

Abstract
Selective estrogen receptor (ER) downregulators (SERDs) reduce ERα protein levels as well as block ER activity, and therefore are promising therapeutic agents for the treatment of hormone refractory breast cancer. Starting with the triarylethylene acrylic acid SERD 4, we have investigated how alterations in both the ligand core structure and the appended acrylic acid substituent affect SERD activity. The new ligands were based on high affinity, symmetrical cyclofenil or bicyclo[3.3.1]nonane core systems, and in these, the position of the carboxyl group was extended from the ligand core, either retaining the vinylic linkage of the substituent or replacing it with an ether linkage. Although most structural variants showed binding affinities for ERα and ERβ higher than that of 4, only the compounds preserving the acrylic acid side chain retained SERD activity, although they could possess varying core structures. Hence, the acrylic acid moiety of the ligand is crucial for SERD-like blockade of ER activities.
Keywords: Selective Estrogen Receptor Downregulators (SERDs), Selective Estrogen Receptor Modulators (SERMs), Estrogen Receptor alpha, endocrine resistance
Introduction
A novel class of compounds capable of modulating the level and activity of the estrogen receptor (ER) are termed selective estrogen receptor downregulators (SERDs). These compounds are mechanistically distinct from ER ligands such as 4-hydroxytamoxifen, which act as either agonists or antagonists, depending upon the target tissue, and are termed selective estrogen receptor modulators (SERMs).1, 2 Although certain SERMs are clinically useful for menopausal hormone replacement and for the prevention and treatment of breast cancer,3 there is a need for new ER ligands that are capable of overcoming acquired endocrine resistance in breast cancer. Current SERDs, such as ICI 182,780 (ICI, Figure 1), have a more antagonistic profile than SERMs, and are capable of inhibiting the growth of tamoxifen-resistant breast cancer cells.4
Figure 1. The Selective Estrogen Receptor Downregulators (SERDs) and Modulators (SERMs), and Symmetrical Ligands.
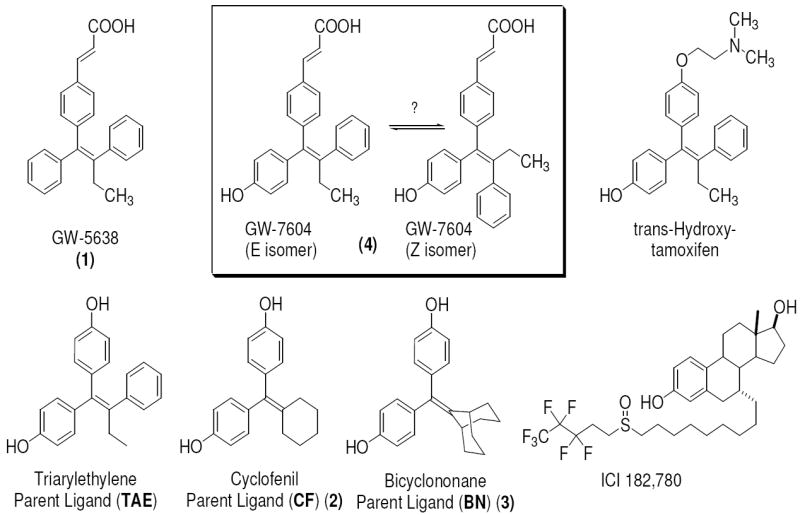
The SERD GW-5638 (1), and its hydroxylated metabolite, GW-7604 (4), can potentially undergo cis-trans isomerization (boxed structures, see text). The GW compounds as well as trans-hydroxytamoxifen, share a triaryethylene (TAE) structural core. The cyclic-core ligands, cyclofenil (CF, 2) and bicyclononane (BN, 3), are symmetrical and thus do not exist as cis-trans isomers. ICI is a known SERD.
SERDs are currently used in the treatment of metastatic breast cancer,4, 5 and their improved clinical activity is thought to derive from their ability to downregulate the ER protein as well as block ER action.6-9 ICI, however, has poor oral bioavailability,10 and a search for alternate drugs led to the identification of GW-5638 (Compound 1, Figure 1), a SERD that shares the triarylethylene ligand core scaffold of tamoxifen but has the basic side chain replaced by an acrylic acid unit. Compound 1 has an agonist profile in bone, but acts as an antagonist in the breast,11-13 and can inhibit the growth of tamoxifen-resistant breast tumors.9 GW-7604 (Compound 4, Figure 1) is a hydroxylated analog of compound 1 with higher ER affinity,11, 12 analogous to 4-hydroxytamoxifen, being a high affinity analog and metabolite of tamoxifen.14
It has been proposed that the SERD activities of both 4 and ICI result from their distortion of the conformation of the ER ligand binding domain (LBD) so that hydrophobic surfaces become exposed, thereby promoting accelerated degradation of the ER protein.15, 16 The X-ray crystallographic structure of an ERα-LBD complex with a structural analog of ICI, shows the ligand bound in an inverted mode relative to estradiol, with the long 7α-substituent projecting outward from the ligand binding pocket, displacing helix-12 of the LBD and occupying the coactivator binding groove.15 A recent structure of 4 with ERα shows that helix-12 is tilted from the normal SERM position through interaction of the carboxylic acid group of the ligand with the positive end of the helix-12 dipole.16 In both cases, fluorescent probes of exposed protein hydrophobic regions have revealed that these ER-SERD structures are more hydrophobic than are complexes with estrogen agonists (estradiol, DES) or SERMs (hydroxytamoxifen and raloxifene).16
Although the distortion of helix-12 in the ERα structure complexed with 4 suggests an important role for the carboxylic acid function of the acrylic acid side chain, it is not clear whether this function is positioned optimally or might be improved by presentation in an alternate manner, through other linkages, or at different distances from the ligand core. Similarly, it is not known whether the triarylethylene core structure is essential or might be replaced by alternate moieties with higher ER binding affinities. The hydroxy-triarylethylene (TAE) core of 4, in particular, might be subject to cis-trans isomerization in solution, because it shares this structural feature with hydroxytamoxifen and diethylstilbestrol, both of which do undergo this isomerization in a facile manner.17, 18
Therefore, to investigate to what extent the SERD activity of 4 is dependent on the hydroxytamoxifen-type core, the vinyl linkage, or the acrylic acid side chain, we prepared and evaluated the SERD activity of a new series of ER ligands having acidic side chains of varying length and linkages and built on different ligand core structures, cyclofenil (Compound 2) and bicyclononane (Compound 3), that are symmetrical and therefore do not exist as stereoisomers (Figures 1 and 2). From our initial series, we selected analogs having high binding affinity for ERα and ERβ, and we further characterized their ability to alter the subcellular localization of ERα and to downregulate its protein levels and inhibit gene stimulation by estrogen (SERD activity). The two new compounds having the most favorable SERD-like activity retained the acrylic acid side chain, although they had different core structures.
Figure 2. Series of SERD Compound 4 Analogs, Based on Cyclofenil (CF) and Bicyclononane (BN) and Other Analogs.
