

Abstract
p38 mitogen-activated protein kinase (MAPK) is rapidly activated by stresses and is believed to play an important role in the stress response. While Chk1 is known to mediate G2 DNA damage checkpoint control, p38 was also reported to have an essential function in this checkpoint control. Here, we have investigated further the roles of p38 and Chk1 in the G2 DNA damage checkpoint in cancer cells. We find that although p38 activation is strongly induced by DNA damage, its activity is not required for the G2 DNA damage checkpoint. In contrast, Chk1 kinase is responsible for the execution of G2 DNA damage checkpoint control in p53-deficient cells. The inhibition of p38 activity has no effect on Chk1 activation and γ-H2AX expression. Global gene expression profiling of cancer cells in response to tumor necrosis factor alpha (TNF-α) revealed that p38 plays a strong prosurvival role through the coordinated downregulation of proapoptotic genes and upregulation of prosurvival genes. We show that the inhibition of p38 activity during G2 DNA damage checkpoint arrest triggers apoptosis in a p53-independent manner with a concurrent decrease in the level of Bcl2 family proteins. Our results suggest that although p38 MAPK is not required for the G2 DNA damage checkpoint function, it plays an important prosurvival role during the G2 DNA damage checkpoint response through the upregulation of the Bcl2 family proteins.
p38 mitogen-activated protein kinase (MAPK) was originally identified as a 38-kDa protein that undergoes rapid tyrosine phosphorylation in response to stress (17). Significant progress has been made in the past decade to understand the p38 signal transduction pathway and the biological processes regulated by p38 MAPK. p38 MAPK is activated in response to stress-related stimuli such as UV light (27), heat (39), osmotic shock (33, 39), endotoxins (18), and inflammatory cytokines like tumor necrosis factor alpha (TNF-α) and interleukin-1 (IL-1) (27, 34). The p38 pathway is implicated in the inflammatory response, as p38 activation induces proinflammatory cytokines and enzymes such as Cox-2, which controls connective tissue remodeling, and inflammation-related adhesion proteins such as VCAM-1 (36), thus making p38 MAPK signaling an attractive therapeutic target for the mitigation of inflammatory diseases (41). This has led to the creation of biochemical inhibitors targeting p38 kinase (7, 20). The latest generation of these inhibitors is highly potent and selective, raising possibilities that therapy involving p38 inhibitors may one day be an effective treatment for inflammatory diseases.
Recently, p38 MAPK activity was reported to be critical for G2 DNA damage checkpoint control in response to DNA damage by UV irradiation (5, 6, 31) or by genotoxic agents (19, 26). The primary mechanism of the p38 involvement in the G2 DNA damage checkpoint is thought to be mediated through the inhibition of CDC25B/C phosphatases, which are required for the activation of CDK1 to initiate mitosis (5, 31). Structural analysis of the p38 binding site, however, suggests that it is unlikely that p38 could interact directly with CDC25B. Instead, its direct downstream target, MAPKAPK2 (MK2), is implicated as the mediator of p38-dependent G2 DNA damage checkpoint control (31).
The ability of cancer cells to establish cell cycle arrest in response to genotoxic agents is one of the reasons for resistance to chemotherapy (8). Cancer cells that undergo reversible cell cycle arrest in response to genotoxic agents such as adriamycin (doxorubicin HCl) and cisplatin have the ability to survive chemotherapy and continue proliferation posttherapy, leading to poor patient outcomes. The implication that p38 activity is necessary for G2 DNA damage checkpoint arrest provides an exciting possibility for a p38 inhibitor (p38i) as a chemosensitizer to enhance the efficacy of chemotherapies by abrogating the G2 DNA damage checkpoint to promote cancer cells to enter mitosis prematurely.
Both p38 and Chk1 are activated by DNA damage in mammalian cells, and both are believed to directly inactivate CDC25 family of protein phosphatases to prevent mitotic entry in the presence of DNA damage (25, 44, 45). Paradoxically, the inhibition of either p38 or Chk1 was shown previously to be sufficient to abrogate the G2 DNA damage checkpoint (5, 21, 44). The role of the p38 MAPK pathway in the G2 DNA damage checkpoint of cancer cells has recently been called into question by the observation that transformed cells do not delay entry into mitosis upon the activation of the p38 stress pathway by anisomycin (32). Furthermore, it was shown recently that the RNA interference (RNAi)-mediated inhibition of Chk1, but not Chk2 or MK2, in HeLa and H1299 cancer cells abrogates DNA damage-induced S-phase or G2-phase arrest (45). The requirement for p38 in G2 DNA damage checkpoint control may be cell type specific or may depend on the type of DNA damage. While p38 is activated by both ionizing and UV radiation, the p38/MK2 pathway was reported to be essential for the G2 DNA damage checkpoint only in the absence of p53 (38). It should be noted that the older generation of small-molecule inhibitors of p38 kinase was used at very high concentrations in many earlier studies, raising the possibility of off-target effects (1, 10). In this study, we revisited the role of p38 activity in G2 DNA damage checkpoint control in response to several types of DNA damage and investigated the relationship between Chk1 and p38 kinases in G2 DNA damage checkpoint control in tumor cells with or without functional p53. We also used a newer generation of small-molecule kinase inhibitors that are more potent and selective at physiologically relevant concentrations and independently confirmed and corroborated the small-molecule kinase inhibitor activity with small interfering RNA (siRNA)-mediated inhibition.
We demonstrate that while p38 is rapidly and strongly induced by DNA damage, the inhibition of p38 activity with a potent and selective inhibitor, LY479754, or siRNA knockdown does not compromise the ability of cancer cells to mount effective, checkpoint-mediated G2 arrest in response to adriamycin, methyl methanesulfonate (MMS), or UV-induced DNA damage. In contrast, the chemical inhibition and siRNA knockdown of Chk1 efficiently abolish the G2 DNA damage checkpoint in cancer cells with deficient p53. Using an unbiased whole-genome transcriptional analysis, we identified a strong link between p38 activation and the suppression of antiapoptotic signaling in TNF-α-treated cells. Extending these findings to the G2 DNA damage checkpoint context, we show that the inhibition of p38 results in a dramatic increase in the level of apoptosis of cells arrested in G2 in response to DNA damage. Based on our observations, we propose that although not required for G2 DNA damage checkpoint control, p38 plays an important cytoprotective role through the regulation of apoptotic and survival pathways to allow cells to recover from DNA damage.
MATERIALS AND METHODS
Cell culture and synchronization.
All cancer cell lines were obtained from the ATCC. HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS); Calu-6 cells were grown in minimal essential medium (MEM) supplemented with 10% FBS, 1% sodium pyruvate, and 1% HEPES; A549 cells were grown in RPMI 1640 medium supplemented with 10% FBS; and U2OS cells were grown in McCoy 5A medium supplemented with 10% FBS. All cell culture media and additives were purchased from Gibco (Invitrogen, Carlsbad, CA). All cells were grown in a cell culture incubator at 37°C and 5% CO2 in T75 or T150 tissue culture flasks (Corning, Lowell, MA). Cells were synchronized at G1 by using double-thymidine block/release or at G2 by using a selective CDK1 inhibitor as previously described (11).
Antibodies, Western blot analysis, and immunofluorescence microscopy.
