

Abstract
We report for the first time a patient with both transient neonatal diabetes mellitus (TNDM) and idiopathic neonatal cholestasis, with both features resolving over a similar time course. Cholestasis was due to paucity of interlobular bile ducts (PILBD). Genetic analysis was consistent with a uniparental disomy of chromosome 6. Paucity of interlobular bile ducts is common in Alagille syndrome but also occurs by unknown mechanisms in a wide spectrum of other diseases. We propose a shared explanation for this patient’s TNDM and PILBD mediated by the noted chromosomal abnormality. We suggest that hepatobiliary function be evaluated in patients with TNDM to determine the prevalence and course of cholestasis of the disease.
Keywords: cholestasis, chromosome 6q24, methylation defect, paucity of interlobular bile ducts, transient neonatal diabetes mellitus, uniparental isodisomy
INTRODUCTION
Neonatal cholestasis is a serious medical condition that often poses a diagnostic dilemma for the clinician. Despite progress in understanding neonatal cholestasis, approximately 25% of cases have idiopathic etiologies [1]. Prompt referral to pediatric gastroenterologists is essential for appropriate diagnosis and therapy.
Neonatal diabetes mellitus is defined as persistent hyperglycemia occurring in infants <6 months of age, lasting >2 weeks, and requiring exogenous insulin therapy [2]. Most affected infants are small for gestational age, volume-depleted, hyperglycemic, glucosuric, and occasionally ketoacidotic. The disease has both transient and permanent forms, which have different clinical courses and underlying etiologies. Transient neonatal diabetes mellitus (TNDM) begins within 6 weeks and remits by 18 months of age, possibly relapsing later. As opposed to the identified etiologies of the permanent form, TNDM etiologies are less clearly understood. However, 71% of patients with transient neonatal diabetes have undermethylated DNA at the chromosome 6q24 locus [3].
We describe for the first time a patient with classic TNDM and neonatal cholestasis associated with paucity of interlobular bile ducts (PILBD) on liver biopsy. Both the cholestasis, which was not secondary to Alagille syndrome, and the transient neonatal diabetes resolved concomitantly, suggesting a shared etiology mediated through the chromosome 6 abnormality commonly found in patients with TNDM. Published reports indicate that biliary dysfunction and neonatal diabetes may coexist but the prevalence in the setting of TNDM remains unknown.
CASE REPORT
A 37-week male infant was small for gestational age at birth in a 19-year-old primiparous mother after an uncomplicated pregnancy. Birth weight was 2100 g (less than 3rd percentile) and physical examination was normal except for macroglossia and extremity edema. The infant fed poorly and had low body temperature (36.2°C). Bedside blood glucose levels were >400 mg/dL; follow-up serum glucose was 476 mg/dL. Urinalysis revealed glucosuria at 500 mg/dL. The insulin level was low normal at 3.1 μU/mL (normal range: 2.0 to 25 μIU/mL). The patient was started on low-dose insulin therapy for the hyperglycemia.
During the 1st month, the direct conjugated bilirubin was mildly elevated (range: 1.2 to 2.5 mg/dL; normal range: 0 to 0.4 mg/dL) while the unconjugated bilirubin remained in the normal range. Serum aspartate amino-transferase was 98 U/L (normal range: 5 to 35 U/L) and alanine aminotransaminase was 72 U/L (normal range: 7 to 56 U/L). The prothrombin time, partial thromboplastin time, and international normalized ratio on day of life 19 were within normal limits. Serum γ-glutamyltransferase was 167 U/L (normal range: 8 to 78 U/L) and alkaline phosphatase was 410 IU/L (normal range: 20 to 160 U/L) at 1 month of age. The serum bile acid level was 138 μmol/L (normal: <57 μmol/L). Urine organic acids, serum amino acids, urine cytomegalovirus (CMV) antigen, and urine culture were normal. Two abdominal ultrasound examinations were normal. A chest radiograph to evaluate for the presence of butterfly vertebrae, often seen in patients with Alagille syndrome, was negative. Abdominal ultrasonograms performed on days 2 and 42 showed a normal-appearing gallbladder and no evidence of intrahepatic or extrahepatic dilatation of the biliary tree.
Further workup of persistent cholestasis revealed a normal α1-antitrypsin phenotype, T4, thyrotropin, sweat chloride, and newborn screen studies. At 7 weeks of life, a liver biopsy revealed severe cytoplasmic and canalicular intralobular cholestasis associated with absent interlobular bile ducts in all 12 portal areas examined. Focal giant cell transformation and persistent extramedullary hematopoiesis were observed (Fig. 1). Anticytokeratin 7 and AE1/AE3 cytokeratin immunostains highlighted the ductal plate and rare ductules but no interlobular bile ducts. Trichrome stain showed a normal pattern of structural collagen without fibrosis. The diagnosis was neonatal hepatitis with PILBD suspicious for Alagille syndrome. By 84 days of age, the conjugated bilirubin was finally within normal limits at 0 mg/dL.
