

Abstract
Dengue virus (DENV) is the most prevalent mosquito-borne viral pathogen that infects humans. Neither a vaccine nor an antiviral therapy is currently available for DENV. Here, we report an adenosine nucleoside prodrug that potently inhibits DENV replication both in cell culture and in a DENV mouse model. NITD449 (2′-C-acetylene-7-deaza-7-carbamoyladenosine) was initially identified as a parental compound that inhibits all four serotypes of DENV with low cytotoxicity. However, in vivo pharmacokinetic studies indicated that NITD449 had a low level of exposure in plasma when dosed orally. To increase the oral bioavailability, we covalently linked isobutyric acids to the 3′- and 5′-hydroxyl groups of ribose via ester linkage to NITD449, leading to the prodrug NITD203 (3′,5′-O-diisobutyryl-2′-C-acetylene-7-deaza-7-carbamoyl-adenosin). Pharmacokinetic analysis showed that upon oral dosing of the prodrug, NITD203 was readily converted to NITD449, resulting in improved exposure of the parental compound in plasma in both mouse and rat. In DENV-infected AG129 mice, oral dosing of the prodrug at 25 mg/kg of body weight reduced peak viremia by 30-fold. Antiviral spectrum analysis showed that NITD203 inhibited various flaviviruses (DENV, yellow fever virus, and West Nile virus) and hepatitis C virus but not Chikungunya virus (an alphavirus). Mode-of-action analysis, using a luciferase-reporting replicon, indicated that NITD203 inhibited DENV RNA synthesis. Although NITD203 exhibited potent in vitro and in vivo efficacies, the compound could not reach a satisfactory no-observable-adverse-effect level (NOAEL) in a 2-week in vivo toxicity study. Nevertheless, our results demonstrate that a prodrug approach using a nucleoside analog could potentially be developed for flavivirus antiviral therapy.
Dengue virus (DENV), the causative agent for dengue fever, is a mosquito-borne pathogen with significant impacts on public health and economy. It was confined to South East Asia in the 1950s, but since then, DENV has increased in its spread in geographic areas and in the number of infected patients (6). It is now estimated that 2.5 billion people are at risk for DENV infection, with about 50 million to 100 million human infections, leading to over 20,000 deaths each year. The disease spectrum ranges from mild undifferentiated fever to life-threatening dengue hemorrhage fever (DHF) and dengue shock syndrome (DSS) (18). There is no clinically approved vaccine or antiviral therapy for DENV. Thus, DENV presents an increasingly unmet medical need to develop anti-DENV therapeutics.
DENV belongs to the family of Flaviviridae, which contains three genera, Hepacivirus, Pestivirus, and Flavivirus. The genus Flavivirus contains many important human pathogens, such as West Nile virus (WNV), yellow fever virus (YFV), Japanese encephalitis virus (JEV), tick-borne encephalitis virus (TBEV), and DENV (4). Dengue virus is further subdivided into four serologically distinct serotypes, named dengue virus serotypes 1 to 4. The DENV genome consists of a positive-strand RNA of approximately 10.7 kb. A single open reading frame, flanked by 5′ and 3′ untranslated regions (UTRs), encodes a long polyprotein, which is processed into three structural (capsid, premembrane or membrane, and envelope) and seven nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins by both viral and host proteases. The NS5 gene encodes at least two enzymatic functions essential for viral propagation, the methyltransferase (MTase) domain at its N terminus and the RNA-dependent RNA polymerase (RdRp) domain at its C terminus. The RdRp, together with other viral and host proteins, is responsible for synthesis of both negative- and positive-strand RNAs. As this enzymatic function is devoid in host cells, the viral RdRp presents an attractive antiviral target.
Inhibitors of viral polymerases are broadly divided into nucleoside and nonnucleoside inhibitors. Nucleoside inhibitors have been the dominant class of antivirals available in the market; approximately half of the antivirals are nucleoside analogs for the treatment of HIV, hepatitis B virus (HBV), and herpes simplex virus (HSV) (3). Most nucleoside inhibitors have to be converted into their corresponding triphosphate form by host enzymes before acting as chain terminators to inhibit viral replication. Nonnucleoside inhibitors usually bind to the allosteric site, which is usually away from the polymerase active site. We recently showed that both the nucleoside and the nonnucleoside approaches could be used for development of DENV inhibitors (19, 20).
Here, we report the discovery of another adenosine nucleoside analog, 2′-C-acetylene-7-deaza-7-carbamoyladenosine (NITD449), that potently inhibits DENV in cell culture. However, NITD449 showed poor exposure when orally (p.o.) dosed in animals. The low level of bioavailability of NITD449 was overcome by adding isobutyric acids to NITD449 via ester linkage, leading to a prodrug, 3′,5′-O-diisobutyryl-2′-C-acetylene-7-deaza-7-carbamoyladenosine (NITD203). The prodrug NITD203 inhibited various members from genus flavivirus as well as hepatitis C virus (HCV), but NIT3203 did not inhibit Chikungunya virus (an alphavirus). Importantly, NITD203 exhibited potent in vivo efficacy in a dengue viremia mouse model. The study has demonstrated that the prodrug approach could potentially be used for development of flavivirus therapeutics.
MATERIALS AND METHODS
Synthesis.
