

Abstract
An efficient Cu-catalyzed method for enantioselective boronate conjugate additions to trisubstituted alkenes of acyclic α,β-unsaturated carboxylic esters, ketones and thioesters is disclosed. All transformations are promoted by 5 mol % of a chiral monodentate NHC–Cu complex, derived from a readily available C1-symmetric imidazolinium salt, and in the presence of commercially available bis(pinacolato)diboron. Reactions are efficient (typically, 60% to >98% yield after purification) and deliver the desired β-boryl carbonyls in up to >98:2 enantiomer ratio (er). Reactions of unsaturated thioesters proceed with higher enantioselectivity (vs carboxylic esters or ketones), and the resulting products can be functionalized by Ag-mediated or Pd-catalyzed reactions that furnish the derived carboxylic ester or various ketones. Routine oxidation affords β-hydroxy ketones or carboxylic esters, ketone aldol products that cannot be otherwise prepared efficiently by an alternative catalytic enantioselective protocol.

Herein, we report the first cases of catalytic enantioselective conjugate boronate additions1 to trisubstituted alkenes of acyclic α,β-unsaturated carboxylic esters, ketones and alkylthioesters, resulting in the formation of B-substituted quaternary carbon stereogenic centers (Scheme 1). Transformations proceed with commercially available bis(pinacolato)diboron (1) and are promoted by a readily accessible (three steps; ~50% overall yield) chiral C1-symmetric Cu-based N-heterocyclic carbene (NHC) complex; the desired β-boryl carbonyls are formed in up to 99:1 enantiomeric ratio (er). Oxidation affords β-hydroxy ketones or carboxylic esters (ketone aldol products; Scheme 1). Reactions of unsaturated thioesters are generally more enantioselective (vs carboxylic esters or ketones); such processes may be followed by Ag-mediated or Pd-catalyzed cross-coupling reactions involving the S-based moiety, allowing conversion to carboxylic esters and unsaturated ketones, respectively.
Scheme 1.

B-Substituted Quaternary Centers by Cu-Catalyzed Conjugate Additions
In spite of advances made regarding catalytic enantioselective conjugate addition reactions,2 protocols that involve acyclic α,β−unsaturated carbonyls (vs cyclic enones) and deliver quaternary carbon stereogenic centers remain scarce. The limited number of existing methods involves additions of alkylmetals to strongly activated acyclic electrophiles such as nitroalkenes3 and Meldrum acid derivatives,4 or pyridylsulfones (with vinylboronic acids).5 Of particular significance are related processes that furnish B-substituted quaternary carbons (Scheme 1);6 subsequent oxidation would deliver the product of a ketone aldol process in high enantiomeric purity.7 We have introduced enantioselective processes involving aryl alkenes8 and terminal alkynes,9 where Cu-based NHC complexes serve as catalysts to generate tertiary B-substituted carbon centers. More recently, we have set out to examine the ability of such NHC–Cu complexes, developed in these laboratories, to promote enantioselective formation of B-substituted quaternary carbon stereogenic centers.10
We began by probing the ability of Cu complexes derived from imidazolinium salts 2–6 to serve as chiral catalysts; unsaturated ester 7 served as the initial substrate. As the data in Table 1 indicate, use of bidentate 211 leads to an efficient process but affords 8 with low enantioselectivity (68.5:31.5 er). Conjugate additions with 3a-b12 are significantly more selective (up to 88:12 er) but relatively low catalytic activity is furnished by the derived NHC–cuprates. A number of structurally distinct monodentate Cu-carbenes were also investigated (entries 4–8, Table 1), and found to offer improved efficiency (95% to >98% conv), with the C1-symmetric Cu complex of 5c delivering the highest er (88:12). Although the C2-symmetric Cu complex of 4 is more effective than the C1-symmetric 5a, through modification of the symmetric N-aryl moiety of the latter system that improvement in enantioselectivity was achieved (cf. entry 5 vs 7). The above observations underline a notable advantage of the monodentate C1-symmetric complexes, which can be more easily modified compared to the more symmetric variants.13
Table 1.
