

Abstract
The pharmacodynamic and pharmacokinetic (PK-PD) properties of amphotericin B (AmB) formulations against invasive pulmonary aspergillosis (IPA) are not well understood. We used an in vitro model of IPA to further elucidate the PK-PD of amphotericin B deoxycholate (DAmB), liposomal amphotericin B (LAmB) and amphotericin B lipid complex (ABLC). The pharmacokinetics of these formulations for endovascular fluid, endothelial cells, and alveolar cells were estimated. Pharmacodynamic relationships were defined by measuring concentrations of galactomannan in endovascular and alveolar compartments. Confocal microscopy was used to visualize fungal biomass. A mathematical model was used to calculate the area under the concentration-time curve (AUC) in each compartment and estimate the extent of drug penetration. The interaction of LAmB with host cells and hyphae was visualized using sulforhodamine B-labeled liposomes. The MICs for the pure compound and the three formulations were comparable (0.125 to 0.25 mg/liter). For all formulations, concentrations of AmB progressively declined in the endovascular fluid as the drug distributed into the cellular bilayer. Depending on the formulation, the AUCs for AmB were 10 to 300 times higher within the cells than within endovascular fluid. The concentrations producing a 50% maximal effect (EC50) in the endovascular compartment were 0.12, 1.03, and 4.41 mg/liter for DAmB, LAmB, and ABLC, respectively, whereas, the EC50 in the alveolar compartment were 0.17, 7.76, and 39.34 mg/liter, respectively. Confocal microscopy suggested that liposomes interacted directly with hyphae and host cells. The PK-PD relationships of the three most widely used formulations of AmB differ markedly within an in vitro lung model of IPA.
Aspergillus fumigatus is an environmentally ubiquitous mold that is a leading cause of morbidity and mortality in immunocompromised patients (18). Despite the advent of newer diagnostic and therapeutic modalities, the mortality rate remains approximately 50% (22). An improved understanding of the pharmacology of existing agents represents an important strategy to improve the outcomes of patients with this rapidly progressive and frequently lethal infectious syndrome.
Amphotericin B (AmB) is a polyene derived from Streptomyces nodosus. This compound was discovered in the mid-1950s and remains a first-line agent for the treatment of invasive aspergillosis and other life-threatening invasive fungal infections (23, 24). Amphotericin B is amphipathic; i.e., it has both hydrophilic and hydrophobic moieties that render it insoluble in water. Aqueous solubility is achieved by formulation with deoxycholate or a variety of lipid carriers. Amphotericin B deoxycholate (DAmB) is a highly potent antifungal formulation, but its clinical utility is limited by a high frequency of adverse effects, such as infusional toxicity and nephrotoxicity (3, 27). Lipid formulations are better tolerated than DAmB and are increasingly used for the treatment of invasive pulmonary aspergillosis (IPA). Three licensed lipid-based formulations have been developed for clinical use: liposomal amphotericin (LAmB), amphotericin B lipid complex (ABLC), and amphotericin B colloidal dispersion (ABCD). These formulations differ significantly in their structures and pharmacological properties (1).
Here, we describe the pharmacokinetics and pharmacodynamics (PK-PD) of the frequently used clinical formulations of amphotericin B by the use of an in vitro model of IPA. This model enabled assessment of the extent of drug penetration into a number of tissue subcompartments that are relevant to the pathogenesis of IPA.
MATERIALS AND METHODS
Construction of the air-liquid model of the human alveolus.
Cell culture models that have been described previously were modified to produce an air-liquid interface model of the human alveolus (6, 15). A cellular bilayer was constructed using human pulmonary artery endothelial cells (HPAEC; Lonza Biologics, Slough, United Kingdom) and human alveolar epithelial cells (A549; LGC Standards, Middlesex, United Kingdom). HPAEC and A549 cells were used in passages 4 and 79 to 86, respectively. HPAECs were grown to near-confluence in endothelial basal medium (EBM-2) supplemented with 2% fetal bovine serum (FBS), ascorbic acid, heparin, hydrocortisone, human endothelial growth factor, vascular endothelial growth factor, human fibroblast growth factor B, and R3-insulin-like growth factor-1 according to the manufacturer's instructions, to produce endothelial growth medium (EGM-2). Amphotericin B and gentamicin, which are ordinarily constituents of EGM-2, were omitted. A549 cells were grown to near-confluence in EBM-2 supplemented with 10% FBS (Lonza Biologics, Slough, United Kingdom) without antimicrobial agents. HPAEC and A549 cells were harvested using warmed 0.25% trypsin-EDTA (Lonza Biologics), centrifuged, and resuspended in warmed fresh media. Final densities of HPAEC and A549 cells of 1 × 106 and 5 × 105 cells/ml, respectively, were obtained by serial dilution in their respective growth media.
One hundred microliters of the HPAEC suspension was seeded onto the bottom of polyester Transwell inserts (6.5-mm-diameter membrane, 3-μm pores; Corning Life Sciences, Lowell, MA). The inverted inserts were incubated for 2 h before being righted and placed in 24-well tissue culture plates containing 600 μl EGM. One hundred microliters of EBM-2 supplemented with 10% FBS was then added to the upper chamber and incubated at 37°C in humidified 5% CO2 for 24 h. To construct the bilayer, spent medium from the upper chamber was removed, and 100 μl of the A549 suspension was added and incubated for 2 h to enable cellular adhesion to the polyester membrane. Medium from the alveolar compartment was then removed to create an air-liquid interface, and the inserts were transferred to trays containing 600 μl EGM-2. Medium in the endovascular compartment was changed daily, and any medium that accumulated in the alveolar compartment was also removed.
