

Abstract
This article presents a microfluidic device which integrates autonomous serial immunofluorocytometry binding reactions of cytometric beads with fluorescence detection and quantification in a continuous flow environment. The microdevice assay is intended to alleviate the extensive benchwork and large sample volumes used when conducting traditional immunoassays, without requiring complex external controls. The technology is based on the miniaturization and automation of the serial processing steps of an antigen sandwich immunoassay, with integrated fluorescence detection using paramagnetic microbeads. The continuous flow design may enable temporal tracking of time-varying protein concentrations in a continuously infused sample for clinical applications, specifically for monitoring inflammation marker proteins in blood produced during cardiac surgeries involving cardiopulmonary bypass (CPB) procedures. The device operation was first validated via a single incubation device which measured the concentration of a fluorescently labeled biotin molecule using streptavidin-coated paramagnetic cytometric beads. Subsequently, a dual incubation device was tested with samples of the anaphylatoxin complement protein C3a, and was shown to be capable of differentiating between samples at typical systemic concentrations of the protein (1–5 μg/ml), with very low sample usage (<6 μl/h). It is believed that this continuous flow, automated microimmunosensor technology will be a platform for high sample rate immunoassays capable of tracking and more thoroughly characterizing the systemic inflammation process, and may aid in the development of better treatment options for systemic inflammation during and after CPB.
Keywords: Immunoassay, Biosensor, Magnetic actuation, Fluorocytometry
1 Introduction
The ultimate goal of this research is to develop microimmunoassays which directly measure protein concentrations in an unmodified sample stream at high sample rates and with low sample usage, specifically for near-continuous clinical blood monitoring applications. The motivating application for this research is systemic inflammation monitoring during and following cardiac surgical procedures, especially those involving cardiopulmonary bypass (CPB). Several studies have shown that cardiac surgery induces complex systemic inflammatory responses, particularly when CPB is used (Moore et al. 1988; Kirklin and Barratt-Boyes 1993). This inflammation is characterized by complement, neutrophil, and platelet activation, and the release of pro-inflammatory cytokines, which are attributed to many surgical and clinical complications during and following the CPB procedures. It is believed that a detailed characterization of a patient’s blood plasma cytokine and complement concentrations while undergoing CPB procedures, with sample intervals of less than 5 min, will be one of the fundamental steps in understanding and eventually reducing the harmful effects of inflammation (Westaby 1987) by allowing clinicians to investigate how changes in surgical procedures, equipment, and intra-operative substitution of pharmacological agents affect CPB-related morbidity. Due to limitations of traditional assays in terms of both required sample volumes and feasibility of processing large numbers of discrete samples, inflammation studies on patients undergoing CPB have used limited number of samples with less frequent intervals. Advanced time-course inflammation studies on pseudo-patients using a mock CPB circuitry, which allow more blood to be drawn than during actual patient studies, have used sample intervals of 15 min and greater over the course of 2 h (Fung et al. 2001). Immunoassays such as the enzyme linked immunosorbent assay (ELISA) and immunofluorocytometry, which are commonly used for these measurements, are inherently discrete due to their benchtop serial processing steps, which limit their usefulness for high sample rate monitoring. These assays are further limited in applications where the volume of sample fluid available for testing is restricted, such as CPB patient blood, since these assays generally require at least 50–100 μl per sample, and the assays used in reported studies collected 3–5 ml of blood per sample (Mainwaring et al. 1998).
Microfluidic devices have been developed which are analogous to traditional immunoassays, but offer benefits such as higher throughput and smaller required sample and reagent volumes due to microchip integration (Sato et al. 2004; Kartalov et al. 2006; Cheng et al. 2007). Many of these devices use optical detection schemes based on fluorescent tags (Kartalov et al. 2006) or chemiluminescent reactions using horseradish peroxidase (Ahn et al. 2007; Bhattacharyya and Klapperich 2007). Other microfluidic biosensor designs have replaced these optical detection schemes with electrochemical detection schemes based on impedance or current measurements (Barton et al. 2008; Wang et al. 2008). These microfluidic detection technologies have resulted in immunoassays with higher throughput and significantly increased sensitivity. Although they use reduced sample volumes as compared to traditional methods, these devices were designed as discrete-sample assays. For applications where the analyte concentration is not time-varying in the short term, such as in the detection of protein markers indicative of disease (Cheng et al. 2007) or environmental toxins (Ahn et al. 2007), these assay technologies are effective. However, in applications where the analyte concentration is changing with time and must be analyzed repeatedly these assays must be processed successively, which limits their feasibility for high sample rate monitoring.
Other technologies have been implemented with the goal of real-time, continuous or semi-continuous biosensing. A design by Engstrom et al. (2006) was able to monitor the concentration of carbohydrates in a sample stream at a very high sample rate, taking one sample every second. Measurements were purely optical, based on total internal reflection fluorescence, allowing direct measurement of the carbohydrate sample without a separate labeling phase. The detection system used the intrinsic change in the autofluorescence intensity of the carbohydrate binding antibody’s tryptophan residues at 295 nm excitation wavelength upon carbohydrate binding to produce an increase in fluorescence emission. However, the flow rate used for the immunoassay was rather high at 250 μl/min, potentially leading to rapid depletion of the sample fluid. Other groups have developed pseudo real-time integrating sensors based on electrochemical (Roper et al. 2003) or fluorescent (Aoyagi et al. 2002) detection. Integrating assays continuously accumulate the analyte, and their direct output is a measure of the total amount of analyte accumulated. The instantaneous sample concentration at any time during the assay is determined based on the rate of change of the sensor’s output. While these assays can have very high sensitivities, they are limited in terms of assay duration due to saturation of the sensing element. They can also be prone to drift due to nonlinearities of the sensing element relative to the total amount of accumulated analyte on the sensor.