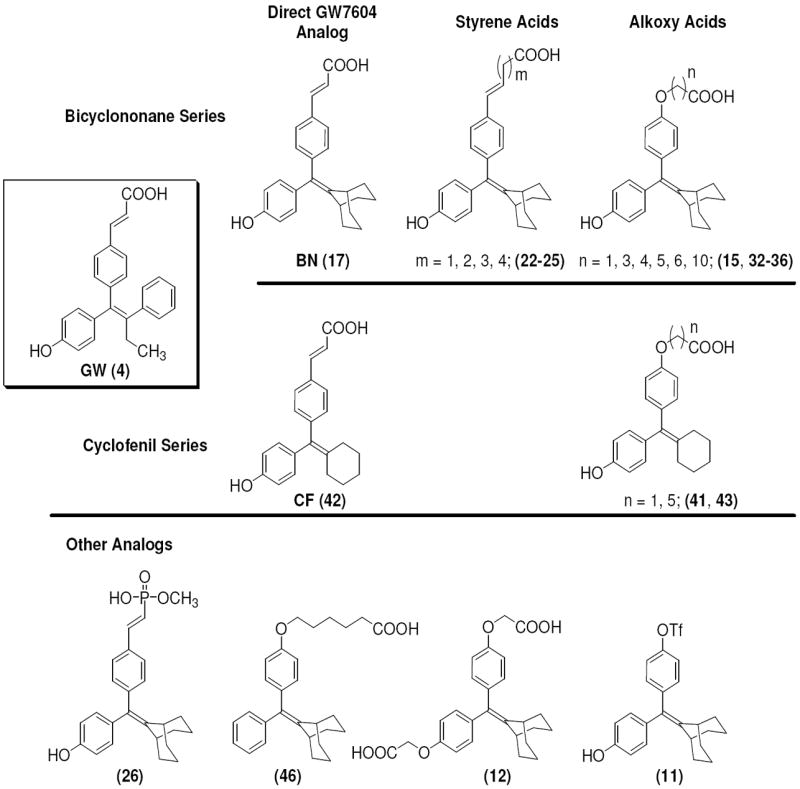
This study expands our current understanding of the optimal pharmacophore requirements for SERD activity and their capabilities in regulating the activity of the estrogen receptor. It also highlighted the importance of the acrylic acid side chain of 4 for engendering SERD-like activity, but revealed that alternate ligand core structures can be used in place of the tamoxifen-like triarylethylene core used in the GW series.
Results
Chemical Synthesis
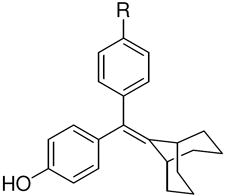
The structures of the SERD 4 analog series that we prepared are summarized in Figure 2, and those studied in greater detail are shown in Table 1 and Table 2 (see compound numbers with underline). Some analogs (42, 43, 44) are based on a well known, high affinity non-steroidal estrogen, cyclofenil (2, Figure 1), but the majority (11, 12, 15, 17, 22-26, 29, 32-36, 46) are based on bicyclononane (3, Figure 1), a related non-steroidal estrogen that has exceptionally high binding affinity for both ERα and ERβ (Table 1a).19 Notably, neither of these symmetrical ligand cores have cis-trans isomers, as does the TAE-based compound (compound 4, Figure 1). In both the cyclofenil and bicyclononane series (designated CF and BN for simplicity), we prepared the direct analog of 4 containing the acrylic acid substituent (17, 42), as well as a series with carboxy-terminated alkoxy substituents (Figure 2, “Alkoxy Acids”). Because members of the bicyclononane series proved to have higher affinity, we prepared additional homologs of the acrylate-substituted system, having longer chain acids linked through a styryl function (Figure 2, “Styrene Acids”), as well as some individual variants, bis-carboxymethyl and a deoxy analog or members with vinylphosphonate or trifluoromethanesulfonyloxy groups (Figure 2, bottom).
Table 1. Estrogen Receptor Binding Assay.
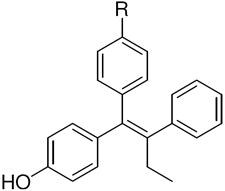
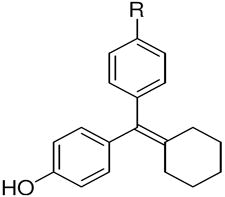
The ERα and ERβ binding affinities of SERDs with the triarylethylene (TAE), cyclofenil (CF) and bicyclononane (BN) core, and with the acrylic acid substituent are presented in the top part of (Table 1a). Some individual variants with the bicyclononane core are presented in the lower part of the (Table 1b).
| a. Structure and Binding Affinity of Basic TAE, CF, and BN Systems | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Triarylethylene (TAE) |
Cyclofenil (CN) |
Bicyclononane (BN) |
||||||||||
 |
 |
 |
||||||||||
| R | lignad | RBAa ERα |
RBAa ERβ |
β/αb | lignad | RBAa ERα |
RBAa ERβ |
β/αb | lignad | RBAa ERα |
RBAa ERβ |
β/αb |
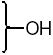 |
TAE | 377 ± 106 | 238 ± 48 | 0.6 | CF, 2 | 68 ± 18 | 334 ± 54 | 4.9 | BN, 3 | 513 ± 17 | 301 ± 54 | 0.6 |
 |
4c,d | 20.3 ± 6 | 28.3 ± 7 | 1.4 | 42c | 2.1 ± 5 | 11.3 ± 3 | 5.4 | 17c | 17.7 ± 4 | 25.9 ± 5 | 1.5 |
| 4’c | 3.6 ± 1 | 4.2 ± 0.6 | 1.2 | -- | -- | -- | -- | -- | -- | -- | -- | |
 |
-- | -- | -- | -- | 44c | 0.63 ± 0.1 | 1.9 ± 0.6 | 3.0 | 15c | 3.6 ± 1 | 3.1 ± 0.3 | 0.9 |
 |
-- | -- | -- | -- | 43c | 8.2 ± 0.5 | 28.8 ± 3 | 3.5 | 34c | 52.0 ± 6 | 89.1 ± 9 | 1.7 |
| b. The ERα and ERβ binding affinity of other ligands with a bicyclononane core | ||||||||||||
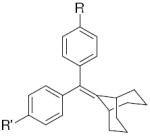 | ||||||||||||
| R’ | R | ligand | RBAa ERα | RBAa ERβ | β/αb | |||||||
| -H | -OH | 46 | 0.14 ± 0.02 | 0.30 ± 0.06 | 2.1 | |||||||
| -O-CH2-CO2H | -O-CH2-CO2H | 12 | 0.26 ± 0.05 | 0.40 ± 0.05 | 1.5 | |||||||
| -OH | -O-(CH2)5-CO2Et | 29 | 15.2 | 30.8 | 2.0 | |||||||
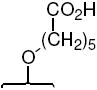
| -OH | 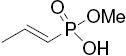 |
26c | 7.5 ± 1 | 7.7 ± 0.7 | 1.0 | |||||||
| -OH | -OSO2CF3 | 11 | 4.2 ± 1 | 8.9 ± 0.7 | 2.1 | |||||||
Relative binding affinity (RBA) values are determined by competitive radiometric binding assays and are expressed as IC50[estradiol] / IC50[compound] × 100 (RBA, estradiol = 100). In these assays the Kd for estradiol is 0.2 nM for ERα and 0.5 nM for ERβ.
For each value, the β/α ratio is calculated such that the ratio is >1 for compounds having higher affinity on ERβ than on ERα.
Compounds selected for further studies are indicated by a bold underline.
This compound, GW-7604 (4), exists as a mixture of E and Z isomers. See Figure 1.
Table 2. Structure and Binding Affinity of the Extended Series of Styryl and Alkoxy Bicyclononanes.
The ERα and ERβ binding affinity of both the styryl and alkoxy bicyclononane series was determined and expressed as outlined in the legend of Table 1.