Cellular confluence was assessed at time points between zero and 120 h by placing 100 μl of 1% (wt/vol) dextran blue (Sigma-Aldrich, Exeter, United Kingdom) in the alveolar compartment and placing inserts in tissue culture plates containing 600 μl of warmed phosphate-buffered saline (PBS; Invitrogen Ltd., Renfrew, United Kingdom) for 2 h. Transgression of dye through the cellular bilayer and into the endothelial compartment containing PBS was measured spectrophotometrically using a wavelength of 620 nm. To ensure that drug penetration was not due to direct cellular toxicity and loss of cellular confluence, the transgression of dextran blue was assessed at the end of experiments using the highest concentration for each formulation of AmB.
Organism, inoculation, and MICs.
An A. fumigatus transformant expressing green fluorescent protein (GFP) was used for all experiments, as previously described (15). Prior to each experiment, the organism was subcultured from beads to a potato dextrose agar slope (Oxoid, Basingstoke, United Kingdom) and incubated at 37°C for 7 days. A conidial suspension was prepared by flooding the slope with 15 ml PBS (without Tween 80). The suspension was centrifuged at 1,000 × g, and the pellet resuspended in PBS; this process was repeated three times. The final inoculum was prepared in cell culture medium EBM-2 without FBS using a hemocytometer and checked with quantitative cultures.
Experiments were performed 5 days after seeding A549 cells. Immediately prior to inoculation, the medium in the endovascular compartment was changed to warmed EBM-2 supplemented with 2% FBS (i.e., without the additional growth factors that constitute EGM-2). One hundred microliters of a suspension containing 1 × 104 conidia/ml was placed in the alveolar compartment. Six hours later, all fluid within the alveolar compartment was removed, and the inserts were transferred to fresh tissue culture plates containing 600 μl of EBM-2 supplemented with 2% FBS along with the desired concentration of DAmB, LAmB, or ABLC (i.e., drug was administered within the endovascular compartment). The 6-h delay in the administration of drug was based on a previous study and mimics the treatment of early IPA (15).
MICs for each of the clinical formulations were determined on three separate occasions using National Committee for Clinical Laboratory Standards M38-A2 methodology (7).
Antifungal compounds.
LAmB (Gilead Sciences, Cambridge, United Kingdom) and DAmB (Bristol-Myers Squibb, Uxbridge, United Kingdom) were reconstituted with sterile water to produce stock solutions of 500 mg/liter. ABLC (Cephalon, Hertfordshire, United Kingdom) was obtained as a lipid complex solution containing 500 mg/liter amphotericin B.
Collection of pharmacokinetic and pharmacodynamic data.
The air-liquid interface model enabled sampling from a number of compartments relevant to the pathogenesis of IPA. The surface of the alveolar cells was washed with 300 μl PBS to mimic a bronchoalveolar lavage, while the endovascular fluid was sampled directly to simulate a blood sample. Endothelial and alveolar epithelial cells were physically removed by abrading the polyester membrane with a microbiological loop and then suspended in 300 μl PBS.
Pharmacokinetic experiments.
The pharmacokinetics of the three formulations within the endovascular fluid endothelial cells and alveolar cells were defined in the presence of infection. The following amphotericin B concentrations for each formulation were studied: DAmB, 0.1, 0.5, 1.0, and 2.0 mg/liter; LAmB, 1, 10, 50, and 150 mg/liter; and ABLC, 1, 10, 50, and 150 mg/liter; these encompassed the known concentration-effect relationships of each compound defined from preliminary experiments. Endovascular fluid, endothelial cells, and alveolar cells were sampled at 3, 6, 12, 18, 24, and 30 h posttreatment for each formulation.
Pharmacodynamics and pharmacodynamic modeling.
The antifungal effect of the three formulations was estimated by measuring the concentrations of galactomannan in alveolar lavage and endovascular fluid using a commercially available double-sandwich enzyme-linked immunosorbent assay (ELISA) (Platelia Aspergillus kit; Bio-Rad Laboratories). The use of the green fluorescent protein transformant also enabled visualization of hyphal invasion using confocal microscopy and therefore provided a complementary measure of fungal burden and antifungal effect. Concentration-response relationships were initially defined following 24 h of drug exposure (i.e., 30 h postinoculation). Based on preliminary dose-finding experiments, the initial concentration ranges of DAmB, LAmB, and ABLC within the endovascular compartment were 0 to 2 mg/liter, 0 to 75 mg/liter, and 0 to 75 mg/liter, respectively.
The concentrations of galactomannan in the alveolar and endovascular compartments were modeled using an inhibitory sigmoid maximal effect (Emax) model that took the following form: Effect = Econ − (Emax × ExpH)/(EC50H + ExpH), where Econ is the fungal burden in the absence of drug, Emax is the asymptotic reduction in fungal burden induced by antifungal drug exposure, Exp is a measure of drug exposure (i.e., initial concentration of drug in endovascular fluid or AUC/MIC ratio), EC50 is the concentration resulting in half-maximal effect, and H is the slope (or Hill) function. The model was implemented within the identification module of the pharmacokinetic program ADAPT II (9), and the data were weighted by the inverse of the observed variance.