Figure 1.
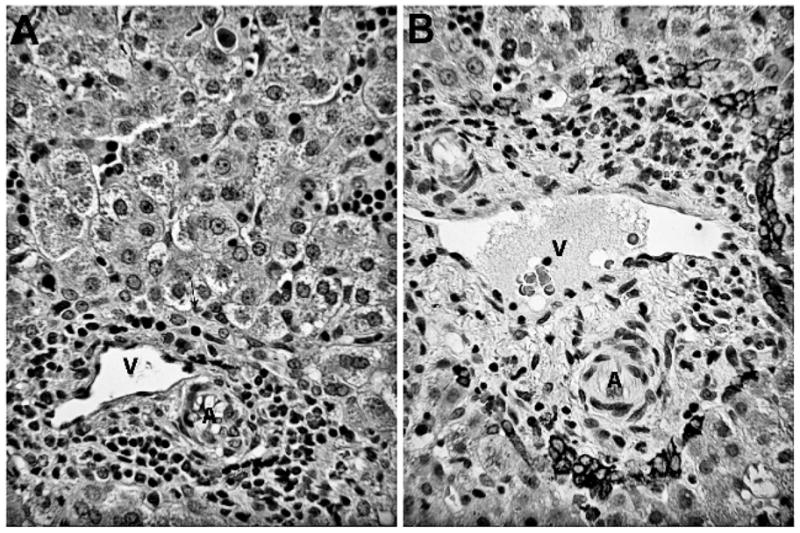
Histopathologic findings of liver biopsy revealing absence of interlobular bile ducts. A. A representative portal zone contains a portal venule (V) and arteriole (A) and myelopoiesis, but no bile duct. A tiny ductule is located along the limiting plate (arrow). Hepatocytes show cytoplasmic and canalicular cholestasis, plus clusters of normoblasts in sinusoids (hematoxylin and eosin, original ×125). B. Cytokeratin 7 immunostain of a portal zone contains normal vascular structure and myelopoiesis but lacks an interlobular bile duct. Remnants of the ductal plate persist in the form of ductules along the portal limiting plate.
During the 1st month of life, the patient’s insulin requirement decreased significantly from a maximum dose of 0.4 U/kg/d and was discontinued by 2 months. Genetic studies were negative for insulin promoting factor-1 and glucokinase mutations. Further testing revealed a methylation defect at the 6q24 locus, consistent with TNDM. Seven microsatellites were concordant, suggesting paternal uniparental isodisomy of chromosome 6. At 5 months of age, the patient presented to the emergency department, and a chest radiograph revealed a right diaphragmatic eventration that had progressed since the initial chest radiograph obtained on day of life 19 (Fig. 2).
Figure 2.
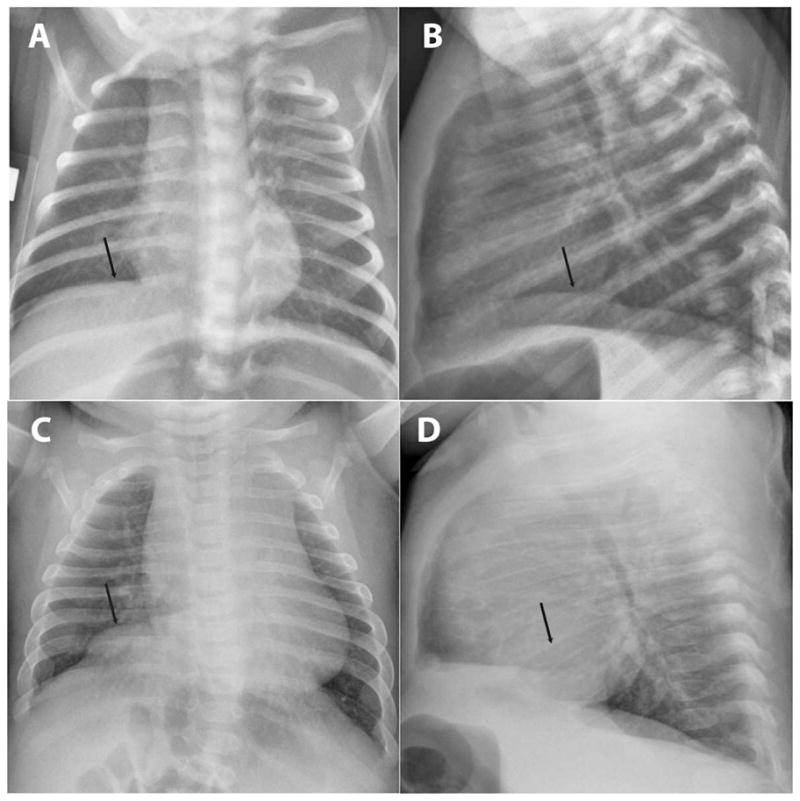
Diaphragmatic eventration revealed on chest radiographs. Postero-anterior radiograph revealing absence of butterfly vertebrae and progression of an initially subtle diaphragmatic eventration (arrows). Posteroanterior (A and C) and lateral (B and D) views on DOL 19 (A and B) and DOL 146 (C and D), respectively.
