoniazid among patients with HIV and tuberculosis. Clin. Infect. Dis. 40:1481-1491. [DOI] [PubMed] [Google Scholar]
- 34.Weiner, M., N. Bock, C. A. Peloquin, W. J. Burman, A. Khan, A. Vernon, Z. Zhao, S. Weis, T. R. Sterling, K. Hayden, and S. Goldberg. 2004. Pharmacokinetics of rifapentine at 600, 900, and 1,200 mg during once-weekly tuberculosis therapy. Am. J. Respir. Crit. Care Med. 169:1191-1197. [DOI] [PubMed] [Google Scholar]
- 35.Wilkins, J. J. 2005. NONMEMory: a run management tool for NONMEM. Comput. Methods Programs Biomed. 78:259-267. [DOI] [PubMed] [Google Scholar]
- 36.Zai, H., M. Kusano, H. Hosaka, Y. Shimoyama, A. Nagoshi, M. Maeda, O. Kawamura, and M. Mori. 2009. Monosodium L-glutamate added to a high-energy, high-protein liquid diet promotes gastric emptying. Am. J. Clin. Nutr. 89:431-435. [DOI] [PubMed] [Google Scholar]
- 37.Zhang, T., M. Zhang, I. M. Rosenthal, J. H. Grosset, and E. L. Nuermberger. 2009. Short-course therapy with daily rifapentine in a murine model of latent tuberculosis infection. Am. J. Respir. Crit. Care Med. 180:1151-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu, M., S. Kaul, P. Nandy, D. M. Grasela, and M. Pfister. 2009. Model-based approach to characterize efavirenz autoinduction and concurrent enzyme induction with carbamazepine. Antimicrob. Agents Chemother. 53:2346-2353. [DOI] [PMC free article] [PubMed] [Google Scholar]