The inhibitory sigmoid Emax model was used to identify concentrations that produced zero, EC20, EC50, and near-maximal effect (0, 0.1, 0.15, and 2.0 mg/liter, respectively, for DAmB; 0, 1, 2, and 75 mg/liter, respectively, for LAmB and ABLC). These values were subsequently used to examine the time course of antifungal effect following a range of effective and ineffective drug concentrations. For these experiments, samples of alveolar lavage and endovascular fluid were collected at 0, 12, 18, 24, 26, and 30 h postinoculation, and the concentration of galactomannan was determined.
HPLC.
Concentrations of amphotericin B were measured using high-performance liquid chromatography (HPLC) as previously described (15). Briefly, 100 μl of each sample was analyzed using a C18 5-μm column (Varian Ltd., Oxford, United Kingdom), Shimadzu SIL20A AutoSampler, LC 20AD pump, and SPD 20A UV/VIS detector (Shimadzu UK Ltd., Milton Keynes, United Kingdom). Amphotericin B powder (Sigma Aldrich) was solubilized in 1:1 (vol/vol) methanol-dimethyl sulfoxide (DMSO), and a four-point standard curve prepared in the respective matrix (i.e., media, PBS, cell suspension). Piroxicam (1 mg/liter) was used as the internal standard. The dynamic range of the assay was 0.01 to 200 mg/liter.
Construction of sulforhodamine-labeled liposomes and confocal microscopy.
Hydrogenated soy phosphatidylcholine (HSPC), cholesterol, distearoylphosphatidylglycerol (DSPG), amphotericin B, and alpha-tocopherol were dissolved in a 2:1:0.8:0.4:0.01 molar ratio in a 1:1 mixture of methanol and chloroform (or, for placebo, the same formula without amphotericin B). Once all components were dissolved, solvents were removed by evaporation under continuous nitrogen flow. Residual solvent was removed by storing the container containing the material in a desiccator under vacuum for at least 48 h. The dried lipid was hydrated in a buffer containing 9% sucrose and 10 mM succinate at desired drug concentrations, and the hydrated material was processed through a high-shear homogenizer to form liposomes. For sulforhodamine-labeled material, the buffer in this step was supplemented with sulforhodamine B at 600 mg/liter. Unentrapped sulforhodamine was removed by ultrafiltration against a Millipore 100-kDa polyethersulfone (PES) membrane. The resulting solution was filtered through a 0.2-μm filter (also PES). Drug-containing preparations had a final pH of 5.4 and were freeze-dried. Formulations without drug were stored as a liquid and had a final pH of 6.4. Samples were confirmed to have a median particle size of <100 nm by dynamic light scattering. Amphotericin B concentration was confirmed by reversed-phase HPLC using a C18 column and isocratic elution against acetonitrile-methanol-2.5 mM EDTA (25:50:30 [vol-vol-vol]) and using the USP standard. Sulforhodamine concentration was determined by UV-visible (Vis) spectroscopy, and preparations contained between 0.1 mM and 1 mM sulforhodamine.
Liposomes were reconstituted in 6 ml sterile water per vial. Further dilutions were prepared in EBM-2 supplemented with 2% FBS. Six hundred microliters of media containing LAmB-containing sulforhodamine-labeled liposomes (LAmB-Rho) or drug-free sulforhodamine-labeled liposomes (LPlac-Rho) was placed in tissue culture plates, and membranes were transferred 6 h after conidial inoculation as described above for other AmB preparations. Samples taken from a range of time points 0 to 24 h posttreatment were fixed using 4% paraformaldehyde (Sigma-Aldrich). Six hundred microliters of 5 μg/ml 4′,6-diamidino-2-phenylindole solution (DAPI; Sigma-Aldrich) was instilled into the alveolar and endovascular compartment, incubated for 30 min at room temperature, and then washed twice with PBS.
Confocal microscopy.
A Nikon Eclipse C1-Plus inverted confocal microscope (Nikon UK Limited, Surrey United Kingdom) with a 20×, 40×, or 100× Apochromat objective lens was used. Image z stacks with 0.3-μm x-y pixel size and an optical slice of 0.34- to 1.00-μm thicknesses were collected and analyzed using EZ-C1 FreeViewer (v3.9) software (Nikon UK Limited).
Mathematical modeling.
The total concentrations of amphotericin B associated with each formulation were modeled using a population methodology which employed the Big version of the program Nonparametric Adaptive Grid (BIG NPAG) (19). The movement of drug from the endovascular fluid into the endothelial and alveolar cells was described using the following three inhomogeneous differential equations:
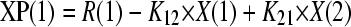 |
(1) |
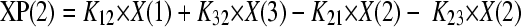 |
(2) |
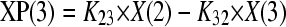 |
(3) |
Equations 1, 2, and 3 describe the movement of drug (XP) into and out of endovascular fluid, endothelial cells, and alveolar cells, respectively. R(1) represents the bolus injection of drug into the endovascular compartment; K12, K21, K23, and K32 are the first-order intercompartmental rate constants between compartment 1 (endovascular fluid), compartment 2 (endothelial cells), and compartment 3 (alveolar cells). X(1), X(2), and X(3) represent the amount of drug (mg) in the respective compartments. The volume of each compartment (liters) was estimated in the output equations that described the time course of concentrations (not shown). The concentration of drug in the lavage fluid from the surface of the (relatively dry) alveolar cells was not modeled because of difficulties in accurately estimating the volume of this compartment.