Two compounds, 2′-C-acetylene-7-deaza-7-carbamoyladenosine (NITD449) and 3′,5′-O-diisobutyryl-2′-C-acetylene-7-deaza-7-carbamoyladenosine (NITD203) (Fig. 1 A), were analyzed in this study. The synthesis of NITD449 (Fig. 1A) was recently reported (1a). Further esterification of NITD449 with isobutyric acid led to the prodrug NITD203. Triethylamine (89 μl, 0.64 mmol, and 4.0 eq) was added to a stirred solution of NITD449 (52 mg, 0.16 mmol, and 1.0 eq) in dimethylformamide (DMF) (2 ml) and cooled to 0°C, after which isobutyric anhydride (0.39 ml, 2.5 mmol, and 2.5 eq) and a catalytic amount of 4-dimethylaminopyridine (DMAP) (1 mg) were added. The reaction mixture was stirred at 0°C for 30 min; formic acid (15 μl) and water (1 ml) were then added to quench the reaction. The crude product was purified by preparative high-performance liquid chromatography (HPLC) with a gradient of acetonitrile in water from 5% to 95% in 30 min. After lyophilization, NITD203 (31.9 mg, 0.067 mmol, and 42.1%) was obtained as a white solid. 1H NMR (300 MHz, deuterated methanol [MeOD]): δ 8.17 (1H, s), 8.12 (1H, s), 6.39 (1H, s), 5.55 (1H, d, J = 6.6 Hz), 4.50 to 4.30 (3H, m), 2.95 (1H, s), 2.79 to 2.60 (2H, m), 1.28 to 1.10 (12H, m). 13C NMR (75 MHz, MeOD): δ 178.5, 177.7, 169.1, 159.6, 153.6, 152.3, 126.9, 112.8, 102.8, 91.8, 81.0, 80.1, 79.2, 77.1, 76.5, 64.8, 35.2, 35.1, 19.5, 19.4. Electrospray ionization-mass spectrometry (ESI-MS): calculated. For C22H27N5O7: found, 474.36. Optical rotation: [α]D = −55.85° (c = 1.0, methanol [MeOH], 26°C).
FIG. 1.

Antiviral activities of NITD449 and NITD203. (A) Compound structure. (B) Cytotoxicity analysis. A549 cells were incubated with NITD449 or NITD203 at various concentrations for 48 h. Cell viability was then measured using an MTT assay and presented as a percentage of colorimetric absorbance derived from the compound-treated cells, compared with that from the mock-treated cells. (C) Antiviral activities in cell culture. CFI assays were performed to determine the EC50s for different DENV strains. Plaque assays were used to determine the EC50 for infection of human PBMC cells. Averages of results from two independent experiments are presented. See Materials and Methods for details. The EC50s for NITD008, previously reported in reference 19, are included for comparison.
In vitro antiviral assays.
Three types of in vitro antiviral assays were performed. Type one assay measured viral titer reduction in the presence of compounds. Vero cells were seeded in a 12-well plate (4 × 105 cells per well). At 24 h postseeding, the cells were infected with the indicated viruses at a multiplicity of infection (MOI) of 0.1 and treated immediately with the compound. For DENV-2 (New Guinea C) and YFV (17D vaccine strain), culture medium was collected at 48 h postinfection. For Chikungunya virus (Ross strain), culture medium was collected at 22 h postinfection. Plaque assays were used to determine the viral titers (11).
A type two assay, known as cell-based flavivirus immunodetection (CFI), was performed to measure the amount of viral E protein in infected cells. A549 cells (human alveolar basal epithelial cells) were seeded to a 96-well plate (2 × 104 cells/well). The cells were infected with DENV-2 (MOI of 0.3) on the following day. During the infection, the cells were incubated with a compound-virus mixture for 1 h with shaking every 10 to 15 min. The culture fluid was then replenished with fresh medium containing compounds. On day 2 postinfection, the cells were washed with phosphate-buffered saline (PBS), fixed with 100% methanol at 4°C for 10 min, and detected for intracellular viral E protein by an enzyme-linked immunosorbent assay (ELISA). The ELISA used mouse monoclonal antibody 4G2 against the DENV E protein and goat anti-mouse IgG conjugated with horseradish peroxidase as the primary and secondary antibodies, respectively.
The type three assay used Huh-7 cells carrying a luciferase-reporting replicon of hepatitis C virus (HCV) (5). The replicon cells were incubated with the compound and assayed for luciferase activities at 48 h posttreatment. The antiviral efficacy against HCV was estimated by the reduction of luciferase signals. Besides the above-mentioned antiviral assays, an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay was performed to estimate compound cytotoxicity. Approximately 1 × 104 cells per well were seeded to a 96-well plate. On the following day, the cells were incubated with various concentrations of the compound for 48 h. Cell viability was then quantified using an MTT assay in accordance with the manufacturer's protocol (American Type Culture Collection).
Analysis of the effect of serum on antiviral activity.
A modified CFI assay was performed to examine the effect of serum on the 50% effective concentrations (EC50s) of compounds. A549 cells were seeded a day prior to the compound treatment, as described for the CFI assay. No compound was added during the 1-h viral inoculation period, after which the cells were replenished with fresh medium. The compounds, together with serum proteins, were added to the medium. EC50 measurement was performed on day 2 postinfection.
In vivo PK studies.
The Institutional Animal Care and Use Committee (IACUC), registered with the Agri-Food and Veterinary Authority (AVA) of Singapore, approved all animal experimental protocols. Mice and rats were allowed to acclimatize before initiation of pharmacokinetic (PK) experiments. Feed and water were given ad libitum to the mice. Compounds were formulated at a concentration of 2.5 mg/ml for a dose of 25 mg/kg of body weight administered orally (p.o.) and at a 1-mg/ml concentration for a dose of 5 mg/kg injected intravenously (i.v.). An optimized formulation was developed for both compounds on the basis of their respective solubility levels. Blood samples were collected from three animals at 0.02 (i.v. only), 0.08 (p.o. only), 0.25, 0.5, 1, 2, 4, 8, 16, and 24 h postdosing. Rats were fasted overnight prior to the study and until 4 h postdosing. Water was given ad libitum. Serial blood samplings were collected from groups of three rats at 0.17, 0.5, 1, 2, 4, 8, 17, 24, 32, and 48 h postdosing. Heparinized blood was centrifuged at 13,000 rpm for 7 min at 4°C, and plasma was harvested and stored at −20°C until analysis.