DISCUSSION
This newborn male infant is the 1st reported to present with a classic picture of TNDM (typical macroglossia, transient insulin requirement, small size for gestational age, and uniparental paternal isodisomy of chromosome 6) as well as an unexpected neonatal cholestasis associated with PILBD. Of note, both the diabetes and the cholestasis resolved over a similar time course. In addition, the patient had left diaphragmatic eventration.
The differential diagnosis of PILBD includes Alagille syndrome, α1-antitrypsin deficiency, Zellweger disease, other rare metabolic diseases, infections such as CMV, and idiopathic nonsyndromic paucity [4,5]. The workup for the other known etiologies of cholestasis in our case proved negative. Importantly, our patient did not have other stigmata of Alagille syndrome (butterfly vertebrae, peripheral pulmonic stenosis, typical facies).
The theoretical etiologies of interlobular bile duct paucity include regression due to direct injury of bile duct epithelium, inadequate hepatic arterial supply, delayed maturation, and abnormal ductal plate development [5]. Our patient had no inflammation or sclerosis in portal areas that lacked interlobular bile ducts. Therefore, the most likely etiology is delayed bile duct development from the ductal plate. Table 1 is a list of disorders mostly derived from published reports in which paucity occasionally, or rarely, has been identified. Many metabolic diseases are on this list but the mechanism by which paucity is associated, whether due to destruction or developmental disturbance, is unknown for all of them. We postulate that the methylation defect at the 6q24 locus, a defect that is known to affect beta cell development and function, may be directly or indirectly involved in delayed bile duct development in our case.
Table 1.
Paucity of the interlobular bile ducts in infants: a classification based on literature citations
|
NS indicates nonsyndromic; PFIC, progressive familial intrahepatic cholestasis.
Within the 6q24 locus, 1 CpG island shows differential paternal undermethylation associated with activation of genes in the area: PLAGL1/ZAC and HYMAI. PLAGL1 encodes for a zinc finger protein associated with apoptosis and cell cycle arrest, which could alter the number and function of beta cells in the neonate or embryo. HYMAI has no known open reading frame and its function remains unknown [6]. It is possible that the apparent shared delay in liver and pancreatic maturation was mediated by the upregulated activity of the cell cycle arrest/apoptosis gene PLAGL1 emanating from this region. Consistent with this, in mouse models of TNDM, the documented initial decrease and subsequent normalization in islet mass suggests a developmental lag. Interestingly, PLAGL1 RNA expression was detected in the livers of these mice [7].
Another possibility for the cholestasis in this clinical setting would be that the isodisomy observed in this patient at the 6q24 region was caused by duplication of the entire paternal chromosome 6. This duplication could have resulted in unmasking of recessive mutations that led to delayed development of interlobular bile ducts independent of the 6q24 defect that caused transient neonatal diabetes. A search for chromosome 6-associated cholestasis anomalies revealed Zellweger syndrome (6q23–24) and autosomal recessive polycystic kidney disease (6p21) [8], neither of which is associated with maturity-onset diabetes of the young (MODY) or neonatal diabetes, and each of which has clinical findings not manifested in this patient [9,10].
Precedents exist for hepatobiliary developmental anomalies associated with pancreatic islet dysfunction in permanent neonatal diabetes. A mutation upstream of the MODY4 gene insulin promoting factor-1 results in an autosomal recessive condition consisting of neonatal diabetes; atresia of the duodenum, jejunum, or gallbladder; absence of islet cells; and death in 4 of 5 patients at <1 year of age [11]. Other evidence supports single genes (hhex and hnf1β) causing perturbed development and function of both liver and pancreas [12]. Until this report, transient neonatal diabetes has not been associated with neonatal cholestasis and concomitant PILBD.