Initial Evaluation of Various Chiral NHC Complexesa
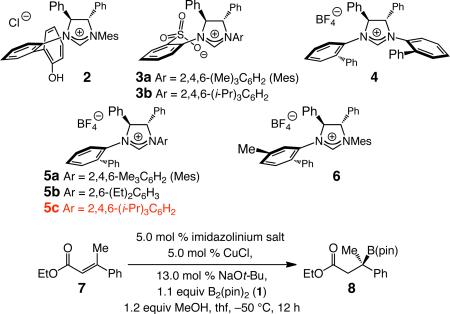 | |||
|---|---|---|---|
| entry | imidazolinium salt | conv (%)b | erc |
| 1 | 2 | 94 | 68.5:31.5 |
| 2 | 3a | 60 | 85.5:14.5 |
| 3 | 3b | 45 | 88:12 |
| 4 | 4 | 95 | 78.5:21.5 |
| 5 | 5a | 96 | 62:38 |
| 6 | 5b | >98 | 69:31 |
| 7 | 5c | >98 | 88:12 |
| 8 | 6 | >98 | 60.5:39.5 |
Reactions performed under N2 atm.
Values determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures.
Values determined by HPLC analysis; see the Supporting Information for details. B(pin) = pinacolatoboron.
With a reasonably effective NHC–Cu complex identified, further optimization of conditions was performed (–78 °C and acidic MeOH to ensure quench at low temperature14), allowing us to establish that various aryl-substituted unsaturated esters undergo highly enantioselective Cu-catalyzed boronate conjugate additions (Table 2). Transformations with substrates bearing an o-bromo- or a 2-naphthyl substituent (entries 2 and 4, Table 2) proceed to afford the desired boronate in 92% and quantitative yield and 95.5:4.5 and 96.5:3.5 er, respectively. In contrast, however, the presence of an o-Me unit results in substantial diminution in reactivity and enantioselectivity (entry 3). Reactions with substrates bearing a m- or p-bromo aryl group (entries 5–6, Table 2) or a trifluoromethyl unit (entry 7) furnish the desired products in 93–96% yield and 97:3 to >98:2 er. Conjugate addition of the unsaturated ester bearing an electron rich p-methoxyphenyl group, affords the desired β-boryl ester in 69% yield and 95:5 er, but is relatively sluggish, likely due to diminished substrate electrophilicity. A substrate that contains an Et-substituted alkene (entry 9, Table 2) undergoes an efficient and equally selective process at –15 °C (vs –78 °C in entry 1, Table 2). With the corresponding unsaturated ester containing a more sizeable i-Pr unit, however, only the hydride conjugate addition product is isolated (51% conv, 33% yield at 22 °C, 24 h). The larger alkyl group may thus disfavor boronate addition at the more hindered carbon, causing Cu–B addition to occur with the same site selectivity as in reactions of disubstituted aryl alkenes (benzylic C–Cu).8 Alternatively, with a less reactive substrate, the catalytically active NHC–Cu–B(pin) might react with MeOH to generate MeOB(pin) and a sterically less demanding NHC–Cu–H, which promotes conjugate hydride addition. The proposed intermediacy of a chiral NHC–Cu complex is consistent with the finding that the saturated ester product is formed in 78:22 er.
Table 2.
NHC–Cu-Catalyzed Enantioselective Boronate Conjugate Additions to Aryl-Substituted α,β-Unsaturated Estersa
 | ||||||
|---|---|---|---|---|---|---|
| entry | aryl | R | time (h) | conv (%)b | yield (%)c | erd |
| 1 | Ph | Me | 24 | >98 | 80 | 96.5:3.5 |
| 2 | o-BrC6H4 | Me | 24 | >98 | 92 | 95.5:4.5 |
| 3 | o-MeC6H4 | Me | 24 | 38 | 31 | 53:47 |
| 4 | 2-naphthyl | Me | 36 | >98 | >98 | 96.5:3.5 |
| 5 | m-BrC6H4 | Me | 18 | >98 | 96 | 97:3 |
| 6 | p-BrC6H4 | Me | 12 | >98 | 93 | 98:2 |
| 7 | p-CF3C6H4 | Me | 24 | >98 | 93 | 99:1 |
| 8 | p-OMeC6H4 | Me | 36 | 73 | 69 | 95:5 |
| 9 | Ph | Et | 18e | >98 | 90 | 94.5:5.5 |
See Table 1.