The microimmunoassay design presented herein builds upon previous work in which streptavidin–biotin binding was used as a model for antibody-antigen interactions (Yang et al. 2007). In this model, streptavidin-coated microbeads are incubated with a fluorescein fluorescently tagged biotin (biotin-FITC) solution within the microdevice. The incubation time is controlled such that the number of biotin molecules bound to the bead surface is dependent on the concentration of biotin in the sample stream. Since the biotin is already labeled with a fluorescent tag, the streptavidin–biotin binding assays are useful as benchmarking studies requiring only a single bead incubation step. The sample concentration is determined based on the fluorescence intensity of the beads following incubation. Such a microdevice must be capable of reliably manipulating the microbeads, in an autonomous fashion, into and out of the sample stream. This requires the trajectory of the bead to be decoupled from the background carrier flow. In earlier work, the decoupling of bead motion was accomplished through an implementation of micro-scale hydrodynamics (Yang et al. 2007) by controlling particle movement at a bifurcating region. While this design proved capable of transferring the beads between adjacent fluid streams, reliability was a problem since the device required precisely tuned flow rates to ensure proper bead trajectories. The design also had a very limited maximum incubation time, since the beads flowed at the same velocity as the background flow, and very low flow rates introduced complications in bead processing due to bead settling and sticking. This design was replaced with a magnetically actuated control scheme. The magnetic design has proven to be more robust than the hydrodynamic one, leading to a less complicated channel design with much simpler tuning.
2 Microdevice assays with magnetic beads
While many types of assays are capable of protein concentration measurement, they are not easily applied to autonomous measurement of protein concentrations with high sample rates, low sample usage, and extended measurement durations. The microimmunofluorocytometry assay presented here, based on autonomous serial processing of paramagnetic microbeads within a microfluidic channel, is capable of meeting these criteria. While magnetic actuation has been applied in various forms within microfluidic devices (Pamme 2006), immunoassay devices utilizing magnetic fields tend to employ microbead-based assays. Bead-based microimmunoassay devices must include some sort of separation method to control the bead incubation process. For magnetic bead assays, these separation schemes generally fall into two categories. First, in bead trapping devices, the beads are held stationary via a magnetic force, while the fluids are manipulated around the beads for the incubation and washing steps. Immunoassays using bead trapping schemes inherently provide batch measurements and are not easily applied to high sample rate monitoring without complex external fluidic controls, which result in lower sample rates (Choi et al. 2002; Herrmann et al. 2006; Do and Ahn 2008). The second type of separation scheme used in magnetic bead-based microimmunoassays is continuous flow operation. In these cases, the fluids containing different bead processing reagents are introduced into the device as steady streams, and the beads are magnetically manipulated to move between the streams. Variations of this basic flow manipulation have previously been shown to be applicable to microimmunoassays. Park et al. presented a magnetic-based detection scheme where antibody conjugated magnetic nanoparticles were used as the secondary label in an antigen sandwich immunoassay (Kim and Park 2005). However, the device did not have an integrated incubation control scheme, and was not capable of analyte detection from a native sample stream. For detection of such samples, a multiple step on-chip incubation scheme can be used to enable an antigen sandwich immunoassay. In the design by Pamme et al., a series of coplanar flows within a common channel provides this type of incubation (Peyman et al. 2008). By pulling paramagnetic beads across the wide channel, perpendicularly to the stratified flow environments, the beads pass through a series of incubation and wash streams. The incubation time in this device depends on the flow rate as well as the magnetic field intensity, and is limited by both the stream width as well as the transverse bead velocity.
In contrast to this type of design where beads are incubated while transferring across the channel through stratified environments, the novel magnetic actuation scheme described herein uses magnets that are placed on both sides of a microchannel so that the bead incubations occur along the walls of the channel, and the beads progress transversely across the channel only to enter or exit reactant streams. The microbeads incubate while rolling along one of the two sidewalls of the main channel. By establishing coplanar flow environments from multiple converging inlets consisting of a bead carrier stream, antigen containing stream and wash stream, with magnets placed on opposite sidewalls, the beads can be pulled into and out of different processing environments enabling autonomous serial bead processing reactions (Fig. 1). The magnetic field is strong enough to pull the beads to the sidewall, but not strong enough to trap them due to the shear stress around the bead from the pressure driven flow. While the absolute position of the external magnets, and hence the magnitude of the magnetic force on the beads affects the speed at which the beads are pulled to the wall of the device, it has been found empirically that when the magnets are placed 2.5 to 3.5 mm adjacent to the microchannels by hand, that small changes in the magnet position over this placement range have a negligible effect on the velocity of the beads while rolling along the wall. Since the bead incubates while rolling along the wall for a much longer time period than it takes to pull the bead to the wall, the exact placement of the magnet is not critical for repeatable incubation times. Consequently, the incubation time can be tailored based on flow rate alone. Since incubation times are relatively insensitive to small changes in the magnetic field strength, the devices are easy to fabricate and operate compared to other magnetically actuated devices which require precise magnetic field intensities. Due to the pressure driven flow in the microdevice and the resulting no-slip boundary condition, the flow outside of the sidewall boundary layer has the highest velocity, and the flow velocity at the wall is near zero. Since the beads are constrained against the channel wall, where the fluid velocity is much lower than toward the center of the channel, they move through the device at a velocity which is significantly slower than the average flow velocity, at 16.5% of the vertical centerline velocity. This effect greatly increases the residence time of the beads in the sample stream relative to the flow rate, achieving much longer incubation times and thus higher sensitivity than a design where the bead incubation occurs, while the beads are at a position farther away from the channel walls. Since the mean antigen stream velocity is also significantly higher than the bead velocity, this arrangement also helps replenish analyte around the bead since the analyte is convectively transported to the surface of the bead not in contact with the sidewall, speeding the incubation process. This design is also critical to the accuracy of the microimmunoassay because it forces every bead to travel through the microchannel at the same velocity, resulting in identical incubation times and minimizing bead to bead variations. Another advantage of this microdevice design is that the bead incubation time can be easily tailored over a wide range of operating condition simply by modifying either the incubation channel length and/or the syringe pump flow rates. Based on these characteristics, the technology presented herein is a favorable platform for the development of high sample rate microimmunoassays for autonomous clinical monitoring applications.
Fig. 1.