The mean drug concentrations from each compartment from three inserts were modeled. The data were weighted by the inverse of the observed variance. The fit of the model to the data was assessed using measures of precision and bias along with the coefficient of determination (r2) and visual inspection of the observed-versus-predicted relationships after the Bayesian step. To assess the extent of drug penetration into each of the pharmacokinetic compartments, the mean parameter values were inserted into the simulation module of ADAPT II (9), and the AUC in each compartment was calculated by integration. The inhibitory sigmoid Emax model was refitted to the data using the AUC/MIC ratio as the independent variable.
RESULTS
Formulation-specific MICs.
The MICs from the three replicate experiments for the three AmB formulations were as follows: DAmB, 0.25, 0.25, and 0.5; ABLC, 0.125, 0.125, and 0.125; LAmB, 0.25, 0.125, and 0.25; and LAmB-Rho, 0.25, 0.25, and 0.25 mg/liter. The MIC for pure amphotericin B was 0.25 mg/liter on three separate occasions.
Air-liquid interface model.
A schematic representation of the in vitro air-liquid model is shown in Fig. 1 A. There was a progressive decline in the extent that dextran blue traversed the cellular bilayer, becoming negligible 96 h post-seeding of A549 cells (Fig. 1B). The addition of DAmB, LAmB, or ABLC did not affect the transgression of dextran blue across the bilayer (data not shown). Progressive hyphal growth and invasion through the cellular bilayer were reflected by changes in galactomannan concentrations in both the alveolar and endovascular compartments (Fig. 1C to F). The kinetics of galactomannan concentrations in the alveolar and endovascular compartments were discordant, reflecting the time required for hyphae to invade across the cellular bilayer (Fig. 1G). The kinetics of galactomannan in the endovascular compartment were mirrored by progressive hyphal invasion observed with confocal microscopy. Both galactomannan concentrations and confocal microscopy suggested that hyphae emerged within the endovascular compartment 14 to 16 h postinoculation (Fig. 1C to F).
FIG. 1.

(A) A schematic representation of the air-liquid interface (x, alveolar compartment; y, endovascular compartment); (B) the decline in the transgression of dextran blue into the endovascular compartment as a function of time, reflecting the attainment of cellular confluence. (C to F) Confocal images at 12, 16, 20, and 24 h postinoculation are shown; the lower panes depict the corresponding cross-sectional images. Scale bar, 10 μm. (G) The kinetics of galactomannan in infected but untreated inserts. The solid line represents the time course of galactomannan in the endovascular compartment, whereas the dashed line is the time course in the alveolar compartment. Data are means ± standard deviations of three inserts.
Pharmacokinetics.
The concentration-time profiles of each of the AmB formulations are shown in Fig. 2. The concentrations of amphotericin B associated with each of the formulations declined in the endovascular compartment throughout the experimental period. Concomitantly, concentrations of amphotericin B in both the endothelial and alveolar cells rose steeply and attained concentrations far in excess of those observed with the endovascular compartment. The highest concentrations were seen with LAmB and ABLC.
FIG. 2.

The pharmacokinetics of amphotericin B deoxycholate (DAmB), liposomal amphotericin (LAmB), and amphotericin B lipid complex (ABLC) in the endovascular fluid (top row), endothelial cells (middle row), and alveolar epithelial cells (bottom row). Four concentrations are displayed in each panel. Data are mean ± standard deviation of three inserts.
Concentration-response relationships for amphotericin B formulations.
The concentration-response relationships for the three formulations were initially examined in detail 24 h posttreatment (30 h postinoculation) (Fig. 3). DAmB induced a steep exposure-response relationship in both the alveolar and endovascular compartments over a very narrow concentration range. In contrast, LAmB and ABLC induced more languid concentration-response relationships, with incomplete suppression of galactomannan in the alveolar compartment, even at high concentrations. Marked differences in the concentration-response and AUC-response relationships were observed for DAmB, LAmB, and ABLC. These differences are reflected in the estimates for EC50 and EC90, despite comparable MICs (Table 1). These findings were further supported by confocal microscopy, which also demonstrated progressive reduction in hyphal penetration with increasing concentrations of all formulations (Fig. 3).
FIG. 3.

Concentration-response relationships at 24 h posttreatment for amphotericin B deoxycholate (DAmB), liposomal amphotericin B (LAmB), and amphotericin B lipid complex (ABLC). Dashed and solid lines represent galactomannan concentrations in the alveolar lavage and endovascular fluid, respectively. The confocal images on the right enable the fungal biomass associated with various concentrations to be visualized. Data are means ± standard deviations of three inserts.
TABLE 1.
The estimates for EC50 and EC90 quantified in terms of the initial concentration and the area under the concentration-time curve/MIC ratio (AUC/MIC) for each amphotericin B formulation
| Parametera | Amphotericin B deoxycholate |
Liposomal amphotericin B |
Amphotericin B lipid complex |
|||
|---|---|---|---|---|---|---|
| Endovascular | Alveolar | Endovascular | Alveolar | Endovascular | Alveolar | |
| Initial concn (mg/liter) | ||||||
| EC50 (95% confidence interval) | 0.12 (0.11-0.13) | 0.17 (0.13-0.19) | 1.03 (0.94-1.12 | 7.76 (2.40-13.19) | 4.41 (4.13-4.70) | 39.34 (35.61-43.03) |
| EC90 | 0.24 | 0.36 | 1.94 | 41.48 | 13.63 | 110.82 |
| AUC/MIC | ||||||
| EC50 (95% confidence interval) | 6.4 (6.14-6.66) | 8.35 (7.99-8.71) | 67.73 (65.72-69.75) | 505.2 (384.7-625.7) | 210.2 (201.6-218.8) | 1,874 (1,751-1,996) |
| EC90 | 12.93 | 17.62 | 127.46 | 2,705.41 | 651.3 | 5,280.56 |
Initial concentration is the concentration of the respective formulations (mg/liter) administered to the endothelial compartment 6 h postinoculation; EC50 and EC90 represent the drug exposure required to induce 50 and 90% of the maximal antifungal effect, respectively.