Rabbit polyclonal antibody to phospho-histone H3 (Ser10) (catalog no. 06-570) was purchased from Upstate Inc. (Charlottesville, VA). Rabbit polyclonal anti-phospho-p38 (catalog no. 9216), anti-phospho-MAPKAPK2 (MK2) (Thr334) (catalog no. 3041), anti-phospho-Chk1 (Ser345) (catalog no. 2341), anti-phospho-HSP27 (Ser17) (catalog no. 2404), anti-cleaved-Casp3 (anti-cl-Casp3; catalog no. 9661), anti-cl-Casp7 (D198) (catalog no. 9491), anti-α/β-tubulin (catalog no. 2148), anti-BCL2 (catalog no. 2872), anti-BCL-xl (catalog no. 2762), anti-γ-H2AX (catalog no. 9718), anti-Fas-associated death domain (anti-FADD; catalog no. 2782), anti-p38α (catalog no. 9218), anti-β-actin (catalog no. 4967), and mouse monoclonal anti-cleaved-poly (ADP-ribose) polymerase (anti-c-PARP) (D214) (catalog no. 9546) were all purchased from Cell Signaling Technologies Inc. (Beverly, MA). Anti-cyclin B1 (catalog no. 610220) was purchased from BD Transduction Laboratories (San Jose, CA). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Amersham (Piscataway, NJ), and Alexa Fluor-linked secondary antibodies were purchased from Invitrogen (Carlsbad, CA).
Protein lysates of cultured cells were prepared in a lysate buffer containing a cocktail of phosphatase and protease inhibitors, and Western blotting was performed as previously described (11). Luminescent substrate detection was performed by using the ECL Advance or ECL Plus chemiluminescent kit (Amersham, Piscataway, NJ). Chemiluminescent signal was detected by using a high-resolution GE Gel-Blot imager.
Cells were plated for confocal microscopy in Lab-Tek 4 chamber slides (Fisher Scientific). Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) and then permeabilized with 0.2% Triton X-100. After blocking for 1 h in 1% bovine serum albumin (BSA) in PBS, the cells were incubated with anti-γ-H2AX (1:200) and anti-cyclin B1 (1:500) antibodies in block solution for 1 h at room temperature. The cells were washed three times in PBS and incubated with secondary antibody (1:1,000) and DNA stain (1:20,000) (Sytox green; Invitrogen) for 1 h at room temperature. The cells were washed three times with PBS and imaged. Cell imaging was acquired with a Zeiss LSM510 confocal microscope.
Use of chemical agents and inhibitors.
The use of biochemical inhibitors and chemical genotoxic compounds in this study was performed as previously described (11, 46). Chemical inhibitors used in this study were synthesized by Lilly chemists. Kinase inhibitors used in this study were p38α/β inhibitor LY479754 (7, 11), MK2 inhibitor (37), and Chk1 inhibitor PF-00477736 (2). CDK1 inhibitor RO-3306 was purchased from Calbiochem (San Diego, CA). All other chemical reagents used in this study were purchased from Sigma-Aldrich (St. Louis, MO).
siRNA transfection.
The transfection of 21-nucleotide siRNA duplexes (Qiagen Sciences, Germantown, MD) for the targeting of endogenous genes was carried out by using Lipofectamine RNAimax (Invitrogen, Carlsbad, CA), as previously described, in low-serum (Opti-Mem) medium (11). The following validated commercial siRNAs from Qiagen were used in this study: SI00300769 and SI00605157 for si-p38α, SI02223697 and SI00288246 for si-MK2, and SI0266000 and SI00299859 for si-Chk1. In addition, an MK2-specific siRNA oligonucleotide described previously by Manke et al. (31) was synthesized by Dharmacon and used.
Acumen Explorer high-content imaging analysis.
HeLa cells were plated into 96-well Beckman Dickinson Biocoat plates at 2,000 cells per well in 100 μl of medium and incubated in 5% CO2 at 37°C for 24 h before treatment with compounds diluted in growth medium with 10% FBS and 0.25% dimethyl sulfoxide (DMSO). All liquids were handled with an automated 96-channel pipette (Multimek 96; Beckman) to process the plates. Cells were fixed with Prefer fixative (Anatech Ltd.) at 25°C for 30 min, permeabilized with 0.1% Triton X-100 in PBS (Gibco, Carlsbad, CA) for 15 min, and then treated with RNase A (50 μg/ml in PBS) (Sigma, St. Louis, MO) at 37°C for 60 min. Immunostaining of cells and counterstaining with propidium iodide (PI) for high-throughput quantitative analysis by Acumen Explorer were similarly done as described previously (3).
UV irradiation and FACS analysis.
UV irradiation was performed at 254 nm (UV-C) by using a Stratalinker 2400 apparatus (Stratagene) with U2OS cells under the same conditions as those described previously by Manke et al. (31). U2OS cells were prepared for fluorescence-activated cell sorter (FACS) analysis also as described previously by Manke et al. (31).
In addition to experiments reproducing the UV damage data described previously by Manke et al., additional UV experiments were performed at 290 nm (UV-B) by using a Bio-Link BLX (Vilber Lourmat) computerized UV cross-linker. For all UV-B experiments, cells were treated with UV-B, as indicated in the figure legends, after the removal of cell growth media, followed immediately by the reintroduction of growth media with the indicated chemical inhibitor treatments. Western blot, FACS, and Acumen high-content imaging experiments were performed as previously described (3, 11, 46).
Gene expression profiling analysis.
Microarray analysis was performed as previously described (29). Briefly, total RNA from Calu-6 cells was isolated with RNA STAT-60 (Tel-Test) according to the manufacturer's protocol. Five micrograms of total RNA was labeled and hybridized to Affymetrix U133plus2 arrays according to the Affymetrix protocol. All samples were assessed for RNA quality such as microarray scaling factors, background levels, percent present calls, β-actin, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) 3′/5′ ratios, etc. Signal intensities as gene expression values were obtained from Microarray Suite, version 5.0 (MAS5), by using the default settings except that the 2% trimmed mean was set to 1,500. To apply statistical analysis, a two-sided t test was used to identify genes differentially expressed between two groups. The P values of the t tests were adjusted for multiple testing by using the false discovery rate (FDR). The adjusted P values, or the FDR, are designated Q values, where Q = P × n/I (where n is the total number of probe sets on the microarrays and I is the sorted rank of P values). The fold change was calculated as the ratio of the two group means based on the observed signal values from MAS5, and the gene expression signal change was calculated as the difference of the two group means. The criteria to define differential gene expression are an FDR of <0.05, a fold change of >1.4, and an absolute change of >250. Differentially expressed genes were mapped to Gene Ontology (GO) biological process categories and KEGG pathways. The significance of GO terms or KEGG pathways overrepresented in differentially expressed genes was tested by using the hypergeometric distribution function adjusted with family-wise error rates for multiple pairwise tests.
RESULTS
p38 MAPK is activated by DNA damage at different stages of the cell cycle.
p38 MAPK is known to be activated in response to DNA damage. We first assessed if p38 activation is associated with G2 arrest induced by different modes of DNA damage. For these experiments, we used different sources of DNA damage that induce a G2 arrest in p53-deficient HeLa cells (Fig. 1 A). In conjunction with the establishment of G2 cell cycle arrest, p38 is strongly activated by increasing doses of UV-B irradiation (Fig. 1B), 0.01% MMS (Fig. 1C), and 160 nM adriamycin (data not shown; also see Fig. 1D) with similar kinetics. To further confirm that the activation of p38 is closely correlated with G2 arrest, we synchronized HeLa cells at G1/S using the double-thymidine block/release protocol before imposing DNA damage by the addition of adriamycin and monitored cell cycle progression by monitoring multiple parameters. Indeed, adriamycin treatment caused G2 arrest and a sustained activation of p38 (Fig. 1D).
FIG. 1.