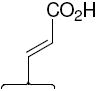
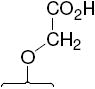
| N [number of atoms: phenyl-carboxyl] | 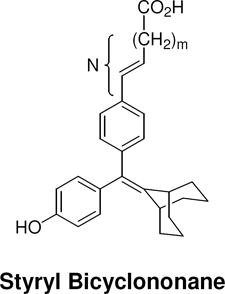 |
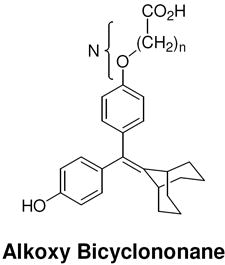 |
||||||
|---|---|---|---|---|---|---|---|---|
| N | lignad | RBAa ERα |
RBAa ERβ |
β/αb | lignad | RBAa ERα |
RBAa ERβ |
β/αb |
| 2 |
17c (m = 0) |
17.7 ± 4 | 25.9 ± 5 | 1.5 |
15c (n = 1) |
3.6 ± 1 | 3.1 ± 0.3 | 0.86 |
| 4 |
22 (m = 2) |
9.0 ± 2 | 23.1 ± 6 | 2.6 |
32 (n = 3) |
5.6 ± 1 | 10.5 ± 1 | 1.9 |
| 5 |
23 (m = 3) |
13.1 ± 2 | 24.6 ± 7 | 1.9 |
33c (n = 4) |
18.3 ± 3 | 38.3 ± 11 | 2.1 |
| 6 |
24c (m = 4) |
38.5 ± 0.2 | 62.8 ± 14 | 1.6 |
34c (n = 5) |
52.0 ± 6 | 89.1 ± 9 | 1.7 |
| 7 |
25c (m = 5) |
87.4 ± 10 | 81.1 ± 14 | 0.93 |
35 (n = 6) |
70.4 ± 20 | 139 ± 0 | 2.0 |
| 11 | -- | -- | -- |
36c (n = 10) |
95.6 ± 10 | 63.9 ± 17 | 0.67 | |
Relative binding affinity (RBA) values are determined by competitive radiometric binding assays and are expressed as IC50[estradiol] / IC50[compound] × 100 (RBA, estradiol = 100). In these assays the Kd for estradiol is 0.2 nM for ERα and 0.5 nM for ERβ.
For each value, the β/α ratio is calculated such that the ratio is >1 for compounds having higher affinity on ERβ than on ERα.
Compounds selected for further studies are indicated by a bold underline.
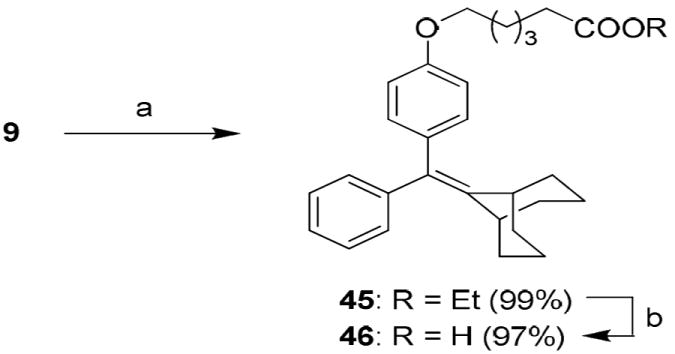
The synthetic routes used to prepare these compounds are shown in Schemes 1-3. McMurry coupling was used to prepare the parent cyclofenil and bicyclononane systems (Compounds 7-9) from their appropriate benzophenone precursors (Compounds 5 and 6, Scheme 1). Members of the alkoxy ether series were prepared from the parent cyclofenil and bicyclononane ligands by Williamson ether synthesis using various ω-bromo esters, followed by ester hydrolysis (Compounds 32-36, Scheme 2 and Compound 43, Scheme 3). Good yields of mono substitution on the bisphenol parent ligands could be obtained in all cases, except with ethyl bromoacetate. In this case, the bisphenols were first protected as the mono-TBDMS ethers (Compounds 10, 37); mono alkylation followed by saponification/deprotection gave the desired mono alkoxymethyl ethers (Compounds 15, 44). The mono-TBDMS ethers were also used to prepare the monotrifluormethane sulfonates (Compounds 11, 38), which where were used in Suzuki coupling to prepare members of the styryl series (Compounds 17, 22-25, Scheme 2 and Compound 42, Scheme 3). A complete description of the syntheses with experimental details and spectroscopic characterization is given in the Supporting Information section.
Scheme 1. Synthesis of Cyclofenil and Bicyclononane Analogs.
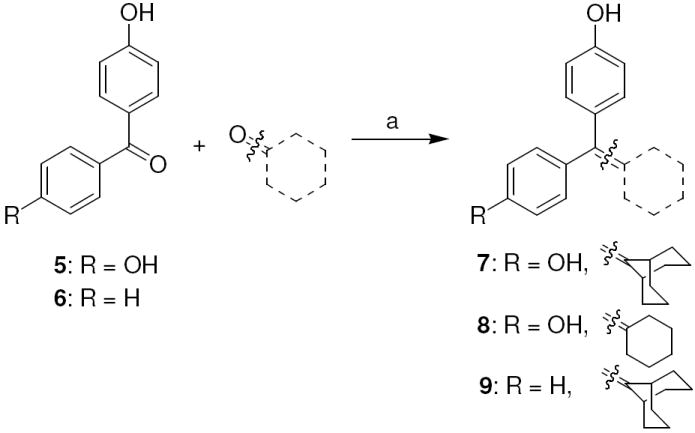
Reagents: (a) TiCl4, Zn, THF, reflux, 10 h.
Scheme 3.
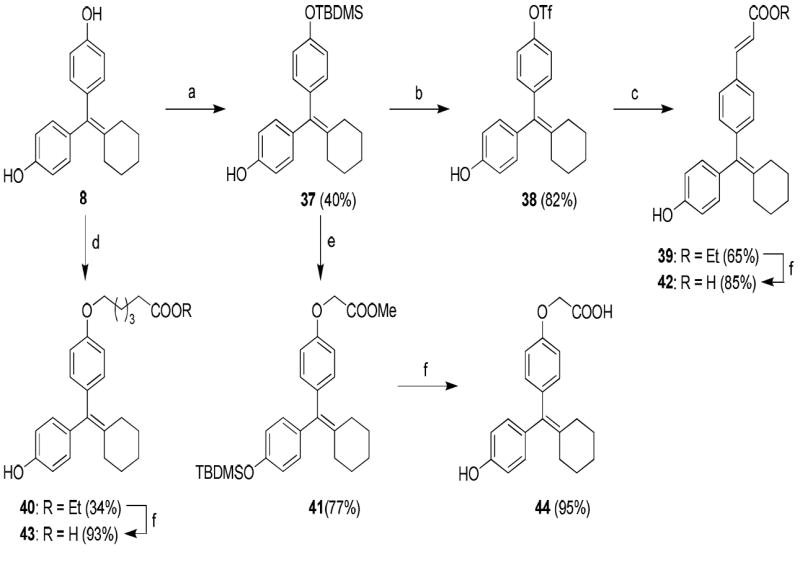
a. Synthesis of Acid Substituted Cyclofenil Analogs. Reagents: (a) TBDMSCl, imidazole, room temp, THF, 24 h; (b) i) Tf2O, triethylamine, 0 °C to room temp, 8 h, ii) TBAF, rt, 15 min; (c) ethyl acrylate, Pd(PPh3)2Cl2, triethylamine, DMF, 120 °C, 24 h; (d) ethyl 6-bromohexanoate, Cs2CO3, CH3CN, room temp, 24 h; (e) BrCH2COOCH3, Cs2CO3, CH3CN, room temp, 24 h; (f) 2 N KOH, MeOH, room temp, 24 h.
b. Synthesis of the Deoxy Analog (46) of Bicyclononane Compound 34 Reagents: (a) ethyl 6-bromohexanoate, Cs2CO3, CH3CN, room temp, 24 h; (b) 2 N KOH, MeOH, room temp, 24 h.
Scheme 2. Synthesis of Acid Substituted Cyclononane Analogs.