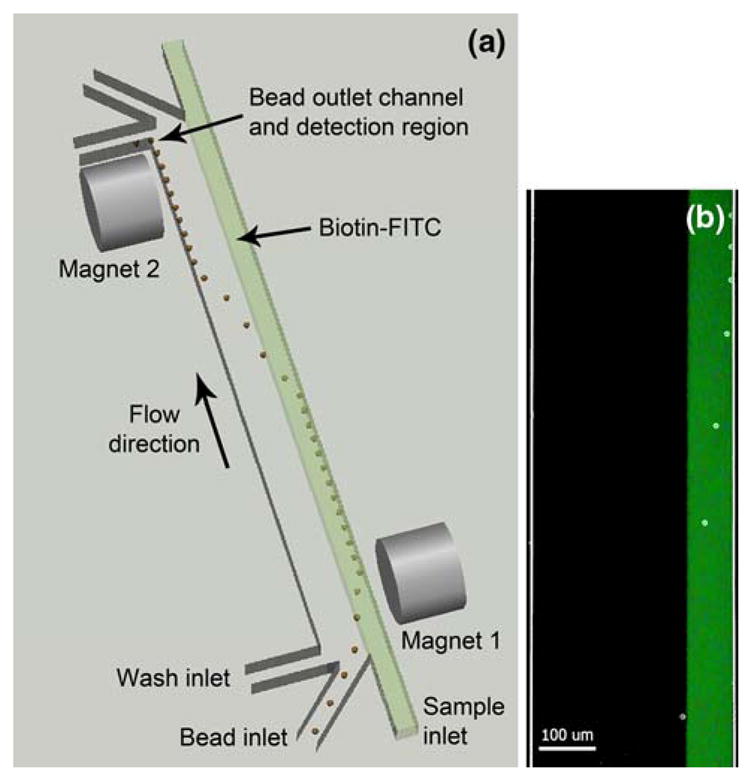
a Top-view conceptual rendering of a single-stage magnetically actuated immunofluorocytometry microdevice. The paramagnetic beads are pulled by the first magnet into the biotin-FITC sample stream, and then pulled by the second magnet across the channel into the wash stream at a downstream location, followed by fluorescence detection. b Composite micrograph of beads being pulled into biotin-FITC sample stream by first magnet. A bright-field image of beads flowing into the channel is overlaid with an epifluorescent image of the same channel with the biotin-FITC sample stream fluorescing
3 Materials and methods
3.1 Chip operation
Both a single incubation and dual incubation device are presented here. The single and dual incubation microdevices both operate on the same principle: a multiple inlet design with laminar flow patterning of reactant streams, with magnets placed on opposite sidewalls to direct the microbead trajectory, followed by integrated fluorescence detection on a microscopy platform. Both the single incubation and dual incubation assays use 8 μm diameter, paramagnetic streptavidin-coated beads containing a 1 μm diameter iron oxide paramagnetic core (Bang’s Laboratories, Inc., Fisher, IN). The first magnet pulls the beads into the antigen stream where they roll along the sidewall while incubation occurs, and the second magnet pulls them out of the sample stream across the channel into a wash stream at a precise downstream location. Figure 1 shows a schematic of this actuation scheme for a single incubation device. The dual incubation version simply adds a second cycle of this scheme after the first incubation to enable a primary incubation of antibody-coated beads with an untagged antigen, and a secondary incubation of the antigen-coated beads with a fluorescently tagged secondary antibody (Fig. 2). The design allows the full sandwich immunoassay to be autonomously processed within the microdevice, relying only on the magnetic force to pull the beads to the proper sidewall, and fluid shear forces to move the bead through the microchannel. As each microbead is pulled into the final wash stream, any unbound fluorescent tag separates from the bead and is diluted in the wash fluid, and then the bead flows past the detection region where optical detection occurs.
Fig. 2.

a Photograph of dual incubation microdevice. b Layout of photolithography mask for dual incubation microdevice. c Conceptual rendering from scaled 3D model of the dual incubation microimmunoassay, with bead state diagrams at device positions 1, 2, and 3. Position 1 The antibody-coated immunofluorocytometry beads are infused into the device and pulled into the sample stream. Position 2 After incubation the beads have antigen bound to them in an amount that is proportional to the sample antigen concentration. The beads are pulled into the first wash stream and transferred to the second stage, where they are pulled into the fluorescently labeled secondary antibody stream. Position 3 After the second incubation all bound antigen is bound by the fluorescent tag. Finally, the beads are pulled into the second wash stream which brings them to the detection region where fluorescence intensity detection occurs
3.2 Microdevice fabrication and design
The microdevices were fabricated by standard soft lithography (Duffy et al. 1998; Xia and Whitesides 1998) using an SU-8 master mold with a feature depth of 20 μm. The PDMS was cast to a thickness of 4 to 5 mm so that the PDMS could mechanically support external magnets placed at the periphery of the microchannels. The device inlet and outlet ports were punched with a sharpened 19 gauge needle. The PDMS was then bonded to a standard 75 mm × 25 mm glass slide. Bonding was accomplished by treating the PMDS and glass with a corona discharge (Electro-Technic Products, Inc., Chicago, IL. BD-10AS) for approximately 10 s, pressing the PDMS to the glass, and placing the device in a 100°C oven for 2 h. The slots for the magnets were cut by hand with a razor blade. Neodymium magnets with a 5,754 Gauss surface field, measuring ¼ inch on each side, (K&J Magnetics, Jamison, PA, No. B444) were pressed into those slots so that their magnetic fields were all aligned in the same direction across the width of the microchannels. The magnet faces were approximately 3 mm from the edge of the incubation microchannel. The main channel width for all microde-vices was 300 μm with 100 μm wide inlet and outlet branches. Both devices had incubation channel lengths of approximately 1 cm (point from bead entering incubation flow to entering wash flow).
3.3 Experimental procedure
The microdevice experiments utilized an epifluorescence microscopy platform with a Nikon TE2000U inverted microscope at 30× magnification. The devices were loaded into a device holder on the microscope, and a 30 mW, 488 nm beam emission from an argon ion laser (Modu-Laser, Stellar-Pro-CE) was focused through the microscope to the detection region of the microdevice. The 30× magnification results in an approximately 20 μm spot size for the focused beam. A photomultiplier tube (PMT) (C&L Instruments, Hershey, PA Model DPC-BA) was mounted to the side camera port of the microscope to measure the emitted light intensity at 200 samples per second. A filter block with a 480 nm bandpass excitation filter and a 488 nm dichroic mirror was used for excitation. For experiments with fluorescein (FITC) as a fluorescent tag (used during single incubation experiments), a 535 nm bandpass emission filter was used. For experiments with Phycoerythrin (PE) (used during dual incubation experiments), the emission filter was replaced with a 580 nm bandpass filter.
