

Abstract
IPEX (Immunodysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome is a rare, recessive disorder in patients with mutations in the foxp3 gene, the normal expression of which is required for the generation of functional regulatory T-cells. Scurfy mice also bear a mutation in the foxp3, and like IPEX patients, spontaneously develop multi-organ inflammation. As reviewed herein, breeding immune response genes into Scurfy mice has provided useful insight into how the inflammatory T-cell response is regulated in the absence of regulatory T-cells and post regulatory T-cell checkpoint. Of particular interest are those that preferentially affect the inflammatory T-cell response in an “apparent” organ-specific manner, implying that specific mechanisms of control exist for individual organs during multi-organ inflammation.
Keywords: Multi-organ inflammation, Scurfy mice, Genetic regulation
Introduction
A major control mechanism for self-tolerance is mediated by a CD4+ T-cell subset termed regulatory T-cells (Treg)2 [1–3]. The expression of Treg is critically dependent on the X-linked gene encoding the transcription factor Foxp3 [4,5]. Patients bearing a mutation in their foxp3 gene invariably develop IPEX syndrome which is characterized by systemic autoimmunity manifested as diarrhea, eczematous dermatitis, insulin-dependent diabetes mellitus, anemia, thrombocytopenia, neutropenia, and tubular nephropathy [6]. However, clinical variation of these symptoms is observed among families and within families [6]. Factors such as the severity of the mutational effect on Foxp3 expression and/or function, genetic background that influences Foxp3 expression or function, and environment are likely reasons for the heterogeneity of IPEX clinical outcome. For this reason, it is difficult to study how the variations are developed in the manifestation of the systemic autoimmunity in IPEX patients.
Scurfy (Sf) mice bear a mutation in foxp3 gene such that they cannot produce functional Foxp3 and are totally devoid of Treg [4]. As a result, spontaneous immune response against host antigens (Ag) develops in an uncontrollable and unabated manner, leading to severe multi-organ inflammation (MOI), and death around 3 to 4 weeks of age. Organs such as skin, tail, lungs and liver are affected first [7]. Unlike IPEX patients, diabetes mellitus and diarrhea are not observed but the inflammation in skin, lung and liver is reproducibly developed due to genetic homogeneity despite the fact that the CD4+ T-cell expansion is stochastic and widespread across the Vβ and Vα families [8]. Theoretically, Sf mice have the potential to develop inflammation in other organs. A low frequency of organ-specific T-cells, a limited supply of Ag, the pre-weaning condition, and the early death are potential reasons that prevent this process from coming to fruition. Transfer of Sf T-cells into Rag1−/− recipients induced inflammation in additional organs [7]. Severe gastrointestinal inflammation was rapidly developed in neonate Rag1−/− recipients just a few days after weaning, suggesting mother's milk and the intestinal microbes play a role in the enteropathy [7]. Moreover, inflammation could be demonstrated in accessory reproductive organs in Sf.faslpr/lpr double mutant mice that lived beyond adulthood [9]. Thus, Sf mice offer a unique system to study how Tinf response against multiple organs is regulated by various immune response genes. Importantly, Tinf response in Sf mice occurs beyond Treg checkpoint and without the confounding factor of the feedback regulation by Treg that are present in conventional immune response [10–12]. A straightforward approach is to breed a specific gene, usually in its mutant form, to Sf mice and then to determine the effect of the gene on Tinf response at the organ, cellular and molecular levels. In this communication, we will review studies that bred various immune response genes into Sf mice to study the specific gene effect on Tinf response (summarized in Table 1).
Table 1.
Summary of breeding experiments that address the genetic control of MOI in Sf mice.
| Gene examined in Sf mice | Cellular change compared with Sf mice | Change in MOI | Lifespan (weeks) |
|---|---|---|---|
| TCR Tg.rag−/− | TCR Tg T-cells only | No MOI | >20 |
| cd4−/− | No CD4+ T-cells | Delayed 1 week | 6 |
| β2m−/− | No CD8+ T-cells | Not delayed | 4 |
| TCR Tg | T-cell number reduced, dual-TCR T-cells expanded | Delayed 2–3 weeks | 7 |
| NOD background | N.D.* | More severe than B6.foxp3sf mice | N.D. |
| BDC2.5 TCR Tg in NOD background | Lymphoproliferation was ameliorated | Rapid development of insulitis and diabetes, MOI was not addressed. | N.D.** |
| aire−/− | N.D. | MOI fastened but did not extend to endocrine organs | 2–3 |
| cd28−/− | Inhibit T-cell activation and lymphokine production | Inhibited | 50% lived >30 |
| stat6−/− | Inhibited IgE and Th2 lymphokine production | Inhibited eosinophilia and lung Goblet cell metaplasia | 5 |
| Igtαε−/− | lymphocyte number decreased by ∼40% | Delayed 2–3 weeks, developed colitis | 6–7 |
| Il2−/− | Lymphocyte number in lymph nodes increased 100%, CD103 and trafficking/chemotaxis receptors inhibited | Delayed 3–5 weeks, no skin inflammation, greatly reduced lung inflammation, liver inflammation remained, developed colitis | 6–10 |
| faslpr/lpr | Slight increase in lymphocyte number in lymph nodes | Not delayed but lifespan prolonged, adult mice developed severe inflammation in accessory reproductive organs and colitis. | 6–18 |
Not described.