See Table 1.
Yields of purified products.
Determined by HPLC analysis. See the Supporting Information for details.
Performed at –15 °C.
Alkyl-substituted unsaturated esters also undergo enantioselective boronate conjugate additions in the presence of the NHC–Cu complex of 5c (Scheme 2). β-Boryl ester R-10 is isolated in 80% yield and 95.5:4.5 er; similar efficiency and selectivity is observed in the formation of S-10 from Z-9 (vs R-10 from E-9). This is in contrast to aryl-substituted substrates. For example, when Z-7 is used, conjugate addition must be performed at –30 °C for appreciable conversion levels (vs –78 °C for E-7 in entry 1, Table 2),15 affording R-8 in 61% yield (72% conv) and 89.5:10.5 er along with ~20% of the conjugate reduction product. The lower reactivity of Z-7 vs Z-9 may be because the larger phenyl unit raises the energy of the substrate conformer where the carbonyl properly overlaps with the alkene [A(1,3)-type interaction]. Such considerations are congruent with the significance of the strong σ-donating ability of NHC–Cu complexes and π Lewis acidity of the unsaturated carbonyl, as suggested by theoretical investigations.16 Formation of β-boronate ester 11 (Scheme 2), containing a β-branched alkyl group is efficient but substantially less selective (73.5:26.5 er) than generation of 10, or the cyclohexyl-bearing 12, which is generated in 94:6 er and quantitative yield.
Scheme 2.

Cu–NHC-Catalyzed Enantioselective Boronate Conjugate Additions to Alkyl-Substituted α,β-Unsaturated Estersa
Unsaturated ketones can be used in the NHC–Cu-catalyzed processes as well, but reactions prove to be somewhat less enantioselective than the aforementioned carboxylic esters (Scheme 3). Thus, β-boryl ketones 14-15 are obtained in 73–89% yield and in 82.5:17.5–92:8 er. The transformation with the sterically demanding phenylketone, which must be performed at –30 °C for appreciable conversion, proceeds with lower selectively than that of the corresponding methyl ketone 13a.
Scheme 3.

Cu–NHC-Catalyzed Enantioselective Boronate Conjugate Additions to an α,β-Unsaturated Ketonesa
To access a wider range of β-boryl carbonyls of high enantiomeric purity, as well as address the diminished selectivity with which certain aforementioned conjugate additions proceed, we turned to the reactions of other substrate classes. First, the suitability of a Weinreb amide was examined; the α,β-unsaturated amide corresponding to 7, however, proved inert under the conditions described in Table 1 or Scheme 2 (<2% conv, 22 °C). Accordingly, we turned to unsaturated thioesters,17 leading us to establish that, as depicted in Scheme 4, NHC–Cu-catalyzed boronate addition to thioester 16 furnishes the derived β-boryl thioester 17 in 87% yield and 99:1 er. Although the reaction of 16 is more sluggish than that of the corresponding carboxylic ester 7 and must be performed at –50 °C (vs –78 °C), the desired product is obtained with higher enantiomeric purity (99:1 er vs 96.5:3.5 er). A similar trend in enantioselectivity is observed in reactions of alkyl-substituted thioesters that deliver 18-20 (Scheme 4; compare to 10-12 in Scheme 2).
Scheme 4.

Cu–NHC-Catalyzed Enantioselective Boronate Conjugate Additions to an α,β-Unsaturated Thioestersa
With a highly enantioselective method for synthesis of β-boryl thioesters available, we turned to exploring selected modes of product functionalization. Carboxylic esters (e.g., R-10), aryl or α,β-unsaturated ketones (e.g., 15) can be obtained via the corresponding alkylthioesters in 60-85% yield through Ag-mediated18 and Pd-catalyzed19 procedures, respectively (Scheme 5). The above two-step protocol thus furnishes products that are of significantly higher enantiomeric purity than is available by transformations of unsaturated esters or ketones.
Scheme 5.