Similar findings were apparent with the temporal galactomannan concentrations for each of the formulations. For each compound, increasing drug concentrations resulted in increased suppression of fungal growth (Fig. 4). Consistent with previous experiments at a single time point, LAmB and ABLC both resulted in incomplete suppression of galactomannan in the alveolar compartment.
FIG. 4.

Serially collected pharmacodynamic data for the endovascular and alveolar compartments following exposure to a range of concentrations of amphotericin B deoxycholate (DAmB), liposomal amphotericin B (LAmB), and amphotericin B lipid complex (ABLC). Data are means ± standard deviations of three inserts.
Interaction of LAmB with human cells and hyphae.
Confocal images of endovascular fluid showed the size and spherical structure of free sulforhodamine-labeled liposomes (LAmB-Rho and LPlac-Rho). Over the subsequent 24 h, there was progressive accumulation of sulforhodamine within the cellular bilayer (Fig. 5). The majority of sulforhodamine was associated with endothelial cells with a less intense signal emanating from the alveolar epithelial cells (Fig. 5). There was an intense signal from sulforhodamine-encapsulated hyphae as they penetrated into the endovascular compartment (Fig. 6).
FIG. 5.

Confocal images of host cell-liposome interactions. (A) Liposomes within the endovascular compartment immediately after administration; scale bar, 1 μm. (B) Endothelial aspect of the cellular bilayer demonstrating the interaction of sulforhodamine-labeled liposomes devoid of amphotericin B with endothelial cells after incubation for 24 h; scale bar, 10 μm. (C) Endothelial aspect of the cellular bilayer demonstrating the interaction of sulforhodamine-labeled liposomes containing amphotericin B; scale bar, 10 μm. The second rows of images in panels B and C represent a cross-sectional view of the cellular bilayer showing a gradient of liposomal deposition from endothelial cells (lower layer) to alveolar cells (upper layer).
FIG. 6.

The interaction of hyphae with sulforhodamine B-labeled liposomal amphotericin B. The lower image represents a cross-sectional image. Scale bar, 10 μm.
Mathematical modeling.
The estimates of the means and dispersion for the model parameters are summarized in Table 2. The fit of the model to the data for all three formulations was acceptable, with r2 values of >99% for the endovascular fluid and endothelial cells, and >89% for the alveolar cells for observed-versus-predicted values, and with acceptable measures of precision and bias. The ratio of AUCs in the endothelial and alveolar cells to those of the endovascular fluid is shown in Fig. 7. For a given concentration, the relative penetration into the endothelial cells was 305.2, 206.0, and 438.1 and into the alveolar cells was 150.1, 14.3, and 21.4 for DAmB, LAmB, and ABLC, respectively.
TABLE 2.
Parameter means and standard deviations from the mathematical model describing the pharmacokinetics of each of the amphotericin B formulations
| Parameter, unitsa | Mean (SD) |
||
|---|---|---|---|
| DAmBb | LAmBc | ABLCd | |
| K12, h−1 | 4.97 (4.31) | 3.01 (1.51) | 5.65 (7.21) |
| K21, h−1 | 19.83 (8.21) | 20.4 (5.36) | 16.79 (8.46) |
| K23, h−1 | 7.13 (10.28) | 1.404 (1.84) | 1.04 (1.15) |
| K32, h−1 | 14.99 (7.83) | 20.07 (6.73) | 20.12 (7.59) |
| Vendo fluid, liters | 7.89 × 10−4 (9.92 × 10−5) | 7.60 × 10−4 (1.15 × 10−4) | 6.9 × 10−4 (1.18 × 10−4) |
| Vendo cells, liters | 6.47 × 10−7 (2.06 × 10−7) | 5.56 × 10−7 (1.13 × 10−7) | 5.29 × 10−7 (2.79 × 10−7) |
| Valv cells, liters | 5.81 × 10−7 (2.80 × 10−7) | 7.47 × 10−7 (2.65 × 10−7) | 6.48 × 10−7 (2.71 × 10−7) |
K12, K21, K23, and K32 are the first-order intercompartmental rate constants describing the movement of drug between compartment 1 (endovascular fluid), compartment 2 (endothelial cells), and compartment 3 (alveolar cells). Vendo fluid is the volume of the endovascular compartment in liters, Vendo cells is the volume of the entire endothelial cell layer, and Valv cells is the volume of the entire alveolar cell layer.
DAmB, amphotericin B deoxycholate.
LAmB, liposomal amphotericin B.
ABLC, amphotericin B lipid complex.
FIG. 7.

The proportional penetration of AmB in the endothelial and alveolar cells compared with endovascular fluid for DAmB, LAmB, and ABLC). The estimates for the area under the concentration-time curve are derived from the mathematical model.