TCR Tg should have prolonged the lifespan.
Sf.cd4−/− mice
One of the earliest experiments to study immune response gene effect on Scurfy phenotype is by breeding cd4−/− and β2m−/− genes into Sf mice [13]. These studies demonstrated that cd4−/− but not β2m−/− gene affected the mortality, organ inflammation, and immunological parameters of Sf mice, thus implicating CD4+ T-cells are critical to the fatal disease. However, the protection by cd4−/− gene was only transient. The lifespan of Sf.cd4−/− mice was extended from 4 weeks to 6 weeks and the mice eventually died of MOI with immunological manifestation similar to that of Sf mice. Although cd4−/− mice lack CD4+ T-cells, class-II-restricted T-cells are still present. The spontaneous T-cell activation in the Sf.cd4−/− mice was likely delayed by the lack of the CD4 co-receptor signal, but the activation of Tinf cells eventually developed and induced Sf-like disease.
TCR transgenic (Tg) Sf mice
Because the MOI in Sf mice is mediated by polyclonal CD4+ T-cells, genetic manipulation that restricts TCR repertoire has a major impact on the fatal disease. Breeding foreign Ag-specific TCR Tg into Sf mice delayed but did not eliminate the fatal MOI [14]. Importantly, TCR Tg Sf mice in Rag−/− background did not develop Sf disease [14]. Although such mice lack Treg, their T-cells express only the Tg TCR and are unable to respond to host and foreign Ag not recognized by the Tg TCR. Because Tg expression of TCR genes greatly reduces but does not totally freeze endogenous TCR gene rearrangement (unless in Rag−/− background), a substantial fraction of their T-cells, estimated to be around 30%, expresses dual-TCR [15,16]. Although there is a loss of diversity, the TCR repertoire derived from endogenous genes is large enough to generate Tinf response and MOI. Also, the endogenous TCR is usually less abundantly expressed than the Tg TCR on a dual-TCR T-cell. These considerations satisfactorily explain the delayed but still fatal MOI in Sf mice bearing foreign Ag-specific TCR Tg.
The power of Treg control of dual-TCR T-cell expansion is displayed in Sf mice bearing the ovalbumin(OVA)323–339-specific Tg TCR (OT-II.foxp3sf) [17]. The expansion of the T-cells bearing dual-TCR begins as early as 6 days after birth and reaches to as much as 85% of the lymphocyte population. By contrast, T-cells bearing only Tg TCR are not activated and remain naïve phenotype even in the total absence of Treg control because they can only be activated by OVA323–339 peptide which is absent in the mice. This expansion occurs in the peripheral lymphoid tissues but not in the thymus. Importantly, transfer of T-cells that had been depleted of the Tg TCRβ into Rag1−/− mice induced MOI, suggesting MOI could be induced by T-cells bearing endogenously derived TCRβ [17].
It is generally believed that autoimmune T-cells are contained by Treg and in the absence of Treg, they expand and MOI develops. Several examples of breeding Tg TCR that has specificity to “natural self-Ag” into Sf mice have been reported. Several such studies are described below.
K/BxN.foxp3sf mice
In an attempt to study thymic T-cell selection, Kouskoff et al. [18] generated a Tg mouse line in B6 background designated KRN mice whose TCRα and TCRβ genes were derived from a B10A.4R T-cell hybridoma specific to RNase and restricted by I-Ak. It was fortuitously discovered that when KRN mice were crossed with NOD mice, the progeny (KRN Tg TCR in [B6xNOD]F1) developed arthritis with severe joint inflammation and swelling [18]. The development of arthritis is critically dependent on the Tg TCR and antibodies with specificity to glucose-6-phosphate isomerase (GPI) [19]. Apparently, a fortuitous cross-reactivity of the Tg TCR with the GPI, processed and presented by the I-Ag7 antigen-presenting cells, initiates the disease process. Although the cross-reactivity deleted thymic T-cells expressing the Tg TCR, some Tg TCR T-cells (likely expressing dual-TCR) emerged in the periphery and initiated the disease process [18].
