

Abstract
The mass balance and pharmacokinetics of telavancin, a semisynthetic lipoglycopeptide antimicrobial agent, were characterized in an open-label, phase 1 study of six healthy male subjects. After a single 1-h intravenous infusion of 10 mg/kg [14C]telavancin (0.68 μCi/kg), blood, urine, and feces were collected at regular intervals up to 216 h postdose. Whole blood, plasma, urine, and fecal samples were assayed for total radioactivity using scintillation counting; plasma and urine were also assayed for parent drug and metabolites using liquid chromatography with tandem mass spectrometry. The concentration-time profiles for telavancin and total radioactivity in plasma were comparable from 0 to 24 h after the study drug administration. Telavancin accounted for >95% and 83% of total radioactivity in plasma at 12 h and 24 h, respectively. By 216 h, approximately 76% of the total administered dose was recovered in urine while only 1% was collected in feces. Unchanged telavancin accounted for most (83%) of the eliminated dose. Telavancin metabolite THRX-651540 along with two other hydroxylated metabolites (designated M1 and M2) accounted for the remaining radioactivity recovered from urine. The mean concentrations of total radioactivity in whole blood were lower than the concentration observed in plasma, and mean concentrations of THRX-651540 in plasma were minimal relative to mean plasma telavancin concentrations. These observations demonstrate that most of an administered telavancin dose is eliminated unchanged via the kidneys. Intravenous telavancin at 10 mg/kg was well tolerated by all subjects.
Telavancin is a once-daily injectable, semisynthetic lipoglycopeptide antibiotic with bactericidal activity in vitro against a broad spectrum of clinically important Gram-positive bacteria including methicillin-resistant Staphylococcus aureus (MRSA), streptococci, and enterococci (4, 5, 9, 10). Telavancin has a dual mode of action, involving inhibition of bacterial cell wall peptidoglycan biosynthesis and disruption of bacterial cell membrane functional integrity (7). In phase 3 trials, telavancin was shown to be noninferior to conventional vancomycin therapy for treatment of complicated skin and skin-structure infections (cSSSIs) and nosocomial pneumonia caused by Gram-positive bacteria and demonstrated an acceptable safety and tolerability profile (13, 15, 16). Telavancin is indicated in the United States and Canada for the treatment of adult patients with cSSSI caused by susceptible Gram-positive bacteria and is under investigation for the treatment of Gram-positive nosocomial pneumonia.
Single- and multiple-dose pharmacokinetic parameters for telavancin have been evaluated in phase 1 studies, in which telavancin displayed linear and time-invariant pharmacokinetics following intravenous administration (6, 14, 17). Since telavancin is largely excreted intact in urine along with small amounts of its primary hydroxylated metabolite THRX-651540 (data on file with Theravance, Inc., South San Francisco, CA), renal clearance is thought to be the major route of elimination. The objectives of the present study were to confirm this hypothesis by characterizing the mass balance and pharmacokinetics of single-dose [14C]telavancin in human subjects.
MATERIALS AND METHODS
Radiolabeled telavancin and dosage form.
Nonradiolabeled telavancin was synthesized by Theravance, Inc. (South San Francisco, CA). [14C]telavancin was synthesized by ViTrax (Placentia, CA). The 14C atom was located on the phosphonate group with specific activity of 17.45 μCi/mg (Fig. 1). Radiochemical purity was 96.1%. Nonradiolabeled telavancin was used as a means of diluting [14C]telavancin prior to intravenous administration. Telavancin for injection was supplied as a sterile, pyrogen-free, lyophilized powder composed of 250 mg of telavancin, 2.5 g of hydroxypropyl-β-cyclodextrin, and 312.5 mg of mannitol. Sodium hydroxide and/or hydrochloric acid was used for pH adjustment after the powder had been reconstituted in 5% dextrose for injection. This formulation was the same as that used in previous clinical studies (6, 14-16). The final infusion solution had a telavancin concentration of 5 mg/ml, which was equivalent to a radioactive concentration of 0.34 μCi/ml. Since the dose of telavancin to be administered was 10 mg/kg infused over 1 h, the dose of radioactivity administered to each subject was 0.68 μCi/kg. The associated radiation exposure falls within International Commission on Radiological Protection Guidelines for category IIa studies (8).
FIG. 1.

Structure of telavancin, showing metabolite hydroxylation sites and location of radiolabel.
Subjects and protocol.