Extraction and LC-MS analysis of the compound.
Plasma samples were extracted with acetonitrile-methanol-acetic acid (90:9.8:0.2) for analytes. Analyte quantification was performed by liquid chromatography-tandem mass spectrometry (LC-MS-MS). Liquid chromatography was performed using an Agilent 1100 HPLC system (Santa Clara, CA), with a Phenomenex Synergi Hydro-RP 100-Å (2.5 μm; 2 by 100 mm) column at an oven temperature of 40°C, coupled with a QTRAP4000 triple quadruple mass spectrometer (Applied Biosystems, Foster City, CA). Instrument control and data acquisition were performed using the Applied Biosystems software program Analyst 1.4.2. The mobile phases used were (i) 20 mM ammonium acetate and (ii) acetonitrile-formic acid (99.9:0.1, vol/vol), and a gradient was used, with a flow rate of 0.7 ml/min and a run time of 6 min. Under these conditions, the retention times of NITD449 and NITD203 were 2.5 and 3.0 min, respectively. Compound detection using the mass spectrometer was performed in electrospray positive-ionization mode, using multiple reaction monitoring (MRM) for specificity (NITD449 transitions 334.1/178.1 and 334.1/161.2 and NITD203 transitions 474.4/178.2 and 474.4/161.1), together with their optimized mass spectrometry (MS) parameters. The lower limits of quantification were 9.2 ng/ml in plasma for both NITD449 and NITD203.
Pharmacokinetic analysis.
The mean value from the three animals at each time point was plotted against time to give the plasma concentration time profile. Pharmacokinetic parameter values were determined using WinNonlin Professional, version 5.0.1 (Pharsight, CA), by noncompartmental modeling using software model 200 for oral dosing and model 201 for intravenous dosing. The oral bioavailability (F) value was calculated as the ratio between the area under the curve (AUC) observed following oral administration and the AUC observed following intravenous administration, corrected for dose (F = AUCp.o. × dosei.v./AUCi.v. × dosep.o.).
In vivo animal efficacy studies.
AG129 mice lacking alpha/beta interferon and gamma interferon receptors were used in our mouse efficacy study (15). DENV-2 (strain TSV01) suspension (a total of 2 × 106 PFU in 0.4 ml) was injected intraperitoneally on the first day. Immediately after viral inoculation, 0.2 ml of compound dissolved in vehicle (ethanol-polyethylene glycol [PEG]-5% dextrose in water [D5W] at a ratio of 10:30:60) was given to AG129 mice via oral gavage twice per day for three consecutive days. On day 4, blood samples were taken, and viral titers were determined using a plaque assay, as previously described (13).
RESULTS
NITD449 as a potent inhibitor of DENV.
During the search for nucleoside inhibitors of DENV, we identified NITD449, an adenosine analog (Fig. 1A). Compared with adenosine, NITD449 contains three modifications: a carbon substitution for N-7 of the purine, an amide at the C-7 position of the purine, and an acetylene at the 2′ position of the ribose. An MTT assay showed that NITD449 was not cytotoxic in cell culture up to 12.5 μM and that cytotoxicity was observed at ≥25 μM (Fig. 1B). The compound inhibited all four serotypes of DENV in a CFI assay (a cell-based ELISA that measured the amount of viral E protein) (see details in Materials and Methods), with EC50s of 1 to 7 μM (Fig. 1C). In addition to A549 cells, NITD449 also showed anti-DENV activity on human peripheral blood mononuclear cells (PBMC), with an EC50 of 5 μM (Fig. 1C). The results demonstrated that NITD449 potently inhibits DENV in cell culture.
Low-level systemic absorption of NITD449 in mouse and rat.
To prepare for an in vivo efficacy study, we determined the pharmacokinetic parameter values of NITD449 in mice. Comparison of different formulation recipes showed that the compound was optimally formulated by dissolving it in 1.5 equimolar of HCl using 0.1 N HCl (pH adjusted to 3.5 with 1 N NaOH) topped off with 100 mM citrate buffer (pH 3.5). After intravenous (i.v.) injection of 5 mg/kg, the compound showed a low volume of distribution at steady state (Vss; 0.26 liters/kg) and a moderate systemic clearance (CL; 21.88 ml/min/kg), with a short to medium elimination half-life (t1/2; 1.31 h) (Fig. 2 A and Table 1). Following oral (p.o.) dosing of 25 mg/kg, NITD449 exhibited a low level of oral bioavailability (F; 1%) and a maximum concentration of drug in plasma (Cmax; 0.27 μM) that was lower than the EC50s (1 to 7 μM). The poor systemic exposure was mainly due to low absorption (AUC extrapolated to infinity [AUC∞] = 0.42 μM/h).
FIG. 2.