The lifespan of K/BxN.foxp3sf mice was prolonged similar to that of other TCR Tg Sf mice. A faster and more aggressive arthritis developed. This change was attributed to the lack of Treg in the affected joints and an increased production of antibodies against the GPI [20] but the expansion of GPI-specific T-cells is also a likely contributing factor. Like other TCR Tg Sf mice, the great majority of CD4+ T-cells in the secondary lymphoid tissues are CD44+. In theory, these cells could be expanded both by GPI-specific activation through the Tg TCR or by endogenous TCR of the dual-TCR T-cells as what has been shown for OT-II.foxp3sf mice [17].
NOD.foxp3sf mice
The NOD.foxp3sf mice developed more severe inflammatory pathology in multiple organs as compared with B6.foxp3sf mice [21]. However, their survival rate appears to be similar to that of B6.foxp3sf mice. In addition, diabetic incidence was not reported, probably because the early death prevented such an analysis [21]. Indeed, the effect of total Treg-deficiency on many chronic autoimmune diseases may not be easily studied because of the dominant and lethal effect of foxp3sf mutation.
As described above, the early mortality of Sf mice can be significantly reduced if TCR Tg was bred into Sf mice. This strategy was used to study Treg effect on type-1 diabetes [22]. In this case, the NOD mice bearing the BDC2.5 TCR Tg specific to an islet Ag was used to generate BDC2.5/NOD.foxp3sf mice. Both MOI and mortality as compared with NOD.foxp3sf mice were significantly ameliorated. Interestingly, diabetes developed as early as 14 days after birth and 100% incidence occurred at 18 days of age, indicating that Ag specificity of T-cells is a critical factor for organ-specific inflammation. BDC2.5/NOD.Rag−/− mice also lacked Treg and developed diabetes earlier than BDC2.5/NOD mice [22]. Diabetes developed at 20 days after birth and 100% incidence occurred at 30 days of age. This “subtle” difference was attributed to the delay of insulitis in BDC2.5/NOD.Rag−/− mice. The T-cells in BDC2.5/NOD.foxp3sf mice but not BDC2.5/NOD.Rag−/− mice contain endogenous TCR. Based on the study of OT-II.foxp3sf mice, dual-TCR T-cells expanded as early as 6 days after birth in an OVA323–339-independent, foreign Ag-dependent manner [17]. Conceivably, such expansion turns naïve T-cells into memory/effector cells and collaterally facilitates the pathogenecity of the dual-TCR T-cells on pancreas. Education and expansion of T-cells that exclusively express BDC2.5 TCR may not be as fast because it occurs only in the pancreatic draining lymph nodes. It will be of interest to determine whether the dual-TCR T-cells are preferentially enriched in the islets of BDC2.5/NOD.foxp3sf mice.
Sf.aire−/− mice
The aire gene is expressed in thymic stromal cells. It is responsible for the synthesis of a family of peripheral tissue Ag whose presence is required for the deletion of the Ag-specific autoimmune T-cells during thymic selection [23]. Indeed, mice with mutant aire gene develop autoimmune diseases directed mostly against endocrine organs [23]. Although naturally occurring Treg develops in the thymus, this process does not seem to be affected by aire. In addition, the effect of Treg occurs in the periphery. Sf.aire−/− mice have a gravely shortened lifespan [21]. However, the affected target organs were not extended; particularly those endocrine organs remained free from inflammation. It could be argued that the inflammation in these organs did not have time to develop due to early death. Nevertheless, it is possible that the role of aire in central tolerance is not limited to Ag of endocrine organs, and as such aire defect may work in synergy with foxp3sf to fasten or augment MOI [21].
Sf.cd28−/− mice
CD28 on CD4+ T-cells interacts with B7 on antigen-presenting cells. This interaction provides the co-stimulation signal required for optimal T-cell activation of CD4+ T-cells. Breeding cd28−/− gene into Sf mice greatly extended the lifespan of the double mutant mice; 50% of which lived more than 200 days [24]. The spontaneous T-cell activation is greatly reduced in Sf.cd28−/− mice as reflected in the presence of a lower fraction of CD44+ T-cells and the inability of their T-cells upon activation to produce high levels of IFN-γ, IL-4 and IL-10 as compared with Sf mice. Paradoxically, IL-2 production upon T-cell activation by anti-CD3 and anti-CD28 was comparable among B6, Sf and Sf.cd28−/− mice. Serum IgE and IL-4, which were high in Sf mice, were reduced significantly in Sf.cd28−/− mice. Consistent with the lack of activation, Sf.cd28−/− mice did not show detectable inflammation in liver and lungs [24]. The effect on skin inflammation was not described in this study but one would expect it was also inhibited based on the strong inhibition of general T-cell activation in these mice.
Sf.Il2−/− mice
IL-2 knockout (Il2−/−) mice develop lymph node enlargement and inflammation in colon, liver and salivary glands [9,25]. Il2−/− mice are deficient in but not totally devoid of Treg [26]. The Treg-deficiency may explain the lympho-proliferation and MOI. However, the MOI in Il2−/− mice is remarkably different from Sf mice that develop severe inflammation in skin and lungs [7]. The phenotype of Il2−/− mice is dominant because Sf.Il2−/− mice also failed to develop skin and lung inflammation whereas their liver inflammation was not affected [25]. Thus, Sf.Il2−/− mice provided the first evidence that the MOI in Sf mice is controlled in an apparent “organ-specific” manner.