Conversion of β-Boryl Thioesters to Esters and Ketonesa
Oxidation of the C–B bond delivers tertiary alcohols in high yield (e.g., 21 in >98% yield, eq 1). The corresponding reactions for efficient C–B to C–N conversion are yet to be disclosed; the present study underlines the significance of such future developments, furnishing routes to enantiomerically enriched Mannich-type products with N-substituted quaternary carbons.
 |
(1) |
Another notable attribute of the catalytic protocol is that the presence of MeOH is not required, allowing access to the corresponding boron enolates with useful efficiency and enantioselectivity.6b As indicated in eq 2, although the absence of the proton source leads to diminution in rate (72% conv, 4 °C vs >98% conv, –78 °C in Scheme 3), the level of enantioselectivity remains unchanged (91.5:8.5 vs 92:8).20
 |
(2) |
Intermediacy of neutral B–Cu complex I (Figure 1, vs cuprates from bidentate ligands) provides a rationale for the levels and trends in selectivity. Alkene coordination likely occurs such that the Cu–B bond is aligned with the substrate π*, while the carbonyl moiety resides proximal to the NHC's mono-substituted N-Ar (vs II). The model accounts for the enhanced selectivity as the size of the substituents of the symmetric N-Ar unit is increased (entries 5–7, Table 1), as well as the inferior enantioselectivity with NHC–Cu complex derived from meta-substituted 6. However, it is difficult to explain the origin of lower enantioselectivity delivered by the C2-symmetric complex derived from 4 (vs 5c); such variations may be the result of subtle conformational changes of the four contiguous aryl units of the NHC, caused by the presence of the 2,4,6-(i-Pr)3-phenyl moiety. The lower er values of a β-branched substrate (e.g., 11 vs 10 or 12) may be due to steric repulsion with methyl groups of the pinacol. The reasons for inactivity of a Weinreb amide or improved selectivities with thioesters, on the other hand, require more detailed mechanistic investigations.
Figure 1.

Proposed models for NHC–Cu-catalyzed boronate conjugate additions.
Supplementary Material
Acknowledgment
Financial support was provided by the NIH (GM-57212) and the NSF (CHE-0715138). J. M. O. is a LaMattina graduate fellow. We thank Dr. S. J. Meek for helpful suggestions. Mass spectrometry facilities at Boston College are supported by the NSF (DBI-0619576).
Footnotes
Supporting Information Available: Experimental procedures and spectral, analytical data for all products (PDF). This material is available on the web: http://www.pubs.acs.org
References
- 1.For Cu-catalyzed enantioselective boronate conjugate additions to unsaturated carbonyls with a disubstituted alkene, see: Lee J-E, Yun J. Angew. Chem., Int. Ed. 2008;47:145. doi: 10.1002/anie.200703699.Lillo V, Prieto A, Bonet A, Díaz-Requejo MM, Ramírez J, Pérez PJ, Fernández E. Organometallics. 2009;28:659.Sim H-S, Feng X, Yun J. Chem. Eur. J. 2009;15:1939. doi: 10.1002/chem.200802150.
- 2.Jerphagnon T, Pizzuti MG, Minnaard AJ, Feringa BL. Chem. Soc. Rev. 2009;38:1039. doi: 10.1039/b816853a. [DOI] [PubMed] [Google Scholar]
- 3.Wu J, Mampreian DM, Hoveyda AH. J. Am. Chem. Soc. 2005;127:4584. doi: 10.1021/ja050800f. [DOI] [PubMed] [Google Scholar]
- 4.For example, see: Wilsily A, Fillion E. J. Org. Chem. 2009;74:8583. doi: 10.1021/jo901559d.
- 5.Mauleón P, Carretero JC. Chem. Commun. 2005:4961. doi: 10.1039/b508142d. [DOI] [PubMed] [Google Scholar]
- 6.For Cu-catalyzed enantioselective boronate conjugate additions to cyclic β-substituted enones with chiral phosphine ligands, see: Chen I-H, Yin L, Itano W, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2009;131:11664. doi: 10.1021/ja9045839. For NHC–catalyzed (metal-free) non-enantioselective conjugate boronate additions to β-substituted cyclic and acyclic unsaturated carbonyls, see: Lee K-s., Zhugralin AR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:7253. doi: 10.1021/ja902889s. For enantioselective synthesis (non-catalytic) of an allyl boronate with a B-substituted quaternary carbon stereogenic center, see: Stymiest JL, Bagutski V, French RM, Aggarwal VK. Nature. 2008;456:778. doi: 10.1038/nature07592.