DNA damage induces G2 arrest and activates p38 MAPK and Chk1 pathways. (A) Cell cycle profiles of control HeLa cells or HeLa cells treated with 160 nM adriamycin, 500 J/m2 UV irradiation, or 0.01% MMS for 24 h. (B and C) Western blot analysis of the activation of p38α and phosphorylation of its substrate MK2 in unsynchronized HeLa cells treated with increasing doses of UV irradiation, with 10 μg/ml anisomycin treatment as a positive control (B) and with 0.01% MMS for various time intervals for up to 10 h (C). (D) Synchronized HeLa cells treated with 160 nM adriamycin 5 h after release from a second thymidine block. Cell cycle progression and p38 activation were analyzed by flow cytometry and Western blotting at the time points indicated after release. (E to G) DNA damage in synchronized cells. HeLa cells were synchronized at G1 by serum starvation (E), at G1/S by double-thymidine block (F), and at G2 with the CDK1 inhibitor RO-3306 (G) and were then released into 0.01% MMS-containing fresh medium. The activation of p38 and Chk1 kinases was followed by Western blotting at the time intervals indicated.
To investigate if p38 activation occurs specifically during G2 DNA damage checkpoint-mediated arrest, HeLa cells were synchronized in G1 phase by serum starvation (Fig. 1E), in early S phase by a double-thymidine block (Fig. 1F), or in G2 phase by use of a CDK1 inhibitor (RO-3306) (Fig. 1G) and then released into fresh growth medium containing 0.01% MMS. Cells were subsequently monitored for the activation status of Chk1, p38, and MAPKAPK-2 (MK2) by using the respective phosphorylation-specific antibodies. As shown in Fig. 1E to G, p38 and Chk1 are rapidly activated after MMS treatment of HeLa cells synchronized at different stages of the cell cycle. The activation of p38 occurred earlier than that of Chk1 in G1- and S-phase cells, whereas p38 and Chk1 activation in G2-phase cells followed similar kinetics (Fig. 1E to G).
Inhibition of p38 does not abrogate G2 DNA damage checkpoint control.
To test whether p38 pathway activity is essential for the G2 DNA damage checkpoint in response to DNA damage, we investigated the effect of the chemical inhibition of the p38 pathway activity with LY479754 (p38i), a highly potent and selective p38 inhibitor (7, 11), on G2 DNA damage checkpoint-mediated arrest in both unsynchronized (Fig. 2 A) and synchronized (Fig. 2B) HeLa cells treated with adriamycin. Nocodazole, a microtubule-depolymerizing agent, was added to the medium to trap in mitosis cells that escape the checkpoint arrest in unsynchronized cells. Despite a strong inhibition of p38 activity, seen as a complete inhibition of the p38-mediated phosphorylation of MK2, HeLa cells were still able to mount effective G2 DNA damage checkpoint control in response to adriamycin treatment. The inhibition of p38 did not lead to any significant increase in the mitotic marker phospho-histone H3 over a 24-h period. Similarly, another small-molecule kinase inhibitor, SB203580, at concentrations above that needed for the completion inhibition of p38, also had no effect on the G2 DNA damage checkpoint, as HeLa cells remained arrested in G2 during a synchronized G2/M progression (data not shown). The inhibition of MK2 also showed no effect on checkpoint activity (Fig. 2B). In contrast, the inhibition of Chk1 with a selective Chk1 inhibitor (Fig. 2A and B) or ATM/ATR inhibition with caffeine (Fig. 2B) in an identical experimental setting led to a dramatic increase in phospho-histone H3 levels, indicating the effective abrogation of the G2 DNA damage checkpoint. Consistent with checkpoint abrogation, the inhibition of Chk1 or ATM/ATR led to a marked decrease in levels of Cdk1 phosphorylation on Tyr15 (Fig. 2B). On the other hand, the inhibition of p38 had no effect on the level of Cdk1 phosphorylation at Tyr15, which remained high (Fig. 2B). Furthermore, the abrogation of the G2 DNA damage checkpoint with either a Chk1 inhibitor or caffeine occurred in the presence of high levels of p38 and MK2 activities (Fig. 2B).
FIG. 2.

Inhibition of Chk1 but not p38 abrogates the G2 DNA damage checkpoint in HeLa cells. (A) Mitotic index of unsynchronized HeLa cells that were pretreated for 20 h with adriamycin prior to the addition of 320 nM p38 inhibitor (p38i) or 1.25μM Chk1 inhibitor (Chk1i) and 150 nM nocodazole. Mitotic indexes were quantified by using the high-content/high-throughput Acumen Explorer for cells expressing phospho-histone H3 at the time points indicated after inhibitor treatment. (B) Western blot analysis of double-thymidine-synchronized HeLa cells treated with various kinase inhibitors in the presence of 160 nM adriamycin and 150 nM nocodazole 20 h after release from the thymidine block. Lane 1, double-thymidine-treated cells; lane 2, untreated HeLa cells; lanes 3 to 9, synchronized cells treated with adriamycin; lane 3, adriamycin alone; lane 4, adriamycin plus 320 nM p38i; lane 5, adriamycin plus 2 μM MK2 inhibitor (MK2i); lane 6, adriamycin plus 1.25 μM Chk1 inhibitor; lane 7, adriamycin plus 6 mM caffeine; lane 8, adriamycin plus 1.25 μM Chk1 inhibitor and 320 nM p38i; lane 9, adriamycin plus 6 mM caffeine and 320 nM p38i. (C) Western blot analysis of the relationship between p38 activation/inactivation and the DNA damage response. Thymidine-synchronized HeLa cells were released into 0.01% MMS, and cells were analyzed at the times indicated. (D) Mitotic index of unsynchronized HeLa cells treated with 500 J/m2 UV-B irradiation for 20 h prior to the addition of 320 nM p38i or 1.25 μM Chk1 inhibitor in the presence of 150 nM nocodazole (Noc). (E) Western blot of HeLa cells treated with 1,000 J/m2 UV-B and the indicated doses of p38i, MK2 inhibitor, or Chk1 inhibitor 24 h after kinase inhibitor treatment.
These analyses were followed by confocal immunofluorescence microscopy of HeLa cells. Cells treated with either adriamycin alone or adriamycin and p38i for 21 h had high levels of γ-H2AX in the nucleus. These cells were arrested at G2 phase, as indicated by the cytoplasmic accumulation of cyclin B1 and 4N DNA content (data not shown; also see Fig. 1A). No mitosis was observed for the p38 inhibitor-treated cells under a microscope. In contrast, HeLa cells that were treated with adriamycin and a Chk1 inhibitor underwent mitosis, as evidenced by mitotic spindles, condensed DNA, and a strong phospho-histone H3 signal, indicating the effective abrogation of the G2 DNA damage checkpoint (data not shown). Western blot analysis further showed that the inhibition of p38 MAPK has no apparent impact on γ-H2AX expression and the activation of Chk1 (Fig. 2C). This shows that despite the potent inhibition of the p38 MAPK pathway, the DNA damage response to adriamycin and MMS is unimpeded, leading to strong G2 DNA damage checkpoint-mediated cell cycle arrest.
Previous reports first implicating p38 as a critical kinase in G2 DNA damage checkpoint function utilized UV irradiation as a source of DNA damage (5). Since p38 activity does not appear to be necessary for adriamycin- or MMS-induced G2 DNA damage checkpoint arrest, we thus wanted to investigate further a role of p38 activity in the response to UV-induced DNA damage. Both synchronous and asynchronous HeLa cell cultures were exposed to UV radiation and incubated with either p38 or Chk1 inhibitors immediately after UV treatment. Nocodazole was added to the cultures to trap in mitosis cells that had escaped from G2 DNA damage checkpoint-mediated arrest. Cells were harvested for analyses of various mitotic markers after 24 h. Again, while the pharmacological inhibition of p38 and MK2 did not lead to any significant increase in the mitotic index over 24 h, the inhibition of Chk1 led to a dramatic increase in the mitotic index and phospho-histone H3 over the same time period (Fig. 2D and E). These results suggest that as in the case of adriamycin treatment, UV damage-induced G2 arrest is not dependent on p38 activity.