This single-center, open-label, nonrandomized phase 1 study was conducted at Charles River Laboratories Clinical Services (Edinburgh, United Kingdom) after approval by the Edinburgh Independent Ethics Committee and in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. Six eligible male subjects who gave written informed consent and were aged between 30 and 55 years, with a body weight of 50 to 100 kg and a body mass index (BMI) of 18 to 29 kg/m2, each received a single 1-h intravenous infusion of 10 mg/kg [14C]telavancin. All subjects were in good health based on screening results of routine clinical laboratory tests, physical examinations, vital signs, 12-lead electrocardiograms (ECGs), and urine drug tests that were performed within 21 days prior to study drug administration. Subjects were not to have a history of drug hypersensitivity or allergic reactions related to glycopeptide antibiotics. Other exclusion criteria included the following: administration of any investigational medicinal product within 12 weeks before entry to the study; a need for any medication within 14 days of study entry; any surgical or medical condition that, in the judgment of the investigator, might interfere with the distribution, metabolism, or excretion of telavancin; history of any drug or alcohol abuse in the past 2 years (or alcohol consumption greater than 21 units per week); administration of radiolabeled substances or exposure to significant radiation within the past 12 months; donation or loss of >400 ml of blood within 12 weeks prior to study entry; and a positive blood test for HIV or hepatitis B or C.
After an overnight fast (water was permitted) in the clinical unit 1 day before dosing, subjects were fed a light breakfast approximately 2 h before administration of the study drug. Thereafter, subjects remained fasted until ≥4 h after the infusion had finished. Smoking tobacco was not permitted from 2 h before dosing until 24 h after dosing. Consumption of caffeine, alcohol, and grapefruit or cranberry juice was not permitted during the study. Subjects were to remain in the clinical unit until 216 h after dosing but could have been discharged after 168 h if >90% of the dose was recovered and/or <1% of the dose was excreted in a 24-h period.
Sample collection.
Blood samples (12 ml) were collected immediately before the infusion and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, 72, 96, 120, 144, 168, 192, and 216 h after the start of infusion into heparinized glass tubes and chilled on ice. One aliquot of heparinized whole blood (2 ml) per time point was stored at −4°C for total radioactivity analysis. The remaining blood (10 ml) was centrifuged, and the harvested plasma was stored at approximately −20°C. One plasma sample (0.5 ml) was used for metabolite profiling while another (0.5 ml) was used for parent drug analysis. The remaining plasma was used for total radioactivity analysis.
Urine samples were collected before [14C]telavancin administration and then during intervals of 0 to 4, 4 to 8, 8 to 12, 12 to 24, 24 to 48, 48 to 72, 72 to 96, 96 to 120, 120 to 144, 144 to 168, 168 to 192, and 192 to 216 h postdose. Samples were refrigerated until the end of the block collection periods, and the total weight and volume of urine were then recorded. At the end of each collection period, two aliquots of urine (20 ml) were taken from the sample and stored in polypropylene tubes at −20°C until analyzed. Urine samples for metabolite profiling were pooled for each subject from 0 to 4, 4 to 8, 8 to 12, 12 to 24, and 24 to 48 h.
All bowel movements were collected preinfusion and at block intervals of 0 to 24, 24 to 48, 48 to 72, 72 to 96, 96 to 120, 120 to 144, 144 to 168 h, 168 to 192, and 192 to 216 h postdose. At the end of each collection period, a portion of the sample (50 g) was retained and stored at approximately −20°C until analyzed. The remaining fecal homogenate was combined with that collected at the end of each time interval and stored at approximately −20°C.
Determination of radioactivity.
All blood, plasma, urine, and fecal samples were assayed for total radioactivity by liquid scintillation counting (LSC). Quench corrections were made by the external standard method. For liquid samples, duplicate portions from each time block interval were diluted with water or compatible solvent (if necessary) and then dissolved in scintillation fluid. All samples prepared in scintillation fluid were subjected to LSC for 5 min together with representative blank samples using a liquid scintillation analyzer with automatic quench correction. Duplicate portions of whole blood and fecal homogenate samples from each time block interval were combusted with a Packard Tri-Carb 307 Automatic Sample Oxidizer, and the resultant 14CO2 was trapped in a suitable absorbent scintillation system. The lower limit of reliable detection of radiation was 30 dpm above background noise. Total radioactivity in plasma and blood was expressed as μg equivalents (eq) of telavancin/ml.