- 7.Generally efficient and highly selective catalytic enantioselective protocols for aldol additions to ketones are yet to be introduced. For an early report, see: Denmark SE, Fan Y, Eastgate MD. J. Org. Chem. 2005;70:5235. doi: 10.1021/jo0506276. For a recent review on Cu-catalyzed ketone aldol processes, see: Shibasaki M, Kanai M. Chem. Rev. 2008;108:2853. doi: 10.1021/cr078340r. For Ag-catalyzed aldol adition to α-ketoesters, see: Akullian LC, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2006;128:6532. doi: 10.1021/ja061166o.
- 8.Lee Y, Hoveyda AH. J. Am. Chem. Soc. 2009;131:3160. doi: 10.1021/ja809382c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee Y, Jang H, Hoveyda AH. J. Am. Chem. Soc. 2009;131:18234. doi: 10.1021/ja9089928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For NHC–Cu-catalyzed allylic substitutions that furnish B-substituted quaternary carbon stereogenic centers, see: Guzman-Martinez A, Hoveyda AH. J. Am. Chem. Soc. 2010:132. doi: 10.1021/ja104254d. in press.
- 11.Lee K.-s., Brown MK, Hird AW, Hoveyda AH. J. Am. Chem. Soc. 2006;128:7182. doi: 10.1021/ja062061o. [DOI] [PubMed] [Google Scholar]
- 12.a Brown MK, May TL, Baxter CA, Hoveyda AH. Angew. Chem., Int. Ed. 2007;46:1097. doi: 10.1002/anie.200604511. [DOI] [PubMed] [Google Scholar]; b May TL, Brown MK, Hoveyda AH. Angew. Chem., Int. Ed. 2008;47:7358. doi: 10.1002/anie.200802910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For development of C1-symmetric NHC–Cu complexes and their utility in conjugate additions of aryl- or vinylsilanes and dimethylphenylsilylpinacolatoboron, respectively, see: Lee K-s., Hoveyda AH. J. Org. Chem. 2009;74:4455. doi: 10.1021/jo900589x.Lee K-s., Hoveyda AH. J. Am. Chem. Soc. 2010;132:2898. doi: 10.1021/ja910989n.
- 14.Control experiments indicate that if reactions are not properly quenched at –78 °C to decompose any remaining B2(pin)2, adventitious conjugate additions can occur as the mixture is allowed to warm to 22 °C, leading to lowering of product enantiomeric purity.
- 15.At –78 °C, reaction of Z-7 proceeds to afford 22% R-8 in 81.5:18.5 er along with 45% of the corresponding conjugate reduction product.
- 16.For related DFT calculations, see: Dang L, Lin Z, Marder TB. Organometallics. 2008;27:4443. and references cited therein.
- 17.Acyclic unsaturated thioesters have been utilized in Cu–phosphinecatalyzed enantioselective conjugate additions with Grignard reagents. See: Mazery RD, Pullez M, López F, Harutyunyan SR, Minnaard AJ, Feringa BL. J. Am. Chem. Soc. 2005;127:9966. doi: 10.1021/ja053020f.
- 18.Masamune S, Hayase Y, Schilling W, Chan WK, Bates GS. J. Am. Chem. Soc. 1977;99:6756. doi: 10.1021/ja00462a049. [DOI] [PubMed] [Google Scholar]
- 19.a Liebeskind LS, Srogl J. J. Am. Chem. Soc. 2000;122:11260. [Google Scholar]; b Prokopcová H, Kappe CO. Angew. Chem., Int. Ed. 2009;48:2276. doi: 10.1002/anie.200802842. [DOI] [PubMed] [Google Scholar]
- 20.The ratio of stereosiomers and identity of the major B-enolate isomer (if any) have not been rigorously determined.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.