To rule out the possibility of off-target effects by chemical inhibitors used in the experiments, we performed a series of siRNA knockdown experiments targeting p38α, MK2, and Chk1 in HeLa cells with two specific siRNA oligonucleotides for each gene. Both siRNA oligonucleotides effectively inhibited their target gene expression as determined by Western blot analysis (Fig. 3 A). Cells were transfected with appropriate siRNA, transferred into fresh growth medium after 48 h, and then treated with adriamycin for an additional 24 h. Consistent with the data obtained by using the small-molecule kinase inhibitors, the knockdown of Chk1 using siRNA also abrogated the G2 DNA damage checkpoint in the presence of high levels of p38 activity, as evidenced by a decrease in the level of CDK1 Tyr15 phosphorylation and an increase in the level of histone H3 phosphorylation and the mitotic index (Fig. 3A and B). Similarly, the siRNA-mediated inhibition of Chk1 also abrogated UV-induced G2 DNA damage checkpoint arrest (Fig. 3B). In contrast, the knockdown of p38α or MK2 did not affect the G2 DNA damage checkpoint arrest induced by adriamycin or UV treatment (Fig. 3A and B).
FIG. 3.

Inhibition of Chk1 but not p38 by small-molecule kinase inhibitors or RNAi abrogates the UV-induced G2 DNA damage checkpoint. (A) Western blot analysis of the response of cells after the inactivation of Chk1, p38, or MK2 by two specific and validated siRNA oligonucleotides directed against each gene to adriamycin. (B) Mitotic index plot of HeLa cells with p38 or Chk1 knockdown and treated with 160 nM adriamycin or 500 J/m2 UV for 24 h. (C) p53-dependent abrogation of the G2 DNA damage checkpoint by Chk1 inhibition. A549, U2OS, and Calu-6 cancer cells were treated with 160 nM adriamycin for 24 h before the addition of kinase inhibitors (320 nM p38i or 1.25 μM Chk1 inhibitor). Mitotic indexes were determined at 3 and 24 h after the addition of kinase inhibitors. (D and E) MK2 is not required for G2 DNA damage checkpoint control following UV-C irradiation. U2OS cells treated with green fluorescent protein (GFP) or MK2 (MAPKAP kinase 2) siRNA were irradiated with 20 J/m2 of UV-C as described previously (31) and then placed into 50 ng/ml nocodazole-containing medium for an additional 16 h. Target protein knockdown, DNA content, and phospho-histone H3 of cells were analyzed by Western blotting (D) and flow cytometry (E).
Lastly, to show that the lack of any effect of p38 inhibition on the G2 DNA damage checkpoint-induced arrest was not a phenomenon specific to HeLa cells, we conducted similar experiments using A549 (lung cancer), U2OS (osteosarcoma), and Calu-6 (lung) cell lines. Similar to the results obtained with HeLa cells, the inhibition of p38 also had no impact on the ability of these cancer cell lines to mount a strong G2 DNA damage checkpoint-imposed cell cycle arrest in response to adriamycin treatment (Fig. 3C). Again, the inhibition of Chk1 was able to abrogate the adriamycin-induced G2 arrest in p53-deficient Calu-6 cells but not in p53-proficient A549 and U2OS cells, as reported previously (22, 28). In addition, we attempted to reproduce the effect of UV-C irradiation in U2OS cells exactly as previously reported (31). We found that two independent siRNA oligonucleotides targeting MK2, one of which was the same siRNA oligonucleotide previously reported (35), effectively inhibited MK2 expression (Fig. 3D). Contrary to that previous report (35), however, the inhibition of MK2 by RNAi had no effect on histone H3 phosphorylation in response to 20 J/m2 UV-C irradiation as monitored by Western blotting or flow cytometry after 18 h in a nocodazole mitotic trap assay (Fig. 3D and E). Consistent with our siRNA results for HeLa cells, these results indicate that MK2 inhibition does not abrogate the G2 DNA damage checkpoint function. In addition, the RNAi-mediated inhibition of MK2 also had no effect on γ-H2AX expression and the activation of p38 MAPK in response to UV-C treatment (Fig. 3D). We also noticed that a significant fraction of U2OS cells lost viability when exposed to 20 J/m2 UV-C. Taken together, these results demonstrate that although the p38 pathway is induced robustly in response to DNA damages, its activity is not required for the execution or maintenance of G2 DNA damage checkpoint control.
Nongenotoxic activation of p38 does not inhibit mitotic entry.
If p38 activity is indeed important for the execution of the G2 DNA damage checkpoint, then the DNA damage-independent activation of p38 would be expected to impede progression into mitosis by the untimely engagement of the G2 DNA damage checkpoint. Therefore, we investigated the effect of the nongenotoxic activation of p38 by anisomycin, a potent antimicrobial agent, on the onset of mitosis. Short-term exposure to anisomycin at 2 μg/ml is not known to cause DNA damage but strongly induces the p38 signaling pathway in our hands (11). HeLa cells were first synchronized at the G2 boundary with a CDK1 inhibitor (42) and then released in the presence or absence of anisomycin. Cell cycle progression from G2 was then monitored up to 6 h after release from the CDK1 inhibitor block. As expected, p38 activation was strongly induced by anisomycin, but high levels of p38 activity had no impact on the ability of synchronized HeLa cells to enter mitosis rapidly (Fig. 4).
FIG. 4.

Nongenotoxic activation of p38 with anisomycin does not impede entry into mitosis in HeLa cells. (A) Mitotic index (phospho-histone H3) of HeLa cells synchronized with the CDK1 inhibitor RO-3306 in the presence or absence of 2 μg/ml anisomycin. (B) Western blot for the p38 activation of HeLa cells synchronized with the CDK1 inhibitor RO-3306 by 2 μg/ml anisomycin.
p38 MAPK plays a pivotal role in the immediate-early stress response and cell survival.
To uncover a new role for p38 activity in the DNA damage response outside the context of the G2 DNA damage checkpoint, we returned to the original context of p38 activation in the stress response. We first demonstrated that the p38i effectively inhibited the TNF-α-induced activation of p38 signaling (data not shown). We then profiled the effects of p38 inhibition on global gene expression in cancer cells induced by TNF-α. Calu-6 lung cancer cells were treated with TNF-α and a p38 inhibitor (LY479754) across a time course. Samples were run on Affymetrix HG-U133plus2 gene chips to enable an unbiased assessment of transcriptional changes in response to TNF-α and p38 inhibition across time.
A total of 853 transcripts showed significant expression changes between TNF-α-treated cells and DMSO-treated controls in at least one of the five time points analyzed (data not shown). To understand the primary effects of TNF-α on gene expression, we focused on transcription changes at the 1-h time point after TNF-α treatment and identified a total of 115 transcripts corresponding to 72 unique genes, which were differentially expressed. Based on their expression patterns across the five time points revealed by hierarchical clustering, they fall into four distinct groups (Fig. 5 A). The first group includes 10 genes; among them, 9 are immediate-early response genes encoding transcription factors (see Table S1 in the supplemental material). Not surprisingly, this group of genes responded most rapidly and transiently to TNF-α treatment (Fig. 5A). The second group is the largest, with 31 genes consisting of cytokines, chemokines, growth factor genes, and genes implicated in the stress response (Table S1). This group also responded to TNF-α rapidly, peaking from 1 to 2 h and then declining more slowly than the genes in the first group (Fig. 5A). The third group includes 22 genes that responded to TNF-α more slowly and at a lower magnitude than the first two groups (Fig. 5A). Most of the genes in this group have functions related to immune regulation (Table S1). The fourth group of nine genes negatively responded to TNF-α treatment (Fig. 5A and Table S1). Taken together, these genes, which were differentially regulated by TNF-α, are associated mostly with stress and immune responses, consistent with the expected function of TNF-α signaling.