The fluids used during all experiments were loaded into 1 ml plastic syringes. The syringes were placed in syringe pumps (PicoPlus 22, Harvard Apparatus), and 0.01 inch ID Tygon Micro Bore PVC tubing (SmallParts Inc., Miramar, FL) was run from each 30 gauge syringe needle to the associated inlet ports (shown in Figs. 1 and 2). The tubing was pressed into each pre-punched port using tweezers with no further sealing necessary. Similarly, tubing was run from each outlet to a glass beaker for waste collection. The syringe pump containing the wash fluid syringe was first turned on and run at 3 μl/min. This flow helps to purge air from the device, and a small amount of bovine serum albumin (BSA) in the wash solution helps to reduce bead sticking in the channel when it is precoated. The remaining syringe pumps were then turned on and run at 3 μl/min to continue purging air from the device. They were then set to the desired infusion flow rate for the experiment, and after a brief settling time the concurrent flows stabilized and measurement began. After running each discrete sample concentration, the sample syringe was removed and replaced with the next sample, the device was again purged, and measurement of the new sample commenced.
3.4 Single incubation microdevice assay
The streptavidin-coated paramagnetic beads were first washed twice with phosphate buffered saline (PBS) (25 μl of bead stock in 1 ml of PBS). The beads were then resuspended in 500 μl of Ficoll-Paque Plus (GE Health-care) for a better density match between the beads and carrier fluid. The final bead density was approximately 1.6 million beads per ml. The analyte for the single incubation assay was prepared by serial dilution of a fluorescein-conjugated biotin molecule (biotin-FITC) stock (Invitrogen, Carlsbad, CA, B-10570) in PBS. The biotin-FITC concentration ranged from 0.025 to 0.75 μg/ml. The three fluids prepared for the single incubation assay included the streptavidin-coated bead carrier solution, the biotin-FITC sample solution, and PBS with 0.01% BSA for the wash stream. The single incubation device experiments used a flow rate of 300 nl/min at each inlet.
3.5 Dual incubation microdevice assay
For the dual incubation assay, 25 μl of the streptavidin-coated paramagnetic beads were washed twice with PBS and then resuspended in 50 μl of PBS. They were then incubated with 5 μl of a 1 mg/ml biotinylated primary monoclonal anti-human C3a antibody stock (Assay Designs Ann Arbor, MI, catalog number GAU017-01B) at room temperature for 1 h in an Eppendorf tube. An excess of antibody was used to ensure complete antibody conjugation to the beads. The beads were then washed again twice in PBS, and resuspended in 500 μl of Ficoll-Paque Plus. The complement C3a samples used in the dual incubation assay were prepared by serial dilution of a stock solution (EMD Chemicals, Gibbstown, NJ) with PBS. The C3a concentration used ranged from 0 to 5 μg/ml. The fluorescently labeled secondary antibody (anti-human C3a antibody) for the dual incubation assay (Assay Designs Ann Arbor, MI, catalog number GAU013-16) was prepared using a phycoerythrin (PE) fluorescent dye conjugation kit according to the manufacturer’s instructions (Prozyme, Inc., San Leandro, CA). After resuspending 50 μg of the fluorescently tagged antibody (assuming 100% recovery after labeling) in 1 ml of PBS, the labeling antibody solution was diluted with PBS to a final concentration of 12.5 μg/ml for use in the microdevice. The five fluids prepared for the dual incubation microimmunoassay included two wash streams (PBS with 0.01% BSA), the antibody coated bead stream, the C3a antigen solution, and the fluorescently labeled secondary antibody solution. The dual incubation assay used a flow rate of 100 nl/min at each inlet, resulting in an empirically determined 45 s incubation time per stage.
3.6 Fluorescence detection
The bead fluorescence intensity is recorded by monitoring the PMT fluorescence counts as the beads pass through the focused laser light within the device detection region. A PMT sample rate of 200 Hz was empirically determined to be sufficient for the flow rates used during the experiments. Each fluorescence sample represents a count of photons within a 5 ms time period. The fluorescence intensity data collected by the PMT was then processed with a 1 Hz Butterworth high-pass filter and a custom peak finding algorithm written with Matlab. The filter shifts the PMT intensity baseline to zero and removes any baseline fluorescence drift. After filtering, peaks with a trailing edge drop-off greater than two standard deviations of the dataset mean were counted as bead fluorescence events. The peak values were then averaged to find the mean bead fluorescence intensity for the sample. Data processing is discussed in detail below.
3.7 Benchtop incubation experiments
Two types of benchtop incubations were performed during development of the dual incubation immunoassay in order to validate the paramagnetic cytometric bead assay and fluorescence detection, and to determine the required incubation times. First, a commercially available Cytometric Bead Array Human Anaphylatoxin Kit (BD Biosciences, San Jose, CA, USA) was used following the manufacturer’s instructions to a create a series of incubated bead standards for complement C4a of varying concentrations. These beads solutions were analyzed for fluorescence intensity using a Becton Dickinson Facscalibur flow cytometer, as well as in a microdevice to generate calibration curves of fluorescence versus C4a concentration, and to compare the measured fluorescence from flow cytometry to that measured using the microscopy setup. The microdevice used for this detection was a 150 μm wide, 20 μm deep straight channel made of PDMS with one inlet and one outlet. The device was used on the microscopy platform, and interfaced to a syringe in the same manner as to the single and dual incubation immunofluorocytometry assay devices. The benchtop incubated bead solutions from the kit were resuspended in PBS and infused through the microchannel at a flow rate of 100 nl/min, excited with a 488 nm argon ion laser, and the PMT was used for fluorescence detection.
In order to verify the binding capability of both the streptavidin-coated and antibody-coated paramagnetic beads, benchtop incubation experiments were performed. This consisted of a benchtop incubation of the streptavidin-coated beads with a biotin-FITC solution of known concentration. The beads were washed twice in PBS, and then incubated for 30 s (within the typical incubation time range for the microdevice assays) with the biotin-FITC sample in an Eppendorf tube. The incubation was ended by adding 1 ml of PBS and immediately vortexing. After this initial wash, the beads were washed a second time in PBS. The bead fluorescence was analyzed in the 150 μm wide, 20 μm deep straight channel as described above.
Subsequently, another benchtop incubation was conducted using the antibody-coated beads and both the antigen and secondary antibody solutions used in the microimmunofluorocytometry assay device. The beads were washed twice in PBS, and then incubated for 45 s with the C3a antigen sample in an Eppendorf tube. The incubation was again ended by adding 1 ml of PBS and vortexing. After this initial wash, the beads were washed a second time in PBS, and a similar 45 s incubation with the fluorescent secondary antibody was begun. This incubation was ended in the same way as the first incubation, again washing the beads twice in PBS. Finally, the beads were resuspended in PBS and a Facsaliber flow cytometer was used to measure the mean fluorescence intensity of each sample.