Interestingly, lymph nodes of Sf.Il2−/− mice are significantly larger than Sf mice and have a higher number of lymphocytes. Apparently, lacking IL-2 did not inhibit T-cell activation and proliferation in vivo. Other lympho-proliferative lymphokines such as IL-4, IL-7, and IL-15 were not obviously higher in Sf.Il2−/− mice as compared with Sf mice (Sharma and Ju, unpublished observation). Reduced FasL (CD178) expression has been implicated in the lymphadenopathy in Il2−/− mice but FasL expression was somewhat higher in Sf.Il2−/− mice than in Sf mice [25].
Inhibition of lymphocyte trafficking, either out of lymph nodes or into peripheral tissues, or both, may cause accumulation of lymphocytes in the lymph nodes. Our unpublished observation suggests that the CD4+ T-cells in Sf.Il2−/− mice fail to up-regulate trafficking/chemotactic/retention receptors necessary for entering into skin and lungs to induce inflammation [25]. Considering the large areas of skin and lungs and the large number of lymphocytes accumulated in these areas in Sf mice, this mechanism cannot be overlooked. This interpretation further implies that the infiltration of the inflammation-inducing T-cells into liver is not affected by the absence of IL-2. Importantly, lymphocyte trafficking to and retention in specific target organs involves many receptors and how IL-2 regulates altogether their expression and controls skin and lung inflammation in such a dominant manner is a challenging issue to address.
Sf.Igtαε−/−
Integrinαε(CD103)β7 is a cell surface receptor that binds to E-cadherin expressed mainly by epithelial cells. As a result, cells expressing CD103 are retained more in tissues like skin and lungs. In Sf mice, the frequency of CD4+CD103+ T-cells in the lymph nodes is significantly higher than their B6 counterpart [27]. An even higher frequency is observed in the skin and lungs. The high frequency of CD4+CD103+ T-cells is dramatically reduced in Sf.Il2−/− mice, demonstrating the requirement of IL-2 for CD103 expression on CD4+ T-cells. In vitro culture experiments demonstrated that optimal expression of CD103 also required TGF-β1 which is not limiting in Sf mice. IL-2 requirement for CD103 expression is specific for CD4+ T-cells including CD4+Foxp3+ Treg. Interestingly, the Treg deficiency in Il2−/− mice is observed mainly in the CD103- Treg [27], suggesting a subtle difference in the regulation of CD103 and Foxp3 expression in Il2−/− mice, perhaps the IL-15- or IL-7-induced Treg does not efficiently express CD103 [26]. CD103 expression on CD8+ T-cells and dendritic cells appears comparable between Sf and Sf.Il2−/− mice [27].
Lacking CD103 expression in Sf.Igtαε−/− mice delayed the skin and lung inflammation for a few weeks, indicating that the IL-2-controlled CD103 expression on CD4+ T-cells contributed to the “organ-specific” development of skin and lung inflammation. However, the lack of CD103 cannot fully explain the lifetime protection from skin and lung inflammation in Sf.Il2−/− mice because the MOI in Sf.Igtαε−/− mice eventually came back with a severity comparable to that observed in Sf mice. Thus, IL-2 must controls additional components of the skin and lung inflammatory process [27].
Sf.stat6−/− mice
IL-4, IL-5, IL-13, and IgE are important components for allergic skin and lung inflammation. The spontaneous development of skin and lung inflammation in Sf mice is associated with a high level of expression of serum IgE and Th2 lymphokines IL-4, IL-5, and IL-13, although Th1 lymphokines such as IL-2 and IFN-γ are also strongly expressed [28]. As STAT6 is the transcription factor critical to Th2 response, breeding stat6−/− gene into Sf mice of Balb/c background was conducted to determine how much the Th2 lymphokines and IgE contributed to the skin and lung inflammation. Serum IgE was greatly reduced and so was the ability of activated Sf.stat6−/− T-cells to produce IL-4, IL-5 and IL-13. Blood eosinophils reached to near normal level. With respect to the immunopathology of the MOI, the expression of mucus in Goblet cells as determined by Periodic Acid-Schiff-staining was inhibited, but severe inflammation in the lungs remained, suggesting a Th2-independent lung inflammation in these mice. Skin inflammation was not described, perhaps due to early death of these mice (T. A. Chatila, personal communication). Sf.stat6−/− mice lived 1 week more than Sf mice, suggesting Th2 regulated allergic inflammatory response in the lungs and skin was not necessary for mortality to occur [28].