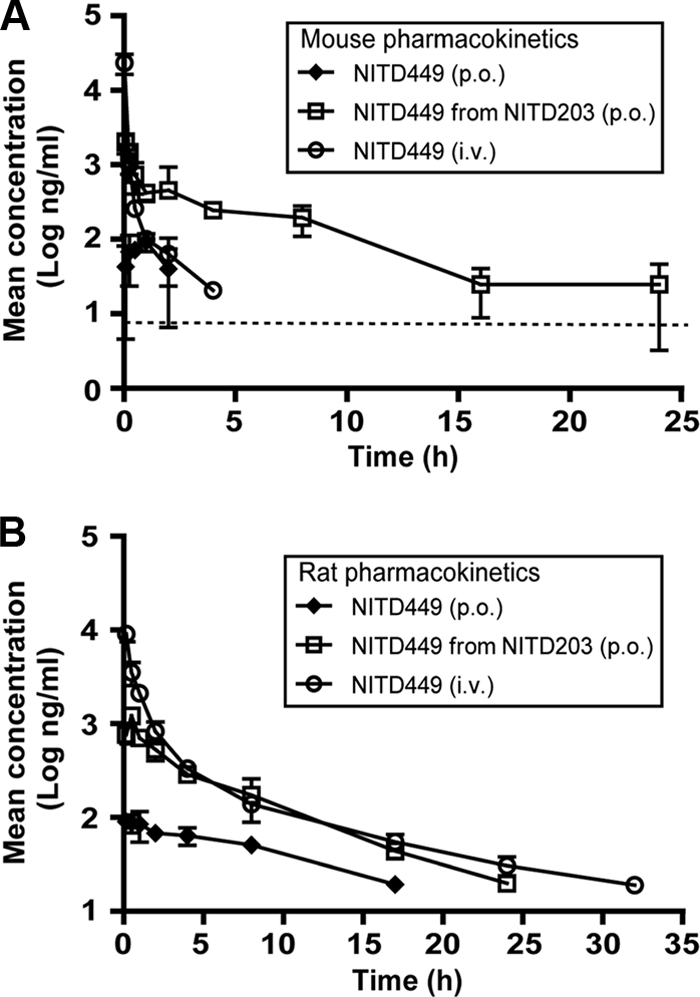
Pharmacokinetic analysis of NITD449 and NITD203. Mice (A) and rats (B) were intravenously injected (i.v.) with 5 mg/kg of NITD449, orally dosed (p.o.) with 25 mg/kg of NITD449, or orally dosed with NITD203. Plasma concentrations of NITD449 (ng/ml) over time are presented. The dotted line in panel A indicates the lower limit of quantification (LOQ); the LOQ for NITD449 and NITD203 was 9.2 ng/ml; values below the LOQ are not included in the graphs. Plasma concentrations are indicated with standard deviations (n = 3).
TABLE 1.
Pharmacokinetic parameter values for NITD449 and its prodrug NITD203 observed following oral and intravenous administration to female CD-1 mice and female Wistar rats
| Species | Compound | Dose(s) (mg/kg) | PK parametera value for indicated route |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Oral |
Intravenousb |
||||||||
| Cmax (μM) | Tmax (h) | AUC∞ (μM/h) | F (%) | Vss (liters/kg) | CL (ml/min/kg) | t1/2 (h) | |||
| Mouse | NITD449e | 25 | 0.27 | 1 | 0.42 | 1 | 0.26 | 21.88 | 1.31 |
| NITD449 from NITD203f | 24.2,c 17.0d | 6.01 | 0.08 | 12.57 | 32 | ||||
| Rat | NITD449e | 24.8 | 0.27 | 0.17 | 3.15 | 2 | 2.71 | 8.98 | 8.33 |
| NITD449 from NITD203f | 26.5, 18.7d | 3.65 | 0.5 | 13.71 | 13 | ||||
| NITD449 from NITD203g | 84.1, 59.2d | 14.2 | 0.5 | 33.45 | 10 | ||||
Cmax, maximum concentration of drug in plasma; Tmax, time to maximum concentration of drug in plasma; AUC∞, area under the curve extrapolated to infinity; Vss, volume of distribution at steady state; CL, clearance; t1/2, elimination half-life; F, oral bioavailability.
The intravenous doses of NITD449 were 5mg/kg in the mouse and 5.9mg/kg in the rat.
The administered dose of the prodrug NITD203 (mg/kg).
The corresponding dose of the conversion product NITD449 (mg/kg).
The solution formulation was 0.1 N HCl (1.5 equimolar amount) and 1 N NaOH (pH adjusted to 3.5), topped off with 100 mM citrate buffer (pH 3.5).
The solution formulation was 10% ethanol, 30% polyethylene glycol 400 (PEG 400), and 60% of 5% dextrose in water (D5W).
The solution formulation was 20% solutol HS15, 30% PEG 300, and 50% of 100 mM citrate buffer (pH 3).
We also analyzed the pharmacokinetic parameter values of NITD449 in rats (Fig. 2B and Table 1). Compared with the results from mice, the compound showed an improved half-life (t1/2 = 8.33 h), a low systemic clearance (CL = 8.98 ml/min/kg), and a moderate volume of distribution (Vss = 2.71 liters/kg). However, it still exhibited a low level of oral bioavailability (F = 2%) and a low maximum plasma concentration (Cmax = 0.27 μM). Collectively, the results indicated that NITD449 was poorly absorbed following oral dosing, resulting in low-level systemic exposure.
The prodrug NITD203 exhibits improved antiviral potency and systemic exposure.
To overcome the limitation of poor exposure of NITD449, we took a prodrug approach and synthesized NITD203. Compared with NITD449, NITD203 contained isobutyric esters at the 3′ and 5′ positions of ribose (Fig. 1A). Such modifications are expected to increase the hydrophobicity of the compound, leading to improvement of gastrointestinal absorption and cell membrane penetration. The isobutyric ester from NITD203 was expected to be removed by host esterases to generate NITD449 (see the figure in the supplemental material). In vitro analysis showed that NITD203 had a good antiviral activity, with EC50s of <1 μM for all four serotypes of DENV in both A549 and human PBMC cells (Fig. 1C). The compound was not cytotoxic up to 25 μM (Fig. 1B). Interestingly, the prodrug NITD203 showed a better potency than the parental compound NITD449, most likely due to the increased cellular uptake of the prodrug.