The overlap of hepatobiliary and pancreatic islet maturation and function echoes the shared ontogeny of these 2 organs. In the mouse, the pancreas and liver are derived from a shared population of bipotential progenitor cells [13]. Therefore, it is perhaps not surprising that a genetic anomaly that may impact beta cell function might also affect biliary development. This hepatopancreas progenitor population evolves to separate liver and pancreas progenitor populations in part by reciprocal signaling between these endoderm progenitors and the mesodermal diaphragm progenitor cells, the septum transversum mesenchyme [14]. This raises the possibility that the uncommon anomaly, the diaphragmatic eventration, observed in this patient, is causally associated.
It is unknown how often patients with TNDM have conjugated hyperbilirubinemia because this is not a regularly sought clinical feature and there is no precedent in the literature to evaluate hepatic function in the setting of TNDM. Measurement of serum bilirubin and consideration of liver biopsy is suggested if cholestasis persists in patients with TNDM [15]. Furthermore, evaluation of patients who present with cholestasis for the uniparental disomy of chromosome 6 anomaly is also worth consideration.
Acknowledgments
The authors thank Drs. Jeffrey Whitsett, Alan Jobe, Aaron Zorn, and James Wells for critical evaluation of this manuscript. We thank Dr. Janet Strife for her counsel related to the patient radiographs.
References
- 1.Stormon MO, Dorney SF, Kamath KR, O’Loughlin EV, Gaskin KJ. The changing pattern of diagnosis of infantile cholestasis. J Paediatr Child Health. 2001;37:47–50. doi: 10.1046/j.1440-1754.2001.00613.x. [DOI] [PubMed] [Google Scholar]
- 2.von Muhlendahl KE, Herkenhoff H. Long-term course of neonatal diabetes. N Engl J Med. 1995;333:704–708. doi: 10.1056/NEJM199509143331105. [DOI] [PubMed] [Google Scholar]
- 3.Flanagan SE, Patch AM, Mackay DJ, et al. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes. 2007;56:1930–1937. doi: 10.2337/db07-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sinha J, Magid MS, VanHuse C, Thung SN, Suchy F, Kerkar N. Bile duct paucity in infancy. Semin Liver Dis. 2007;27:319–323. doi: 10.1055/s-2007-985076. [DOI] [PubMed] [Google Scholar]
- 5.de Tommaso AM, Santos DS, Hessel G. Caroli’s disease: 6 case studies. Acta Gastroenterol Latinoam. 2003;33:47–51. [PubMed] [Google Scholar]
- 6.Temple IK, Shield JP. Transient neonatal diabetes, a disorder of imprinting. J Med Genet. 2002;39:872–875. doi: 10.1136/jmg.39.12.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma D, Shield JP, Dean W, et al. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J Clin Invest. 2004;114:339–348. doi: 10.1172/JCI19876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKusick V. A Catalog of Human Genes and Genetic Disorders. Baltimore, MD: Johns Hopkins University Press; 2009. Online Mendelian Inheritance in Man. [Google Scholar]
- 9.Hadchouel M. Paucity of interlobular bile ducts. Semin Diagn Pathol. 1992;9:24–30. [PubMed] [Google Scholar]
- 10.Waters K, Howman-Giles R, Rossleigh M, Lam A, Uren R, Knight J. Intrahepatic bile duct dilatation and cholestasis in autosomal recessive polycystic kidney disease: demonstration with hepatobiliary scintigraphy. Clin Nucl Med. 1995;20:892–895. doi: 10.1097/00003072-199510000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell J, Punthakee Z, Lo B, et al. Neonatal diabetes, with hypoplastic pancreas, intestinal atresia and gall bladder hypoplasia: search for the aetiology of a new autosomal recessive syndrome. Diabetologia. 2004;47:2160–2167. doi: 10.1007/s00125-004-1576-3. [DOI] [PubMed] [Google Scholar]
- 12.Beckers D, Bellanne-Chantelot C, Maes M. Neonatal cholestatic jaundice as the first symptom of a mutation in the hepatocyte nuclear factor-1beta gene (HNF-1beta) J Pediatr. 2007;150:313–314. doi: 10.1016/j.jpeds.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Deutsch G, Jung J, Zheng M, Lora J, Zaret KS. A bipotential precursor population for pancreas and liver within the embryonic endoderm. Development. 2001;128:871–881. doi: 10.1242/dev.128.6.871. [DOI] [PubMed] [Google Scholar]
- 14.Rossi JM, Dunn NR, Hogan BL, Zaret KS. Distinct mesodermal signals, including BMPs from the septum transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev. 2001;15:1998–2009. doi: 10.1101/gad.904601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shield JP. Neonatal diabetes. Horm Res. 2007;68(Suppl 5):32–36. doi: 10.1159/000110471. [DOI] [PubMed] [Google Scholar]