4 Hydrodynamics and physical design considerations
An important feature of the microchannel design is the bead injection region. In order to achieve the most consistent bead incubation possible and thus more consistent bead-to-bead response, the beads must be pulled into the sample stream at exactly the same downstream position. Figures 3a and b show the bead injection channel design, as well as a composite image of beads entering the sample stream. The wall between the parallel portions of the sample inlet and bead inlet allows the beads to organize against the side of the channel before being pulled into the sample stream. Previous designs that did not include this parallel region before the merging of the two channels resulted in beads scattered across the channel, entering the sample stream at significantly different locations. The special injection region design ensures that all beads enter the sample stream at the same location, therefore experiencing a consistent incubation time.
Fig. 3.

a Conceptual rendering of the bead injection region. The wall between the two inlets ensures that the beads always enter the main channel from the same point. b Composite micrograph of a bead entering the main channel and being pulled against the sidewall in the antigen stream. c Composite micrograph of a bead transferring from incubation stage 1 to stage 2
The intended magnet position was approximately 3 mm from the edge of the microchannel for all four magnets. Placement was done by eye without direct measurement. As long as the magnets were placed within a range of approximately 2.5 to 3.5 mm from the channel wall, the bead rolling velocity was similar between difference devices. While variations in the magnetic field strength within this range have an effect on the time, it takes to bring the beads to the wall, this time is relatively short compared to the total incubation time, and these variations do not have a significant effect on the downstream velocity of the beads once the beads reach the wall.
The magnetic force acting on the bead can be approximated as , where B is the magnetic field from the magnet, μ0 is the magnetic permeability of free space, χ is the magnetic susceptibility of the bead (χ = 0.170) (Shevkoplyas et al. 2007) and Rc is the radius of the bead magnetic core (0.5 μm). Since the exact configuration of the magnetic field and gradient is complicated to model or measure on this length scale, the magnetic force on the bead is estimated by balancing the magnetic force with the Stokes drag force F⃗D = 6πμUR, where μ is the viscosity of water, U is the bead velocity toward the wall, and R is the bead radius. After measuring the bead velocity to the wall to be 100 μm/s where the magnetic force on the bead is estimated to be approximately 7.5 pN for this system and falls off quickly as the bead rolls along the sidewall down the channel beyond the magnet face. Conversely, the fluid shear force on an 8 μm diameter bead at the channel sidewall is approximated by calculating the centerline shear stress at the bead centroid (4 μm from the wall) considering the laminar flow profile in a rectangular duct as τ = 0.111 Pa, and multiplying by the bead surface area to estimate the drag force on the bead as 22.3 pN. If the bead is off the centerline along the depth of the channel, the calculated force increases to a maximum of 28.9 pN when the centroid is 4 μm from the top or bottom boundary. Based on these estimates, the fluid drag force is expected to be 3.0 to 3.9 times greater than the magnetic force on the beads. It should also be noted that along the face of the magnet these forces are orthogonal to each other (the magnetic force pulling the bead toward the sidewall and the shear forces propelling the beads forward), so the actual drag force on the bead as it rolls along the wall will depend on the wall dynamic frictional force (which will be a function of the orthogonal magnetic force) but is expected to be smaller in magnitude than the magnetic force. Beyond the magnet face, the magnetic force will fall off appreciably, so the shear force on the bead remains dominant as it rolls along the wall downstream of each magnet. Additionally, if the flow rate is decreased significantly (<50 nl/min) then the magnetic and shear force magnitudes approach each other and beads are more prone to being captured at the channel sidewall. In this case, the magnets need to be moved further from the channel boundaries to keep the beads from sticking.
Thus, the bead incubation time of the microdevice at the flow rates used is significantly more sensitive to changes in the flow rate than it is to device to device variations in magnet placement, which allows for incubation rate tuning based on flow rate alone. The bead velocity, which was 220 μm/s, was found empirically by measuring the time taken for the beads to traverse a certain distance during incubation. As the beads passed the end of each magnet, there was no discernable change in their velocity due to the magnetic force pulling back on them along the length of the channel.
When designing the dual incubation device, pressure matching must also be considered between the first incubation stage outlet and second incubation stage inlet. In Fig. 2 it can be seen that the first stage outlet runs back along the length of the device further than would seem necessary. This additional channel length is included to balance the flow resistance so that two thirds of the fluid in the channel exits through the outlet, and one third continues into the second stage. The two thirds exiting represents the original bead carrier stream and the antigen stream together. Without this hydrodynamic resistance balancing, too much fluid would exit through the first outlet leaving very little flow in the second stage. The required channel length for flow balancing for the device presented was determined using a computational fluid dynamics model (Comsol Multiphysics), but it is also possible to estimate with sufficient accuracy using simplified flow resistance calculations. Figure 3c shows bead transfer from the first to the second incubation stage while the second magnet pulls the beads across the channel.
The outlet channel containing the detection region is also designed for accurate control of microbead trajectories, in this case to reduce bead aggregation. Bead aggregates in the detection region artificially increase the effective bead fluorescence intensity seen by the detector, and therefore must be avoided. These aggregates are separated when the beads experience a large increase in flow velocity, which can be easily accomplished by reducing the relative channel width. The design presented uses a doubling of the flow velocity as the beads enter the bead outlet channel to separate aggregated beads. The flow velocity is subsequently reduced once again when the channel widens briefly for fluorescence detection; however, the beads do not aggregate in this brief time.
For the microdevice designs described, there is minimal mixing between the adjacent fluid streams since the flow is laminar at this scale. The Reynolds number for these channels with the flow rates used for experiments is approximately 5 × 10−2, and the Peclet number is estimated to be in the range of 103 for biotin-FITC to 104 for complement C3a. In a longer incubation channel, diffusional mixing could become a significant factor, diluting the sample and slowing incubation. In such a case, the channel arrangement would have to include an outlet for the bead carrier fluid after the beads are transferred into the sample stream. This design would allow the sample stream to flow independently without the possibility of dilution. The wash stream can be introduced at the end of the incubation region before the beads transfer back across the device.
A uniform suspension of beads in the carrier fluid is also very important for reliable bead injection into the device. If the beads begin to settle in the syringe, tubing, or microchannel during the experiment, the flow of beads will be reduced or stopped, inhibiting the assay. To mitigate this problem, the beads are suspended in Ficoll-Paque Plus, which has a density of 1.077 g/ml. This density is similar to the microbeads’ mean density of 1.07 g/ml, providing a uniform suspension for many hours.