DISCUSSION
Despite intensive efforts, IPA remains a rapidly progressive and frequently lethal infectious syndrome for which there are relatively few therapeutic options. The lipid preparations of amphotericin B have an established role for the treatment of proven and suspected Aspergillus infection (8, 25), and they are consistently less toxic than DAmB (3, 27). The air-liquid interface model of the human alveolus provides an ideal construct to examine the extent of penetration for each of the amphotericin B formulations into various subcompartments of the lung that are relevant for events in the pathogenesis of early IPA.
The formulation of amphotericin B has a profound effect on the disposition, elimination, and activity of the pure compound in both laboratory animal models and in humans (1, 2). Our results also suggest that the specific formulation influences the pharmacokinetic and pharmacodynamic relationships at a cellular level. Consistent with a previous study, the MICs of amphotericin B and lipid formulations of amphotericin B were comparable (17). Despite this, the exposure-response relationships for DAmB versus LAmB and ABLC were strikingly different, with estimates for EC50 for both lipid formulations being significantly higher than those for DAmB. These results suggest that the MIC of lipid preparations transmits relatively little information that can be used to predict exposure-response relationships in experimental systems and in humans.
The formulation-specific pharmacodynamic relationships likely result from thermodynamic constraints that govern the release and transfer of pure amphotericin B from its lipid carrier. Despite the likely interaction between liposomes and human cells, the majority of pure amphotericin B probably remains preferentially complexed within the liposome, rather than engaging with mammalian lipids, because the former is a more energetically favorable state. Indeed, this phenomenon is used for quality control processes for the manufacture of LAmB. The incubation of defective liposomes with human erythrocytes results in the release of amphotericin B at relatively low drug concentrations, and this causes potassium leakage from damaged erythrocyte membranes (16). In the presence of a higher-affinity target (i.e., ergosterol in fungal membranes), it becomes energetically more favorable for amphotericin B to disengage from the liposome and aggregate within fungal membranes. Presumably these conclusions are also (qualitatively) applicable to ABLC, although this is difficult to confirm in the absence of a similarly labeled preparation.
Our results suggest two potential cellular mechanisms by which amphotericin B within LAmB engages hyphae and conidia of Aspergillus within the lung during invasive pulmonary aspergillosis: (i) the liposome directly engages with hyphae (without initially interacting with host cells) as they invade into the lumen of blood vessels and (ii) LAmB initially associates with mammalian cell membranes and is then able to engage with conidia or hyphae as they, in turn, interact with host cells in the course of invasive infection. Both points of cellular interaction permit the selective transfer of amphotericin B from liposomes to fungal cell membranes. Amphotericin B remains strongly associated with the liposome of LAmB, such that only a small fraction of “free” drug (<1%) is detectable in biological media (4). These hypotheses require an assumption that the sulforhodamine used to visualize liposomal distribution largely remains preferentially complexed within the liposomal structure and does not itself freely distribute. The location of sulforhodamine is within the enclosed aqueous space of the LAmB liposome structure, and one expects sulforhodamine to be carried with the liposome so long as the liposome remains intact.
All three formulations achieve significantly higher concentrations in the cells than in the contiguous endovascular fluid. The proportional concentrations in endothelial and alveolar cells relative to endovascular fluid are similar to those observed with epithelial lining fluid and pulmonary alveolar macrophages relative to serum in rabbits (11). The potential significance of drug concentrations in tissues is inextricably linked with the pathogenesis of the infectious diseases (13). IPA begins with the inhalation of conidia into the lung, a proportion of which contact alveolar epithelial cells and undergo phagocytosis (12). The resulting phagolysosome is enveloped by the cell membrane. If this membrane contains amphotericin B, then this may contribute to the killing of conidia within the phagolysosome. Similarly, following germination, hyphae invade through cell membranes of alveolar and endothelial cells, at which time they are exposed to high concentrations of drug, which may lead to hyphal damage and death.
The immune status of the host and innate immunological effectors are critical determinants of the outcome of invasive fungal diseases (14). A limitation of this in vitro model is that the potential antifungal effect of immune effector cells, such as circulating monocytes or pulmonary alveolar macrophages, cannot be estimated. Furthermore, the reticuloendothelial system may influence the serum concentration-time profile of the lipid formulations of amphotericin B (26). Relatively large concentrations of amphotericin B from LAmB and ABLC accumulate within pulmonary alveolar macrophages. This additional compartment provides another point of potential cellular interaction between fungal elements and amphotericin within the alveolus. Empty liposomes have an antifungal effect in laboratory animal models of IPA that presumably reflects immunomodulation that is favorable for the host (20). A further possibility is that immune effector cells laden with drug may traffic into the alveolar space and deliver drug to the fungal target. This so-called “dump truck” phenomenon also has been postulated to account for the action of the macrolides because this class of compounds does not achieve high concentrations within the epithelial lining fluid of the lung (10). Mehta and colleagues found that an alternative formulation of amphotericin B accumulated in inflammatory peritoneal cells after intravenous administration of fluorescence-labeled L-AmB, suggesting that macrophages play an important role in the transport of the intravenously administered lipid formulation to inflammatory sites (21). This transport process is likely during invasive pulmonary aspergillosis but has not been well studied to any extent. The extent to which these kinetics may be altered in neutropenic versus nonneutropenic hosts, in which the inflammatory responses differ markedly (5), merits further study.
