

Abstract
As multidrug resistance increases alarmingly, polymyxin B and colistin are increasingly being used in the clinic to treat serious Pseudomonas aeruginosa infections. In this opportunistic pathogen, subinhibitory levels of polymyxins and certain antimicrobial peptides induce resistance toward higher, otherwise lethal, levels of these antimicrobial agents. It is known that the modification of lipid A of lipopolysaccharide (LPS) is a key component of this adaptive peptide resistance, but to date, the regulatory mechanism underlying peptide regulation in P. aeruginosa has remained elusive. The PhoP-PhoQ and PmrA-PmrB two-component systems, which control this modification under low-Mg2+ conditions, do not appear to play a major role in peptide-mediated adaptive resistance, unlike in Salmonella where PhoQ is a peptide sensor. Here we describe the identification and characterization of a novel P. aeruginosa two-component regulator affecting polymyxin-adaptive resistance, ParR-ParS (PA1799-PA1798). This system was required for activation of the arnBCADTEF LPS modification operon in the presence of subinhibitory concentrations of polymyxin, colistin, or the bovine peptide indolicidin, leading to increased resistance to various polycationic antibiotics, including aminoglycosides. This study highlights the complexity of the regulatory network controlling resistance to cationic antibiotics and host peptides in P. aeruginosa, which has major relevance in the development and deployment of cationic antimicrobials.
Pseudomonas aeruginosa is a Gram-negative ubiquitous environmental bacterium that is capable of infecting a wide range of plant and animal hosts (40). In humans, it is responsible for eventually fatal chronic lung infections in patients with cystic fibrosis (CF), as well as serious acute infections in immunocompromised and injured individuals, where it is a serious problem in ventilator-associated pneumonia and septic burn wounds (32, 43). Indeed, P. aeruginosa is the third leading cause of nosocomial infections (2).
The ability of this microorganism to adapt to different environments is, to a large extent, due to intricate regulatory networks. P. aeruginosa PAO1 has one of the largest collections of regulatory proteins found in bacteria, representing just less than 10% of all open reading frames (ORFs) (47), and a major subset of these ORFs corresponds to two-component regulatory systems. These systems classically consist of a histidine kinase sensor protein, located in the cytoplasmic membrane, and a cytoplasmic response regulator. When the periplasmic domain of the sensor kinase detects and interacts with an effector molecule, the protein autophosphorylates at a conserved cytoplasmic histidine residue. This phosphate is subsequently transferred to an aspartate residue of the response regulator, and the phosphorylated regulator will then activate and/or repress transcription of its target genes (41). The P. aeruginosa genome encodes 64 response regulators and 63 histidine kinases, as well as 16 atypical kinases (41). Although some of these regulatory systems have been shown to control key aspects of P. aeruginosa antibiotic resistance and virulence (19), the function of many of them remains undetermined.
Infections caused by P. aeruginosa are very difficult to treat because of its high level of intrinsic resistance to drugs (24). In addition, P. aeruginosa can also exhibit adaptive resistance, a phenomenon by which certain environmental cues, including subinhibitory concentrations of antibiotics, can transiently induce resistance to otherwise lethal doses of antimicrobial agents (33, 3). This is one of the reasons why in vitro analysis of antibiotic resistance does not necessarily reflect the clinical effectiveness of a specific drug. In P. aeruginosa, adaptive resistance to polymyxins and cationic antimicrobial peptides is known to occur in response to limiting extracellular concentrations of divalent Mg2+ and Ca2+ cations (33, 36). This adaptation is controlled by the two-component regulators PhoP-PhoQ and PmrA-PmrB which upregulate the expression of the arnBCADTEF lipopolysaccharide (LPS) modification operon (36). The products of these arn genes participate in the addition of 4-aminoarabinose to lipid A of LPS, thereby reducing the net negative charge of LPS and limiting the interaction and self-promoted uptake of polycationic antibiotics, such as polymyxins, host defense peptides, and aminoglycosides, with the outer membrane. Nevertheless, this adaptation to low divalent cations is unlikely to be clinically meaningful, as the human body contains 1 to 2 mM divalent cations. More alarmingly, peptides themselves are also able to induce adaptive resistance via LPS modification (36). Subinhibitory concentrations of peptides can induce the expression of the pmrAB and arnBCADTEF operons in P. aeruginosa. However, neither PhoP-PhoQ nor PmrA-PmrB seems to be essential for this upregulation (36). Thus, an unidentified regulatory system has been proposed to respond to peptides and promote the activation of the arnBCADTEF and pmrAB operons independently of PhoP-PhoQ and PmrA-PmrB, consequently increasing resistance to antimicrobial peptides (36, 19).
The polymyxins, a group of cationic antimicrobial lipopeptides synthesized by bacteria, are being used increasingly to treat infections by multiresistant P. aeruginosa strains (31). Despite the fact that polymyxins have been used for decades, the pharmacokinetics and pharmacodynamics of polymyxins are still poorly understood. Consequently, there is the possibility that bacteria are exposed to sublethal concentrations during treatment.
Cationic antimicrobial peptides are widely present in nature, being produced by bacteria and complex organisms, such as plants and animals. In the latter, peptides constitute key parts of their host defense mechanisms. As such, P. aeruginosa comes into contact with the human cationic host defense (antimicrobial) peptides produced by epithelial surfaces (e.g., β-defensins and cathelicidins), during infection. The combination of antimicrobial and immunomodulatory activity has made host defense peptides very attractive for the design of new drugs to combat bacterial infections (23). Therefore, it is desirable to develop peptides with a poor ability to induce an adaptive bacterial response while maintaining good activity. All these factors make it important to attain a better understanding of the molecular mechanisms behind adaptive resistance to cationic antimicrobial peptides in P. aeruginosa.
The aim of this study was the identification of the major sensor/regulator controlling polymyxin- and cationic-peptide-mediated adaptive resistance in P. aeruginosa. We demonstrate that the novel two-component regulator ParR-ParS is a key component in the cascade that leads to the induction of the LPS modification operon in response to subinhibitory concentrations of peptides. Additionally, analysis of the transcriptome of a parR mutant in the presence of subinhibitory indolicidin (a bovine host defense peptide) provided new insights into the genes involved in the regulatory pathway activated by this peptide.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and antimicrobial agents.
The bacterial strains and plasmids used in this study are described in Table 1. Cultures were routinely grown in Luria-Bertani (LB) broth containing 2.0% (wt/vol) Difco agar (Becton-Dickinson Co.) when appropriate. The defined minimal medium used was BM2-glucose [62 mM potassium phosphate buffer (pH 7), 7 mM (NH4)2SO4, 10 μM FeSO4, 0.4% (wt/vol) glucose] containing 2 mM MgSO4 (high Mg2+) or 20 μM MgSO4 (low Mg2+). The antibiotics and concentrations used for selection in P. aeruginosa were as follows: tetracycline, 50 μg/ml; carbenicillin, 500 μg/ml; and gentamicin, 30 μg/ml. The antibiotics and concentrations used for selection in Escherichia coli were as follows: ampicillin, 100 μg/ml; and kanamycin, 50 μg/ml. All antibiotics were purchased from Sigma. Peptides were synthesized by 9-fluorenylmethoxy carbonyl (Fmoc) methods and were 95% pure by high-performance liquid chromatography (HPLC) and mass spectrometry (MS).