Plasma assays.
Plasma samples were transferred frozen on dry ice to Covance Laboratories, Inc. (Indianapolis, IN). Determination of telavancin and THRX-651540 concentrations was performed using a validated bioanalytical assay with liquid chromatography with tandem mass spectrometry (LC-MS/MS) (Theravance, Inc., internal report) using a Merck Chromolith SpeedROD RP-18e column and positive electrospray ionization on a Perkin-Elmer Sciex API 3000. The method was linear over the range of 0.250 to 100 μg/ml in human plasma, and the lower limit of quantification (LLOQ) was 0.250 μg/ml for both analytes. The overall accuracy for the calibration standards and quality control samples ranged from 85% to 115%.
Metabolite profiling and identification.
Urine samples were pooled for each subject from 0 to 48 h postdose by combining an equal percentage (by weight) from each time point. For metabolite profiling, an Agilent 1100 high-performance liquid chromatography (HPLC) system equipped with a Prodigy ODS3 (250 by 4.6 mm; 5-μm particle size) LC column and a flow rate of 1.0 ml/min were used. The HPLC mobile phase A was 0.1% formic acid in water, and mobile phase B was acetonitrile. The gradient was as follows: 5% phase B for 0 to 3 min, 5% to 80% phase B for 3 to 43 min, and then a 5-min hold followed by 5% phase B. The total run time was 57 min. Centrifuged urine samples were profiled for metabolites by Packard Radiomatic 150 Series Flow Scintillation Analyzers.
Urine samples from subject 0003 M, collected over 0 to 4, 4 to 8, 8 to 12, 12 to 24, and 24 to 48 h postdose, were used for metabolite isolation (this subject had the largest amount of metabolites excreted over the 48-h period). Human urine samples acidified with 2% phosphoric acid were first extracted with a polymeric weak cation exchanger solid-phase extraction (SPE) column (Phenomenex Strata-XCW). The metabolites were eluted with 5% trifluoroacetic acid (TFA) in 50% acetonitrile. An aliquot of 100 μl of SPE-purified sample was injected into an Agilent 1100 HPLC system equipped with a Phenomenex LUNA (5-μm particle size; 4.6 by 150 mm) LC column with a flow rate of 1.5 ml/min. The HPLC mobile phase A was 0.05% TFA in 5% acetonitrile, and mobile phase B was 0.05% TFA in 95% acetonitrile. The gradient was as follows: 10% phase B for 0 to 15 min, 10% to 20% phase B for 15 to 25 min, 20% to 40% phase B for 25 to 45 min, and then immediately to 90% phase B, a hold for 5 min, and then back to 10% phase B. The total run time was 60 min. Extracted samples were profiled for metabolites using UV spectrophotometry (214 nm) or mass spectrometry (API-3000; Perkin-Elmer Sciex Instruments, Norwalk, CT). Fractions containing each metabolite were manually collected and then pooled for a second HPLC purification. The same HPLC system, column, flow rate, and mobile phases were used as for the first HPLC purification. For the purification of metabolite M1, an isocratic gradient was used with 15% phase B and a run time of 20 min. For the purification of metabolite M2, the same gradient as described in the first HPLC purification was used. For the purification of metabolite M3, an isocratic gradient was used with 25% phase B and a run time of 10 min. A complete nuclear magnetic resonance (NMR) data set was acquired on telavancin and THRX-651540 as well as on the purified metabolites (M1, M2, and M3), which included 1H spectrum, correlation spectroscopy (COSY), total correlation spectroscopy (TOCSY), rotating frame Overhauser effect spectroscopy (ROESY), heteronuclear single-quantum coherence (HSQC), and heteronuclear multiple-bond correlation (HMBC). Telavancin and THRX-651540 or purified metabolites were placed in amber vials and dissolved with 750 μl of dry dimethyl sulfoxide (DMSO)-d6. The resulting individual solutions were transferred into dry, nitrogen gas-purged, 5-mm NMR tubes. All NMR experiments were acquired at 303 K on a Bruker Avance 600 operating at a 1H frequency of 600.13 MHz and a 13C frequency of 150.91 MHz. All chemical shifts were referenced to DMSO-d6 multiplet, 2.5 ppm for 1H and 39.5 ppm for 13C. The resulting data were transferred to a personal computer and processed using Bruker Xwin3.5 software.