A final consideration is the continuous flow operation of the device. If the device continuously samples an antigen containing fluid where the concentration varies with time, the ultimate bead fluorescence should also vary accordingly. Since the assay analyzes multiple beads with a sampling time of 5 min or less to make a measurement, tracking the bead fluorescence should allow antigen concentration tracking for time-varying antigen concentrations, such as during the systemic inflammation process in blood. This detection time is in contrast to traditional assays such as ELISA and bench-top immunofluorocytometry, which use multiple incubation steps of 1 h or longer requiring multiple hours of total assay time. Temporal tracking measurements would represent a significant improvement over inflammation studies in the literature in terms of both the sample interval, as well as the analysis time, since concentration changes would be detected almost immediately using the autonomous microimmunoassay.
5 Results and discussion
5.1 Single incubation device
Complete on-chip incubation and detection experiments were conducted using the single incubation microdevice at a flow rate of 300 nl/min from each syringe for the bead carrier fluid, biotin-FITC sample, and PBS wash solution. The results are shown in Fig. 4a. The bead incubation time in the biotin-FITC stream in the microdevice was 18 s at this flow rate. Approximately, 300 bead fluorescence events per minute were recorded for this assay. Results from this assay show a linear fluorescence versus concentration response provided that the incubation remains in the linear portion of the streptavidin–biotin binding curve. Figure 4b shows a calibration curve of bead fluorescence versus concentration for streptavidin–biotin binding over an extended concentration range. This data was obtained by the benchtop incubation of the beads with a biotin-FITC solution of known concentration as described. Following incubation, the beads were resuspended and run through a microchannel for fluorescence detection using the argon ion laser and PMT. These data show that there is a linear response region where bead fluorescence intensity is proportional to the biotin-FITC concentration, and a saturation region at higher solution concentrations. In order to provide meaningful data, the incubation time of the microdevice must be tuned for the expected concentration range so that the beads are not saturated at the highest concentration tested. The benchtop experiments such as this one were used as a simple method to determine the required incubation times in order to design the microchannel incubation length and appropriate syringe pump flow rates for the microdevice assay.
Fig. 4.

a Bead fluorescence versus biotin-FITC concentration from on-chip incubation and detection using a single-stage incubation device. b Mean fluorescence intensity versus biotin-FITC concentration response curve from microdevice detection following benchtop incubation for 30 s to study streptavidin–biotin binding over an extended concentration range. Bead fluorescence saturation is evident at biotin-FITC concentrations higher than 300 ng/ml
5.2 Dual incubation device
For autonomous fluorescence detection of a protein in a native biological sample such as whole blood, two incubation steps are required to complete an antigen sandwich immunoassay. In the first step, antibody coated beads are incubated with the sample fluid to bind the antigen of interest. In the second step, the beads are incubated with a fluorescently labeled secondary antibody also specific to the antigen of interest. In this work, two compatible monoclonal antibodies were used, each binding a different epitope on the antigen of interest. By continuously infusing fresh immunofluorocytometry beads at the device inlet with autonomous bead manipulation within the device, the amount of antigen bound to the bead surface following incubation varies with the antigen concentration. Thus, the total bead fluorescence following the secondary labeling step is also proportional to the antigen concentration. A concentration measurement is made by detecting the fluorescence intensity of each bead after the second incubation step and comparing the fluorescence intensity to a previously determined calibration curve. Fluorescence intensity versus C3a antigen concentration data from the dual incubation microimmunoassay are plotted in Fig. 5a. The limit of detection for the device as tested was 625 ng/ml. Therefore, the immunoassay is capable of producing a fluorescence output curve for concentrations in the clinically expected systemic range of 1–5 μg/ml.
Fig. 5.

a Results from the dual incubation microimmunoassay with fully autonomous on-chip incubation and detection, showing fluorescence intensity in PMT counts versus C3a antigen concentration. b Partial PMT trace from the dual incubation experiment showing the original detected signal (top) and signal after high-pass filtering (bottom). The dots represent each peak detected by the peak finding algorithm. c Overlayed fluorescence intensity histograms of three datasets from the dual incubation microimmunoassay experiments, using C3a sample concentrations of 5, 1.25, and 0.625 μg/ml. At the lowest concentration, the intensity histogram merges completely with the baseline noise, thus representing the detection limit of the assay
5.3 Data processing
The PMT was sampled at 200 Hz in order to capture light quickly enough from any passing bead, while it is near the center of the field of view of the detection region. If the sample rate is too low relative to the velocity of the beads in the detection region, multiple beads may pass the detector during the sampling time. As the sample rate increases, the amount of light captured in each sample is lower, reducing the signal to noise ratio.
As noted, the fluorescence intensity collected by the PMT was filtered by a 1 Hz Butterworth high-pass filter and custom peak finding algorithm. Figure 5b shows a representative PMT trace using a 5 μg/ml C3a sample before and after filtering. The Butterworth filter shifts the PMT intensity baseline to zero and removes baseline drift before the data is processed by a peak finding algorithm. The high-pass filter together with the peak finding algorithm remove the background fluorescence from the bead fluorescence measurements. Peaks with a trailing edge drop-off greater than two standard deviations of the dataset mean are counted as bead fluorescence events. These are shown by the dots in Fig. 5b. The measured fluorescence intensity shown in Fig. 5a for each tested sample is determined by finding the mean value of the peak maxima, with the error bars representing the standard error about the mean for each sample. Fluorescence histograms of three datasets after filtering are shown in Fig. 5c. For samples with moderate to high antigen concentrations, the dataset histograms show a Gaussian distribution around the mean bead fluorescence value, and low values of standard error. This distribution is similar to that of a traditional benchtop incubated immunofluorocytometry assay using a standard flow cytometer for detection (Fig. 5c). At concentrations closer to the lower limit of the device’s detection range, the bead intensity histogram peak has a lower signal to noise ratio and begins to merge with the baseline noise. At this point, a Gaussian peak is no longer discernable and the standard error increases. At even lower concentrations, the mean peak value is equal to that of the system noise alone, which was around 12 counts, or 1% of the 10-bit measurement range of the PMT, for the experimental parameters used. For the device parameters used here, the dual incubation microdevice assay was unable to differentiate a sample at a C3a concentration of 625 ng/ml from background noise, and if sample concentrations below this were to be processed a longer incubation channel and/or lower flow rate would be used to increase the incubation times and achieve a sufficient bead fluorescence signal.