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial straina or plasmid | Relevant genotype or characteristicsb | Reference(s) or source |
|---|---|---|
| Bacterial strains | ||
| P. aeruginosa | ||
| WT | Wild-type P. aeruginosa PAO1; strain H103 | 3, 47 |
| UW-WT | UW wild-type P. aeruginosa PAO1 | 26 |
| UW-PA1799 | PA1799::ISlacZ/hah-Tcr; insertion at 492 (708) bp in PA1799; derived from strain UW-WT | 26 |
| UW-PA1798 | PA1798::ISlacZ/hah-Tcr; insertion at 307 (1,287) bp in PA1798; derived from UW-WT | 26 |
| parR mutant | PA1799::ISlacZ/hah-Tcr, H103 background; Tetr | This study |
| parS mutant | PA1798::ISlacZ/hah-Tcr, H103 background; Tetr | This study |
| parR (Tn7-parRS+) complemented strain | PA1799 mutant with Tn7-PA1798-1799+ integrated; Tcr Gmr | This study |
| UW-PA1797 | PA1797::ISlacZ/hah-Tcr; insertion at 423 (1,833) bp in PA1797; derived from UW-WT | 26 |
| UW-PA1559 | PA1559::ISlacZ/hah-Tcr; insertion at 406 (732) bp in PA1559; derived from UW-WT | 26 |
| UW-pagL | pagL::ISphoA/hah-Tcr; insertion at 196 (522) bp in pagL; derived from UW-WT | 26 |
| UW-PA4782 | PA4782::ISphoA/hah-Tcr; insertion at 147 (246) bp in PA4782; derived from UW-WT | 26 |
| UW-pmrB | pmrB::ISphoA/hah-Tcr; insertion at 67 (1,434) bp in pmrB; derived from UW-WT | 26 |
| UW-arnB | arnB::ISphoA/hah-Tcr; insertion at 327 (1,149) bp in arnB; derived from UW-WT | 26 |
| UW-arnC | arnC::ISlacZ/hah-Tcr; insertion at 408 (1,020) bp in arnC; derived from UW-WT | 26 |
| E. coli TOP10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZM15 ΔlacX74recA1araΔ139 Δ(ara-leu)7697galUgalKrpsL (Strr) endA1nupG | Invitrogen |
| Plasmids | ||
| pCR-Blunt II-TOPO | PCR cloning vector; Kanr | Invitrogen |
| pCR-PA1798-1799+ | pCR-Blunt II-TOPO harboring 2.3-kb PA1798-PA1799 amplicon | This study |
| pUC18-mini-Tn7T-Gm | Suicide plasmid; Gmr Ampr | 5 |
| pUC-Tn7-PA1798-1799+ | pUC18-mini-Tn7T-Gm with 2.3-kb PA1798-PA1799 fragment from pCR-PA1798-PA1799 | This study |
| pTNS2 | Transposition helper plasmid; Ampr | 5 |
| pUCPlux-pPA3552 | pUCP23 containing a transcriptional fusion between the arnB promoter and luxCDABE | 36 |
UW, University of Washington Genome Science Center.
Antibiotic resistance phenotypes: Ampr, ampicillin resistance for E. coli and carbenicillin resistance for P. aeruginosa; Gmr, gentamicin resistance; Kanr, kanamycin resistance; Tcr, tetracycline resistance. An “insertion at 492 (708) bp” means that the insertion is at nucleotide 492 within a 708-bp-long gene. “hah” is a 63-codon insertion which encodes an influenza hemagglutinin epitope together with a hexahistidine metal affinity purification tag.
Transferring the UW-PA1798 and UW-PA1799 transposon mutations into a different P. aeruginosa PAO1 background.
The UW-PA1798 and UW-PA1799 transposon mutations (Table 1) were first confirmed to be correct by colony PCR as recommended by the University of Washington Genome Science Center (UW) (26). These mutations were then transferred into our laboratory wild-type (WT) P. aeruginosa PAO1 strain (H103) as described previously (6, 17). Briefly, genomic DNA was isolated from each of the UW mutants, and 1 μg was electroporated into competent cells of strain H103. After cells recovered for 1 h at 37°C, aliquots were plated onto LB agar plates containing 50 μg/ml tetracycline. The following day, tetracycline-resistant colonies were analyzed by PCR to verify the correct site of transposon insertion.
Genetic complementation.
In order to complement the mutation in parR (PA1799), the operon parRS (PA1798-1799+) was cloned together with 185 bp of upstream DNA (native promoter) and 50 bp of downstream DNA. This fragment was amplified from P. aeruginosa WT PAO1 genomic DNA using Platinum Pfx DNA polymerase (Invitrogen), and the PCR product was purified with the Qiagen PCR purification kit. Forward and reverse primers for PA1798-PA1799 were designed from the P. aeruginosa PAO1 genome sequence (Pseudomonas Genome Database v2 at www.pseudomonas.com) using Primer3 (44). These primers were as follows: PA1799-F (F for forward) (5′-GAATCAAGGGCATGCATTCTA-3′) and PA1798-R (R for reverse) (5′-GTCCTTACGGCTTTGTCAGC-3′). The generated amplicon was then cloned into pCR-Blunt II-TOPO, using the Zero Blunt TOPO PCR cloning kit (Invitrogen), and transformed into One Shot TOP10 E. coli cells (Invitrogen), creating pCR-PA1798-1799+. An NsiI fragment containing PA1798-PA1799+ was excised from pCR-PA1798-1799 and subcloned into pUC18-mini-Tn7T-Gm, generating pUC-Tn7-PA1798-1799+. Next, pUC-Tn7-PA1798-PA1799+ was coelectroporated with pTNS2 (transposition helper plasmid) into electrocompetent cells of the parR mutant prepared by the sucrose method (6). Gentamicin-resistant transformants were analyzed by colony PCR using primers PglmS-up and PTn7L to determine the correct transposon integration of mini-Tn7 into the chromosome as previously described (5).
Luminescence gene expression assays.
Induction of the luxCDABE fusion under different conditions was carried out by the method of McPhee et al. (36). Different P. aeruginosa strains carrying plasmid pUCPlux-pPA3552 were grown overnight in LB broth containing carbenicillin. The following day, the cells were pelleted and resuspended in BM2-glucose liquid medium containing either high or low Mg2+ concentration. These cell suspensions were then diluted 1:20 into the same medium. When necessary, different peptides or aminoglycosides were added to the medium. The production of luminescence at different time points was measured using a SPECTRAFluorPlus luminometer (Tecan, San Jose, CA). Luminescence was corrected for growth by measuring the optical density at 620 nm (OD620).
Killing curves.
P. aeruginosa cultures were grown overnight in BM2-glucose (high Mg2+) and diluted 1:50 the next morning into fresh BM2-glucose (high Mg2+) with or without subinhibitory indolicidin (4 μg/ml). Upon reaching the mid-logarithmic phase of growth (OD600 ∼ 0.5), 1 ml of culture was harvested, centrifuged, and resuspended in 1 ml of 1× BM2 salts. This cell suspension was diluted 1:10 into 1× BM2 salts, and killing was then initiated by the addition of 10.0 μg/ml or 1.0 μg/ml polymyxin B sulfate (Sigma) for the analysis of adaptive and intrinsic resistance, respectively. The tubes were continuously shaken at room temperature using a benchtop nutator (BD Clay Adams nutator, model 1105). To assay for survivors, 50-μl aliquots were withdrawn at the designated times and serially diluted 10-fold, and 100-μl portions from serial dilutions of these aliquots were plated onto LB agar plates.
MIC determination.
MICs were assessed according to the standard broth microdilution method (7) as described by CLSI/NCCLS guidelines (M7-A7 [7] and M100-S17 [7a]) using BM2-glucose minimal medium containing 2 mM or 20 μM Mg2+. The MIC was determined following 24 h of growth at 37°C and represented the minimal concentration at which no growth could be visually observed with the naked eye. For MICs using cationic antimicrobial peptides, polypropylene microtiter plates were used to prevent peptide binding to the plate surface and artificially high MICs (50). The amino acid sequences of the peptides used in this study are included in Table 2. To determine the adaptive MICs to peptides and aminoglycosides, cells were grown before and during MIC determination in medium supplemented with 4 μg/ml indolicidin.
TABLE 2.
Antimicrobial peptides used in this study
| Peptide | Sequencea | Reference |
|---|---|---|
| Polymyxin B | fa-BL.TL.B(BL.BL.FLL.BL.BL.TL.) | 49 |
| Colistin | fa-BL.TL.B(BL.BL.LLL.BL.BL.TL.) | 49 |
| CP11N | ILKKWPWWPWRRK-NH2 | 15 |
| Indolicidin | ILPWKWPWWPWRR-NH2 | 46 |
| CP26 | KWKSFIKKLTSAAKKVVTTAKPLISS | 16 |
| CP28 | KWKLFKKIGIGAVLKVLTTGLPALKLTK | 39 |
| Bac2A | RLARIVVIRVAR-NH2 | 50 |
| Bactenecin | RLCRIVVIRVCR | 42 |
| LL-37 | LLGDFFRKSKEKIFKEFKRIVQRIKDFLRNLVPRTES | 20 |
| Polyphemusin | RRWC1FRVC2YRGFC2YRKC1R | 51 |
Sequences are shown in the one-letter amino acid code. In the first two sequences, the sequences for polymyxin B and colistin, fa indicates a 6-methyloctanoyl or 6-methylheptanoyl fatty acid chain, B indicates α,γ-diaminobutirate, and a superscripted L indicates that the amino acid is the L-enantiomer. For the remaining sequences, -NH2 indicates amidation of the carboxyl terminus and numbered cysteines represent residues joined by disulfide bonds.
Assessment of antimicrobial interactions between peptides.