No metabolite profiling was performed on fecal samples as fecal excretion of radioactive material was negligible.
Pharmacokinetic and statistical analyses.
Pharmacokinetic parameters of telavancin and THRX-651540 in plasma and total radioactivity in blood and plasma were determined by noncompartmental analysis using WinNonlin pharmacokinetic software (version 4.1; Pharsight, Mountain View, CA). For the purpose of calculating pharmacokinetic parameters, telavancin and THRX-651540 concentrations below the LLOQ were defined as 0 μg/ml. Summary statistics (i.e., mean, standard deviation [SD], and range) were performed for all pharmacokinetic parameters. For all subjects, peak concentrations (Cmax) and time to Cmax (Tmax) were taken directly from plasma or whole-blood concentration-time data. The terminal elimination rate constant (λz) was estimated by linear regression of the natural logarithms of plasma concentrations versus time during the terminal phase. The terminal-phase elimination half-life (t1/2) was calculated as ln(2)/λz. The areas under the plasma or whole-blood concentration-time curve from time zero to the last measurable concentration (Clast), AUC(0-t), and from time zero to infinity, AUC(0-∞) [sum of AUC(0-t) and Clast/λz], were estimated by the linear trapezoidal rule. Total apparent body clearance (CL) of telavancin from the plasma after intravenous administration was calculated as dose/AUC0-∞. The area under the first moment curve (AUMC) was calculated using the trapezoidal rule, and the mean residence time (MRT) was defined as AUMC/AUC(0-∞). The volume of distribution at the steady state (Vss) was calculated as the product of CL and MRT. The total amounts of radioactivity excreted in urine or feces up to 216 h (Ae and Fe, respectively) were calculated according to the following equations: 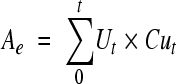 or
or 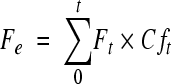 , where Ut or Ft and
, where Ut or Ft and 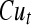 or
or 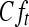 are the urine volume or fecal weight and concentration based on total radioactivity in urine or feces, respectively, for time t (up to 216 h). The percent dose excreted in urine or feces based on total radioactivity was calculated according to the following equation: 100% × Ae/administered dose or 100% × Fe/administered dose.
are the urine volume or fecal weight and concentration based on total radioactivity in urine or feces, respectively, for time t (up to 216 h). The percent dose excreted in urine or feces based on total radioactivity was calculated according to the following equation: 100% × Ae/administered dose or 100% × Fe/administered dose.
The total recovery (percent excretion in urine plus percent excretion in feces) was also calculated. Ae 0-48 represents the total amount of telavancin excreted in urine during the 48-h postdose interval. Ae 0-48 was calculated by multiplying the percentage of dose recovered in urine as telavancin over 0 to 48 h based on data from metabolite profiling in urine by the total amount (mg) of telavancin administered. Dividing this value by the total radioactive count gave the proportion of dose recovered as telavancin over this time period. Telavancin renal clearance (CLr) was calculated as the ratio of Ae 0-48 to AUC(0-∞).
Safety and tolerability.
Safety was assessed from adverse event (AE) reports, physical examinations, 12-lead and continuous ECGs, vital signs, and laboratory evaluations performed at specified times during the study. All AEs were recorded from the time of dosing on day 1 until study discharge. The study investigators assessed all AEs for type, frequency, severity, and relationship to telavancin.
RESULTS
Subjects.
All six healthy male subjects who entered the study completed single-dose administration of [14C]telavancin (10 mg/kg) and were included in the pharmacokinetic analyses. The mean ± SD age was 44.8 ± 6.5 years, and the mean ± SD BMI was 23.30 ± 3.23 kg/m2. No serious AEs were observed, and no subject was withdrawn from the study because of an AE. A total of eight AEs in five subjects were reported during the study. All AEs were mild in intensity and had resolved by study end. The most frequently reported AE was dysgeusia (reported by three subjects); all episodes were considered to be possibly or probably related to treatment. All results of routine clinical laboratory testing were within the normal ranges. There was a small mean change from baseline increase in the QT and corrected QT (QTc) intervals (31.8 and 14.2 ms, respectively) from predose to 1 h postinfusion, but these observations were not considered to be clinically significant, based on the results of a previous study investigating the effect of telavancin on cardiac repolarization in healthy subjects (1).
Whole-blood and plasma pharmacokinetics.