Next, the pharmacokinetic parameter values of NITD203 in both mice and rats were analyzed. In these experiments, NITD449 plasma levels were quantified following oral dosing of the animals with the prodrug NITD203 (Fig. 2). As summarized in Table 1, the pharmacokinetic profile of NITD449 (derived from its prodrug NITD203) revealed a much improved exposure and oral bioavailability, indicating that the prodrug approach provides a clear benefit to the overall pharmacokinetics of this nucleoside analog. When normalized to dose, peak plasma concentration and systemic exposure were >30-fold increased when NITD449 was administered as the prodrug NITD203 compared to the values observed following dosing of NITD449. Similar results were obtained for rats, confirming that the pharmacokinetic parameter values of NITD449 were improved when NITD449 was administered as the prodrug NITD203 (Table 1). The rat experiments also showed that the Cmax and AUC∞ values increased proportionally as the dose of NITD203 increased from 26.5 to 84.1 mg/kg, indicating that neither absorption nor elimination processes were saturated at these doses. Throughout the above-mentioned experiments, the levels of NITD203 were below the detection limit in both animal species and at all time points, even immediately following administration, indicating a rapid conversion of the prodrug to NITD449. Overall, the results clearly demonstrate that the prodrug improves the antiviral potency and pharmacokinetic properties.
Antiviral spectrum of NITD203.
To examine the antiviral spectrum of NITD203, we tested whether the compound inhibits other members of the Flavivirus genus. A viral titer reduction assay showed that the compound suppressed both DENV-2 and YFV in a dose-responsive manner (Fig. 3 A). At 12.5 μM, NITD203 suppressed the viral titers of DENV-2 and YFV by 1,000- and 100-fold, respectively. To examine the antiviral activity against WNV, we tested the compound in cells infected with WNV replicon-packaged particles. The WNV replicon particles were prepared by trans packaging a luciferase-reporting replicon with homologous viral structural proteins (10). The compound inhibited the luciferase activity derived from the WNV replicon, indicating that NITD203 inhibits WNV RNA replication. Next, we tested the compound in cells bearing a luciferase-reporting replicon of HCV (genotype 1b), a member from genus Hepacivirus. Remarkably, the compound suppressed the luciferase signal from the HCV replicon, with an EC50 of 0.03 μM. In contrast, it did not inhibit Chikungunya virus (a plus-strand RNA alphavirus). These results indicate that NITD203 selectively inhibits viruses within the family Flaviviridae.
FIG. 3.

Antiviral spectrum and mode-of-action analysis. (A) Vero cells were infected with the indicated viruses at an MOI of 0.1 and treated immediately with NITD203. For DENV-2 and YFV, culture medium was collected at 48 h postinfection and measured for viral titers using plaque assays. For Chikungunya virus, culture medium was collected at 22 h postinfection and measured for viral titer. For WNV, Vero cells were infected with virus-like particles (containing a luciferase replicon) at an MOI of about 0.1; the infected cells were assayed for luciferase activity at 24 h postinfection; relative luciferase activities were presented, with the level for the mock-treated replicon cells set as 100%. For HCV, Huh-7 cells carrying a luciferase replicon HCV (5) were incubated with NITD203 and assayed for luciferase activities at 48 h posttreatment. Average results and standard deviations (n = 3) are presented. (B) Transient DENV-2 replicon assay. A luciferase reporter replicon of DENV-2 was transfected into BHK-21 cells. The transfected cells were immediately incubated with the indicated concentrations of the compound and measured for luciferase activities at various time points posttransfection (p.t.). The effects of compound (1 μM and 3 μM) on viral translation and RNA synthesis were quantified by the luciferase signals at 2 to 4 h p.t. and at 24 to 40 h p.t., respectively. Average results and standard deviations (n = 3) are presented.
Mode-of-action analysis.
A luciferase-reporting replicon of DENV-2 was used to analyze the mechanism of inhibition of NITD203. The luciferase reporter was engineered into the replicon, where the viral structural genes were deleted (11). After electroporation of the replicon RNA into BHK-21 cells, the transfected cells were treated with NITD203 and assayed for luciferase activities at various time points. As shown in Fig. 3B, NITD203 did not affect the 2- to 4-h luciferase signals (representing input replicon translation) but significantly suppressed the luciferase activities at 24 to 40 h posttransfection (representing translation of newly synthesized replicon RNA). The results suggest that the compound inhibits DENV through suppression of viral RNA synthesis.
As an alternative means to study the mode of action, we tried to select resistant viruses by passaging DENV on Vero and other cells in the presence of increasing concentrations of inhibitors. To our surprise, no resistance virus could be obtained after multiple attempts; some of the passaging experiments lasted more than 2 months (data not shown). For example, the wild-type virus was continuously cultured for four rounds at each of the following concentrations: 0.4 μM, 0.8 μM, 1.2 μM, 1.6 μM, 2.0 μM, and 2.4 μM. The passaging either eliminated the virus or yielded lower titers of viruses that were not resistant to the inhibitor. These results suggest a high barrier to resistance selection in vitro.
Effect of human plasma proteins on antiviral efficacy of NITD449 and NITD203.
The in vivo efficacy of the compound is often confounded by the binding of the compound to plasma proteins. To address this question, we examined the effect of human plasma proteins on the efficacies of NITD449 and NITD203 against DENV. Addition of the human serum proteins human serum albumin (HSA; 40 mg/ml) and alpha-1-acid glycoprotein (AGP; 2 mg/ml) did not increase the EC50 by more than 1-fold (Table 2). The results suggest that human plasma proteins have minimal effect on compound potency.
TABLE 2.