To determine whether there was synergistic action between certain peptides, the checkerboard method was used (8). This technique is a modification of the broth microdilution method, in which two drugs are diluted in a two-dimensional fashion. As a result of this, each well in the microtiter plate contains a unique combination of concentrations of the two drugs. The fractional inhibitory concentration (FIC) for each drug was determined by dividing the concentration necessary to inhibit growth in a specific row or column by the MIC of the bacterium for that drug by itself (10). The FIC index (FICi), calculated as the sum of the FICs, was interpreted as follows: a FICi of ≤0.5 represents synergy, a FICi of ≥4.0 represents antagonism, and a FICi of> 0.5 to 4 represents no interaction. As in the MIC determinations, the medium used was BM2-glucose containing 2 mM Mg2+, and polypropylene microtiter plates were used. Results were read after 24 h of incubation at 37°C.
Microarray analysis.
Microarray analysis was carried out as previously described (35). In overview, four independent cultures of each P. aeruginosa strain, WT and the parR mutant, were grown with shaking in BM2-glucose medium containing 2 mM MgSO4 at 37°C for 18 h and then diluted 1:100 into fresh medium supplemented with 4 μg/ml indolicidin. Cultures were grown to the mid-logarithmic phase of growth (OD600 ∼ 0.5) at 37°C with shaking (250 rpm), and then total RNA was isolated using the RNeasy Midi RNA isolation kit (Qiagen) following the manufacturer's instructions. Contaminating genomic DNA was removed by treatment with the DNA-free kit (Ambion Inc., Austin, TX). RNA was stored at −80°C with 0.2 U μl−1 of SUPERase-In RNase inhibitor (Ambion Inc.). The quality of the RNA was assessed spectrophotometrically and by agarose gel electrophoresis. RNA was converted to cDNA, hybridized, and analyzed as previously described (35). P. aeruginosa PAO1 microarray slides were provided by the J. Craig Venter Institute (JCVI) (formerly The Institute for Genomic Research [TIGR]) Pathogenic Functional Genomics Resource Center (http://pfgrc.jcvi.org/). Images of slides were quantified using ImaGene 6.0, standard edition, software (BioDiscovery, Inc., El Segundo, CA). ArrayPipe, version 1.7, was used for assessment of slide quality, normalization, detection of differential gene expression, and statistical analysis, using available genome annotation from the Pseudomonas Genome Database v2 at www.pseudomonas.com. The four biological replicates were averaged to obtain overall changes for the parR mutant relative to the WT, and two-sided one-sample Student's t test was applied to determine significant changes in gene expression. Changes of 2-fold or greater with a Student t test P value of ≤0.05 were used as the cutoffs for reporting expression changes.
Reverse transcription-quantitative PCR (RT-qPCR).
Three independent cultures of the P. aeruginosa WT strain and the parR mutant were grown to the mid-logarithmic phase in BM2-glucose minimal medium containing 2 mM Mg2+ with or without 4 μg/ml indolicidin. Total RNA was then isolated, using RNeasy minicolumns (Qiagen). DNase treatment of RNA samples, cDNA synthesis, and real-time PCR (qPCR) were carried out as described previously (35). cDNA was diluted 1:100, and 2.5 μl was used as a template for each reaction, using 1× SYBR green PCR master mix (Applied Biosystems, Foster, CA) and an ABI Prism 7000 instrument (Applied Biosystems). Internal forward and reverse primers for each gene were designed using PrimerExpress (Applied Biosystems). Experiments were repeated with three independent cultures, each tested in duplicate. Comparison to the rpsL gene, encoding the 30S ribosomal protein S12, allowed calculation of the fold change by the threshold cycle (CT) method (31a).
Microarray data accession number.
The MIAMExpress accession number is E-FPMI-21.
RESULTS
Initial identification of ParR-ParS.
Indolicidin, a bovine host defense peptide (Table 2), is known to be a strong inducer of the arnBCADTEF LPS modification operon. Examination of the promoter regions of genes that were dysregulated by indolicidin indicated a motif similar to that of PmrA and PhoQ (35). We sought to identify regulators involved in this induction. To do so, we utilized plasmid pUCPlux-PPA3552, which carries a transcriptional fusion of the arnB (PA3552) promoter with a promoterless luxCDABE cassette (36). This reporter construct allows quantification of the expression from the arnB promoter by measuring luminescence. Plasmid pUCPlux-PPA3552 was electroporated into a panel of UW mutants (UW for University of Washington Genome Science Center) (26) that harbored transposon insertions in different response regulator genes that were most similar to PmrA and/or PhoP, namely, PA0756, PA1799, PA2479, PA2523, PA2657, PA2809, PA3204, PA3205, and PA4885. These strains were grown with and without 4 μg/ml indolicidin, and luminescence production was determined. This permitted the identification of an intragenic transposon mutation, located within the response regulator-encoding gene PA1799 (Table 1), which rendered the cells unresponsive to arnB induction by indolicidin (Fig. 1A); no other mutants showed any effect on peptide-mediated induction. The same unresponsive phenotype could be observed in the cognate sensor kinase-encoding mutant UW-PA1798. The transposon mutations were then transferred into a new P. aeruginosa PAO1 background corresponding to our routine laboratory wild-type (WT) strain H103 (Table 1), resulting in new PA1798 and PA1799 mutant strains that were used for all subsequent experiments in this paper. These genes will hereafter be referred to as parR (PA1799) and parS (PA1798) for peptide adaptive resistance regulator and sensor, respectively.
FIG. 1.

Identification of the parRS operon. (A) Fold induction of pPA3552::lux fusion in BM2-glucose minimal medium supplemented with 2 mM Mg2+ in the presence of 4 μg/ml indolicidin compared to induction in the absence of indolicidin in the WT (H103) and mutant strains UW-PA1798 (UW-1798) and UW-PA1799 (UW-1799). The pPA3552::lux fusion contained a promoterless lux cassette fused to the promoter of the arnB gene and reported on the transcription of the eight-gene operon encoding the arn LPS modification operon. The results provided correspond to one representative experiment of at least three independent assays which displayed the same trends. (B) Schematic representation showing the genomic context of the parRS operon. The black arrows represent ORFs.
The parR and parS genes are predicted to be cotranscribed according to the database of prokaryotic operons (34) and their sequential location in the P. aeruginosa PAO1 genome (Fig. 1B). Analysis of the amino acid sequence of ParS with program SOSUI 1.11 (25) revealed the presence of two putative transmembrane domains (residues 8 to 30 and 132 to 154) flanking a periplasmic loop (residues 28 to 134), which probably corresponds to the sensor domain of the protein. The charge of this periplasmic loop at pH 7.0 was estimated to be −6.6, using the program Protein Calculator v3.3 (http://www.scripps.edu/∼cdputnam/protcalc.html), a result consistent with a putative function in polycation binding.
Given that the best-characterized adaptive peptide resistance mechanism thus far is that of Salmonella, the sequence of parR was compared with the Salmonella enterica serovar Typhimurium genome, using the tool BLAST Microbial Genomes from the National Center for Biotechnology Information website. The results showed that, among the P. aeruginosa response regulators, ParR had the highest similarity to Salmonella PhoP (51%); however, the highest homologue of ParR was actually the response regulator RstA (62% similarity). Likewise, protein ParS is 58% similar to the S. enterica sensor kinase RstB. This resemblance only concerned the intracellular domain, whereas no significant similarity could be found between the sensor domain of ParS and any protein in the Salmonella genome.
Mutations in parR and parS affect peptide-mediated expression of arnB, but not induction in low Mg2+.
The plasmid pUCPlux-PPA3552 was transformed into the parR and parS mutants, and the production of luminescence under different conditions was assayed. The induction of this operon by indolicidin and low Mg2 was tested in the parR and parS mutants, the WT, and a complemented parR mutant [parR-Tn7 (parRS+)]. Unlike the WT, neither mutant showed a significant induction of reporter luminescence in the presence of indolicidin (Fig. 2A). This phenotype could be complemented by the introduction of a single copy of the WT parRS+ operon into the chromosome of the parR mutant by means of mini-Tn7 integration (6) (Fig. 2A).
FIG. 2.

ParR-ParS mediates the upregulation of the LPS modification operon in response to certain antimicrobial peptides. (A) Induction of pPA3552::lux fusion in bacteria grown in BM2-glucose minimal medium supplemented with 20 μM Mg2+, 2 mM Mg2+, or 2 mM Mg2+ and 4 μg/ml indolicidin. The WT (H103), parR and parS mutants, and the complemented parR mutant were studied. The pPA3552::lux fusion contained a promoterless lux cassette fused to the promoter of the arnB gene and reported on the transcription of the eight-gene operon encoding the arn LPS modification operon. Luminescence is shown in relative light units per optical density (RLU/OD). The results provided correspond to one representative experiment of at least four independent assays which displayed the same trends. (B) Fold induction of pPA3552::lux fusion in the WT (H103) and the parR mutant grown in BM2-glucose minimal medium supplemented with 2 mM Mg2+ and different antimicrobial compounds. The values shown here correspond to the means plus standard deviations (error bars) of four independent experiments at the concentration of each antibiotic leading to the highest induction level. These antibiotics and concentrations were as follows: tobramycin, 0.5 μg/ml; polymyxin B, 1 μg/ml; indolicidin, 4 μg/ml; colistin, 1 μg/ml; CP28, 3 μg/ml; polyphemusin, 2 μg/ml; and LL-37, 32 μg/ml.