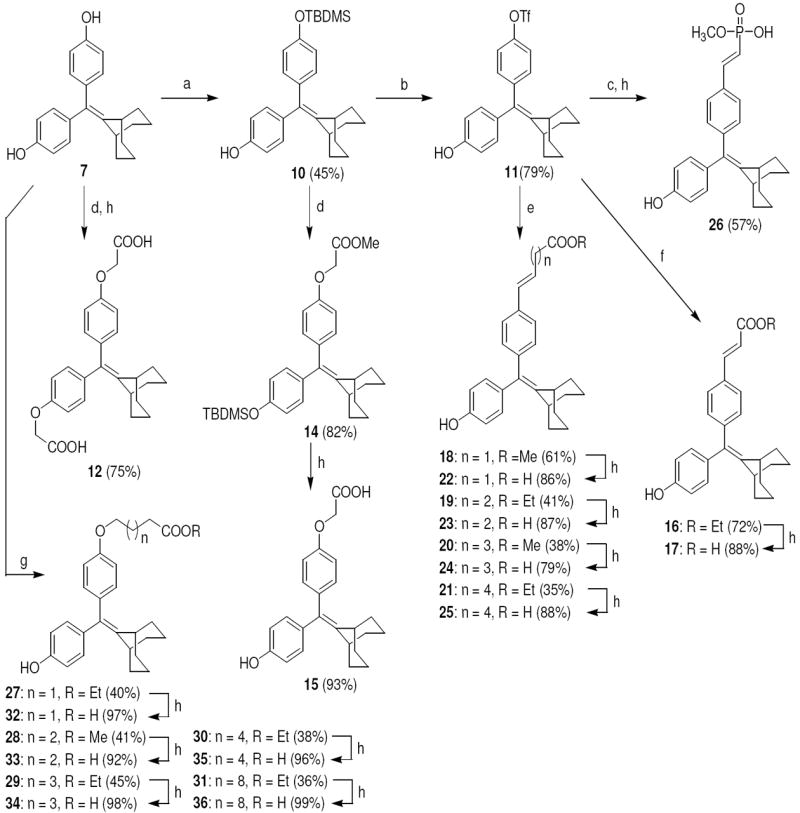
Reagents: (a) TBDMSCl, imidazole, room temp, THF, 24 h; (b) i) Tf2O, triethylamine, 0 °C to room temp, 8 h, ii) TBAF, rt, 15 min; (c) dimethyl vinylphosphonate, Pd(PPh3)2Cl2, triethylamine, DMF, 120 °C, 24 h; (d) BrCH2COOCH3, Cs2CO3, CH3CN, room temp, 24 h; (e) CH2=CH(CH2)nCOOR, Pd(PPh3)2Cl2, triethylamine, DMF, 120 °C, 24 h; (f) ethyl acrylate, Pd(PPh3)2Cl2, triethylamine, DMF, 120 °C, 24 h; (g) BrCH2(CH2)nCH2COOR, Cs2CO3 or K2CO3, CH3CN, room temp, 24 h; (h) 2 N KOH, MeOH, room temp, 24 h.
Biological Results
Estrogen Receptor Binding Assays and Structure-Affinity Relationships
The compounds were assayed for their binding affinity for human ERα and ERβ in a radiometric competitive binding assay using [3H]estradiol as tracer and estradiol as the reference standard.20, 21 Binding affinities are expressed as relative binding affinity (RBA) values, with estradiol set at 100. Results are given in Table 1 (comparison of triarylethylene (TAE), cyclofenil (CF) and bicyclononane (BN) series) and Table 2 (extended BN styryl and alkoxy series).
The binding affinities of the parent phenols in the triarylethylene (TAE), cyclofenil (CF) and bicyclononane (BN) systems are presented in Table 1a. All three parent compounds have very high binding affinity for ERα and ERβ, but the highest affinity is shown by the parent bicyclononane (3). As with hydroxytamoxifen,14 compound 4 exists as a cis-trans isomer mixture. After careful recrystallization, we were able to obtain only the Z isomer in pure form (4’). The lower affinity of the pure Z isomer (Table 1a) suggests that the E isomer is predominant in binding to the ERs. This is consistent with the X-ray crystal structures of this compound,16 as well as binding affinity measurements and X-ray structures for hydroxytamoxifen and diethylstilbestrol, where the isomer with the corresponding trans-4-hydroxystilbene isomer is the active one.14, 17, 22
In the acrylic acid series (Table 1), the affinity of the cyclofenil analog (42) is considerably lower than that of the GW compound (4); the bicyclononane analog (17), however, binds with comparable affinity. The binding affinities of two alkoxy analogs prepared in both the cyclofenil and bicyclononane series (compounds 44 and 43, and compounds 15 and 34, respectively) provided additional evidence for the higher affinity of members of the bicyclononane series; therefore, further synthetic efforts focused on this series. Also of note are the binding affinities of certain other bicyclononane analogs (Tables 1a and 1b, Figure 2, bottom) designed to test other side chain functional groups. Compounds in which the OH is missing (46) or substituted with a second alkoxy group (12) have poor binding affinities. The ester (29) has lower affinity than the corresponding acid 34; the methyl phosphonate 26 binds less well than the corresponding carboxylate 17, and the trifluoromethane sulfonate (11) binds only moderately well.
Binding affinities of the extended bicyclononane series of styryl and alkoxy analogs are given in Table 2. In both series and for both receptors, binding affinities (with but one exception) increase with increasing chain length, reaching affinities much greater than that of 4 and even rivaling that of estradiol. This is shown graphically in Figure 3. None of the compounds in either Table 1 or 2 showed significant selectivity for either ERα or ERβ, although those in the cyclofenil series (Table 1a) tended to be more ERβ selective, with the cyclofenil acrylic acid (42) being ca 5-fold in favor of ERβ.
Figure 3. Dependence of Ligand Relative Binding Affinity on Acid Chain Length.
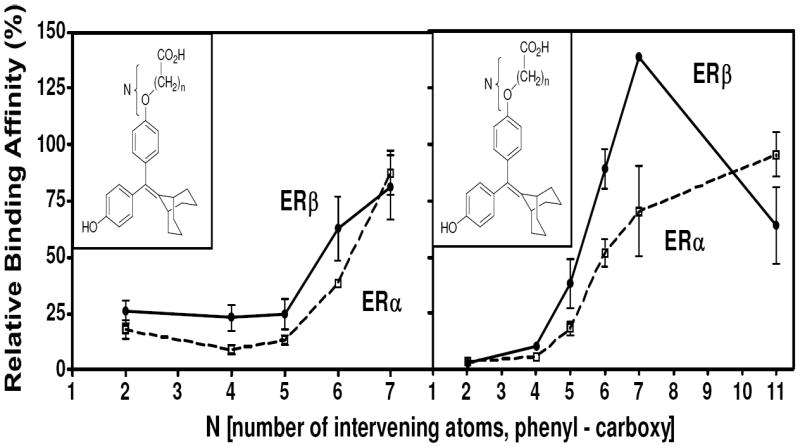
N represents the number of intervening atoms between the phenyl and the carboxyl moieties, irrespective of whether they are carbon or oxygen.
Western Immunoblotting
We selected ten compounds with good binding affinity for ER for further analysis of their SERD character (see underlined compound numbers in Tables 1 and 2). These compounds were the initial SERD compound (4), the direct analogs of 4 in both the cyclofenil (42) and bicyclononane (17) series, the high affinity analogs in the bicyclononane styryl (24 and 25) and alkoxy (15, 33, 34, and 36) series, and the BN vinyl methyl phosphonate (26). The well established SERD ICI was also included for comparison.
MCF-7 breast cancer cells were treated with these compounds, and the ERα protein was extracted and analyzed by SDS-PAGE and Western immunoblotting. Because SERDs are known not only to lower ER levels in cells but also to affect the subcellular distribution of ER,6-8 we monitored ER levels in whole cell extracts (WC), and in cytoplasmic (C), nuclear (N), matrix (M), and insoluble (I) fractions after treatment with these various compounds. The procedures used to obtain these extracts are given in the Methods, and representative Western immunoblots are shown in Figure 4.
Figure 4. Subcellular Distribution and Downregulation of ERα Protein by ICI and compounds 36, 25, 4, 42, 17 (Panel A) and 33, 34, 24, 15, and 26 (Panel B).