FIG. 5.

Inactivation of p38 inhibits immediate-early response and antiapoptotic pathways in TNF-α-treated Calu-6 cells. (A) Cluster analysis of genes differentially expressed 1 h after TNF-α treatment and effect of 320 nM p38i LY479754 on these expression changes. Gene expression values are represented by colors, with red and green indicating high and low expressions, respectively. (B) Box plots of TNF-α response of immediate-early response genes and members of apoptosis pathway component genes at the 1-h time point in the presence or absence of p38 kinase inhibitor. (C) Western blots of p38 activation, apoptosis, and cell death/survival pathway protein expression in Calu-6 cells treated with TNF-α in the presence of 320 nM p38i LY479754. (D) Apoptotic index (percentage of cells expressing cleaved PARP) of Calu-6 cells treated with 25 ng/ml TNF-α and various concentrations of p38i using an Acumen Explorer high-content imaging assay. CTRL, control with no treatment.
The treatment of Calu-6 cells with a selective p38 kinase inhibitor (p38i), LY479754, alone caused expression changes only in three genes across time compared to the DMSO-treated controls, further demonstrating the extraordinary selectivity of this kinase inhibitor (data not shown). One of the genes that was downregulated is COX2, a known p38 target gene (15), while FADD, a proapoptotic component of the fatty acid synthase (FAS) receptor pathway, was upregulated at the early time points (Fig. 5B). Among the 853 transcripts regulated by TNF-α, the p38 kinase inhibitor completely blocked the expression changes of 260 transcripts and also significantly inhibited changes in the expression levels of another 185 transcripts induced by TNF-α (see Table S2 in the supplemental material). Together, 445 (52%) TNF-α-regulated genes responded to the inactivation of p38, providing strong evidence for a significant role of p38 MAPK in the TNF-α-induced stress response. Furthermore, the inactivation of p38 abolished >70% of the expression changes induced by TNF-α at the 1-h time point. As shown in Fig. 5A, expression changes in cluster 1 and 2 genes that responded most rapidly to TNF-α were also most strongly inhibited by the p38i, whereas genes in cluster 3 that responded more slowly to TNF-α and at a lower magnitude were much less affected by the p38 inactivation. The data further demonstrate that p38 MAPK plays a pivotal role in early cellular responses to TNF-α.
Among many genes, networks, and canonical pathways that were affected by the p38 inhibition of TNF-α treatment, we found a significant representation of genes modulating the antiapoptosis and cell survival pathway (Fig. 5A; see Table S2 in the supplemental material). In response to TNF-α, Calu-6 cells immediately activated a potent cell survival response, including the upregulation of prosurvival pathways such as BCL-xl, IL-6, Myc, and EGR (13, 40, 43) and also the downregulation of proapoptotic signaling components such as TRADD and FADD (Fig. 5B). The inhibition of p38 significantly reversed these prosurvival responses, resulting in a recovery of TRADD and FADD and a significant decrease in levels of BCL-xl, IL-6, EGR, and Myc (Fig. 5B).
Gene expression changes were confirmed at the protein level by Western blotting. The inhibition of the p38 pathway with LY479754 indeed led to a significant decrease in the levels of BCL2 and BCL-xl and the reversal of decreased FADD expression in the TNF-α-treated cells, in line with results from the gene expression study (Fig. 5C). Concurrently, the inhibition of p38 also led to an early induction of PARP cleavage, a cellular marker for apoptotic cell death. To further confirm and quantify apoptotic cell death, we determined the apoptosis index (percentage of cells expressing cleaved PARP) of TNF-α-treated cells in the presence of p38i. We found that p38i in combination with TNF-α indeed led to increased apoptosis compared to TNF-α alone as early as 3 h after treatment (Fig. 5D). Together, these results strongly suggest that p38 signaling plays an important role in the immediate-early response and in the induction of prosurvival/antiapoptotic signaling in response to TNF-α stress.
Inhibition of p38 in response to DNA damage leads to cell death via inhibition of BCL2 family proteins.
The discovery that p38 inhibition results in a strong dampening of antiapoptotic gene expression in response to TNF-α led us to reason that p38 activity may play a role in modulating apoptotic induction in the context of DNA damage. If so, then the inhibition of p38 should result in the induction of apoptosis of cells treated with DNA-damaging agents. To test this hypothesis, both synchronous and asynchronous HeLa (p53-deficient) and A549 (p53-proficient) cells were treated with adriamycin or MMS in the presence of the p38i LY479754 for up to 48 h and assayed for apoptotic markers, namely, the cleavage of caspase 3 or 7 and PARP. A dose escalation experiment with the p38 inhibitor in combination with adriamycin showed a corresponding increase in cleaved-caspase-3 levels measured as the apoptotic index at 48 h posttreatment (Fig. 6 A). Consistent with this, additional experiments with siRNA targeting p38α and MK2 in HeLa cells also showed a marked increase in levels of apoptotic markers in combination with adriamycin but not in cells treated with adriamycin alone or nonspecific siRNA in the presence of adriamycin (Fig. 6B). The inhibition of p38 with LY479754 also led to a dramatic increase in PARP cleavage in p53-positive A549 cells after DNA damage by adriamycin (Fig. 6C).
FIG. 6.

Inhibition of p38 MAPK sensitizes cells to adriamycin and induces cell death. (A) Apoptosis index (percentage of cells expressing cleaved caspase 3) of HeLa cells treated with increasing doses of the p38i LY479754 in the presence of 160 nM adriamycin for 48 h. (B) Western blot for apoptosis of HeLa cells treated with siRNA targeting p38α, MK2, or Chk1 in the presence or absence of 160 nM adriamycin for 48 h. NS-si, nonspecific/scramble siRNA. (C) Western blot analysis of synchronized A549 cells by serum starvation for 48 h after treatment with 1 μM p38i LY479754 in the presence of DNA damage induced by 160 nM adriamycin for 48 h. (D) Western blot of A549 cells treated with 0.01% MMS and 1 μM p38i LY479754 for 48 h. (E) Apoptosis induced by p38 inhibition in the presence of DNA damage is associated with G2 arrest. The apoptosis index and cell cycle state were determined for A549 cells treated with 160 nM adriamycin and 1 μM p38i LY479754 using Acumen Explorer.
Since we observed a strong inhibition of BCL2 family gene expression upon p38 inhibition in TNF-α-treated cells, we wanted to test if the inhibition of BCL2 family proteins may provide a mechanistic explanation for a role of p38 in the regulation of apoptosis following DNA damage. We find that p38 inhibition in response to both adriamycin (Fig. 6C) and MMS (Fig. 6D) damage leads to a dramatic decrease in BCL-xl protein levels, matched with a concordant increase in the level of PARP cleavage. Finally, using multiparametric cytometry, we also find that the inhibition of p38 induced the apoptosis of cells that were largely arrested in the G2 phase in the presence of DNA damage (Fig. 6E). Taken together, these observations suggest that p38 activity is an integral part of the prosurvival signaling network induced in response to DNA damage.