The mean plasma concentration-versus-time profiles for telavancin and total radioactivity were comparable over the first 24 h after study drug administration (Fig. 2). Thus, values of Cmax, Tmax, AUC(0-t), and CL in the plasma compartment were similar for telavancin (as determined by LC-MS/MS) and total radioactivity (Table 1). However, after 24 h the telavancin and radioactivity profiles began to deviate, resulting in large differences in mean estimates of plasma t1/2 (7.08 h versus 94.2 h) and Vss (150 ml/kg versus 478 ml/kg), respectively (Table 1). Of note, the mean t1/2 for total radioactivity was based on limited data since its value was nearly as long as the interval over which it was calculated.
FIG. 2.

Mean (± standard deviation) concentrations of total radioactivity, telavancin, and THRX-651540 in plasma and whole blood following a 1-h intravenous infusion of [14C]telavancin (10 mg/kg) to healthy male volunteers (n = 6).
TABLE 1.
Noncompartmental pharmacokinetics of telavancin and THRX-651540 in plasma and whole blood following intravenous administration of [14C]telavancin to healthy male volunteersa
| Parameter | Value for telavancin |
Value for THRX-651540 in plasma by LC-MS/MS | ||
|---|---|---|---|---|
| LC-MS/MS of plasma | Total radioactivity in: |
|||
| Plasma | Whole blood | |||
| Cmax (μg/ml or μg-eq/ml) | 93.6 ± 10.2 | 89.6 ± 11.8 | 43.7 ± 7.0 | 0.493 ± 0.107 |
| Tmax (h) | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | 13.8 ± 8.5 |
| AUC(0-t) (μg/h/ml or μg-eq/h/ml) | 620 ± 125 | 764 ± 119 | 371 ± 67 | 11.6 ± 6.2 |
| AUC(0-∞) (μg/h/ml or μg-eq/h/ml) | 649 ± 103 | 827 ± 120 | 396 ± 56 | NR |
| t1/2 (h) | 7.08 ± 0.50 | 94.2 ± 17.8 | 9.89 ± 1.21 | NR |
| CL (ml/h/kg) | 15.7 ± 2.5 | 12.3 ± 1.8 | 25.7 ± 3.8 | NR |
| Vss (ml/kg) | 150 ± 19 | 478 ± 102 | 301 ± 28 | NR |
| CLr (ml/h/kg) | 9.00 ± 1.32 | NA | NA | NR |
| % Excreted in urine as telavancin (0 to 48 h) | 82.3 ± 7.4 | NA | NA | NR |
[14C]telavancin was administered at a dose of 10 mg/kg to six subjects. All values are presented as arithmetic mean ± SD. NA, not applicable; NR, not reported.
Radioactivity was evident in whole blood 1 h following intravenous infusion of [14C]telavancin and exhibited a qualitatively similar concentration-time profile to that of telavancin in plasma. The mean concentrations of radioactivity in whole blood were substantially lower than those observed in plasma (Fig. 2), with mean whole blood-to-plasma total radioactivity ratios per time point ranging from 0.491 to 0.608 up to 24 h postdose. Radioactivity in whole blood persisted at least up to 96 h postdose, and mean concentrations in whole blood were similar to those in plasma after 48 h postdose (Fig. 2).
As shown in Fig. 2, mean concentrations of THRX-651540 in plasma were minimal relative to mean plasma telavancin concentrations: mean ± SD Cmax and AUC ratios of THRX-651540 versus telavancin were 0.00527 ± 0.00090 and 0.0185 ± 0.0068, respectively. The mean Cmax for THRX-651540 (0.493 μg/ml) occurred approximately 13.8 h after study drug administration and remained close to this concentration at 24 h postdose (Table 1).
Concentrations of telavancin in plasma following intravenous administration of [14C]telavancin accounted for >95% of the total plasma radioactivity for up to 12 h postdose and for 83% of the total plasma radioactivity for up to 24 h postdose (Fig. 2). At 48 h postdose, unchanged telavancin accounted for 13% of the total radioactivity in plasma. While plasma radioactivity was still quantifiable after 48 h postdose, plasma telavancin concentrations were below the LLOQ of LC-MS/MS detection. Mean CLr of telavancin (based on cumulative recovery of telavancin in urine over 0 to 48 h) was 9.00 ml/h/kg (Table 1).
Mass balance.