Effect of human serum albumin and alpha-1-acid glycoprotein on EC50s of NITD449 and NITD203
| Assay conditiona | EC50 (μM) |
|
|---|---|---|
| NITD449 | NITD203 | |
| No HSA or AAG | 2.61 | 0.69 |
| 40 mg/ml HSA | 4.76 | 0.79 |
| 2 mg/ml AAG | 2.52 | 0.76 |
| 40 mg/ml HSA and 2 mg/ml AAG | 4.64 | 0.85 |
A549 cells were infected with DENV-2 (New Guinea C) at an MOI of 0.3 for 1 h without the compound. After removal of the viral inoculum, one set of the infected cells was incubated with medium containing various concentrations of NITD449 or NITD203, and another set of cells was incubated with medium containing the compound plus human serum albumin (HSA) and/or alpha-1-acid glycoprotein (AAG). On day 2 postinfection, viral titers in culture fluids were determined by a CFI assay. See Materials and Methods for details.
In vivo efficacy of NITD203.
We used a dengue viremia mouse model to examine the in vivo efficacy of NITD203. Infection of AG129 mice (lacking alpha/beta interferon and gamma interferon receptors) with DENV-2 (strain TSV01) develops viremia that peaks on day 3 postinfection (13). Using this model, we previously found that the peak viremia could be used to indicate the in vivo antiviral activity of inhibitors (13). As shown in Fig. 4, oral dosing of infected mice with NITD203 at 3, 10, and 25 mg/kg twice a day, starting immediately after infection, reduced peak viremia by 2.7-, 4.4-, and 30-fold, respectively. As a positive control, we also treated the infected mice with NITD008, a different adenosine nucleoside inhibitor of DENV that we have described earlier (19). Treatment with NITD008 at 25 mg/kg suppressed peak viremia by about 10-fold, indicating that NITD203 is 3-fold more potent than NITD008 in vivo. However, a Student t test showed that, at a P value of 0.08, the difference in viremia between the NITD203-treated mice and the NITD008-treated mice did not reach the statistical significance threshold represented by a P value of ≤0.05. Nevertheless, the results demonstrate that NITD203 has potent in vivo efficacy.
FIG. 4.

In vivo efficacy of NITD203. AG129 mice were intraperitoneally inoculated with 2 × 106 PFU of DENV-2 (strain TSV01) on day 0. The infected mice (6 or 8 animals per group) were immediately treated with the indicated doses of NITD203 orally twice per day for three consecutive days. The peak viremia on day 4 postinfection was quantified by a plaque assay. For comparison, the infected mice were treated with 25 mg/kg of NITD008, and the reduction of peak viremia is presented.
Toxicity and safety analyses of NITD203.
To estimate the in vitro toxicity of NITD203, we assessed the compound in more than 110 biochemical assays, including the Ames test for gene toxicity, the hERG channel for cardiovascular toxicity, CYP450 inhibition for drug-drug interaction, and the micronucleus assay for mutagenicity as well as assays for various receptors, ion channels, and kinase profiles. The compound did not show significant inhibition in any of these assays (data not shown).
Next, we tested the in vivo toxicity by orally dosing male Wistar rats (10 weeks old) at 10, 30, and 75 mg/kg/day of NITD203 for 2 weeks. Five animals were used for each dosing group and were monitored for clinical signs, body weight, and mortality. For the 10-mg/kg/day group, none of the three animals showed any clinical signs or body weight loss. For the 30-mg/kg/day group, one animal had to be prematurely killed on day 8 due to poor health condition, one animal was found dead on day 10, and all three other animals were sacrificed on day 10 because of body weight loss. For the 75-mg/kg/day group, two and three animals had to be prematurely sacrificed on days 6 and 7, respectively, due to severe body weight loss. Clinical signs were observed for the 30- and 75-mg/kg dosing groups, including decreased motor activity, pale appearance, piloerection, and chromodacryorrhea (dried material [red] around the nose). Overall, the results showed that NITD203 was clinically well tolerated at 10 mg/kg/day for 2 weeks but was not tolerated at 30 and 75 mg/kg/day.
DISCUSSION
A number of approaches have been taken to develop small molecule inhibitors of DENV and other flaviviruses (7). Although many inhibitors have been reported to possess anti-DENV activities in vitro, in vivo efficacy has been achieved only by two adenosine analog inhibitors (13, 19) and iminosugar inhibitors of host α-glucosidase (e.g., castanospermine) (17). Among the three in vivo-active compounds, NITD008 (7-deaza-2′-C-acetylene-adenosine; EC50, 0.5 to 2.6 μM) (Fig. 1C) was more potent than the other nucleoside inhibitor (7-deaza-2′-C-methyl-adenosine; EC50, 15 μM) (8, 13) and castanospermine (EC50, 1 to 86 μM, depending on cell types) (17). Here, we report another nucleoside analog, NITD203, which appears to have a better potency than NITD008 both in vitro (Fig. 1C) and in vivo (Fig. 4). These results, together with the fact that about half of the antivirals in clinical use are nucleoside analogs (3), clearly suggest that the nucleoside approach has the potential for development of flavivirus therapy.
During the course of the study, NITD449 was initially identified as a potent inhibitor of DENV in cell culture. However, NITD449 suffered from a low level of oral bioavailability (F = 1 to 2%). We took a prodrug approach to overcome this problem by synthesizing NITD203, an isobutyric ester derivative of NITD449. The prodrug molecules for nucleosides are usually amino acid- or aliphatic acid-ester derivatives of parental compounds. The ester prodrug efficiently enters cells via a peptide transport mechanism for amino acid-prodrug or via a better passive penetration for aliphatic acid-prodrug and is rapidly converted to the parental compound by enzymatic hydrolysis, leading to a significant increases in oral bioavailability and the cellular uptake of the parental compound. This approach has been well established during the development of the prodrug of acyclovir (16). For HCV, three nucleoside inhibitors of NS5B under clinical development are all ester prodrugs: NM283 (valopicitabine) is a 3′-O-valinyl ester of 2′-C-methylcytidine (9), R1626 is a tri-isobutyric ester prodrug of R1479 (4′-C-azidocytidine) (12), and R7128 is a di-isobutyric ester prodrug of PSI-6130 (β-d-2′-deoxy-2′-fluoro-2′-C-methyulcitidine) (1). In this study, we showed that the isobutyric ester prodrug of NITD449 improved the oral bioavailability from 1% to 32% in mouse and from 2% to 10 to 13% in rat (Table 1). In agreement with the prodrug mechanism, we found that after oral dosing, the amount of the prodrug NITD203 was below the detection limit in animals at all times, even immediately after dosing, indicating a rapid conversion to the parental compound NITD449.