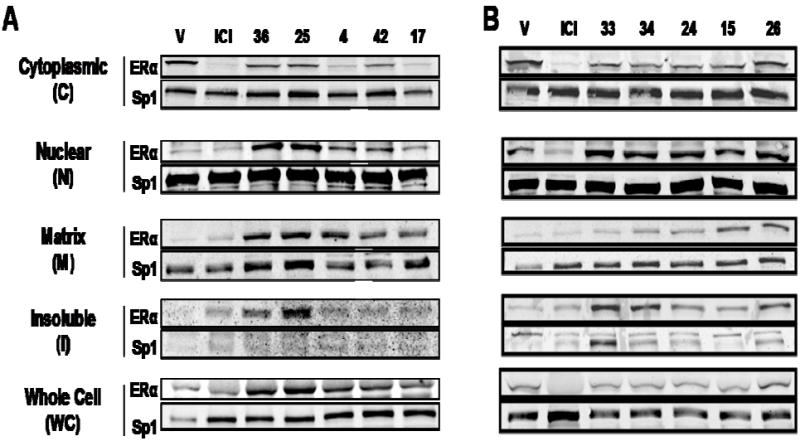
MCF-7 cells were treated with 1 μM compounds for 16h, and the level of cellular ERα in the indicated subcellular fraction and in whole cell extracts was measured by Western immunoblotting. Sp1 was monitored as an internal reference for gel loading.
The well characterized SERD ICI markedly reduced ERα protein levels in the cytoplasmic fraction and did not raise ERα levels in the nuclear fraction; the slight elevation of ERα in the matrix and insoluble fractions is consistent with reports that this compound does redistribute ERα into the less soluble cell fractions (Figure 4A).6-8 The known SERD 4 also reduces cytoplasmic ERα protein, but ERα levels in the nuclear, matrix and insoluble fractions are elevated, consistent with what others have shown.23 The bicyclononane analog (17) redistributed and downregulated of ER in a manner very similar to that of the SERD 4, as did the cyclofenil analog (compound 42), though the latter to a somewhat lesser degree (Figure 4A).
Surprisingly, although they have higher ERα binding affinities than the three compounds discussed above, none of the bicyclononane acrylate homologs (styryl compounds 24 and 25) nor the carboxyalkoxy analogs (compounds 15, 33, 34, and 36) that we tested demonstrated substantial SERD-like activity (Figure 4A and B). While cytoplasmic ERα levels were somewhat lower, nuclear ER, and in some cases also matrix and insoluble fractions, showed substantially increased ERα levels. These compounds all lack the acrylic acid side chain moiety which is present in compounds 4, 17, and 42.24
Western immunoblot analysis of MCF-7 cells treated with ICI and compounds 4, 42, and 17, alone or with E2, is shown in Figure 5. In the presence of E2, all of the SERDs tested were capable of lowering ERα protein levels. This downregulation was seen particularly in the whole cell lysate, the cytoplasmic and the nuclear fractions. The cyclofenil acrylic acid (42) showed lower activity (i.e., less ER protein reduction), especially in the cytoplasmic fraction. The sequestration of ERα protein in the insoluble fraction increased upon concomitant E2 treatment. This may represent ligand-dependent turnover of the receptor,25 as it is well known that E2 enhances ER turnover.26
Figure 5. Western Immunoblot Analysis of of ERα Protein Level and Subcellular Distribution with Compound and Estradiol Treatment.
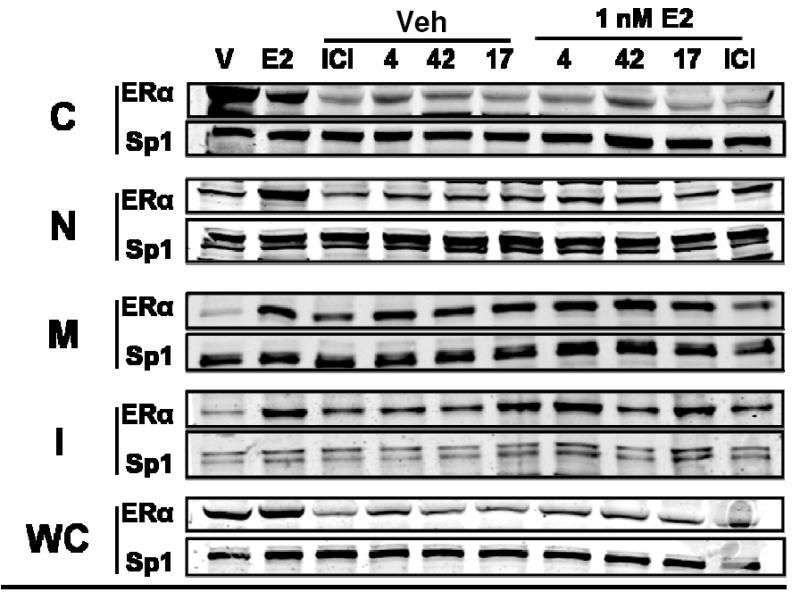
MCF-7 cells were treated with 1 μM compound for 16h and then ±1 nM estradiol for 4h prior to cell fractionation and monitoring of ERα by Western immunoblots; leftmost two lanes show vehicle or E2 treatment for 20 h. Sp1 was monitored as an internal reference for gel loading.
Analysis of ERα Mediated Transcription Regulation
Transcriptional analysis of established ERα target genes was carried out on MCF-7 cells treated with the best three compounds in the absence or presence of E2. The RNA levels of three E2-upregulated genes (pS2, progesterone receptor, carbonic anhydrase 12) were in most cases reduced by the SERDs 4, 17, and 42 (Figure 6A). Compound 4 and 17 significantly blocked the E2-mediated stimulation of the pS2 and PR genes, but compound 42 was ineffective, consistent with its less effective downregulation of the ER protein. However, 42 did block the E2-mediated activation of the CA12 gene even more so than did compound 17. Compound 4 was the most effective in blocking estrogen action on all three of these E2-stimulated genes.
Figure 6. Impact of Different Compounds on ERα Mediated Transcriptional Activity.
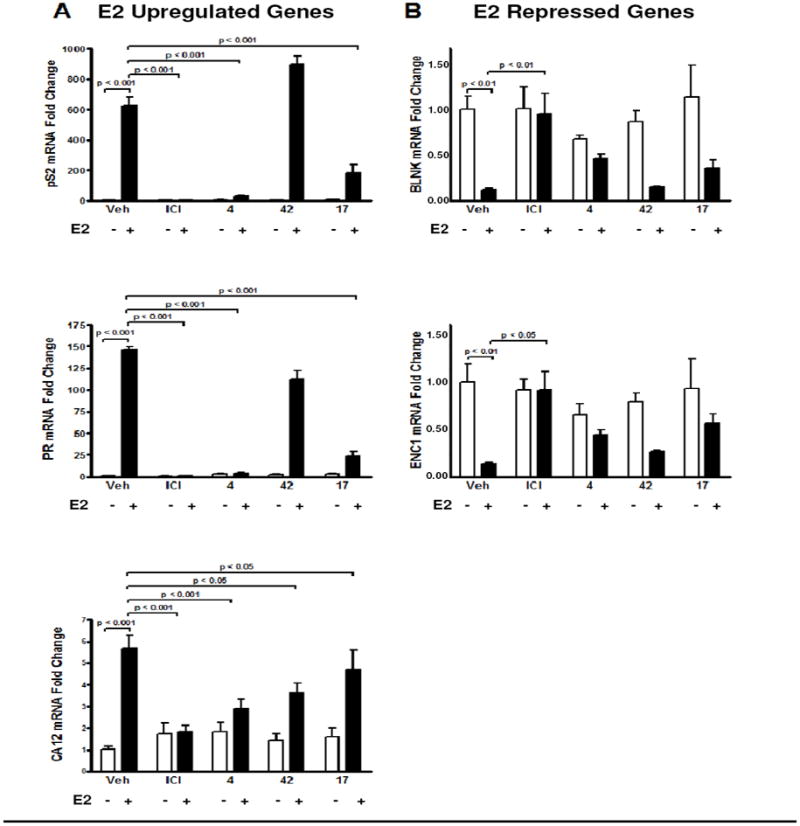
MCF-7 cells were treated with 1 μM compounds for 16h and then with vehicle (0.1% ethanol) or 1 nM estradiol for 4h. Real-time PCR analysis was performed for E2-stimulated ERα-target genes (Panel A) or E2-repressed ERα-target genes (Panel B), as a measure of compound ability to antagonize estradiol action.