Sf.faslpr/lpr mice
Fas (CD95)/FasL interaction has been considered as a major mechanism that regulates T-cell homeostasis but its role in organ damage is less appreciated. As compared with Sf mice, the number of lymphocyte in the lymph nodes of Sf.faslpr/lpr mice is only mildly increased by about 20% whereas 100% increase was observed for Sf.Il2−/− mice [25]. By contrast, the average lifespan of Sf.faslpr/lpr mice (12–14 weeks old) is comparable to Sf.Il2−/− mice and both lived significantly longer than Sf mice. In addition to the fully developed inflammation in the skin and lungs, the spontaneous inflammation in Sf.faslpr/lpr mice was extended to other organs not observed in Sf mice. Unlike Sf mice, Sf.faslpr/lpr and Sf.Il2−/− mice developed inflammation in colon and accessory reproductive organs (9, unpublished observation). These observations strongly suggest that Fas/FasL system has little to do with “organ-specific” inflammation and that FasL-dependent organ damage is an important factor for mortality induced by inflammation. FasL-induced organ damage could be due to its direct cytotoxicity or due to its ability to recruit inflammatory cells through Fas/FasL interaction [25,29].
Discussion/Summary
Participation of Th1 and Th2 responses in MOI
Most allergic inflammation conditions including IPEX are attributed to a preferential Th2 response and IgE over-expression. IL-2 is critically important in IL-4 induction and Th2 development under Th2 induction condition in vitro [30,31]. The Sf.Il2−/− mice represents the first animal model for which the role of IL-2 in Th2-mediated allergic inflammation can be explored. Sf mice displayed highly up-regulated Th1 and Th2 responses. Interestingly, expression of IL-4, IL-5, and IL-13 in CD4+ T-cells and serum IgE level were up-regulated both in Sf and Sf.Il2−/− mice as compared with B6 control (Sharma and Ju, unpublished observation), yet skin and lung inflammation in the latter group was strongly inhibited. This is interesting in light of the fact that the hyper-production of Th2 lymphokines (IL-4, IL-5, and IL-13) and IgE in Sf mice was reduced to normal levels in Sf.stat6−/− mice and yet, lung (and perhaps skin) inflammation persists in the latter group. This comparison suggests that the skin and lung inflammation in Sf mice also involves Th1 response and that a critical step shared by both Th1 and Th2 responses for skin and lung inflammation must have been inhibited in Sf.Il2−/− mice.
How IL-2 controls skin and lung inflammation in Sf mice?
To address this issue, genes differentially expressed between lymph node CD4+ T-cells of Sf and Sf.Il2−/− mice were determined (Sharma and Ju, unpublished observation). Genes encoding receptors for trafficking/chemotaxis/retention were highly differentially-expressed in Sf lymph node CD4+ T-cells as compared with Sf.Il2−/− samples. Among them, many skin-homing receptors such as Cysteinyl Leukotriene Receptor 1, Leukotriene β4 Receptor 1, CD103, CCR8, and others are the most differentially expressed (Fig. 1, Also see review in reference 32 for T-cell trafficking). These observations suggest that T-cell entering skin and lungs is a critical step preceding the T-cell activation in these organs and the subsequent inflammatory response. Consequently, even a strong expansion of potential inflammation-inducing T-cells in the lymph nodes cannot induce skin inflammation when the expression of these trafficking receptors is inhibited. The chemotactic factors for T-cell entrance to skin and lungs are likely produced by mast cells, basophils (for leukotrienes) and dermal microvessels, melanocytes, and Langerhans cells (for CCL1) [32,33]. IL-2, by regulating the receptors for these ligands, enables T-cell infiltration into skin and lungs to induce clinical symptoms (Fig. 1). How IL-2 controls the expression of these receptors is critical to the understanding how “organ-specific” control of MOI in Sf mice is developed.
Figure 1.