However, there was no significant difference in luminescence production among the four strains when incubated under inducing low-Mg2+ or noninducing high-Mg2+ conditions (Fig. 2A). This indicated that the upregulation of the arn LPS modification operon mediated by parRS was a specific response to the presence of the antimicrobial peptide.
Next, the ability of different peptides to induce the LPS modification operon in the WT and the parR mutant strain was determined. The peptides tested were indolicidin, polymyxin B, colistin, CP28, polyphemusin, and LL-37 (see Table 2 for peptide sequences). The aminoglycoside tobramycin was also assayed as a negative control, as it is known that it does not upregulate the arn operon, even though LPS modification increases resistance to aminoglycosides (36). First, the concentration that elicited the highest increase in luminescence production relative to the nonantibiotic control was determined. Of note, the upregulation caused by doses around normally inhibitory concentrations could be tested because the inoculum size was much higher than that used for routine MIC determination. The effect of the selected concentrations on the WT and the mutant was then assessed (Fig. 2B). As previously indicated by McPhee et al. (36), not all peptides were able to induce upregulation of the arn genes involved in the adaptive response to the same degree (Fig. 2B). The strongest inducer of luminescence in the WT was indolicidin at a concentration of 4 μg/ml, leading to a 31-fold increase. Conversely, only a 4-fold increase could be observed in the parR mutant. The peptides of bacterial origin, polymyxin B and colistin, showed patterns similar to that of indolicidin, although the degree of induction in the WT was marginally lower. The presence of 1 μg/ml of polymyxin B or colistin induced the expression of the arnB promoter in the WT by 23- and 17-fold, respectively, whereas the increase in the mutant was negligible. Intriguingly, the results obtained with the α-helical peptide CP28 seem to indicate that there could be distinct regulatory pathways for different peptides, since both the WT and the mutant experienced an upregulation of about 28-fold when 3 μg/ml CP28 was added to the medium. In a similar manner, a concentration of 2 μg/ml polyphemusin induced 24- and 17-fold in the WT and the parR mutant, respectively. According to this, ParR-ParS was not necessary to detect and respond to these two peptides. The human peptide LL-37 did not trigger a major response in either strain even at a concentration of 32 μg/ml, with only 6- and 4-fold increases in expression of the arn operon in the WT and the parR mutant, respectively (Fig. 2B). Statistical comparison of the data corresponding to the WT and the mutant with Student's t test confirmed the existence of differential induction in the parR mutant by indolicidin, polymyxin B, and colistin (P values < 0.05), whereas the relative induction values found for CP28, polyphemusin, LL-37, and tobramycin were not significantly different between the parR mutant and its parent strain (P values > 0.05).
The parR and parS mutants display reduced adaptive resistance to polymyxin B.
Killing curves were carried out in order to investigate the adaptive and intrinsic polymyxin B resistance of the parR mutant. To prevent the stimulatory effect of a low Mg2+ concentration (20 μM) on peptide resistance, the cells were grown in BM2-glucose medium with a high Mg2+ concentration (2 mM) before they were killed with polymyxin B. Additionally, the medium was supplemented with subinhibitory indolicidin (4.0 μg/ml) to test adaptive resistance. Under noninducing conditions (high Mg2+ alone), the parR mutant and the WT exhibited similar levels of killing with 1 μg/ml polymyxin B, approximately 3 log units in 10 min (Fig. 3A). The results obtained in the adaptive resistance assay were, however, quite different for the two strains. After preincubation in the presence of indolicidin, the WT cells showed only 1 log unit of killing within 10 min when challenged with 10 μg/ml polymyxin B, which indicates that they had become adaptively resistant (Fig. 3B). In contrast, the parR mutant was supersusceptible to polymyxin B under the same conditions, with 4-log-unit reduction in survivors after 10 min of killing (Fig. 3B). Adaptive peptide killing assays performed with the complemented strain, the parR (Tn7-parRS+) strain, showed levels of adaptive resistance equivalent to those of the WT (Fig. 3B). This indicates that the parRS genes are required for the adaptive peptide resistance response in P. aeruginosa.
FIG. 3.

Comparison of intrinsic (A) and adaptive (B) polymyxin B resistance in the WT and parR mutant. Sensitivity was analyzed by means of killing curves. Cells from the different strains were grown to mid-log phase in BM2-glucose minimal medium with 2 mM Mg2+ (A) or the same medium supplemented with 4 μg/ml indolicidin (B) and exposed to 1 μg/ml (A) or 10 μg/ml (B) of polymyxin B. Diluted aliquots were then plated to assay for survivors. The results presented correspond to one representative experiment for each condition out of four independent experiments which produced identical trends.
Susceptibility of parRS mutants to different classes of antibiotics.
MICs were determined to establish whether the parR and parS mutants presented increased susceptibility or resistance to different classes of drugs. As can be seen in Table 3, both mutant strains displayed supersusceptibility (4-fold) toward polymyxin B and colistin compared to the WT in BM2-glucose medium with high Mg2+. No difference could be observed under these conditions with any of the other antibiotics tested, including antimicrobial peptides (indolicidin, CP11CN, CP26, CP28, Bac2A, bactenecin, LL-37, and polyphemusin), aminoglycosides (tobramycin and gentamicin), quinolones (ciprofloxacin), and β-lactams (ceftazidime) (Table 3).
TABLE 3.
MICs of different antibiotics toward P. aeruginosa WT, parR and parS mutants, and complemented parR/Tn7-parRS+ straina
| Medium and antibiotic | MIC (μg/ml) (mode) in: |
|||
|---|---|---|---|---|
| WT | parR mutant | parS mutant | parR/Tn7- parRS+ strain | |
| Minimal medium with high Mg2+ (2 mM) | ||||
| Polymyxin B | 1 | 0.25 | 0.25 | 1 |
| Colistin | 1 | 0.25 | 0.25 | 1 |
| Indolicidin | 64 | 64 | 64 | 64 |
| CP11CN | 64 | 64 | 64 | 64 |
| CP26 | 32 | 32 | 32 | 32 |
| CP28 | 3 | 3 | 3 | 3 |
| Bac2A | 128 | 128 | 128 | NTb |
| Bactenecin | 128 | 128 | 128 | NT |
| LL-37 | 16 | 16 | 16 | 16 |
| Polyphemusin | 2 | 2 | 2 | 2 |
| Tobramycin | 2 | 2 | 2 | 2 |
| Gentamicin | 2 | 2 | 2 | NT |
| Ceftazidime | 1 | 1 | 1 | 1 |
| Ciprofloxacin | 0.1 | 0.1 | 0.1 | 0.1 |
| Minimal medium with high Mg2+ (2 mM) and supplemented with 4 μg/ml indolicidinc | ||||
| Polymyxin Bc | 8 | 0.125 | 0.125 | 8 |
| Colistinc | 8 | 0.125 | 0.125 | 8 |
| CP28c | 3 | 3 | 3 | 3 |
| LL-37c | 64 | 16 | 16 | 64 |
| Polyphemusinc | 0.5 | 0.25 | 0.25 | 0.5 |
| Tobramycinc | 4 | 2 | 2 | 4 |
| Gentamicinc | 16 | 2 | 2 | NT |
| Minimal medium with low Mg2+ (20 μM) | ||||
| Polymyxin B | 16 | 4 | 4 | 16 |
MICs were determined by serial 2-fold dilutions in BM2-glucose minimal medium with 2 mM Mg2+, unless indicated otherwise. The MIC represents the concentration at which no growth was visually observed after 24 h of incubation at 37°C. The values shown are the modes of 4 to 8 independent experiments.
NT, not tested.
The cultures used to inoculate the MIC plates were also grown in the presence of indolicidin.