The three SERDs were also effective in reversing estrogen action on two genes that are repressed by E2, namely, BLNK and ENC1 (Figure 6B).27 Compounds 4 and 17 did, in fact, reduce the E2-mediated repression of BLNK and ENC1 gene expression (Figure 6B), increasing RNA levels for these genes relative to the E2-repressed state, but the repression was not fully reversed to control, basal level of expression.
Discussion
In this study, we have examined a range of compounds structurally related to the selective estrogen receptor downregulator (SERD), compound 4. In all of these analogs, the isomerization-prone hydroxyl-triarylethylene ligand core of 4 was replaced by two symmetrical cyclic systems, cyclofenil and bicyclononane, that are known to engender high ligand binding affinity for both estrogen receptors, ERα and ERβ. In addition, homologs were prepared in which the carboxylic acid group on the acrylic acid side chain was extended (keeping the styrene linkage) or analogs in which it was replaced by an ether-linked carboxyalkyl substituent. While in both analog series, extension of the acid function away from the ligand core increased ER binding affinity substantially, it did not result in compounds having SERD activity. In fact, the only compounds capable of reducing levels of ERα protein, bicyclononane 17 and cyclofenil 42, were ones that retained the acrylic acid group present in 4. Of the two new SERDs, 17 and 42, the higher affinity bicyclononane compound (17) proved to be the better, and in most respects it closely replicated the activity of 4. The marked dampening of ER action shown by compounds 4 and 17 indicates an obliteration of transactivation from both AF-1 and AF-2, a known effect of pure antagonist/SERD binding to the ER.28-30
Structural studies have suggested that the acrylic acid side chain of 4 down regulates ERα protein levels by interactions with specific amino acids, namely D351 in the ligand binding domain of the receptor.31 This charge-charge repulsion blocks the proper positioning of the critical helix 12,31, 32 preventing it from adopting either the folded-back orientation found in agonist structures or the extended conformation found in SERM structures, where helix 12 occludes the hydrophobic groove of the AF-2 function, blocking interaction with coregulators.16 With helix 12 thus mal-disposed, hydrophobic patches are exposed, and the receptor is thought to become a target for ubiquitination and processing by the ubiquitin-proteasome pathway.33 A related mechanism is proposed for the activity of ICI-type SERDs,16 although in this case, helix-12 appears to be completely disordered,34 with even greater exposure of hydrophobic regions for targeting ubiquitination.16
Remarkably, the SERD activity of the compounds in this series appears to be uniquely associated with the acrylic acid unit that is common to compound 4, cyclofenil 42, and bicyclononane 17. Even replacing it with a carboxymethyl ether, which enables the carboxyl group to access the same space as it occupies in the acrylic acid, did not suffice to engender SERD activity. This suggests that the rigidity of the acrylate unit, in fact, might be essential for maintaining the critical charge-charge repulsion with D351. Thus, the higher binding affinity found when the carboxylic acid group is spaced further from the aromatic ring might indicate relief of this ligand carboxylate-ERα D351 coulombic repulsion, and it suggests that the critical interaction needed to distort the position of helix 12 and induce ER degradation—the very essence of SERD activity—comes with a cost in binding energy. Relevant to this point, others have found that maintaining the double bond but replacing the carboxylic acid group with a carboxamide or methyl ketone, which would maintain rigidity but remove the charge, also results in loss of SERD activity.24
While activation of the ubiquitin-proteasome pathway is a plausible explanation for the downregulation of ERα protein, Wijayaratne and McDonnell did not find a large increase in ERα ubiquitination upon treatment with compound 4.35 There are, however, other reports implicating the proteasome-ubiquitin pathway in ER turnover. Duong et al. described a ternary complex between ERα, p53, and the oncogenic ubiquitin-ligase Mdm2 in MCF-7 cells.36 Also, receptor downregulation is blocked by proteasome-inhibitor MG132 treatment.37-39 It has been suggested that the 26S proteasome is necessary for the cycling of ERα protein needed to maintain transcription of target genes.25 These studies all suggest the involvement of the ubiquitin-proteasome pathway in the downregulation of ERα protein.
As others have seen, SERDs such as ICI not only reduce ERα protein levels overall, but also redistribute it from the most soluble cellular compartments, the cytoplasm and nucleus, to less soluble compartments, the matrix and insoluble fractions. Presumably, at these locations it cannot function in its role as a ligand-modulated transcription factor. In fact, the movement of ER into these compartments might be a consequence of its ubiquitination, as others have noted.40-42 Our observations of such downregulations and alterations in intracellular distribution of ER after treatment with these new SERDs are consistent with their prevention of estrogen-regulated gene expression and highlight the importance of the acrylic acid side chain in enabling SERD activity.
Conclusion
Compounds capable of suppressing estrogen action by reducing estrogen receptor levels appear to have particular promise for novel endocrine therapies in breast cancer. It is hoped that they might not succumb to the development of acquired resistance that typically follows treatment with SERMs, and this appears to be the case in model systems.43 Our study has demonstrated that other ligand core structures, in addition to the triarylethylene ligand core in traditional SERMs like tamoxifen and in the original GW SERD (4), can be used to produce SERD compounds (namely, the cyclofenil compound 42 and the bicyclononane compound 17) that are still very effective at moderating the activity of ER. Our biological analyses of these novel SERDs underscore the importance of the acrylic side chain in effecting the downregulation of ERα and the suppression of ER-regulated gene expression.
Experimental
Synthesis Materials and Methods
See Supporting Information for details on synthesis, spectroscopic characterizations, and purity determination by normal and reversed phase HPLC analysis. All compounds assayed were >95% pure in both HPLC systems.
Biological Materials and Methods
Compounds
E2 and ICI 182,780 (ICI) were purchased from Sigma Chemical Co. (St. Louis, MO).
Cell Culture
The human breast cancer MCF-7 cell line was maintained in culture as previously described.44 For Western analysis, cells were steroid depleted in phenol red-free media supplemented with 5% charcoal-dextran-treated calf serum for five days prior to the indicated ligand treatment. Media was changed on day 2 of culture, and cells were then treated with ligands. For real-time PCR gene expression analysis, cells were steroid depleted in phenol red-free media supplemented with 5% charcoal-dextran-treated calf serum for seven days prior to the indicated ligand treatment. Media was changed on d 2 and 4 of culture, and cells were then treated with ligands.
Cellular Fractionation
MCF-7 cells were plated on 10 cm2 plates and grown to ca. 80% confluency. Cells were treated with either vehicle (0.1% EtOH) or 1 μM SERDs for 16 h ± 1 nM E2 for 4 h and harvested by scraping in 100 μL of buffer. Cellular fractionation was carried out as previously described.23, 45 Whole cell lysates were obtained by harvesting in 100 μL of whole cell lysis buffer [50 mM Tris (pH 8), 1 mM EDTA, 150 mM NaCl, 1% IGEPAL, 1% SDS, 5% glycerol]. Protein concentrations of the whole cell lysates, cytoplasmic and nuclear extracts were determined by BCA Protein Assay (Pierce, Rockford, IL).