One challenge for development of nucleoside analogs for therapeutics is the unpredictable toxicity. NITD203 showed potent efficacy in a dengue mouse model but failed in achieving a no-observable-adverse-effect level (NOAEL) in the 2-week toxicity test when dosed at 30 and 75 mg/kg/day. Although the compound did not show side effects at 10 mg/kg/day, only a 4-fold reduction of viremia was observed when mice were treated at this dose (Fig. 4). Similar in vivo toxicity results were found for NITD008 (19). These results indicate that these compounds have very marginal therapeutic windows, which have prohibited their further development. A common side effect of nucleoside compounds is mitochondrial toxicity, which usually involved inhibition of mitochondrial polymerase γ, leading to a gradual decrease in mitochondrial DNA (2). However, most nucleoside analogs that cause mitochondrial toxicity are deoxynucleoside analogs. The nucleoside analogs for DENV and other RNA viruses are ribose nucleosides and, thus, are less likely to cause mitochondrial toxicity. Indeed, treatment of human cells with NITD008 did not reduce the mitochondria DNA content, nor did the treatment increase the lactic acid production in mitochondria; furthermore, the triphosphate form of NITD008 did not inhibit human DNA polymerase α, β, or γ in vitro (unpublished results). We are currently investigating the cause of toxicity and trying to overcome the side effects.
With regard to toxicity, two major points should be considered during development of DENV therapeutics. First, the AG129 mouse used in the current in vivo efficacy experiments lacks alpha/beta interferon and gamma interferon receptors (14). The defect in the innate immunity of the AG129 mouse may underestimate the real in vivo efficacy of compounds. Since NITD203 inhibited other flaviviruses in cell culture (Fig. 3), it would be informative to determine its potency against the flaviviruses that have well-established animal models using immunocompetent mice (e.g., WNV, JEV, and YFV). Second, dengue is an acute disease with a fever duration of less than a week (4), so the length of therapeutic treatment is expected to be less than a week. This is in contrast to the antiviral therapy of chronic diseases, such as HIV infection, which requires long-term treatment. The difference between the acute and chronic diseases should be considered during preclinical development of DENV inhibitors.
In summary, we have identified a new adenosine nucleoside inhibitor of DENV. An isobutyric ester prodrug was successfully used to improve the bioavailability of the parental compound. The ester prodrug showed a good potency in a dengue viremia mouse model. Besides DENV, the compound inhibited a broad spectrum of flaviviruses as well as HCV. However, the compound could not reach a satisfactory NOAEL in rats when they were dosed at ≥30 mg/kg/day. Further studies are needed to understand the nature of the toxicity and to improve the therapeutic window of the current compound.
Supplementary Material
Acknowledgments
We thank Gang Wang, Wai Ling Chan, Chin Chin Lim, Wei Liu, Feng Gu, Qing-Yin Wang, Kai Lin, Kang Min Low, and Arkadius Pichota for technical support and discussions during the course of the study. We also thank Subhash G. Vasudevan, Shamala Devi, and Ooi Eng Eong for providing different strains of dengue virus and Chikungunya virus for compound testing.
Footnotes
Published ahead of print on 1 June 2010.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Ali, S., V. Leveque, S. Le Pogam, H. Ma, F. Philipp, N. Inocencio, M. Smith, A. Alker, H. Kang, I. Najera, K. Klumpp, J. Symons, N. Cammack, and W. R. Jiang. 2008. Selected replicon variants with low-level in vitro resistance to the hepatitis C virus NS5B polymerase inhibitor PSI-6130 lack cross-resistance with R1479. Antimicrob. Agents Chemother. 52:4356-4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1a.Chen, Y.-L., Z. Yin, J. Duraiswamy, W. Schul, C. C. Lim, B. Liu, H. Y. Xu, M. Qing, A. Yip, G. Wang, W. L. Chan, H. P. Tan, M. Lo, S. Liung, R. R. Kondreddi, R. Rao, H. Gu, H. He, T. H. Keller, and P.-Y. Shi. 2010. Inhibition of dengue virus RNA synthesis by an adenosine nucleoside. Antimicrob. Agents Chemother. 54:2932-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cote, H. C., Z. L. Brumme, K. J. Craib, C. S. Alexander, B. Wynhoven, L. Ting, H. Wong, M. Harris, P. R. Harrigan, M. V. O'Shaughnessy, and J. S. Montaner. 2002. Changes in mitochondrial DNA as a marker of nucleoside toxicity in HIV-infected patients. N. Engl. J. Med. 346:811-820. [DOI] [PubMed] [Google Scholar]
- 3.De Clercq, E., and J. Neyts. 2009. Antiviral agents acting as DNA or RNA chain terminators. Handb. Exp. Pharmacol. 2009:53-84. [DOI] [PubMed] [Google Scholar]