To explain these data, we hypothesized that the presence of certain concentrations of polymyxin B in MIC assay wells can elicit an adaptation response in the WT, but not in the mutants. To test this, WT cells were grown in different concentrations of polymyxin B (0.125, 0.25, and 0.5 μg/ml) and then challenged with a lethal dose (1 μg/ml) of the same antibiotic. The results obtained showed that these concentrations of polymyxin B made the cells more resistant to the killing effect of the same compound in a dose-dependent manner (data not shown). This hypothesis also agrees with the data obtained in the luminescence assay (Fig. 2B). Also, when the MICs were determined for cells grown in low-Mg2+ conditions, both mutant strains underwent an increase in resistance to polymyxin B equivalent to that of the WT, while remaining 4-fold more susceptible (Table 3), indicating that this difference in MIC was insensitive to low-Mg2+ regulation.
Since the ParR-ParS system seemed to be specifically required for peptide-induced adaptive resistance, the MICs in the presence of subinhibitory indolicidin (4.0 μg/ml) (adaptive MICs) were also determined. The same concentration of indolicidin was added to the medium during growth of the culture used as inoculum for the MIC test. The difference in MICs between the WT or parR mutant complemented with a single copy of the parRS operon and the parR or parS mutant increased from 4-fold in the absence of indolicidin to 64-fold, a result that reflected in part peptide-induced adaptive resistance. Interestingly, while indolicidin increased by 8-fold the polymyxin B resistance of the WT and complemented mutant, as expected due to induction of adaptive resistance, the parR and parS mutants showed a slight 2-fold increase in susceptibility (Table 3). This was likely due to interactions between indolicidin and polymyxin B, as these antibiotics demonstrated an average fractional inhibitory concentration index (FICi) of 0.127 ± 0.033 over four independent experiments. Presumably, in the WT and complemented strains, the induction of polymyxin B resistance by subinhibitory indolicidin masked any synergy between the two peptides. Adaptive MICs to colistin followed trends identical to those observed for polymyxin B (Table 3).
The adaptive MICs of other peptides were also determined with various results (Table 3). For example, the activity of CP28 was not altered by the presence of indolicidin or by the mutation of parRS, as the MIC values were the same for all strains under inducing and noninducing conditions. For the human cathelicidin LL-37 as well as the aminoglycosides tobramycin and gentamicin, the WT cells became 2- to 8-fold more resistant when indolicidin was present in the medium (Table 3), and this response was abolished in the parR and parS mutants (Table 3). The presence of indolicidin led to 4- and 8-fold-increased polyphemusin susceptibility in the WT or complemented strain and the parR and parS mutants, respectively (Table 3). This could be explained by interaction assays showing synergy between polyphemusin and indolicidin with an average FIC of 0.057 ± 0.009 over four independent experiments.
Transcriptional analysis.
Microarrays were performed in order to compare the transcriptional responses of the parR mutant and the WT in the presence of a subinhibitory concentration of indolicidin (4 μg/ml). Under these conditions, the mutant exhibited an altered expression (≥2-fold) of 114 genes, of which 66 were upregulated and 48 were downregulated. A list of selected genes from the microarray can be seen in Table 4 (for the full list, see Table S1 in the supplemental material). The microarray data confirmed that the expression of the LPS modification operon in the presence of indolicidin was lower in the mutant than in the WT, consistent with the luminescence assay results. In addition to induction of the LPS modification operon, which is also induced under low-Mg2+ conditions, further overlap with the Mg2+ regulon could be observed (35). Thus, the following genes known to be dysregulated under Mg2+ limitation, PA0282, PA1559-1560, PA3446, PA3920, PA4773-4775-pmrAB-PA4778, and PA4782 (35), were found here to be downregulated in the parR mutant. Furthermore, all but PA3920 are positively regulated by PmrA-PmrB (35). The operon containing pmrAB is known to be modulated by both Mg2+ and the presence of peptides (36), and PmrA-PmrB is able to induce the expression of the LPS modification operon when Mg2+ is limiting in the medium, leading to increased resistance to peptides. However, peptide regulation of the arn and PA4774-pmrAB operons occurs, at least in part, independently of PmrA-PmrB (or PhoP-PhoQ) (36). Therefore, the overlaps between the PmrA-PmrB and ParR-ParS regulons might reflect similar DNA binding site motifs as well as action through peptide regulation of PmrA-PmrB.
TABLE 4.
Selected genes from the microarray analysis of genes expressed differently in the presence of 4 μg/ml indolicidin in the parR mutant compared to the WTa
| GeneID | Gene name | Fold changeb | P value | Description |
|---|---|---|---|---|
| PA0282 | cysT | −2.3 | 0.03 | Sulfate transport protein CysT |
| PA0517 | nirC | 2.7 | 0.04 | Probable c-type cytochrome precursor |
| PA0523 | norC | 6.0 | 0.01 | Nitric oxide reductase subunit C |
| PA0524 | norB | 10.7 | 0.009 | Nitric oxide reductase subunit B |
| PA1559 | −4.2 | <0.001 | Hypothetical protein | |
| PA1560 | −2.9 | <0.001 | Hypothetical protein | |
| PA1797 | −3.3 | <0.001 | Hypothetical protein | |
| PA3392 | nosZ | 2.1 | 0.04 | Nitrous oxide reductase precurser |
| PA3396 | nosL | 2.0 | 0.04 | NosL protein |
| PA3446 | −5.8 | 0.0007 | Hypothetical protein | |
| PA3552 | arnB | −1.6 | 0.0009 | Hypothetical protein |
| PA3553 | arnC | −2.0 | <0.001 | Probable glycosyltransferase |
| PA3554 | arnA | −2.5 | <0.001 | Hypothetical protein |
| PA3555 | arnD | −2.9 | <0.001 | Hypothetical protein |
| PA3556 | arnT | −2.1 | 0.003 | Inner membrane l-Ara4N transferase ArnT |
| PA3559 | pmrE | −3.3 | <0.001 | Probable nucleotide sugar dehydrogenase |
| PA3920 | 2.3 | 0.0059 | Membrane proteins/transport of small molecules | |
| PA4661 | pagL | −2.5 | <0.001 | Lipid A 3-O-deacylase |
| PA4773 | −5.1 | <0.001 | Hypothetical protein | |
| PA4774 | −7.4 | <0.001 | Hypothetical protein | |
| PA4775 | −3.3 | <0.001 | Hypothetical protein | |
| PA4776 | pmrA | −2.1 | 0.003 | Two-component system response regulator |
| PA4777 | pmrB | −1.7 | 0.002 | Two-component system signal sensor kinase |
| PA4782 | −6.7 | <0.001 | Hypothetical protein |
The full list of dysregulated genes is given in Table S1 in the supplemental material.
Fold change in gene expression in the presence of 4 μg/ml indolicidin in the parR mutant compared to gene expression in the presence of 4 μg/ml indolicidin in the WT.
Other genes of note included downregulated pagL, which encodes a lipid A 3-O-deacylase situated in the outer membrane. This finding is particularly relevant, as the presence of a specific LPS structure, deacylated at position 3, was previously demonstrated in isolates from infants with cystic fibrosis (CF) (11). The PA1797 gene, located just downstream of the parRS operon (but not in the parRS operon itself), was transcribed at a lower level in the parR mutant than in the WT. The product of the PA1797 gene is predicted to be a lipoprotein (30) of unknown function, which shares similarity with β-lactamases according to PFAM prediction. It is also worth mentioning that several genes involved in the denitrification route, nirC, norC, norB, nosZ, and nosL, were upregulated in the parR mutant compared to the WT, as some of these genes are known to be involved in aminoglycoside susceptibility (45).
Microarray data often underestimate the extent of dysregulation due to the very low concentrations of nucleic acids used for hybridization. To validate the data obtained in the microarray, the involvement of the Par system and indolicidin in the regulation of several genes (PA1559, PA1797, arnB, pagL, PA4774, pmrB, and PA4782) was confirmed using the more accurate method of reverse transcription-quantitative PCR (RT-qPCR) (Table 5). These genes were found to be suppressed between 5- and 100-fold in the parR mutant, indicating that this regulator and indolicidin were involved in the activation of gene expression; indeed, when the indolicidin peptide was omitted, there was no effect of parR on the expression of the three genes tested. Additionally, RT-qPCR results showed that pmrB, PA1797, and pagL were upregulated in the presence of indolicidin in the WT, but not in the parR mutant (Table 5). In contrast to these results, parR was not autoregulatory, as the expression of the parRS operon did not change in the presence of subinhibitory indolicidin in either the mutant or the WT (Table 5).
TABLE 5.