SDS-PAGE and Western Analysis
20 μg of whole cell lysate and 40 μg of cytoplasmic and nuclear extracts were loaded on Tris-HCl SDS-PAGE gels (Bio-Rad, Hercules, CA). Equivalent fraction volumes of the detergent-extractable nuclear matrix and insoluble fractions were loaded. Proteins were transferred to nitrocellulose membranes (Pall Corporation, Pensacola, FL). Membranes were blocked in Odyssey Blocking Buffer (Li-Cor Biosciences, Lincoln, NE). Overnight primary antibody (α-F10 [Santa Cruz Biotechnology, Santa Cruz, CA] and α-Sp1 [Upstate/Millipore, Bedford, MA] at 1:1000 dilution) and secondary antibody (Li-Cor Biosciences, Lincoln, NE at 1:15,000 dilution) incubations were done in the presence of 0.1% Tween 20. PBST (PBS buffer with Tween) was used to wash off unbound antibody, and subsequent PBS washes were performed to limit the cross-reactivity of Tween 20 with the LiCor infrared imaging system.
Total RNA Isolation and Real-Time PCR Analysis
MCF-7 cells were plated in six-well plates and grown to ca. 80% confluency. Cells were pretreated with 1 μM SERDs or 0.1% EtOH vehicle for 1h, and then treated with SERDs alone or concomitantly with 1 nM E2 for 16h. Total RNA was extracted, reverse transcribed, and analyzed via real-time PCR as previously described.44 Primers used are as follows: ENC1 forward (f), GGCCTCCCCCTCAGTCTCT; ENC1 reverse (r), GCACTCACTACTGCGGCGT. The primers used for pS2, progesterone receptor, CA12, and BLNK were those previously described.27, 44, 46, 47 Statistical analysis was performed with repeated measures one-way ANOVA using the Tukey’s t-test.
Estrogen Receptor Binding Affinity Assays
Relative binding affinities were determined by a competitive radiometric binding assay, as previously described,20, 21 using 10 nM [3H]estradiol as tracer (GE Healthcare, Piscataway, NJ), and purified full-length human ERα and ERβ (PanVera/InVitrogen, Carlsbad, CA). Incubations were for 18-24 h at 0 °C, then the receptor-ligand complexes were absorbed onto hydroxyapatite (BioRad, Hercules, CA) and unbound ligand was washed away. The binding affinities are expressed as relative binding affinity (RBA) values, with the RBA of estradiol set to 100. The values given are the average ± range or SD of two or more independent determinations. Estradiol binds to ERα with a Kd of 0.2 nM and to ERβ with a Kd of 0.5 nM.
Supplementary Material
Acknowledgments
We thank Zeynep Madak-Erdogan for helpful experimental advice. This work was supported by grants from the National Institutes of Health [NIH CA 018119 (B.S.K.), NIH DK 015556 (J.A.K.)] and a Breast Cancer Research Foundation grant (B.S.K.)
Abbreviation List
- BN
bicyclononane
- CF
cyclofenil
- DES
diethylstilbestrol
- E2
estradiol
- ER
estrogen receptor
- SERD
selective estrogen receptor downregulators
- SERM
selective estrogen receptor modulators
- TAE
triarylethylene
Footnotes
Supporting Information Available: Synthesis and spectroscopic characterization (9-12, 14-46), 1H and 13C NMR spectra (9-12, 14-46), normal and reversed phase HPLC chromatograms (11, 15, 17, 22-26, 29, 31-36, 42-44, 46). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor transcription and transactivation: Estrogen receptor alpha and estrogen receptor beta: regulation by selective estrogen receptor modulators and importance in breast cancer. Breast Cancer Res. 2000;2:335–344. doi: 10.1186/bcr78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grese TA, Dodge JA. Selective estrogen receptor modulators (SERMs) Curr Pharm Des. 1998;4:71–92. [PubMed] [Google Scholar]
- 3.Eng-Wong J, Zujewski JA. Raloxifene and its role in breast cancer prevention. Expert Rev Anticancer Ther. 2004;4:523–532. doi: 10.1586/14737140.4.4.523. [DOI] [PubMed] [Google Scholar]
- 4.Chia S, Gradishar W. Fulvestrant: expanding the endocrine treatment options for patients with hormone receptor-positive advanced breast cancer. Breast. 2008;17(Suppl 3):S16–21. doi: 10.1016/j.breast.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Vergote I, Abram P. Fulvestrant, a new treatment option for advanced breast cancer: tolerability versus existing agents. Ann Oncol. 2006;17:200–204. doi: 10.1093/annonc/mdj047. [DOI] [PubMed] [Google Scholar]
- 6.Dauvois S, Danielian PS, White R, Parker MG. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc Natl Acad Sci U S A. 1992;89:4037–4041. doi: 10.1073/pnas.89.9.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dauvois S, White R, Parker MG. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J Cell Sci. 1993;106(Pt 4):1377–1388. doi: 10.1242/jcs.106.4.1377. [DOI] [PubMed] [Google Scholar]
- 8.Lupien M, Jeyakumar M, Hebert E, Hilmi K, Cotnoir-White D, Loch C, Auger A, Dayan G, Pinard GA, Wurtz JM, Moras D, Katzenellenbogen J, Mader S. Raloxifene and ICI182,780 increase estrogen receptor-alpha association with a nuclear compartment via overlapping sets of hydrophobic amino acids in activation function 2 helix 12. Mol Endocrinol. 2007;21:797–816. doi: 10.1210/me.2006-0074. [DOI] [PubMed] [Google Scholar]
- 9.Connor CE, Norris JD, Broadwater G, Willson TM, Gottardis MM, Dewhirst MW, McDonnell DP. Circumventing tamoxifen resistance in breast cancers using antiestrogens that induce unique conformational changes in the estrogen receptor. Cancer Res. 2001;61:2917–2922. [PubMed] [Google Scholar]
- 10.Yoneya T, Taniguchi K, Tsunenari T, Saito H, Kanbe Y, Morikawa K, Yamada-Okabe H. Identification of a novel, orally bioavailable estrogen receptor downregulator. Anticancer Drugs. 2005;16:751–756. doi: 10.1097/01.cad.0000171515.27439.de. [DOI] [PubMed] [Google Scholar]
- 11.Willson TM, Henke BR, Momtahen TM, Charifson PS, Batchelor KW, Lubahn DB, Moore LB, Oliver BB, Sauls HR, Triantafillou JA, Wolfe SG, Baer PG. 3-[4-(1,2-Diphenylbut-1-enyl)phenyl]acrylic acid: a non-steroidal estrogen with functional selectivity for bone over uterus in rats. J Med Chem. 1994;37:1550–1552. doi: 10.1021/jm00037a002. [DOI] [PubMed] [Google Scholar]
- 12.Willson TM, Norris JD, Wagner BL, Asplin I, Baer P, Brown HR, Jones SA, Henke B, Sauls H, Wolfe S, Morris DC, McDonnell DP. Dissection of the molecular mechanism of action of GW5638, a novel estrogen receptor ligand, provides insights into the role of estrogen receptor in bone. Endocrinology. 1997;138:3901–3911. doi: 10.1210/endo.138.9.5358. [DOI] [PubMed] [Google Scholar]
- 13.McDonnell DP. The Molecular Pharmacology of SERMs. Trends Endocrinol Metab. 1999;10:301–311. doi: 10.1016/s1043-2760(99)00177-0. [DOI] [PubMed] [Google Scholar]
- 14.Robertson DW, Katzenellenbogen JA, Long DJ, Rorke EA, Katzenellenbogen BS. Tamoxifen antiestrogens. A comparison of the activity, pharmacokinetics, and metabolic activation of the cis and trans isomers of tamoxifen. J Steroid Biochem. 1982;16:1–13. doi: 10.1016/0022-4731(82)90137-6. [DOI] [PubMed] [Google Scholar]