An improved understanding of the intrapulmonary pharmacokinetics and pharmacodynamics of AmB is required for the design of optimal (and innovative) dosing regimens for the prevention and treatment of IPA. Furthermore, novel formulations of existing antifungal compounds may provide a way to extend and improve their clinical utility and serve as a valuable mechanism to further improve the safety and efficacy for a persistently lethal infectious syndrome.
Acknowledgments
This work was funded, in part, by the Fungal Research Trust and Gilead Sciences. William Hope is supported by a National Institute of Health Research (NIHR) Clinician Scientist Fellowship. This study was supported, in part, by the intramural research program of the National Cancer Institute, National Institutes of Health.
We thank David Perlin and Jill Adler-Moore for their comments and insights. We also thank Robert Fernandez (Biomedical Imaging, Faculty of Life Sciences, University of Manchester, United Kingdom) and Huy Pham and Tark Bunch (Gilead Sciences, San Dimas) for their outstanding technical assistance.
Footnotes
Published ahead of print on 3 May 2010.
REFERENCES
- 1.Adler-Moore, J. P., and R. T. Proffitt. 2008. Amphotericin B lipid preparations: what are the differences? Clin. Microbiol. Infect. 14(Suppl. 4):25-36. [DOI] [PubMed] [Google Scholar]
- 2.Andes, D., N. Safdar, K. Marchillo, and R. Conklin. 2006. Pharmacokinetic-pharmacodynamic comparison of amphotericin B (AMB) and two lipid-associated AMB preparations, liposomal AMB and AMB lipid complex, in murine candidiasis models. Antimicrob. Agents Chemother. 50:674-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bates, D. W., L. Su, D. T. Yu, G. M. Chertow, D. L. Seger, D. R. Gomes, E. J. Dasbach, and R. Platt. 2001. Mortality and costs of acute renal failure associated with amphotericin B therapy. Clin. Infect. Dis. 32:686-693. [DOI] [PubMed] [Google Scholar]
- 4.Bekersky, I., R. M. Fielding, D. E. Dressler, J. W. Lee, D. N. Buell, and T. J. Walsh. 2002. Plasma protein binding of amphotericin B and pharmacokinetics of bound versus unbound amphotericin B after administration of intravenous liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate. Antimicrob. Agents Chemother. 46:834-840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berenguer, J., M. C. Allende, J. W. Lee, K. Garrett, C. Lyman, N. M. Ali, J. Bacher, P. A. Pizzo, and T. J. Walsh. 1995. Pathogenesis of pulmonary aspergillosis. Granulocytopenia versus cyclosporine and methylprednisolone-induced immunosuppression. Am. J. Respir. Crit. Care Med. 152:1079-1086. [DOI] [PubMed] [Google Scholar]
- 6.Bermudez, L. E., F. J. Sangari, P. Kolonoski, M. Petrofsky, and J. Goodman. 2002. The efficiency of the translocation of Mycobacterium tuberculosis across a bilayer of epithelial and endothelial cells as a model of the alveolar wall is a consequence of transport within mononuclear phagocytes and invasion of alveolar epithelial cells. Infect. Immun. 70:140-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.CLSI/NCCLS. 2002. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi. Approved standard M38-A. National Committee for Clinical Laboratory Standards, Wayne, PA.
- 8.Cornely, O. A., J. Maertens, M. Bresnik, R. Ebrahimi, A. J. Ullmann, E. Bouza, C. P. Heussel, O. Lortholary, C. Rieger, A. Boehme, M. Aoun, H. A. Horst, A. Thiebaut, M. Ruhnke, D. Reichert, N. Vianelli, S. W. Krause, E. Olavarria, and R. Herbrecht. 2007. Liposomal amphotericin B as initial therapy for invasive mold infection: a randomized trial comparing a high-loading dose regimen with standard dosing (AmBiLoad trial). Clin. Infect. Dis. 44:1289-1297. [DOI] [PubMed] [Google Scholar]
- 9.D'Argenio, D. Z., and A. Schumitzky. 1997. ADAPT II. A program for simulation, identification, and optimal experimental design. User manual. Biomedical Simulations Resource, University of Southern California, Los Angeles, CA. http://bmsr.esc.edu/.