5.4 Dual incubation assay validation
Development of the dual incubation immunoassay began with benchtop testing of all binding reactions. The components used in the dual incubation microimmunoassay are similar to those in commercially available immunofluorocytometry bead assay kits. However, the beads used for this microimmunoassay are paramagnetic, which is required for the autonomous processing within the microdevice. This assay also differs from the commercially available kits in that it is intended to directly measure samples at systemic antigen concentrations without any dilution factors. The immunofluorocytometry bead assay kits detect lower antigen concentrations in the range of a few ng/ml with incubation times of over an hour. The microimmunoassay is intended for detecting higher concentrations in the μg/ml range, the target human systemic sample concentration range seen before and during CPB (Ascione et al. 2000), with incubation times of under five minutes. In order to validate the on-chip fluorescence detection for the dual incubation assay, the microbeads included with a commercial analphylatoxin cytometric bead assay kit (BD Biosciences, San Jose, CA, USA) were incubated with complement C4a at varying concentrations, according to the manufacturer’s instructions, and analyzed with a microfluidic channel on the described microscopy platform with PMT detection. By comparing the on-chip analysis with the published flow cytometry standards (BD Biosciences 2007), it was evident that on-chip detection could yield a similar fluorescence response to that seen with a standard flow cytometer (Fig. 6a).
Fig. 6.

a Comparison plot of commercial anaphylatoxin kit published C4a standard calibration curve with fluorescence measurements of the same beads after benchtop C4a incubation according to manufacturer’s instructions followed by infusion through a microdevice with PMT fluorescence detection. b Benchtop-incubated C3a antigen sandwich immunoassay fluorescence results using the paramagnetic immunofluorocytometry beads measured with a Facscaliber flow cytometer. The primary and secondary incubation times were 45 s each. c Representative histogram from flow cytometer for a 5 μg/ml C3a sample with a 2 min bench top incubation. An ideal dataset is expected to show a Gaussian distribution around the mean fluorescence intensity
A second validation test that was performed tested the feasibility of detecting C3a concentration using much shorter incubation times than those used in the commercial kit. Since the concentrations that will be seen by the microimmunoassay are approximately 100–1,000 times higher than those used in the benchtop kit; it was expected that a shorter incubation could result in similar fluorescence intensities. A benchtop dual incubation was performed using the antibody-conjugated paramagnetic beads used in the microimmunoassay, with 45 s incubations for both antigen binding and secondary labeling at concentrations in the expected systemic range (1–5 μg/ml). These beads were then analyzed using the Facscalibur flow cytometer. The results of this experiment showed that a shorter incubation time with a more concentrated analyte provides fluorescence intensities within the detection range of the flow cytometer (Fig. 6b). Based on these experiments comparing on-chip detection to detection with standard flow cytometry, it was expected that a microim-munoassay with 45 s primary and secondary incubation time would be able to detect complement C3a in the systemic concentration range.
6 Conclusions
A continuous flow microimmunoassay technology based on autonomous immunofluorocytometry with a novel bead manipulation scheme has been presented. It has been validated with both a single incubation between streptavidin and biotin-FITC, as well as in a full dual incubation assay for conducting an autonomous antigen sandwich immunoassay. Both assays have had tailored incubation times to produce calibration curves of bead fluorescence as a function of antigen concentration. It is believed that the technology presented for a continuous flow autonomous microimmunoassay is a stepping stone toward the development of high sample rate clinical monitoring assays.
This technology has implications for the development of personalized patient treatments. It is believed that the device presented will be directly applicable to monitoring complement C3a concentration during cardiac surgeries involving CPB. Ultimately, similar microdevice assays will be developed based on this technology in order to conduct a multiplexed detection to measure other biomarkers of systemic inflammation. The combined assay will offer a perspective into systemic inflammation during CPB that is currently unavailable. The high sample rate that is possible with this type of microimmunoassay will offer a thorough profile of the inflammation process. Near continuous monitoring may allow treatment to be adjusted during the surgery to reduce inflammation and its negative effects.
Acknowledgments
This work was funded by grants from the National Institutes of Health Heart Lung Blood Institute grant No. 1R21HL084367-01A1 and the Wallace H. Coulter Foundation Early Career Translational Research Awards in Biomedical Engineering. LAS is supported by a NIH Biotechnology Fellowship Grant No. T32 GM008339-19.
Contributor Information
Lawrence A. Sasso, Email: Lsasso@eden.rutgers.edu, BioMEMS Laboratory, Department of Biomedical Engineering, Rutgers, The State University of New Jersey, Room 370, 599 Taylor Road, Piscataway, NJ 08854, USA
Akif Ündar, Email: aundar@psu.edu, Department of Pediatrics, Surgery, and Bioengineering, Penn State College of Medicine, Penn State Children’s Hospital, Penn State Hershey Pediatric Cardiovascular Research Center, Penn State Milton S. Hershey Medical Center, Room C6525, Hershey, PA 17033-0850, USA.