RT-qPCR analysis of the expression of selected genes in the parR mutant and WT cells grown in BM2-glucose minimal medium with or without indolicidin peptide
| Gene | Fold change (mean ± SD) in gene expressiona |
|||
|---|---|---|---|---|
| WT/parR mutant |
WT with indolicidin/WT without indolicidin | parR mutant with indolicidin/parR mutant without indolicidin | ||
| With indolicidin | Without indolicidin | |||
| PA1559 | 16.46 ± 6.13 | NTb | NT | NT |
| PA1797 | 114.8 ± 50.01 | 1.56 ± 0.70 | 25.3 ± 5.8 | 0.38 ± 0.26 |
| PA1799 (parR) | 0.66 ± 0.08 | NT | 1.05 ± 0.015 | NT |
| PA3552 (arnB) | 16.34 ± 7.32 | NT | NT | NT |
| PA4661 (pagL) | 5.26 ± 0.53 | 1.33 ± 0.44 | 4.06 ± 0.64 | 1.00 ± 0.20 |
| PA4774 | 23.34 ± 14.66 | NT | NT | NT |
| PA4777 (pmrB) | 4.88 ± 1.86 | 1.13 ± 0.50 | 2.27 ± 0.39 | 0.54 ± 0.24 |
| PA4782 | 8.73 ± 1.92 | NT | NT | NT |
Fold change in gene expression comparing gene expression in the WT to gene expression in the parR mutant (WT/parR mutant) in the absence and presence of 4 μg/ml indolicidin or comparing gene expression in the presence and absence of 4 μg/ml indolicidin in the WT or parR mutant.
NT, not tested.
Polymyxin B MICs of selected P. aeruginosa mutants.
After determining the genes that were differentially expressed in the parR mutant, we attempted to investigate which ones might be involved in the adaptive response to peptides. With that objective, polymyxin B intrinsic and adaptive MIC tests were performed with mutants carrying transposon insertions in selected genes from the microarray. As shown in Table 6, the PA1559 and pagL mutants were as resistant to polymyxin B as the WT was. Some differences could be seen, however, in the PA1797, pmrB, and PA4782 mutants. The MICs of these mutants were similar to that of the WT under noninducing conditions, but the adaptive MICs of these strains were 4-fold lower for the PA1797 and pmrB mutants and 2-fold lower for the PA4782 mutant (Table 6), consistent with the possibility that these genes modulate the development of adaptive resistance. Strains with mutations in the arn LPS modification operon, specifically in the arnB and arnC genes, exhibited phenotypes similar to those of the parR and parS mutants (Table 6). This is consistent with the conclusion that the major effect of the parRS mutations was through the lack of induction of the LPS modification operon. Of note, none of the mutations in mutant strains with phenotypes different from that of the WT are predicted to have polar effects due to the transposon insertion.
TABLE 6.
Polymyxin B MICs of selected P. aeruginosa mutantsa
| Strain | MIC (μg/ml) (mode) |
|
|---|---|---|
| Without indolicidin | With indolicidin | |
| WT | 1 | 8 |
| parR mutant | 0.25 | 0.125 |
| UW-PA1559 | 1 | 8 |
| UW-PA1797 | 0.5-1b | 1-2b |
| UW-pagL | 1 | 8 |
| UW-pmrB | 0.5-1b | 2 |
| UW-PA4782 | 1 | 4 |
| UW-arnB | 0.25 | 0.25 |
| UW-arnC | 0.25 | 0.125 |
MICs were determined by serial 2-fold dilutions in BM2-glucose minimal medium with 2 mM Mg2+ without indolicidin or with 4 μg/ml indolicidin. The MIC represents the concentration at which no growth was visually observed after 24 h of incubation at 37°C. Results are the mode of 5 or 6 independent experiments.
The MICs varied over the 5 or 6 different experiments.
DISCUSSION
A better understanding of the mechanisms involved in modulating antibiotic susceptibility inside the host is essential to achieve greater success in the battle against bacterial infections. This paper reports the identification of a two-component regulator, ParR-ParS, that plays a decisive role in the acquisition of peptide-induced adaptive resistance to antimicrobial peptides and aminoglycosides in P. aeruginosa.
Previous studies have demonstrated the involvement of PhoP-PhoQ and PmrA-PmrB in cationic peptide resistance in P. aeruginosa under Mg2+-deficient growth conditions (36). In contrast, neither of these two-component regulators has a major influence when antimicrobial peptides are the activating signal (36). We showed here that while ParR-ParS is necessary for the induction of the LPS modification arn operon by polymyxins and the antimicrobial peptide indolicidin, it is not required for the activation of this operon under Mg2+-limiting conditions. Hence, this regulator seems to be specific to adaptive resistance mediated by certain peptides. Our results show analogies but also striking differences with the situation in S. enterica serovar Typhimurium. In this organism, PhoP-PhoQ is able to sense both low Mg2+ and antimicrobial peptides and, through the upregulation of PmrA-PmrB, induces the expression of genes involved in the addition of aminoarabinose to LPS. This, in turn, leads to an increase in polymyxin B resistance (12, 21). Thus, peptides interact with and activate the Salmonella sensor PhoQ by displacing the Mg2+ cations from their binding site in the sensor domain. Conversely, the sensor domain of P. aeruginosa PhoQ cannot substitute for that of Salmonella and does not bind peptides (1).
In Pseudomonas, it is clear that there are two Mg2+ sensory systems, PhoP-PhoQ and PmrA-PmrB, and at least one additional peptide sensor, ParS. Although the specific mechanism by which ParS senses peptides is still to be determined, it seems possible that the mechanism might be analogous to that of Salmonella PhoQ, since both proteins have a negatively charged sensor domain; however, the basis for the selectivity observed here remains to be elucidated.
The closest homologues of ParR and ParS found in the Salmonella genome correspond to RstB and RstA, respectively. Nevertheless, the periplasmic sensor domain of ParR does not share any significant similarity with that of RstA or with any other protein from S. enterica. This emphasizes the differences between the specific mechanisms by which these two microorganisms sense peptides, even though the triggered responses are analogous. In Salmonella, RstB-RstA is activated by PhoP-PhoQ in low Mg2+ and at acidic pH, and it upregulates the expression of iron acquisition genes like feoAB (4). A similar regulatory hierarchy between PhoP-PhoQ and RstB-RstA was found in Escherichia coli (9). In contrast, expression of the parRS operon is not under the control of the P. aeruginosa PhoP-PhoQ counterparts, as indicated by previous microarray analysis (35).
The abilities of P. aeruginosa to sense and respond to the clinically important antibiotics polymyxin B and colistin are dependent on ParR-ParS. This finding is relevant in a clinical setting, since colistin is one of the antibiotics used in the treatment of CF patients (27). The fact that Pseudomonas exposure to polymyxins can induce increased resistance to selected antimicrobial peptides and aminoglycosides is of particular concern. The ability of polymyxin B to upregulate the pmrAB operon was first demonstrated by McPhee et al. (36). More recently, it was shown that colistin can upregulate the arn operon in a subpopulation of biofilm cells and induce tolerance (22, 38). The human cathelicidin LL-37 was, however, a weak inducer of the LPS modification operon. In fact, the transcriptional responses that occurred in the presence of this host defense peptide do not include the arn operon or any of the PmrA-dependent genes upregulated by indolicidin (37). Surprisingly, not all antimicrobial peptides seem to require the participation of ParR-ParS in order to induce the LPS modification operon. A clear example is the upregulation of the arnB promoter by CP28 and polyphemusin. According to this, it appears that different peptides and/or different concentrations of a peptide may activate distinct sensing mechanisms corresponding to convergent regulatory pathways. The nature of the alternative sensor/regulator is yet to be identified, although the partial involvement of PhoQ and/or PmrB cannot be discarded.
The data gathered in this study indicate that ParR-ParS contributes exclusively to adaptive resistance to peptides and aminoglycosides, but not to intrinsic resistance. Among peptides, polymyxins are affected the most by this mechanism of adaptive resistance. Some host defense peptides, like LL-37 and polyphemusin, show some modest effects, whereas the action of the α-helical peptide CP28 is independent of this bacterial adaptive response. We also found evidence for very strong synergistic interactions between some antimicrobial peptides, particularly polyphemusin and indolicidin. This suggests that it might be possible to counteract the effects of adaptive resistance by designing therapeutic programs based on combinations of peptides with synergistic action. Moreover, it would be of particular interest to develop peptides that exhibit such an interaction with human host defense peptides. The presence of a sublethal concentration of indolicidin increased the resistance to the tobramycin and gentamicin aminoglycosides in a ParR-ParS-dependent manner. This effect is probably due to a reduced ability of aminoglycosides, which are cationic molecules, to interact with the more positively charged cell surface when LPS modification is present. The fact that sublethal doses of certain peptides can induce Pseudomonas cells to become more resistant not only to other peptides but also to aminoglycosides is quite alarming, as both types of antibiotics are used in the treatment of CF patients. On the other hand, awareness of this phenomenon will be advantageous so that more efficient therapeutic programs can be implemented.