- 10.Drusano, G. L. 2005. Infection site concentrations: their therapeutic importance and the macrolide and macrolide-like class of antibiotics. Pharmacotherapy 25:150S-158S. [DOI] [PubMed] [Google Scholar]
- 11.Groll, A. H., C. A. Lyman, V. Petraitis, R. Petraitiene, D. Armstrong, D. Mickiene, R. M. Alfaro, R. L. Schaufele, T. Sein, J. Bacher, and T. J. Walsh. 2006. Compartmentalized intrapulmonary pharmacokinetics of amphotericin B and its lipid formulations. Antimicrob. Agents Chemother. 50:3418-3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hope, W. W. 2009. Invasion of the alveolar-capillary barrier by Aspergillus spp.: therapeutic and diagnostic implications for immunocompromised patients with invasive pulmonary aspergillosis. Med. Mycol. 47(Suppl. 1):S291-S298. [DOI] [PubMed] [Google Scholar]
- 13.Hope, W. W., and G. L. Drusano. 2009. Antifungal pharmacokinetics and pharmacodynamics: bridging from the bench to bedside. Clin. Microbiol. Infect. 15:602-612. [DOI] [PubMed] [Google Scholar]
- 14.Hope, W. W., G. L. Drusano, C. B. Moore, A. Sharp, A. Louie, T. J. Walsh, D. W. Denning, and P. A. Warn. 2007. Effect of neutropenia and treatment delay on the response to antifungal agents in experimental disseminated candidiasis. Antimicrob. Agents Chemother. 51:285-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hope, W. W., M. J. Kruhlak, C. A. Lyman, R. Petraitiene, V. Petraitis, A. Francesconi, M. Kasai, D. Mickiene, T. Sein, J. Peter, A. M. Kelaher, J. E. Hughes, M. P. Cotton, C. J. Cotten, J. Bacher, S. Tripathi, L. Bermudez, T. K. Maugel, P. M. Zerfas, J. R. Wingard, G. L. Drusano, and T. J. Walsh. 2007. Pathogenesis of Aspergillus fumigatus and the kinetics of galactomannan in an in vitro model of early invasive pulmonary aspergillosis: implications for antifungal therapy. J. Infect. Dis. 195:455-466. [DOI] [PubMed] [Google Scholar]
- 16.Jensen, J. M., C. R. Skenes, T. H. Bunch, N. Weissman, N. Amirghahari, A. Satorius, K. L. Moynihan, and C. G. S. Eley. 1999. Determination of the relative toxicity of amphotericin B formulations: a red blood cell potassium release assay. Drug Delivery 6:81-88. [Google Scholar]
- 17.Lass-Flörl, C., A. Mayr, S. Perkhofer, G. Hinterberger, J. Hausdorfer, C. Speth, and M. Fille. 2008. Activities of antifungal agents against yeasts and filamentous fungi: assessment according to the methodology of the European Committee on Antimicrobial Susceptibility Testing. Antimicrob. Agents Chemother. 52:3637-3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Latgé, J. P. 1999. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 12:310-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leary, R., R. Jelliffe, A. Schumitzky, and M. van Guilder. 2001. An adaptive grid, non-parametric approach to pharmacokinetic and dynamic (PK/PD) models, p. 389-394. In Proceedings of the 14th IEEE Symposium on Computer-Based Medical Systems. IEEE Computer Society, Bethesda, MD.
- 20.Lewis, R. E., G. Chamilos, R. A. Prince, and D. P. Kontoyiannis. 2007. Pretreatment with empty liposomes attenuates the immunopathology of invasive pulmonary aspergillosis in corticosteroid-immunosuppressed mice. Antimicrob. Agents Chemother. 51:1078-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehta, R. T., T. J. McQueen, A. Keyhani, and G. Lopez-Berestein. 1994. Phagocyte transport as mechanism for enhanced therapeutic activity of liposomal amphotericin B. Chemotherapy 40:256-264. [DOI] [PubMed] [Google Scholar]
- 22.Nivoix, Y., M. Velten, V. Letscher-Bru, A. Moghaddam, S. Natarajan-Ame, C. Fohrer, B. Lioure, K. Bilger, P. Lutun, L. Marcellin, A. Launoy, G. Freys, J. P. Bergerat, and R. Herbrecht. 2008. Factors associated with overall and attributable mortality in invasive aspergillosis. Clin. Infect. Dis. 47:1176-1184. [DOI] [PubMed] [Google Scholar]
- 23.Pappas, P. G., C. A. Kauffman, D. Andes, D. K. Benjamin, Jr., T. F. Calandra, J. E. Edwards, Jr., S. G. Filler, J. F. Fisher, B. J. Kullberg, L. Ostrosky-Zeichner, A. C. Reboli, J. H. Rex, T. J. Walsh, and J. D. Sobel. 2009. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 48:503-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh, T. J., E. J. Anaissie, D. W. Denning, R. Herbrecht, D. P. Kontoyiannis, K. A. Marr, V. A. Morrison, B. H. Segal, W. J. Steinbach, D. A. Stevens, J. A. van Burik, J. R. Wingard, and T. F. Patterson. 2008. Treatment of aspergillosis: clinical practice guidelines of the Infectious Diseases Society of America. Clin. Infect. Dis. 46:327-360. [DOI] [PubMed] [Google Scholar]
- 25.Walsh, T. J., R. W. Finberg, C. Arndt, J. Hiemenz, C. Schwartz, D. Bodensteiner, P. Pappas, N. Seibel, R. N. Greenberg, S. Dummer, M. Schuster, and J. S. Holcenberg. 1999. Liposomal amphotericin B for empirical therapy in patients with persistent fever and neutropenia. National Institute of Allergy and Infectious Diseases Mycoses Study Group. N. Engl. J. Med. 340:764-771. [DOI] [PubMed] [Google Scholar]
- 26.Walsh, T. J., J. L. Goodman, P. Pappas, I. Bekersky, D. N. Buell, M. Roden, J. Barrett, and E. J. Anaissie. 2001. Safety, tolerance, and pharmacokinetics of high-dose liposomal amphotericin B (AmBisome) in patients infected with Aspergillus species and other filamentous fungi: maximum tolerated dose study. Antimicrob. Agents Chemother. 45:3487-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wingard, J. R., P. Kubilis, L. Lee, G. Yee, M. White, L. Walshe, R. Bowden, E. Anaissie, J. Hiemenz, and J. Lister. 1999. Clinical significance of nephrotoxicity in patients treated with amphotericin B for suspected or proven aspergillosis. Clin. Infect. Dis. 29:1402-1407. [DOI] [PubMed] [Google Scholar]