DISCUSSION
In this study, we show that p38 activation is strongly induced by DNA damage and is correlated with G2 arrest. Contrary to data from previous reports (5, 26, 31), our data strongly suggest that p38 pathway activity is not necessary for the G2 DNA damage checkpoint function. Furthermore, the inhibition of Chk1 or ATM/ATR kinase abrogates the G2 DNA damage checkpoint in the presence of high levels of p38 activity. While HeLa cells were the primary cell model used in this study, we also show that the inhibition of p38 activity was unable to abrogate G2 DNA damage checkpoint control in the Calu-6, A549, and U2OS cell lines. In concordance with data from previous reports (24, 28), we find that the pharmacological inhibition of Chk1 alone with a selective small-molecule kinase inhibitor or siRNA knockdown was not sufficient to abrogate the G2 DNA damage checkpoint in p53-proficient cells. The corroboration of pharmacological inhibition using small-molecule kinase inhibitors with siRNA knockdown rules out the possibility that the observations may be due to an off-target activity of the chemical kinase inhibitors. Conversely, the nongenotoxic activation of p38 by anisomycin in G2 was not sufficient to activate the G2 DNA damage checkpoint. Taken together, our results strongly suggest that neither the suppression of p38 activity nor its nongenotoxic activation has an impact on G2 DNA damage checkpoint activity.
The inhibition of CDC25B/C phosphatase activity is believed to be the primary mechanism through which the p38 pathway participates in G2 DNA damage checkpoint control (5, 31). This prevents the formation of an active CDK1/cyclin B complex, thus blocking progression into mitosis. We find that the effective inhibition of p38 activity had no discernible impact on the level of CDK1 Tyr15 phosphorylation in response to adriamycin treatment. This lack of an effect of p38 inhibition on CDK1 activation through Tyr15 dephosphorylation by CDC25 provides further biochemical evidence in support of the proposition that p38 does not play an important role in G2 DNA damage checkpoint control. Alternatively, as Chk1 kinase is activated in a very similar manner in response to DNA damage, potential pathway redundancies may mitigate the effect of p38 inhibition on CDC25B activity. In p53-deficient cells, however, we find that the inactivation of Chk1 alone effectively abrogated the G2 DNA damage checkpoint. Furthermore, the abrogation of the G2 DNA damage checkpoint by Chk1 inactivation occurs in the presence of high levels of p38 kinase pathway activities. Therefore, in agreement with data from many previous publications (4, 9), our data suggest that the Chk1 signaling pathway is primarily responsible for the inactivation of CDK1 in response to DNA damage to prevent cells' progression into mitosis.
As we were interested in the exciting possibility of using potent and selective p38 kinase inhibitors as chemosensitizers to enhance the anticancer efficacy of chemotherapies, the inability of a highly selective and potent p38 kinase inhibitor to abrogate the G2 DNA damage checkpoint comes as a surprise. A closer examination of previous reports, however, reveals a certain degree of discrepancies concerning the role of p38 in G2 DNA damage checkpoint control in response to different types of DNA damage and the function of p53 (5, 31, 38). In addition, earlier studies used an older generation of p38 kinase inhibitors at very high concentrations (5, 38). At such high concentrations, it is likely that these p38 kinase inhibitors may have off-target activities, as shown recently (1, 10). Our data are consistent with a more recent report that demonstrated that using the RNAi approach, only Chk1 but not Chk2 or MK2 is responsible for G2 DNA damage checkpoint control in cancer cells (45). Furthermore, it was also recently shown that the p38 pathway response at the G2 DNA damage checkpoint is strongly attenuated in transformed cells (32). Earlier studies that implicated p38 activity in G2 DNA damage checkpoint control were performed with untransformed human cells and mouse embryonic fibroblasts (5, 35, 38). However, untransformed mammalian cells have intact p53 and Chk1 functions. Thus, it is unconceivable that normal, untransformed mammalian cells with functional p53 and Chk1 would depend on p38 alone for G2 DNA damage checkpoint function but not cancer cells, which are frequently deficient in p53 function. Indeed, similar to the findings for p53-proficient cancer cells, we find that the inhibition of p38 activity by the small-molecule inhibitor LY479754 was unable to abrogate the G2 DNA damage checkpoint in human umbilical vein endothelial cells (HUVECs) in response to adriamycin treatment (data not shown). Together, our results therefore rule out the feasibility of developing a p38 inhibitor as a chemosensitizer to enhance the efficacy of chemotherapies.
To identify a new role for p38 activity in the DNA damage response outside cell cycle checkpoint control, we conducted a genome-wide gene expression profiling analysis of the effect of p38 inhibition on the response to TNF-α stress. We find that the inhibition of p38 dramatically dampens the immediate-early transcriptional response and the ability of cancer cells to mount an effective antiapoptotic/prosurvival response to TNF-α. Moreover, the prosurvival signaling induced immediately after exposure to TNF-α consisted of the downregulation of proapoptotic factors such as FADD and TRADD and the upregulation of antiapoptosis components, including antiapoptosis BCL2 family proteins.
Testing the hypothesis derived from the analysis of transcriptional data in the context of DNA damage, we find that the inhibition of p38 in combination with adriamycin leads to a strong induction of apoptosis. Increased apoptosis was observed for both p53-deficient HeLa cells as well as p53-proficient A549 cells, implying that the link between p38 activity and prosurvival signaling does not depend on the p53 status. Further mechanistic studies in the context of DNA damage show that p38 may confer its prosurvival effect in response to DNA damage through the regulation of antiapoptotic BCL2 family proteins. Consistent with this notion, we find that the chemical inhibition or siRNA knockdown of p38 in the presence of adriamycin or MMS treatment leads to a dramatic decrease in levels of BCL2 and BCL-xl. The data suggest that p38 activity, while not connected directly with the proper functioning of the G2 DNA damage checkpoint, plays a pivotal role in response to DNA damage.
We note that the link between p38 activity, prosurvival signaling in response to DNA damage, and stress may be unexpected, given the strong association of p38 activation with Fas ligand (FasL)- and TNF-α-induced apoptosis (14). The behavior of DNA-damaged cells in which the checkpoint has been abrogated may be of some relevance. We have observed that the Chk1 inhibitor- or caffeine-mediated abrogation of the G2 DNA damage checkpoint occurs with high levels of p38 activity. This implies that while the inhibition of p38 in conjunction with DNA damage leads to increased apoptosis, high p38 activity alone does not prevent apoptosis. Thus, in the case of Chk1 inhibition-mediated mitotic catastrophe, other apoptosis-inducing factors may override the cytoprotective effects of p38 activity. Although the underlying mechanistic rationale for this observation is unclear, these observations suggest that there may be a more complex and context-specific relationship between p38 and apoptosis induction. From a teleological perspective, it can be argued that in an early response to stress, p38 signaling promotes cell survival to facilitate the evaluation of the extent of damage to the cell. Once the G2 DNA damage checkpoint is breached, p38-mediated prosurvival signaling is no longer required or sufficient, as the elimination of cells undergoing mitotic catastrophe would be in the best interest of multicellular organisms.
Our assertion that p38 plays a role in cell survival is supported by a number of recent reports linking this signaling pathway to increased levels of BCL2 and BCL-xl in response to DNA damage and stress (12, 23). Furthermore, the chemical inhibition of p38 has been strongly associated with increased chemosensitivity in cancer cells (16, 30). Based on our study and correlative evidence from other reports, we propose a new role for p38 in the context of the response to DNA damage (Fig. 7). According to this scheme, while p38 is activated in response to DNA damage, resulting in G2 DNA damage checkpoint-mediated cell cycle arrest, its activity is not required for the activation or maintenance of the G2 DNA damage checkpoint. Instead, p38 activity in response to DNA damage induces prosurvival signaling to prevent the onset of premature apoptosis in the immediate aftermath of the stress of DNA damage and allows recovery from DNA damage. This antiapoptosis response likely allows cells to ascertain the extent of damage and to respond accordingly. It appears that the role of p38 in the regulation of apoptosis is context dependent and may switch from prosurvival to proapoptosis depending on both the timing and the physiological context of the stress induction. Clearly, an elucidation of the full mechanism of p38 in the regulation of apoptosis would require further investigations.
FIG. 7.