The transcriptome of the parR mutant in the presence of a subinhibitory concentration of indolicidin was substantially different from that of the WT under the same conditions. Although ParR-ParS does not influence the response to limiting Mg2+, a significant number of genes that can be upregulated at low Mg2+ concentrations were actually downregulated in the parR mutant compared to the WT (35). These genes include the arn LPS modification operon (PA3552-3559), the operon containing pmrAB (PA4773-4777), PA0282, PA1559-1560, PA3446, PA3920, and PA4782. Most of these overlapping genes are under the control of PmrA-PmrB and have typical PmrA recognition sites in their promoters (35). McPhee et al. (35) also demonstrated that many of these genes are induced in the presence of subinhibitory CP11N, a peptide related to indolicidin. Lipid A modifications, including aminoarabinose addition, have been observed in isolates from cystic fibrosis patients (14, 13). These modifications can be induced in vitro under low Mg2+ conditions (36), but to date, all the information available points toward other host factors, such as peptides, as being responsible for the appearance of these modifications during infection. The results in this study suggest that ParS could be one protein capable of sensing the presence of antimicrobial peptides during infection.
The pagL gene encodes a deacylase involved in LPS modification. Lipid A 3-O deacylation and palmitoylation reduce the ability of lipid A to activate the host Toll-like receptor 4, suggesting that these modifications help pathogens to avoid innate immune recognition (29). The expression of pagL in S. enterica is activated directly by PhoP-PhoQ (48). It has been shown recently that in S. enterica pmrA and pmrE mutants, the protein PagL is released from a latency state and compensates for the loss of inducible polymyxin B resistance (28). In P. aeruginosa, pagL is not part of the PhoP-PhoQ regulon (35), and it is upregulated by ParR-ParS in the presence of subinhibitory concentrations of peptides. The pagL gene is constitutively expressed in isolates from CF patients and in laboratory-adapted strains, but not in isolates from environmental samples or from patients with acute infections (11). Our current data, however, indicate that pagL is not necessary for peptide-induced resistance.
Despite the importance of ParR-ParS in modulating transcriptional changes that occur in the presence of subinhibitory indolicidin, the parRS operon itself is not regulated by this peptide. Nor is its expression altered by the lack of a functional ParR product. On the other hand, the PA1797 gene, located downstream of but transcribed independently of the parRS operon, is significantly induced by indolicidin in a ParR-dependent manner. Moreover, a PA1797 mutant seems to have a reduced ability to acquire adaptive resistance compared to the WT. It is tempting to speculate that the product of PA1797 might play an intermediary role in the cascade initiated by ParR-ParS or that its product has some direct function in resistance. However, the specific function of the PA1797 product still needs to be elucidated. Even though PA1797 is not dysregulated during growth under low-Mg2+ conditions or in a PhoP mutant (35), its expression is induced 4-fold in a phoQ mutant (18). It would be interesting to determine whether this reflects the existence of some kind of interaction between PhoP-PhoQ and ParR-ParS.
In conclusion, the data presented here constitute a substantial step forward toward understanding adaptive resistance to polymyxin B and cationic antimicrobial peptides in P. aeruginosa. A greater insight into the molecular mechanisms involved will be essential for the optimum design of new peptides for therapeutic purposes. In that sense, it will be important to evaluate the ability of specific peptides to induce adaptive resistance in order to select those that do not significantly trigger this response. Neglect to do so would lead to decreased susceptibility to subsequent treatment with peptides or aminoglycosides. In addition, our results indicate that adaptive resistance can be counteracted in some cases by taking advantage of synergistic interactions between peptides. Thus, the identification of parRS is of great significance within this context.
Supplementary Material
Acknowledgments
This work was funded by a grant from the Canadian Cystic Fibrosis Foundation (CCFF). W.J.G. was supported by a CCFF studentship, and L.F. received a postdoctoral fellowship from the Fundacion Alfonso Martin Escudero (Spain). I.W. was supported by the Juergen Manchot Foundation and the Mukoviszidose e.V. (Bonn, Germany) (German Cystic Fibrosis Association). R.E.W.H. holds a Canada Research Chair in Microbiology.
We also thank Herbert Schweizer (Colorado State University) for kindly providing the Tn7 plasmids.
Footnotes
Published ahead of print on 14 June 2010.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Bader, M. W., S. Sanowar, M. E. Daley, A. R. Schneider, U. Cho, W. Xu, R. E. Klevit, H. Le Moual, and S. I. Miller. 2005. Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell 122:461-472. [DOI] [PubMed] [Google Scholar]
- 2.Bonomo, R. A., and D. Szabo. 2006. Mechanisms of multidrug resistance in Acinetobacter species and Pseudomonas aeruginosa. Clin. Infect. Dis. 43:SS49-SS56. [DOI] [PubMed] [Google Scholar]
- 3.Brazas, M. D., E. B. M. Breidenstein, J. Overhage, and R. E. W. Hancock. 2007. Role of Lon, an ATP-dependent protease homolog, in resistance of Pseudomonas aeruginosa to ciprofloxacin. Antimicrob. Agents Chemother. 51:4276-4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi, E., E. A. Groisman, and D. Shin. 2009. Activated by different signals, the PhoP/PhoQ two-component system differentially regulates metal uptake. J. Bacteriol. 191:7174-7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi, K. H., J. B. Gaynor, K. G. White, C. Lopez, C. M. Bosio, R. R. Karkhoff-Schweizer, and H. P. Schweizer. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2:443-448. [DOI] [PubMed] [Google Scholar]
- 6.Choi, K. H., A. Kumar, and H. P. Schweizer. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64:391-397. [DOI] [PubMed] [Google Scholar]
- 7.Clinical and Laboratory Standards Institute. 2006. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 7th ed. Approved standard M7-A7. Clinical and Laboratory Standards Institute, Wayne, PA.
- 7a.Clinical and Laboratory Standards Institute. 2007. Performance standards for antimicrobial susceptibility testing. CLSI approved standard M100-S17. Clinical and Laboratory Standards Institute, Wayne, PA.
- 8.Dougherty, P. F., D. W. Yotter, and T. R. Matthews. 1977. Microdilution transfer plate technique for determining in vitro synergy of antimicrobial agents. Antimicrob. Agents Chemother. 11:225-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eguchi, Y., T. Okada, S. Minagawa, T. Oshima, H. Mori, K. Yamamoto, A. Ishihama, and R. Utsumi. 2004. Signal transduction cascade between EvgA/EvgS and PhoP/PhoQ two-component systems of Escherichia coli. J. Bacteriol. 186:3006-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elion, G. B., S. Singer, and G. H. Hitchings. 1954. Antagonists of nucleic acid derivatives. VIII. Synergism in combinations of biochemically related antimetabolites. J. Biol. Chem. 208:477-488. [PubMed] [Google Scholar]
- 11.Ernst, R. K., K. N. Adams, S. M. Moskowitz, G. M. Kraig, K. Kawasaki, C. M. Stead, M. S. Trent, and S. I. Miller. 2006. The Pseudomonas aeruginosa lipid A deacylase: selection for expression and loss within the cystic fibrosis airway. J. Bacteriol. 188:191-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ernst, R. K., T. Guina, and S. I. Miller. 2001. Salmonella typhimurium outer membrane remodeling: role in resistance to host innate immunity. Microbes Infect. 3:1327-1334. [DOI] [PubMed] [Google Scholar]
- 13.Ernst, R. K., S. M. Moskowitz, J. C. Emerson, G. M. Kraig, K. N. Adams, M. D. Harvey, B. Ramsey, D. P. Speert, J. L. Burns, and S. I. Miller. 2007. Unique lipid A modifications in Pseudomonas aeruginosa isolated from the airways of patients with cystic fibrosis. J. Infect. Dis. 196:1088-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ernst, R. K., E. C. Yi, L. Guo, B. Lim Kheng, J. L. Burns, M. Hackett, and S. I. Miller. 1999. Specific lipopolysaccharide found in cystic fibrosis airway Pseudomonas aeruginosa. Science 286:1561-1565. [DOI] [PubMed] [Google Scholar]