Mean total recovery of the administered radioactivity across all subjects was 77.0% (range, 71.2% to 82.4%) at 216 h postdose (Fig. 3). Around 50% (range, 45.1% to 56.5%) of the administered radiation was recovered within 12 h postdose, and 75% (range, 69.5% to 81.0%) of the administered dose was recovered 72 h postdose. After 72 h postdose, all subjects excreted <1% of the administered dose during each subsequent 24-h collection period. The overall dose recovered in urine during the 0- to 216-h collection period was 76.3% (range, 70.8% to 81.9%). There was negligible excretion of radioactivity in feces, which accounted for a mean of 0.7% (range, 0.4% to 0.9%) of the administered dose by the end of the last collection interval.
FIG. 3.

Mean cumulative urinary and fecal excretion of radioactivity after a single intravenous dose of [14C]telavancin (10 mg/kg) to healthy male volunteers (n = 6). Whole-blood concentrations from 48 h to 216 h postdose were <30 dpm above background and were not shown in the plot.
Telavancin and metabolite profiling in urine.
Urine sample pools (0 to 48 h) used for radiochemical profiling accounted for 73.2% (range, 68.0% to 79.4%) of the administered dose and in all subjects accounted for >95% of the total radioactivity excreted via urine over the 216-h collection period. A representative UV chromatogram is shown in Fig. 4. Most of the eliminated dose (approximately 82%) during this period was identified as unchanged telavancin. Three metabolites, designated M1, M2, and M3, were identified and confirmed by LC-MS/MS as hydroxylated metabolites of telavancin (Fig. 5). M3 had the same retention time as the 7-hydroxy metabolite standard, THRX-651540, and M2 and M1 had the same retention times as the 8-hydroxy and 9-hydroxy metabolite standards, respectively (metabolite hydroxylation sites are indicated in Fig. 1).
FIG. 4.

Representative UV chromatogram of solid-phase extraction of a purified human urine sample belonging to subject 0003 M at 12 to 24 h after receiving [14C]telavancin (10 mg/kg).
FIG. 5.

Representative mass spectra of telavancin and three hydroxylated conjugates after solid-phase extraction of a purified human urine sample. (a) Telavancin. (b) Metabolite M1 retention time, 25.13 min. (c) Metabolite M2 retention time, 25.49 min. (d) Metabolite M3 retention time, 26.19 min.
The metabolite distribution based on peak area for each metabolite is summarized in Table 2. Only 17% of the total administered dose was excreted in urine as telavancin metabolites. M3 was the major metabolite present in urine, accounting for approximately 50% of the total peak area for all three metabolites (based on UV absorbance).
TABLE 2.
Distribution of radioactivity in pooled urine samples from six healthy male subjects 48 h after intravenous administration of 10 mg/kg [14C]telavancin
| Analyte | Retention time (min) | % of total administered dosea | Metabolite distribution (% analyte in recovered dose)a |
|---|---|---|---|
| Telavancin | 42.7 | 60.4 ± 7.2 | 82.3 ± 7.4 |
| THRX-651540 or M3 (7-OH) | 27.8 | 6.45 ± 2.37 | 8.88 ± 3.58 |
| M2 (8-OH) | 26.9 | 3.12 ± 1.27 | 4.31 ± 1.95 |
| M1 (9-OH) | 26.6 | 3.23 ± 1.25 | 4.46 ± 1.90 |
| All | 73.2 ± 3.8 | 100 |
Values are mean ± SD.
DISCUSSION
The present investigation has demonstrated that telavancin is mostly excreted as unchanged drug in urine. The study has further characterized the concentration-time profile for telavancin in healthy male subjects, showing that most of a 10 mg/kg intravenous dose is present as intact parent drug at 12 h (>95%) and 24 h (83%) postdose. These data are consistent with the observed low systemic exposure to the primary metabolite, THRX-651540, which represents oxidation on the alkyl side chain of telavancin.
Analysis of telavancin concentrations (based on total radioactivity) in whole blood and plasma suggests a low degree of red blood cell partitioning for this compound. Specifically, whole blood-to-plasma systemic exposure ratios for Cmax, AUC(0-t), and AUC(0-∞) ranged from 0.48 to 0.49, which approximates to 1-hematocrit in healthy males (range, 0.41 to 0.53) (3). There were no notable changes in any hematological indices assessed between baseline and any time point until 168 h postdose.