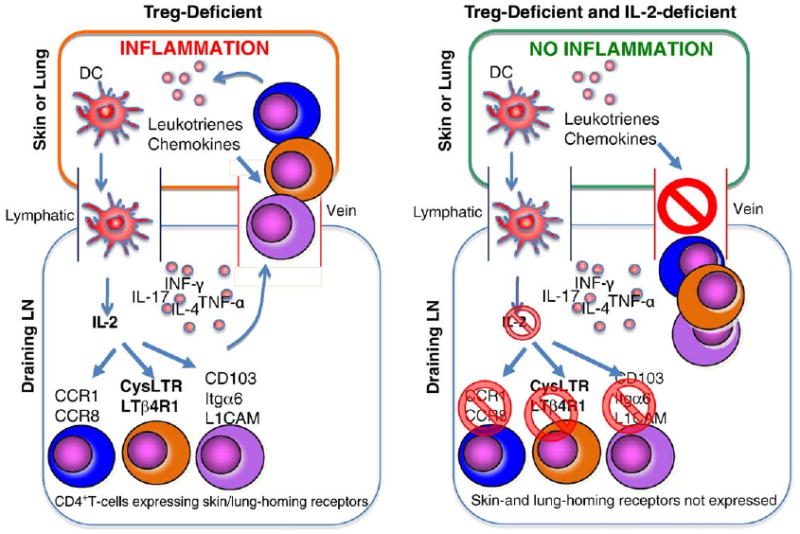
IL-2 controls trafficking receptor expression for skin and lung inflammation. In the Treg-deficient Sf mice (left panel), both Th1 and Th2 responses are strongly developed in the draining lymph nodes (LN). In the presence of IL-2, receptors for trafficking, chemotaxis, and retention for skin and lungs are induced on CD4+ T-cells, allowing them to enter target organs and induce inflammation. In the absence of IL-2 as in Sf.Il2−/− mice (right panel), these receptors are not expressed and despite having an up-regulated Tinf response, the CD4+ T-cells cannot enter the target organs to induce inflammation.
Another example that organ-specific control of inflammation exists in the Sf mice is supported by the rstudy of submandibular gland (SMG) [9]. Sf mice do not develop inflammation in SMG but contain CD4+ T-cells capable of inducing SMG inflammation in Rag1−/− recipients upon adoptive transfer. Interestingly, oral treatment of Sf mice with LPS or poly-I:C induces SMG inflammation [9]. The SMG in Sf mice is developmentally arrested at the stage equivalent to <2 weeks old normal B6 male mice [9]. This developmental arrest is in part caused by the lack of hormones such as testosterone and thyroxine. Even in 10–16 weeks old Sf.faslpr/lpr mice, the SMG remained undeveloped due to the severe inflammation damage of the accessory reproductive organs that produce testosterone. These observations suggest that the production of chemokines/chemotactic factors in the undeveloped SMG is limited, thus, preventing entrance of the inflammation-inducing T-cells and rendering the SMG resistant to the attack of inflammation-inducing T-cells (Fig. 2).
Figure 2.
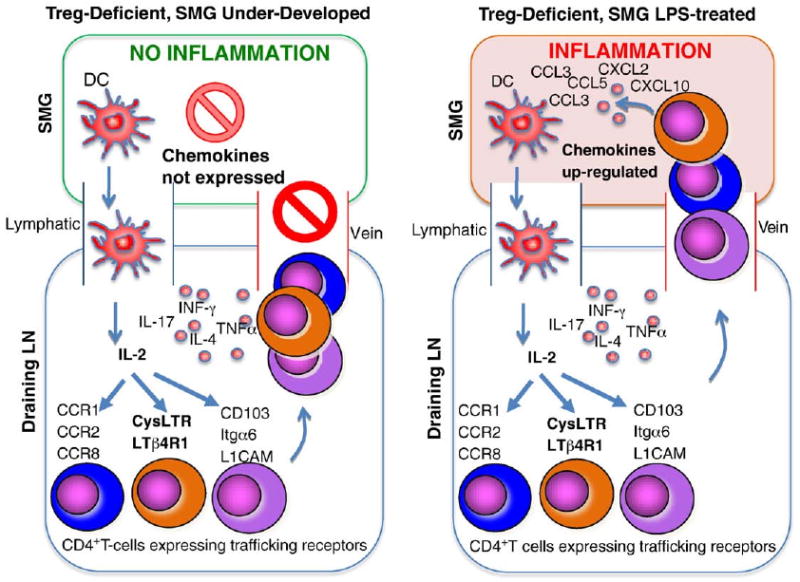
Chemokine expression controls SMG inflammation. The SMG in Sf mice is free of inflammation despite the fact that the Sf lymph nodes (LN) contain CD4+ T-cells capable of inducing SMG inflammation upon transferring into Rag1−/− recipients. The SMG of Sf is severely under-developed and may not produce sufficient chemokines to attract CD4+ T-cells to induce inflammation (left panel). Oral application of LPS or poly-I:C up-regulates chemokine expression in SMG, allowing entrance of the CD4+ T-cells to induce inflammation (right panel).
Implication
Sf defect can be cured by transfer of Treg in newborns and by partial bone marrow transplantation [34,35]. When performed before organ damage, bone marrow transplantation using non-myeloablative conditioning regimens can inhibit clinical symptoms in IPEX patients [36]. The genetic breeding experiments using Sf mice do not address how to cure Treg defect but rather aim at understanding how Tinf response and MOI are developed and controlled. These studies revealed that MOI of Sf mice is controlled by many critical components of the inflammation process. As summarized in Table 1, regulation occurs at many levels, from as early as those involved in T-cell development to those inflammation-inducing molecules involved in the latter stage of the inflammation process. Several studies such as those using BDC2.5 TCR Tg, stat6−/−, Il2−/−, and Igtαε genes, demonstrated that MOI in Sf mice can be controlled in an “organ-specific” manner, through Ag-specific TCR, cytokines, trafficking receptors, and integrin receptors. These checkpoints may be useful targets for the treatment of organ inflammation. Among them, the IL-2 regulated “organ-specific” inflammation process is highly significant considering the fact that numerous immune diseases occur in the skin and lungs. It seems that inhibiting Ag-specific production of IL-2 or blocking the major signaling component of IL-2 (i.e., STAT5) can be a useful approach to treat these diseases.