- 15.Friedrich, C. L., D. Moyles, T. J. Beveridge, and R. E. W. Hancock. 2000. Antibacterial action of structurally diverse cationic peptides on Gram-positive bacteria. Antimicrob. Agents Chemother. 44:2086-2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedrich, C., M. G. Scott, N. Karunaratne, H. Yan, and R. E. W. Hancock. 1999. Salt-resistant alpha-helical cationic antimicrobial peptides. Antimicrob. Agents Chemother. 43:1542-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gooderham, W. J., M. Bains, J. B. McPhee, I. Wiegand, and R. E. W. Hancock. 2008. Induction by cationic antimicrobial peptides and involvement in intrinsic polymyxin and antimicrobial peptide resistance, biofilm formation, and swarming motility of PsrA in Pseudomonas aeruginosa. J. Bacteriol. 190:5624-5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gooderham, W. J., S. L. Gellatly, F. Sanschagrin, J. B. McPhee, M. Bains, C. Cosseau, R. C. Levesque, and R. E. W. Hancock. 2009. The sensor kinase PhoQ mediates virulence in Pseudomonas aeruginosa. Microbiology 155:699-711. [DOI] [PubMed] [Google Scholar]
- 19.Gooderham, W. J., and R. E. W. Hancock. 2009. Regulation of virulence and antibiotic resistance by two-component regulatory systems in Pseudomonas aeruginosa. FEMS Microbiol. Rev. 33:279-294. [DOI] [PubMed] [Google Scholar]
- 20.Gudmundsson, G. H., B. Agerberth, J. Odeberg, T. Bergman, B. Olsson, and R. Salcedo. 1996. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur. J. Biochem. 238:325-332. [DOI] [PubMed] [Google Scholar]
- 21.Gunn, J. S. 2008. The Salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol. 16:284-290. [DOI] [PubMed] [Google Scholar]
- 22.Haagensen, J. A., M. Klausen, R. K. Ernst, S. I. Miller, A. Folkesson, T. Tolker-Nielsen, and S. Molin. 2007. Differentiation and distribution of colistin- and sodium dodecyl sulfate-tolerant cells in Pseudomonas aeruginosa biofilms. J. Bacteriol. 189:28-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hancock, R. E. W., and H. G. Sahl. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24:1551-1557. [DOI] [PubMed] [Google Scholar]
- 24.Hancock, R. E. W., and D. P. Speert. 2000. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and impact on treatment. Drug Resist. Updat. 3:247-255. [DOI] [PubMed] [Google Scholar]
- 25.Hirokawa, T., S. Boon-Chieng, and S. Mitaku. 1998. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378-379. [DOI] [PubMed] [Google Scholar]
- 26.Jacobs, M. A., A. Alwood, I. Thaipisuttikul, D. Spencer, E. Haugen, S. Ernst, O. Will, R. Kaul, C. Raymond, R. Levy, L. Chun-Rong, D. Guenthner, D. Bovee, M. V. Olson, and C. Manoil. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 100:14339-14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen, T., S. S. Pedersen, S. Garne, C. Heilmann, N. Høiby, and C. Koch. 1987. Colistin inhalation therapy in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. J. Antimicrob. Chemother. 19:831-838. [DOI] [PubMed] [Google Scholar]
- 28.Kawasaki, K., K. China, and M. Nishijima. 2007. Release of the lipopolysaccharide deacylase PagL from latency compensates for a lack of lipopolysaccharide aminoarabinose modification-dependent resistance to the antimicrobial peptide polymyxin B in Salmonella enterica. J. Bacteriol. 189:4911-4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawasaki, K., R. K. Ernst, and S. I. Miller. 2004. 3-O-deacylation of lipid A by PagL, a PhoP/PhoQ-regulated deacylase of Salmonella typhimurium, modulates signaling through Toll-like receptor 4. J. Biol. Chem. 279:20044-20048. [DOI] [PubMed] [Google Scholar]
- 30.Lewenza, S., J. L. Gardy, F. S. Brinkman, and R. E. W. Hancock. 2005. Genome-wide identification of Pseudomonas aeruginosa exported proteins using a consensus computational strategy combined with a laboratory-based PhoA fusion screen. Genome Res. 15:321-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, J., R. L. Nation, J. D. Turnidge, R. W. Milne, K. Coulthard, C. R. Rayner, and D. L. Paterson. 2006. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 6:589-601. [DOI] [PubMed] [Google Scholar]
-
31a.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the
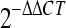 method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar] - 32.Lyczak, J. B., C. L. Cannon, and G. B. Pier. 2000. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect. 2:1051-1060. [DOI] [PubMed] [Google Scholar]
- 33.Macfarlane, E. L. A., A. Kwasnicka, M. M. Ochs, and R. E. W. Hancock. 1999. PhoP-PhoQ homologues in Pseudomonas aeruginosa regulate expression of the outer-membrane protein OprH and polymyxin B resistance. Mol. Microbiol. 34:305-316. [DOI] [PubMed] [Google Scholar]
- 34.Mao, F., P. Dam, J. Chou, V. Olman, and Y. Xu. 2009. DOOR: a database for prokaryotic operons. Nucleic Acids Res. 37:D459-D463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McPhee, J. B., M. Bains, G. Winsor, S. Lewenza, A. Kwasnicka, M. D. Brazas, F. S. Brinkman, and R. E. W. Hancock. 2006. Contribution of the PhoP-PhoQ and PmrA-PmrB two-component regulatory systems to Mg2+-induced gene regulation in Pseudomonas aeruginosa. J. Bacteriol. 188:3995-4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McPhee, J. B., S. Lewenza, and R. E. W. Hancock. 2003. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 50:205-217. [DOI] [PubMed] [Google Scholar]
- 37.Overhage, J., A. Campisano, M. Bains, E. C. Torfs, B. H. Rehm, and R. E. W. Hancock. 2008. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 76:4176-4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pamp, S. J., M. Gjermansen, H. K. Johansen, and T. Tolker-Nielsen. 2008. Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol. Microbiol. 68:223-240. [DOI] [PubMed] [Google Scholar]
- 39.Piers, K. L., M. H. Brown, and R. E. W. Hancock. 1994. Improvement of outer membrane-permeabilizing and lipopolysaccharide-binding activities of an antimicrobial cationic peptide by C-terminal modification. Antimicrob. Agents Chemother. 38:2311-2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rahme, L. G., E. J. Stevens, S. F. Wolfort, J. Shao, R. G. Tompkins, and F. M. Ausubel. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899-1902. [DOI] [PubMed] [Google Scholar]
- 41.Rodrigue, A., Y. Quentin, A. Lazdunski, V. Méjean, and M. Foglino. 2000. Two-component systems in Pseudomonas aeruginosa: why so many? Trends Microbiol. 8:498-504. [DOI] [PubMed] [Google Scholar]
- 42.Romeo, D., B. Skerlavaj, M. Bolognesi, and R. Gennaro. 1988. Structure and bactericidal activity of an antibiotic dodecapeptide purified from bovine neutrophils. J. Biol. Chem. 263:9573-9575. [PubMed] [Google Scholar]
- 43.Rowe, S. M., S. Miller, and E. J. Sorscher. 2005. Cystic fibrosis. N. Engl. J. Med. 352:1992-2001. [DOI] [PubMed] [Google Scholar]
- 44.Rozen, S., and H. J. Skaletsky. 2000. Primer3 on the WWW for general users and for biologist programmers, p. 365-386. In S. Krawetz and S. Misener (ed.), Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, NJ. [DOI] [PubMed]
- 45.Schurek, K. N., A. K. Marr, P. K. Taylor, I. Wiegand, L. Semenec, B. K. Khaira, and R. E. W. Hancock. 2008. Novel genetic determinants of low-level aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 52:4213-4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Selsted, M. E., M. J. Novotny, W. L. Morris, Y. Q. Tang, W. Smith, and J. S. Cullor. 1992. Indolicidin, a novel bactericidal tridecapeptide amide from neutrophils. J. Biol. Chem. 267:4292-4295. [PubMed] [Google Scholar]
- 47.Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J. Hickey, F. S. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L. Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G. K. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E. W. Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959-964. [DOI] [PubMed] [Google Scholar]
- 48.Trent, M. S., W. Pabich, C. R. Raetz, and S. I. Miller. 2001. A PhoP/PhoQ-induced lipase (PagL) that catalyzes 3-O-deacylation of lipid A precursors in membranes of Salmonella typhimurium. J. Biol. Chem. 276:9083-9092. [DOI] [PubMed] [Google Scholar]
- 49.Windholz, M., S. Budavari, L. Stroumtsos, and N. Fertig. 1976. The Merck index. Merck, Rahway, NJ.
- 50.Wu, M., and R. E. W. Hancock. 1999. Improved derivatives of bactenecin, a cyclic dodecameric antimicrobial cationic peptide. Antimicrob. Agents Chemother. 43:1274-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang, L., M. G. Scott, H. Yan, L. D. Mayer, and R. E. W. Hancock. 2000. Interaction of polyphemusin I and structural analogs with bacterial membranes, lipopolysaccharide, and lipid monolayers. Biochemistry 39:14504-14514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.