Jeffrey D. Zahn, Email: jdzahn@rci.rutgers.edu, Department of Biomedical Engineering, Rutgers, The State University of New Jersey, Room 311, 599 Taylor Road, Piscataway, NJ 08854, USA
References
- Ahn KC, Lohstroh P, Gee SJ, Gee NA, Lasley B, Hammock BD. High-throughput automated luminescent magnetic particle-based immunoassay to monitor human exposure to pyrethroid insecticides. Anal Chem. 2007;79:8883–8890. doi: 10.1021/ac070675l. [DOI] [PubMed] [Google Scholar]
- Aoyagi S, Miyasaka T, Yoshimi Y, Sakai K. A new reagentless immunosensor for measuring IgG concentration in human plasma based on fluorescence-enhancement immunoassay. Japn Soc Artif Organs. 2002;5:60–63. [Google Scholar]
- Ascione R, Lloyd CT, Underwood MJ, Lotto AA, Pitsis AA, Angelini GD. Inflammatory response after coronary revascularization with or without cardiopulmonary bypass. Ann Thorac Surg. 2000;69:1198–1204. doi: 10.1016/s0003-4975(00)01152-8. [DOI] [PubMed] [Google Scholar]
- Barton AC, Davis F, Higson SPJ. Labeless immunosensor assay for prostate specific antigen with picogram per milliliter limits of detection based upon an ac impedance protocol. Anal Chem. 2008;80:6198–6205. doi: 10.1021/ac800491m. [DOI] [PubMed] [Google Scholar]
- BD Biosciences. Human Anaphylatoxin Kit instruction manual. BD Cytometric Bead Array (CBA); 2007. [Google Scholar]
- Bhattacharyya A, Klapperich CM. Design and testing of a disposable microfluidic chemiluminescent immunoassay for disease biomarkers in human serum samples. Biomed Microdevices. 2007;9:245–251. doi: 10.1007/s10544-006-9026-2. [DOI] [PubMed] [Google Scholar]
- Cheng XH, Irimia D, Dixon M, Sekine K, Demirci U, Zamir L, Tompkins RG, Rodriguez W, Toner M. A microfluidic device for practical label-free CD4+ T cell counting of HIV-infected subjects. Lab Chip. 2007;7:170–178. doi: 10.1039/b612966h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Oh KW, Thomas JH, Heineman WR, Halsall HB, Nevin JH, Helmicki AJ, Henderson HT, Ahn CH. An integrated microfluidic biochemical detection system for protein analysis with magnetic bead-based sampling capabilities. Lab Chip. 2002;2:27–30. doi: 10.1039/b107540n. [DOI] [PubMed] [Google Scholar]
- Do J, Ahn CH. A polymer lab-on-a-chip for magnetic immunoassay with on-chip sampling and detection capabilities. Lab Chip. 2008;8:542–549. doi: 10.1039/b715569g. [DOI] [PubMed] [Google Scholar]
- Duffy DC, McDonald JC, Schueller OJA, Whitesides GM. Rapid prototyping of microfluidic systems in poly(dimethylsiloxane) Anal Chem. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- Engstrom HA, Andersson PO, Ohlson S. A label-free continuous total-internal-reflection-fluorescence-based immunosensor. Anal Biochem. 2006;357:159–166. doi: 10.1016/j.ab.2006.03.058. [DOI] [PubMed] [Google Scholar]
- Fung M, Loubser PG, Undar A, Mueller M, Sun C, Sun WN, Vaughn WK, Fraser CD. Inhibition of complement neutrophil, and platelet activation by an anti-factor D monoclonal antibody in simulated cardiopulmonary bypass circuits. J Thorac Cardiovasc Surg. 2001;122:113–122. doi: 10.1067/mtc.2001.114777. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Veres T, Tabrizian M. Enzymatically-generated fluorescent detection in micro-channels with internal magnetic mixing for the development of parallel microfluidic ELISA. Lab Chip. 2006;6:555–560. doi: 10.1039/b516031f. [DOI] [PubMed] [Google Scholar]
- Kartalov EP, Zhong JF, Scherer A, Quake SR, Taylor CR, Anderson WF. High-throughput multi-antigen microfluidic fluorescence immunoassays. Biotechniques. 2006;40:85–90. doi: 10.2144/000112071. [DOI] [PubMed] [Google Scholar]
- Kim KS, Park JK. Magnetic force-based multiplexed immunoassay using superparamagnetic nanoparticles in microfluidic channel. Lab Chip. 2005;5:657–664. doi: 10.1039/b502225h. [DOI] [PubMed] [Google Scholar]
- Kirklin JW, Barratt-Boyes BG. Hypothermia, circulatory arrest, and cardiopulmonary bypass. In: Kirklin JW, Barratt-Boyes BG, editors. Cardiac surgery. New York: Churchill Livingstone; 1993. [Google Scholar]
- Mainwaring RD, Lamberti JJ, Hugli TE. Complement activation and cytokine generation after modified Fontan procedure. Ann Thorac Surg. 1998;65:1715–1720. doi: 10.1016/s0003-4975(98)00068-x. [DOI] [PubMed] [Google Scholar]
- Moore FD, Warner KG, Assousa S. The effects of complement activation during cardiopulmonary bypass: attenuation by hypothermia, heparin, and hemodilution. Ann Surg. 1988;208:95–103. doi: 10.1097/00000658-198807000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamme N. Magnetism and microfluidics. Lab Chip. 2006;6:24–38. doi: 10.1039/b513005k. [DOI] [PubMed] [Google Scholar]
- Peyman SA, Iles A, Pamme N. Rapid on-chip multi-step (bio)chemical procedures in continuous flow—manoeuvring particles through co-laminar reagent streams. Chem Commun. 2008:1220–1222. doi: 10.1039/b716532c. [DOI] [PubMed] [Google Scholar]
- Roper MG, Shackman JG, Dahlgren GM, Kennedy RT. Microfluidic chip for continuous monitoring of hormone secretion from live cells using an electrophoresis-based immunoassay. Anal Chem. 2003;75:4711–4717. doi: 10.1021/ac0346813. [DOI] [PubMed] [Google Scholar]
- Sato K, Yamanaka M, Hagino T, Tokeshi M, Kimura H, Kitamori T. Microchip-based enzyme-linked immunosorbent assay (microELISA) system with thermal lens detection. Lab Chip. 2004;4:570–575. doi: 10.1039/b411356j. [DOI] [PubMed] [Google Scholar]
- Shevkoplyas SS, Siegel AC, Westervelt RM, Prentiss MG, White-sides GM. The force acting on a superparamagnetic bead due to an applied magnetic field. Lab Chip. 2007;7:1294–1302. doi: 10.1039/b705045c. [DOI] [PubMed] [Google Scholar]
- Wang HX, Meng S, Guo K, Liu Y, Yang PY, Zhong W, Liu BH. Microfluidic immunosensor based on stable antibody-patterned surface in PMMA microchip. Electrochem Commun. 2008;10:447–450. [Google Scholar]
- Westaby S. Organ dysfunction after cardiopulmonary bypass: a systemic inflammatory reaction initiated by the extracorporeal circuit. Intensive Care Med. 1987;13:89–95. doi: 10.1007/BF00254791. [DOI] [PubMed] [Google Scholar]
- Xia YN, Whitesides GM. Soft lithography. Annu Rev Mater Sci. 1998;28:153–184. [Google Scholar]
- Yang S, Undar A, Zahn JD. Continuous cytometric bead processing within a microfluidic device for bead based sensing platforms. Lab Chip. 2007;7:588–595. doi: 10.1039/b703808a. [DOI] [PubMed] [Google Scholar]