Acknowledgments
We thank Dr. M. Okusa and S. M. Fu for their critical comments and help for the preparation of the manuscript.
Abbreviations
- Ag
antigen
- foxp3sf
foxp3 gene mutation in Sf mice
- GPI
Glucose-6-phosphate Isomerase
- MOI
Multi-organ Inflammation
- OVA323–339
peptide #323 to 339 of ovalbumin
- Sf
Scurfy mice
- SMG
submandibular gland
- Tg
transgenic
- Tinf
inflammation-inducing T-cells
- Treg
regulatory T-cells
Footnotes
Supported by NIH grants AR-051203 and DE-017579.
References
- 1.Wing K, Fehervari Z, Sakaguchi S. Emerging possibilities in the development and function of regulatory T cells. Int Immunol. 2006;18:991–1000. doi: 10.1093/intimm/dxl044. [DOI] [PubMed] [Google Scholar]
- 2.Bluestone JA, Tang Q. How do CD4+CD25+ regulatory T cells control autoimmunity? Curr Opin Immunol. 2005;17:638–642. doi: 10.1016/j.coi.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Piccirillo CA, Shevach EM. Naturally-occurring CD4+CD25+ immunoregulatory T cells: central players in the arena of peripheral tolerance. Semin Immunol. 2004;16:81–88. doi: 10.1016/j.smim.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 5.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 6.Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. J Med Genet. 2002;39:537–545. doi: 10.1136/jmg.39.8.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma R, Jarjour WN, Zheng L, Gaskin F, Fu SM, Ju ST. Large functional repertoire of regulatory T-cell suppressible autoimmune T-cells in scurfy mice. J Autoimm. 2007;29:10–19. doi: 10.1016/j.jaut.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng L, Sharma R, Kung JT, Fu SM, Ju ST. Pervasive and stochastic changes in the TCR repertoire of regulatory T-cell-deficient mice. Int Immunol. 2008;20:517–523. doi: 10.1093/intimm/dxn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma R, Deshmukh U, Zheng L, Fu SM, Ju ST. X-linked Foxp3 (Sf) mutation dominantly inhibits submandibular gland development and inflammation by adaptive and innate mechanisms. J Immunol. 2009;183:3212–3218. doi: 10.4049/jimmunol.0804355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Gorman WE, Dooms H, Thorne SH, Kuswanto WF, Simonds EF, Krutzik PO, Nolan GP, Abbas AK. The initial phase of an immune response functions to activate regulatory T cells. J Immunol. 2009;183:332–339. doi: 10.4049/jimmunol.0900691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor JJ, Mohrs M, Pearce EJ. Regulatory T cell responses develop in parallel to Th responses and control the magnitude and phenotype of the Th effector population. J Immunol. 2006;176:5839–5847. doi: 10.4049/jimmunol.176.10.5839. [DOI] [PubMed] [Google Scholar]
- 12.Suffia IJ, Reckling SK, Piccirillo CA, Goldszmid RS, Belkaid Y. Infected site-restricted Foxp3+ natural regulatory T cells are specific for microbial antigens. J Exp Med. 2006;203:777–788. doi: 10.1084/jem.20052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blair PJ, Bultman SJ, Haas JC, Rouse BT, Wilkinson JE, Godfrey VL. CD4+CD8- T cells are the effector cells in disease pathogenesis in the Scurfy (sf) mouse. J Immunol. 1994;153:3764–3774. [PubMed] [Google Scholar]
- 14.Zahorsky-Reeves JL, Wilkinson JE. The murine mutation scurfy (sf) results in an antigen-dependent lymphoproliferative disease with altered T cell sensitivity. Eur J Immunol. 2001;31:196–204. doi: 10.1002/1521-4141(200101)31:1<196::AID-IMMU196>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 15.Heath WR, Carbone FR, Bertolino P, Kelly J, Cose S, Miller JFAP. Expression of two T cell receptor β chains on the surface of normal murine T cells. Eur J Immunol. 1995;25:1617–1623. doi: 10.1002/eji.1830250622. [DOI] [PubMed] [Google Scholar]