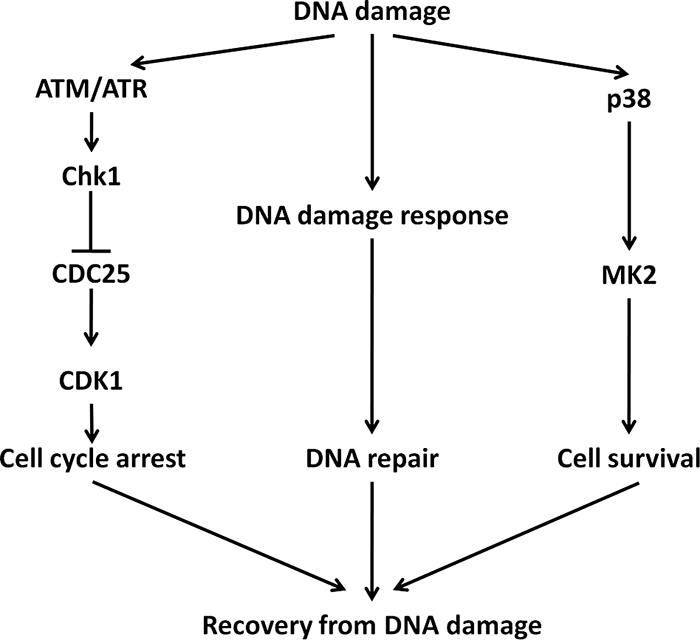
New model for a novel role of p38 MAPK in the DNA damage response in cancer cells. DNA damage elicits the activation of cell cycle checkpoint, DNA repair, and cell survival signaling pathways. In response to DNA damage, the Chk1-mediated G2 DNA damage checkpoint is activated to prevent entry into mitosis with damaged DNA, whereas the p38-mediated cell survival pathway is activated to keep cells alive. Together, these two pathways afford cell the time to repair DNA damage and ultimately to allow recovery from DNA damage.
Supplementary Material
Acknowledgments
We thank Li Fan of Ye's laboratory and Chris Sim Kim Siew and Connie Er Poh Nee at Systems Biology, Lilly Singapore Center for Drug Discovery, Pte Ltd., for their technical assistance. We thank Song Qing Na and Mark Marshall for providing the MAPKAPK2 (MK2) inhibitor and the Chk1 inhibitor, respectively. We acknowledge Xi Lin for her assistance in running the microarrays and Shuguang Huang for initial gene expression profiling analysis. We also thank Gunaretnam (Guna) Rajagopal (NJ Cancer Institute) for his help in this work.
Footnotes
Published ahead of print on 1 June 2010.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Bain, J., L. Plater, M. Elliott, N. Shpiro, C. J. Hastie, H. McLauchlan, I. Klevernic, J. S. Arthur, D. R. Alessi, and P. Cohen. 2007. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408:297-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blasina, A., J. Hallin, E. Chen, M. E. Arango, E. Kraynov, J. Register, S. Grant, S. Ninkovic, P. Chen, T. Nichols, P. O'Connor, and K. Anderes. 2008. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer Ther. 7:2394-2404. [DOI] [PubMed] [Google Scholar]
- 3.Bowen, W. P., and P. G. Wylie. 2006. Application of laser-scanning fluorescence microplate cytometry in high content screening. Assay Drug Dev. Technol. 4:209-221. [DOI] [PubMed] [Google Scholar]
- 4.Bucher, N., and C. D. Britten. 2008. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer 98:523-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulavin, D. V., Y. Higashimoto, I. J. Popoff, W. A. Gaarde, V. Basrur, O. Potapova, E. Appella, and A. J. Fornace, Jr. 2001. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411:102-107. [DOI] [PubMed] [Google Scholar]
- 6.Bulavin, D. V., S. Saito, M. C. Hollander, K. Sakaguchi, C. W. Anderson, E. Appella, and A. J. Fornace, Jr. 1999. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 18:6845-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Dios, A., C. Shih, B. Lopez de Uralde, C. Sanchez, M. del Prado, L. M. Martin Cabrejas, S. Pleite, J. Blanco-Urgoiti, M. J. Lorite, C. R. Nevill, Jr., R. Bonjouklian, J. York, M. Vieth, Y. Wang, N. Magnus, R. M. Campbell, B. D. Anderson, D. J. McCann, D. D. Giera, P. A. Lee, R. M. Schultz, L. C. Li, L. M. Johnson, and J. A. Wolos. 2005. Design of potent and selective 2-aminobenzimidazole-based p38alpha MAP kinase inhibitors with excellent in vivo efficacy. J. Med. Chem. 48:2270-2273. [DOI] [PubMed] [Google Scholar]
- 8.Didier, C., C. Cavelier, M. Quaranta, M. O. Galcera, C. Demur, G. Laurent, S. Manenti, and B. Ducommun. 2008. G2/M checkpoint stringency is a key parameter in the sensitivity of AML cells to genotoxic stress. Oncogene 27:3811-3820. [DOI] [PubMed] [Google Scholar]
- 9.Enders, G. H. 2008. Expanded roles for Chk1 in genome maintenance. J. Biol. Chem. 283:17749-17752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fabian, M. A., W. H. Biggs III, D. K. Treiber, C. E. Atteridge, M. D. Azimioara, M. G. Benedetti, T. A. Carter, P. Ciceri, P. T. Edeen, M. Floyd, J. M. Ford, M. Galvin, J. L. Gerlach, R. M. Grotzfeld, S. Herrgard, D. E. Insko, M. A. Insko, A. G. Lai, J. M. Lelias, S. A. Mehta, Z. V. Milanov, A. M. Velasco, L. M. Wodicka, H. K. Patel, P. P. Zarrinkar, and D. J. Lockhart. 2005. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23:329-336. [DOI] [PubMed] [Google Scholar]
- 11.Fan, L., X. Yang, J. Du, M. Marshall, K. Blanchard, and X. Ye. 2005. A novel role of p38 alpha MAPK in mitotic progression independent of its kinase activity. Cell Cycle 4:1616-1624. [DOI] [PubMed] [Google Scholar]
- 12.Flacke, J. P., S. Kumar, S. Kostin, H. P. Reusch, and Y. Ladilov. 2009. Acidic preconditioning protects endothelial cells against apoptosis through p38- and Akt-dependent Bcl-xL overexpression. Apoptosis 14:90-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gatti, G., G. Maresca, M. Natoli, F. Florenzano, A. Nicolin, A. Felsani, and I. D'Agnano. 2009. MYC prevents apoptosis and enhances endoreduplication induced by paclitaxel. PLoS One 4:e5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grethe, S., M. P. Ares, T. Andersson, and M. I. Porn-Ares. 2004. p38 MAPK mediates TNF-induced apoptosis in endothelial cells via phosphorylation and downregulation of Bcl-xL. Exp. Cell Res. 298:632-642. [DOI] [PubMed] [Google Scholar]
- 15.Guan, Z., S. Y. Buckman, A. P. Pentland, D. J. Templeton, and A. R. Morrison. 1998. Induction of cyclooxygenase-2 by the activated MEKK1→SEK1/MKK4→p38 mitogen-activated protein kinase pathway. J. Biol. Chem. 273:12901-12908. [DOI] [PubMed] [Google Scholar]
- 16.Hamanoue, M., K. Sato, and K. Takamatsu. 2007. Inhibition of p38 mitogen-activated protein kinase-induced apoptosis in cultured mature oligodendrocytes using SB202190 and SB203580. Neurochem. Int. 51:16-24. [DOI] [PubMed] [Google Scholar]
- 17.Han, J., J. D. Lee, L. Bibbs, and R. J. Ulevitch. 1994. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265:808-811. [DOI] [PubMed] [Google Scholar]
- 18.Han, J., J. D. Lee, P. S. Tobias, and R. J. Ulevitch. 1993. Endotoxin induces rapid protein tyrosine phosphorylation in 70Z/3 cells expressing CD14. J. Biol. Chem. 268:25009-25014. [PubMed] [Google Scholar]
- 19.Hirose, Y., M. Katayama, M. S. Berger, and R. O. Pieper. 2004. Cooperative function of Chk1 and p38 pathways in activating G2 arrest following exposure to temozolomide. J. Neurosurg. 100:1060-1065. [DOI] [PubMed] [Google Scholar]