- 4.Gubler, D., G. Kuno, and L. Markoff. 2007. Flaviviruses, p. 1153-1253. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 1. Lippincott William & Wilkins, Philadelphia, PA. [Google Scholar]
- 5.Krieger, N., V. Lohmann, and R. Bartenschlager. 2001. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J. Virol. 75:4614-4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morens, D. M., and A. S. Fauci. 2008. Dengue and hemorrhagic fever: a potential threat to public health in the United States. JAMA 299:214-216. [DOI] [PubMed] [Google Scholar]
- 7.Noble, C. G., Y. L. Chen, H. Dong, F. Gu, S. P. Lim, W. Schul, Q. Y. Wang, and P. Y. Shi. 2010. Strategies for development of dengue virus inhibitors. Antiviral Res. 85:450-462. [DOI] [PubMed] [Google Scholar]
- 8.Olsen, D. B., A. B. Eldrup, L. Bartholomew, B. Bhat, M. R. Bosserman, A. Ceccacci, L. F. Colwell, J. F. Fay, O. A. Flores, K. L. Getty, J. A. Grobler, R. L. LaFemina, E. J. Markel, G. Migliaccio, M. Prhavc, M. W. Stahlhut, J. E. Tomassini, M. MacCoss, D. J. Hazuda, and S. S. Carroll. 2004. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob. Agents Chemother. 48:3944-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pierra, C., A. Amador, S. Benzaria, E. Cretton-Scott, M. D'Amours, J. Mao, S. Mathieu, A. Moussa, E. G. Bridges, D. N. Standring, J. P. Sommadossi, R. Storer, and G. Gosselin. 2006. Synthesis and pharmacokinetics of valopicitabine (NM283), an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. J. Med. Chem. 49:6614-6620. [DOI] [PubMed] [Google Scholar]
- 10.Puig-Basagoiti, F., T. S. Deas, P. Ren, M. Tilgner, D. M. Ferguson, and P.-Y. Shi. 2005. High-throughput assays using luciferase-expressing replicon, virus-like particle, and full-length virus for West Nile virus drug discovery. Antimicrob. Agents Chemother. 49:4980-4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puig-Basagoiti, F., M. Tilgner, B. Forshey, S. Philpott, N. Espina Wentworth, S. Goebel, P. S. Masters, B. Falgout, P. Ren, Ferguson, and P. Y. Shi. 2006. Triaryl pyrazoline compound inhibits flavivirus RNA replication. Antimicrob. Agents Chemother. 50:1320-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts, S. K., G. Cooksley, G. J. Dore, R. Robson, D. Shaw, H. Berns, G. Hill, K. Klumpp, I. Najera, and C. Washington. 2008. Robust antiviral activity of R1626, a novel nucleoside analog: a randomized, placebo-controlled study in patients with chronic hepatitis C. Hepatology 48:398-406. [DOI] [PubMed] [Google Scholar]
- 13.Schul, W., W. Liu, H. Y. Xu, M. Flamand, and S. G. Vasudevan. 2007. A dengue fever viremia model in mice shows reduction in viral replication and suppression of the inflammatory response after treatment with antiviral drugs. J. Infect. Dis. 195:665-674. [DOI] [PubMed] [Google Scholar]
- 14.Shresta, S., K. L. Sharar, D. M. Prigozhin, P. R. Beatty, and E. Harris. 2006. Murine model for dengue virus-induced lethal disease with increased vascular permeability. J. Virol. 80:10208-10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Broek, M. F., U. Muller, S. Huang, M. Aguet, and R. M. Zinkernagel. 1995. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J. Virol. 69:4792-4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weller, S., M. R. Blum, M. Doucette, T. Burnette, D. M. Cederberg, P. de Miranda, and M. L. Smiley. 1993. Pharmacokinetics of the acyclovir pro-drug valaciclovir after escalating single- and multiple-dose administration to normal volunteers. Clin. Pharmacol. Ther. 54:595-605. [DOI] [PubMed] [Google Scholar]
- 17.Whitby, K., T. Pierson, B. Geiss, K. Lane, M. Engle, Y. Zhou, R. Doms, and M. Diamond. 2005. Castanospermine, a potent inhibitor of dengue virus infection in vitro and in vivo. J. Virol. 79:8698-8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitehead, S. S., J. E. Blaney, A. P. Durbin, and B. R. Murphy. 2007. Prospects for a dengue virus vaccine. Nat. Rev. Microbiol. 5:518-528. [DOI] [PubMed] [Google Scholar]
- 19.Yin, Z., Y.-L. Chen, W. Schul, Q.-Y. Wang, F. Gu, J. Duraiswamy, K. R. Reddy, P. Niyomrattanakit, S. B. Lakshminarayana, A. Goh, H. Y. Xu, W. Liu, B. Liu, J. Y. H. Lim, C. Y. Ng, M. Qing, C. C. Lim, A. Yip, G. Wang, W. L. Chan, H. P. Tan, K. Lin, B. Zhang, G. Zou, K. A. Bernard, C. Garrett, B. Beltz, M. Dong, M. Weaver, H. He, X. Han, A. Pichota, V. Dartois, T. H. Keller, and P.-Y. Shi. 2009. An adenosine nucleoside inhibitor of dengue virus. Proc. Natl. Acad. Sci. U. S. A. 106:20435-20439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin, Z., Y. L. Chen, R. R. Kondreddi, W. L. Chan, G. Wang, R. H. Ng, J. Y. Lim, W. Y. Lee, D. A. Jeyaraj, P. Niyomrattanakit, D. Wen, A. Chao, J. F. Glickman, H. Voshol, D. Mueller, C. Spanka, S. Dressler, S. Nilar, S. G. Vasudevan, P. Y. Shi, and T. H. Keller. 2009. N-Sulfonylanthranilic acid derivatives as allosteric inhibitors of dengue viral RNA-dependent RNA polymerase. J. Med. Chem. 52:7934-7937. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.