- 16.Padovan E, Giachino C, Cella M, Valitutti S, Acuto O, Lanzavecchia A. Normal T lymphocytes can express two different T cell receptor β chains: Implications for the mechanism of allelic exclusion. J Exp Med. 1995;181:1587–1591. doi: 10.1084/jem.181.4.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma R, Ju ACY, Kung JT, Fu SM, Ju ST. Rapid and selective expansion of nonclontypic T cells in regulatory T cell-deficient, foreign antigen-specific TCR-transgenic scurfy mice: antigen-dependent expansion and TCR analysis. J Immunol. 2008;181:6934–6941. doi: 10.4049/jimmunol.181.10.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kouskoff V, Korganow AS, duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286:1732–1735. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen LT, Jacobs J, Mathis D, Benoist C. Where Foxp3-dependent regulatory T cells impinge on the development of inflammatory arthritis. Arthritis Rheum. 2007;56:509–520. doi: 10.1002/art.22272. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Benoist C, Mathis D. How defects in central tolerance impinge on a deficiency in regulatory T cells. Proc Nat Acad Sci. 2005;102:14735–14740. doi: 10.1073/pnas.0507014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z, Herman AE, Matos M, Mathis D, Benoist C. Where CD4+CD25+ Treg cells impinge on autoimmune diabetes. J Exp Med. 2005;202:1387–1397. doi: 10.1084/jem.20051409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Villasenor J, Benoist C, Mathis D. AIRE and APECED: molecular insights into an autoimmune disease. Immunol Rev. 2005;204:156–164. doi: 10.1111/j.0105-2896.2005.00246.x. [DOI] [PubMed] [Google Scholar]
- 24.Nagendra S, Chandler PR, Seki Y, Baban B, Takezaki M, Kahler DJ, Munn DH, Larsen CP, Mellor AL, Iwashima M. Role of CD28 in fatal autoimmune disorder in scurfy mice. Blood. 2007;110:1199–1206. doi: 10.1182/blood-2006-10-054585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng L, Sharma R, Gaskin F, Fu SM, Ju ST. A novel role of IL-2 in organ-specific autoimmune inflammation beyond regulatory T cell check-point: both IL-2 knockout and Fas mutation prolong lifespan of scurfy mice but by different mechanisms. J Immunol. 2007;179:8035–8041. doi: 10.4049/jimmunol.179.12.8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kieng BV, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA. IL-2, -7 and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol. 2008;181:3285–3290. doi: 10.4049/jimmunol.181.5.3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma R, S-S J, Abaya Sung CE, Ju ACY, Fu SM, Ju ST. IL-2 regulates CD103 expression on CD4+ T cells in scurfy mice that display both CD103-dependent and independent inflammation. J Immunol. 2009;183:1065–1073. doi: 10.4049/jimmunol.0804354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin W, Truong N, Grossman WJ, Haribhai d, Williams CB, Wang J, Martin MG, Chatila TA. Allergic dysregulation and hyperimmunoglobulinemia E in Foxp3 mutant mice. J Allergy Clin Immunol. 2005;116:1106–1115. doi: 10.1016/j.jaci.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 29.Hohlbaum AM, Gregory MS, Ju ST, Marshak-Rothstein A. Fas ligand engagement of resident peritoneal macrophages in vivo induces apoptosis and the production of neutrophil chemotactic factors. J Immunol. 2001;167:6217–6214. doi: 10.4049/jimmunol.167.11.6217. [DOI] [PubMed] [Google Scholar]
- 30.Yamane H, Zhu J, Paul WE. Independent roles for IL-2 and GATA-3 in stimulating naïve CD4+ T cells to generate a Th2-inducing cytokine environment. J Exp Med. 2005;202:793–804. doi: 10.1084/jem.20051304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, Zhu J, Paul WE. Interleukin 2 plays a central role in Th2 differentiation. Proc Nat Acad Sci USA. 2004;101:3880–3885. doi: 10.1073/pnas.0400339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Medoff BD, Thomas SY, Luster AD. T cell trafficking in allergic asthma: the ins and outs. Annual Rev Immunol. 2008;26:205–232. doi: 10.1146/annurev.immunol.26.021607.090312. [DOI] [PubMed] [Google Scholar]
- 33.Schaerli P, Ebert L, Willimann K, Blaser A, Roos RS, Loetscher P, Moser B. A skin-selective homing mechanism for human immune surveillance T cells. J Exp Med. 2004;199:1265–1275. doi: 10.1084/jem.20032177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 35.Smyk-Pearson SK, Bakke AC, Held PK, Wildin RS. Rescue of the autoimmune scurfy mouse by partial bone marrow transplantation or by injection with T-enriched splenocytes. Clin Exp Immunol. 2003;133:193–199. doi: 10.1046/j.1365-2249.2003.02217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rao A, Kamani N, Filipovich A, Lee SM, Davies SM, Dalal J, Shenoy S. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood. 2007;109:383–385. doi: 10.1182/blood-2006-05-025072. [DOI] [PubMed] [Google Scholar]