- 20.Hynes, J., Jr., and K. Leftheri. 2005. Small molecule p38 inhibitors: novel structural features and advances from 2002-2005. Curr. Top. Med. Chem. 5:967-985. [DOI] [PubMed] [Google Scholar]
- 21.Jackson, J. R., A. Gilmartin, C. Imburgia, J. D. Winkler, L. A. Marshall, and A. Roshak. 2000. An indolocarbazole inhibitor of human checkpoint kinase (Chk1) abrogates cell cycle arrest caused by DNA damage. Cancer Res. 60:566-572. [PubMed] [Google Scholar]
- 22.Jurvansuu, J., M. Fragkos, C. Ingemarsdotter, and P. Beard. 2007. Chk1 instability is coupled to mitotic cell death of p53-deficient cells in response to virus-induced DNA damage signaling. J. Mol. Biol. 372:397-406. [DOI] [PubMed] [Google Scholar]
- 23.Kim, M. J., S. Y. Choi, I. C. Park, S. G. Hwang, C. Kim, Y. H. Choi, H. Kim, K. H. Lee, and S. J. Lee. 2008. Opposing roles of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase in the cellular response to ionizing radiation in human cervical cancer cells. Mol. Cancer Res. 6:1718-1731. [DOI] [PubMed] [Google Scholar]
- 24.Koniaras, K., A. R. Cuddihy, H. Christopoulos, A. Hogg, and M. J. O'Connell. 2001. Inhibition of Chk1-dependent G2 DNA damage checkpoint radiosensitizes p53 mutant human cells. Oncogene 20:7453-7463. [DOI] [PubMed] [Google Scholar]
- 25.Kramer, A., N. Mailand, C. Lukas, R. G. Syljuasen, C. J. Wilkinson, E. A. Nigg, J. Bartek, and J. Lukas. 2004. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat. Cell Biol. 6:884-891. [DOI] [PubMed] [Google Scholar]
- 26.Kurosu, T., Y. Takahashi, T. Fukuda, T. Koyama, T. Miki, and O. Miura. 2005. p38 MAP kinase plays a role in G2 checkpoint activation and inhibits apoptosis of human B cell lymphoma cells treated with etoposide. Apoptosis 10:1111-1120. [DOI] [PubMed] [Google Scholar]
- 27.Kyriakis, J. M., and J. Avruch. 1996. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J. Biol. Chem. 271:24313-24316. [DOI] [PubMed] [Google Scholar]
- 28.Levesque, A. A., A. A. Fanous, A. Poh, and A. Eastman. 2008. Defective p53 signaling in p53 wild-type tumors attenuates p21waf1 induction and cyclin B repression rendering them sensitive to Chk1 inhibitors that abrogate DNA damage-induced S and G2 arrest. Mol. Cancer Ther. 7:252-262. [DOI] [PubMed] [Google Scholar]
- 29.Li, S., H. Y. Zhang, C. C. Hu, F. Lawrence, K. E. Gallagher, A. Surapaneni, S. T. Estrem, J. N. Calley, G. Varga, E. R. Dow, and Y. Chen. 2008. Assessment of diet-induced obese rats as an obesity model by comparative functional genomics. Obesity (Silver Spring) 16:811-818. [DOI] [PubMed] [Google Scholar]
- 30.Lim, S. J., Y. J. Lee, and E. Lee. 2006. p38MAPK inhibitor SB203580 sensitizes human SNU-C4 colon cancer cells to exisulind-induced apoptosis. Oncol. Rep. 16:1131-1135. [PubMed] [Google Scholar]
- 31.Manke, I. A., A. Nguyen, D. Lim, M. Q. Stewart, A. E. Elia, and M. B. Yaffe. 2005. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 17:37-48. [DOI] [PubMed] [Google Scholar]
- 32.Mikhailov, A., D. Patel, D. J. McCance, and C. L. Rieder. 2007. The G2 p38-mediated stress-activated checkpoint pathway becomes attenuated in transformed cells. Curr. Biol. 17:2162-2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moriguchi, T., H. Kawasaki, S. Matsuda, Y. Gotoh, and E. Nishida. 1995. Evidence for multiple activators for stress-activated protein kinase/c-Jun amino-terminal kinases. Existence of novel activators. J. Biol. Chem. 270:12969-12972. [DOI] [PubMed] [Google Scholar]
- 34.Ono, K., and J. Han. 2000. The p38 signal transduction pathway: activation and function. Cell. Signal. 12:1-13. [DOI] [PubMed] [Google Scholar]
- 35.Pedraza-Alva, G., M. Koulnis, C. Charland, T. Thornton, J. L. Clements, M. S. Schlissel, and M. Rincon. 2006. Activation of p38 MAP kinase by DNA double-strand breaks in V(D)J recombination induces a G2/M cell cycle checkpoint. EMBO J. 25:763-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pietersma, A., B. C. Tilly, M. Gaestel, N. de Jong, J. C. Lee, J. F. Koster, and W. Sluiter. 1997. p38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. Biochem. Biophys. Res. Commun. 230:44-48. [DOI] [PubMed] [Google Scholar]
- 37.Reinhard, E., S. Kolodziej, D. Anderson, N. Stehle, W. Vernier, L. Lee, and S. Hegde. July 2004. U.S. patent US2004/0127519.
- 38.Reinhardt, H. C., A. S. Aslanian, J. A. Lees, and M. B. Yaffe. 2007. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 11:175-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rouse, J., P. Cohen, S. Trigon, M. Morange, A. Alonso-Llamazares, D. Zamanillo, T. Hunt, and A. R. Nebreda. 1994. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 78:1027-1037. [DOI] [PubMed] [Google Scholar]
- 40.Sakamoto, K. M., and D. A. Frank. 2009. CREB in the pathophysiology of cancer: implications for targeting transcription factors for cancer therapy. Clin. Cancer Res. 15:2583-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schieven, G. L. 2009. The p38alpha kinase plays a central role in inflammation. Curr. Top. Med. Chem. 9:1038-1048. [DOI] [PubMed] [Google Scholar]
- 42.Vassilev, L. T. 2006. Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle 5:2555-2556. [DOI] [PubMed] [Google Scholar]
- 43.Wegiel, B., A. Bjartell, Z. Culig, and J. L. Persson. 2008. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int. J. Cancer 122:1521-1529. [DOI] [PubMed] [Google Scholar]
- 44.Xiao, Z., Z. Chen, A. H. Gunasekera, T. J. Sowin, S. H. Rosenberg, S. Fesik, and H. Zhang. 2003. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J. Biol. Chem. 278:21767-21773. [DOI] [PubMed] [Google Scholar]
- 45.Xiao, Z., J. Xue, T. J. Sowin, and H. Zhang. 2006. Differential roles of checkpoint kinase 1, checkpoint kinase 2, and mitogen-activated protein kinase-activated protein kinase 2 in mediating DNA damage-induced cell cycle arrest: implications for cancer therapy. Mol. Cancer Ther. 5:1935-1943. [DOI] [PubMed] [Google Scholar]
- 46.Yang, H., T. Burke, J. Dempsey, B. Diaz, E. Collins, J. Toth, R. Beckmann, and X. Ye. 2005. Mitotic requirement for aurora A kinase is bypassed in the absence of aurora B kinase. FEBS Lett. 579:3385-3391. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.