The mean t1/2 of telavancin (based on LC-MS/MS analysis) was approximately 7 h, similar to previously reported values (6, 14). The mean t1/2 of telavancin based on the total radioactivity measurement was longer (approximately 94 h). Whereas the total radioactivity measurement represents the amount of radiolabel in the sample, the LC-MS assay specifically detects the parent molecule and metabolites. At 48 h postdose, the mean telavancin concentration based on the LC-MS/MS measurement was 0.221 μg/ml; however, the total radioactivity in plasma at 48 h postdose was 1.42 μg-eq/ml, with telavancin accounting for only 13% of the total radioactivity. Other minor metabolites or impurities may account for the remaining radioactivity in plasma. We postulate that the longer mean t1/2 as measured by the total radioactivity may potentially result from emanation from tissues or low clearance of metabolites. Estimates of telavancin t1/2 based on total radioactivity were approximately 10-fold longer in plasma compared than in whole blood. This long t1/2 in plasma may reflect release of [14C]telavancin from tissue, low clearance rates of metabolites, or possible differences in assay sensitivity between blood and plasma. It should also be noted that the plasma and whole-blood concentration-time intervals from which telavancin t1/2 values were calculated were different.
The typical recovery of radiolabel in human mass balance studies is ≥80% (12). In this study, 77% of the total dose was excreted at 216 h according to our mass balance calculations (76.3% in urine and 0.7% in feces). It is not unusual for there to be incomplete recovery of antimicrobial-associated radioactivity in human mass balance studies such as this. In similar studies with other 14C-labeled antimicrobials, the recovery of vancomycin, ertapenem, and daptomycin in urine was comparable with that of telavancin in this study (75% to 80%). Urine recovery of tigecycline was lower, around 33%, with fecal recovery accounting for 59%. Approximate total recovery in these studies was 83% for daptomycin, 90% for ertapenem, and 92% for tigecycline (2, 11, 18, 19).
Following intravenous administration of [14C]telavancin in rats and dogs, urinary recovery of radiation by 24 h postdose was 75% and 62%, respectively. The total recovery of the administered radioactivity in organs, tissues, and excreta across all animals was >90%, indicating that nonexcreted radioactivity was trapped outside the plasma compartment (Theravance, Inc., data on file). As in humans, dogs excreted <1% of the administered dose in feces.
In summary, the results of this study demonstrate that most of a single telavancin dose is excreted unchanged in urine within 48 h after intravenous administration. The vast majority of radioactive component in plasma over the 24-h dosing interval was microbiologically active parent telavancin. Our mass balance calculations allow for a maximum of 30% to 37% of dose to be metabolized to multiple hydroxylated conjugates within 216 h of administration, but this range does not account for proven distribution of intact telavancin to tissue and intracellular compartments of the immune system. Telavancin at 10 mg/kg was well tolerated in all six healthy male subjects.
Acknowledgments
The research and publication process were supported jointly by Theravance, Inc., and Astellas Pharma Global Development, Inc.
All authors were employees of Theravance, Inc., during the conduct of this study and preparation of the manuscript but have since left the company.
Editorial support was provided by medical writers at Envision Scientific Solutions.
Footnotes
Published ahead of print on 1 June 2010.
REFERENCES
- 1.Barriere, S., F. Genter, E. Spencer, M. Kitt, D. Hoelscher, and J. Morganroth. 2004. Effects of a new antibacterial, telavancin, on cardiac repolarization (QTc interval duration) in healthy subjects. J. Clin. Pharmacol. 44:689-695. [DOI] [PubMed] [Google Scholar]
- 2.Baxter Healthcare Corporation. 2003. Vancocin package insert. Baxter Healthcare Corporation, Deerfield, IL.