- 15.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JA, Carlquist M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. Embo J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu YL, Yang X, Ren Z, McDonnell DP, Norris JD, Willson TM, Greene GL. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell. 2005;18:413–424. doi: 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 17.Katzenellenbogen JA, Carlson KE, Katzenellenbogen BS. Facile geometric isomerization of phenolic non-steroidal estrogens and antiestrogens: limitations to the interpretation of experiments characterizing the activity of individual isomers. J Steroid Biochem. 1985;22:589–596. doi: 10.1016/0022-4731(85)90210-9. [DOI] [PubMed] [Google Scholar]
- 18.Airy SC, Sinsheimer JE. Isomerization of trans-diethylstilbestrol to pseudo-diethylstilbestrol. Steroids. 1981;38:593–603. doi: 10.1016/0039-128x(81)90057-x. [DOI] [PubMed] [Google Scholar]
- 19.Muthyala RS, Sheng S, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. Bridged bicyclic cores containing a 1,1-diarylethylene motif are high-affinity subtype-selective ligands for the estrogen receptor. J Med Chem. 2003;46:1589–1602. doi: 10.1021/jm0204800. [DOI] [PubMed] [Google Scholar]
- 20.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry. 1997;36:14897–14905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 21.Katzenellenbogen JA, Johnson HJ, Jr, Myers HN. Photoaffinity labels for estrogen binding proteins of rat uterus. Biochemistry. 1973;12:4085–4092. doi: 10.1021/bi00745a010. [DOI] [PubMed] [Google Scholar]
- 22.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 23.Wittmann BM, Sherk A, McDonnell DP. Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators. Cancer Res. 2007;67:9549–9560. doi: 10.1158/0008-5472.CAN-07-1590. [DOI] [PubMed] [Google Scholar]
- 24.Fan M, Rickert EL, Chen L, Aftab SA, Nephew KP, Weatherman RV. Characterization of molecular and structural determinants of selective estrogen receptor downregulators. Breast Cancer Res Treat. 2007;103:37–44. doi: 10.1007/s10549-006-9353-2. [DOI] [PubMed] [Google Scholar]
- 25.Lonard DM, Nawaz Z, Smith CL, O’Malley BW. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol Cell. 2000;5:939–948. doi: 10.1016/s1097-2765(00)80259-2. [DOI] [PubMed] [Google Scholar]
- 26.Eckert RL, Mullick A, Rorke EA, Katzenellenbogen BS. Estrogen receptor synthesis and turnover in MCF-7 breast cancer cells measured by a density shift technique. Endocrinology. 1984;114:629–637. doi: 10.1210/endo-114-2-629. [DOI] [PubMed] [Google Scholar]
- 27.Frasor J, Stossi F, Danes JM, Komm B, Lyttle CR, Katzenellenbogen BS. Selective estrogen receptor modulators: discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res. 2004;64:1522–1533. doi: 10.1158/0008-5472.can-03-3326. [DOI] [PubMed] [Google Scholar]
- 28.McDonnell DP, Norris JD. Connections and regulation of the human estrogen receptor. Science. 2002;296:1642–1644. doi: 10.1126/science.1071884. [DOI] [PubMed] [Google Scholar]
- 29.Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 30.Johnston SR. Endocrinology and hormone therapy in breast cancer: selective oestrogen receptor modulators and downregulators for breast cancer - have they lost their way? Breast Cancer Res. 2005;7:119–130. doi: 10.1186/bcr1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Regan RM, Jordan VC. The evolution of tamoxifen therapy in breast cancer: selective oestrogen-receptor modulators and downregulators. Lancet Oncol. 2002;3:207–214. doi: 10.1016/s1470-2045(02)00711-8. [DOI] [PubMed] [Google Scholar]
- 32.Bentrem D, Dardes R, Liu H, MacGregor-Schafer J, Zapf J, Jordan V. Molecular mechanism of action at estrogen receptor alpha of a new clinically relevant antiestrogen (GW7604) related to tamoxifen. Endocrinology. 2001;142:838–846. doi: 10.1210/endo.142.2.7932. [DOI] [PubMed] [Google Scholar]
- 33.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 34.Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, Carlquist M. Structural insights into the mode of action of a pure antiestrogen. Structure. 2001;9:145–153. doi: 10.1016/s0969-2126(01)00568-8. [DOI] [PubMed] [Google Scholar]
- 35.Wijayaratne AL, McDonnell DP. The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem. 2001;276:35684–35692. doi: 10.1074/jbc.M101097200. [DOI] [PubMed] [Google Scholar]
- 36.Duong V, Boulle N, Daujat S, Chauvet J, Bonnet S, Neel H, Cavailles V. Differential regulation of estrogen receptor alpha turnover and transactivation by Mdm2 and stress-inducing agents. Cancer Res. 2007;67:5513–5521. doi: 10.1158/0008-5472.CAN-07-0967. [DOI] [PubMed] [Google Scholar]
- 37.Nawaz Z, Lonard DM, Dennis AP, Smith CL, O’Malley BW. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci U S A. 1999;96:1858–1862. doi: 10.1073/pnas.96.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alarid ET, Bakopoulos N, Solodin N. Proteasome-mediated proteolysis of estrogen receptor: a novel component in autologous down-regulation. Mol Endocrinol. 1999;13:1522–1534. doi: 10.1210/mend.13.9.0337. [DOI] [PubMed] [Google Scholar]
- 39.Laios I, Journe F, Nonclercq D, Vidal DS, Toillon RA, Laurent G, Leclercq G. Role of the proteasome in the regulation of estrogen receptor alpha turnover and function in MCF-7 breast carcinoma cells. J Steroid Biochem Mol Biol. 2005;94:347–359. doi: 10.1016/j.jsbmb.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 40.Stenoien DL, Patel K, Mancini MG, Dutertre M, Smith CL, O’Malley BW, Mancini MA. FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent. Nat Cell Biol. 2001;3:15–23. doi: 10.1038/35050515. [DOI] [PubMed] [Google Scholar]
- 41.Stenoien DL, Mancini MG, Patel K, Allegretto EA, Smith CL, Mancini MA. Subnuclear trafficking of estrogen receptor-alpha and steroid receptor coactivator-1. Mol Endocrinol. 2000;14:518–534. doi: 10.1210/mend.14.4.0436. [DOI] [PubMed] [Google Scholar]
- 42.Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol Cell. 2003;11:695–707. doi: 10.1016/s1097-2765(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 43.Osborne CK, Coronado-Heinsohn EB, Hilsenbeck SG, McCue BL, Wakeling AE, McClelland RA, Manning DL, Nicholson RI. Comparison of the effects of a pure steroidal antiestrogen with those of tamoxifen in a model of human breast cancer. J Natl Cancer Inst. 1995;87:746–750. doi: 10.1093/jnci/87.10.746. [DOI] [PubMed] [Google Scholar]
- 44.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 45.Coutts AS, Davie JR, Dotzlaw H, Murphy LC. Estrogen regulation of nuclear matrix-intermediate filament proteins in human breast cancer cells. J Cell Biochem. 1996;63:174–184. doi: 10.1002/(sici)1097-4644(19961101)63:2<174::aid-jcb5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 46.Stossi F, Likhite VS, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen-occupied estrogen receptor represses cyclin G2 gene expression and recruits a repressor complex at the cyclin G2 promoter. J Biol Chem. 2006;281:16272–16278. doi: 10.1074/jbc.M513405200. [DOI] [PubMed] [Google Scholar]
- 47.Barnett DH, Sheng S, Charn TH, Waheed A, Sly WS, Lin CY, Liu ET, Katzenellenbogen BS. Estrogen receptor regulation of carbonic anhydrase XII through a distal enhancer in breast cancer. Cancer Res. 2008;68:3505–3515. doi: 10.1158/0008-5472.CAN-07-6151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.