- 3.Davies, B., and T. Morris. 1993. Physiological parameters in laboratory animals and humans. Pharm. Res. 10:1093-1095. [DOI] [PubMed] [Google Scholar]
- 4.Draghi, D. C., B. M. Benton, K. M. Krause, C. Thornsberry, C. Pillar, and D. F. Sahm. 2008. Comparative surveillance study of telavancin activity against recently collected gram-positive clinical isolates from across the United States. Antimicrob. Agents Chemother. 52:2383-2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Draghi, D. C., B. M. Benton, K. M. Krause, C. Thornsberry, C. Pillar, and D. F. Sahm. 2008. In vitro activity of telavancin against recent Gram-positive clinical isolates: results of the 2004-05 Prospective European Surveillance Initiative. J. Antimicrob. Chemother. 62:116-121. [DOI] [PubMed] [Google Scholar]
- 6.Gotfried, M. H., J. P. Shaw, B. M. Benton, K. M. Krause, M. R. Goldberg, M. M. Kitt, and S. L. Barriere. 2008. Intrapulmonary distribution of intravenous telavancin in healthy subjects and effect of pulmonary surfactant on in vitro activities of telavancin and other antibiotics. Antimicrob. Agents Chemother. 52:92-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Higgins, D. L., R. Chang, D. V. Debabov, J. Leung, T. Wu, K. M. Krause, E. Sandvik, J. M. Hubbard, K. Kaniga, D. E. Schmidt, Jr., Q. Gao, R. T. Cass, D. E. Karr, B. M. Benton, and P. P. Humphrey. 2005. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 49:1127-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.International Commission on Radiological Protection. 1993. ICRP publication 62: radiological protection in biomedical Research, 62. Pergamon Press, Oxford, United Kingdom.
- 9.Krause, K. M., M. Renelli, S. Difuntorum, T. X. Wu, D. V. Debabov, and B. M. Benton. 2008. In vitro activity of telavancin against resistant gram-positive bacteria. Antimicrob. Agents Chemother. 52:2647-2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leuthner, K. D., C. M. Cheung, and M. J. Rybak. 2006. Comparative activity of the new lipoglycopeptide telavancin in the presence and absence of serum against 50 glycopeptide non-susceptible staphylococci and three vancomycin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 58:338-343. [DOI] [PubMed] [Google Scholar]
- 11.Merck and Co. 2008. Invanz package insert. Merck and Co., Whitehouse Station, NJ.
- 12.Roffey, S. J., R. S. Obach, J. I. Gedge, and D. A. Smith. 2007. What is the objective of the mass balance study? A retrospective analysis of data in animal and human excretion studies employing radiolabeled drugs. Drug Metab. Rev. 39:17-43. [DOI] [PubMed] [Google Scholar]
- 13.Rubinstein, E., G. R. Corey, M. E. Stryjewski, H. W. Boucher, R. N. Daly, F. C. Genter, S. L. Barriere, M. M. Kitt, and H. D. Friedland. 2008. Telavancin for hospital-acquired pneumonia, including ventilator-associated pneumonia: the ATTAIN studies. Clin. Microbiol. Infect. 14:S14. [Google Scholar]
- 14.Shaw, J. P., J. Seroogy, K. Kaniga, D. L. Higgins, M. Kitt, and S. Barriere. 2005. Pharmacokinetics, serum inhibitory and bactericidal activity, and safety of telavancin in healthy subjects. Antimicrob. Agents Chemother. 49:195-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stryjewski, M. E., V. H. Chu, W. D. O'Riordan, B. L. Warren, L. M. Dunbar, D. M. Young, M. Vallée, V. G. Fowler, Jr., J. Morganroth, S. L. Barriere, M. M. Kitt, and G. R. Corey. 2006. Telavancin versus standard therapy for treatment of complicated skin and skin structure infections caused by gram-positive bacteria: FAST 2 study. Antimicrob. Agents Chemother. 50:862-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stryjewski, M. E., D. R. Graham, S. E. Wilson, W. O'Riordan, D. Young, A. Lentnek, D. P. Ross, V. G. Fowler, A. Hopkins, H. D. Friedland, S. L. Barriere, M. M. Kitt, and G. R. Corey. 2008. Telavancin versus vancomycin for the treatment of complicated skin and skin-structure infections caused by gram-positive organisms. Clin. Infect. Dis. 46:1683-1693. [DOI] [PubMed] [Google Scholar]
- 17.Sun, H. K., K. Duchin, C. H. Nightingale, J. P. Shaw, J. Seroogy, and D. P. Nicolau. 2006. Tissue penetration of telavancin after intravenous administration in healthy subjects. Antimicrob. Agents Chemother. 50:788-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodworth, J. R., E. H. Nyhart, Jr., G. L. Brier, J. D. Wolny, and H. R. Black. 1992. Single-dose pharmacokinetics and antibacterial activity of daptomycin, a new lipopeptide antibiotic, in healthy volunteers. Antimicrob. Agents Chemother. 36:318-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wyeth Pharmaceuticals Inc. 2009. Tygacil package insert. Wyeth Pharmaceuticals Inc., Philadelphia, PA.