

Abstract
Purpose
Hypoxic tumor cells overexpressing hypoxia-inducible factor 1alpha (HIF-1α) are generally resistant to chemo/radiotherapy. We have reported that Se-methylselenocysteine (MSC) therapeutically enhances the efficacy and selectivity of irinotecan against human tumor xenografts. The aim of this study was to delineate the mechanism responsible for the observed efficacy targeting on HIF-1α and its transcriptionally regulated genes VEGF and CAIX.
Methods
We investigated the mechanism of HIF-1α inhibition by MSC and its critical role in the therapeutic outcome by generating HIF-1α stable knockdown (KD) human head and neck squamous cell carcinoma, FaDu by transfecting HIF-1α short hairpin RNA.
Results
While cytotoxic efficacy in combination with methylselenic acid (MSA) with SN-38 (active metabolites of MSC and irinotecan) could not be confirmed in vitro against normoxic tumor cells, the hypoxic tumor cells were more sensitive to the combination. Reduction in HIF-1α either by MSA or shRNA knockdown resulted in significant increase in cytotoxicity of SN38 in vitro against hypoxic, but not the normoxic tumor cells. Similarly, in vivo, either MSC in combination with irinotecan treatment of parental xenografts or HIF-1α KD tumors treated with irinotecan alone resulted in comparable therapeutic response and increase in the long-term survival of mice bearing FaDu xenografts.
Conclusions
Our results show that HIF-1α is a critical target for MSC and its inhibition was associated with enhanced antitumor activity of irinotecan. Inhibition of HIF-1α appeared to be mediated through stabilization of PHD2, 3 and downregulation of ROS by MSC. Thus, our findings support the development of MSC as a HIF-1α inhibitor in combination chemotherapy.
Keywords: HIF-1α, Se-methylselenocysteine, Irinotecan, Hypoxic tumor cells, PHD
Introduction
Human tumors, including those of the head and neck, often display regions of hypoxia and are typically resistant to chemo and radiation therapy. An agent that decreases the hypoxic tumor microenvironment may enhance the therapeutic efficacy of anticancer drugs. We have demonstrated the ability of a non-toxic dose and schedule of selenium (MSC) to enhance the therapeutic efficacy and selectivity of irinotecan against several human tumor xenografts, including FaDu human head and neck squamous cell carcinoma (HNSCC) [1]. However, combining SN38 (an active metabolite of irinotecan) with MSA (an active metabolite of MSC) in vitro under normoxic culture conditions did not demonstrate cytotoxic efficacy. This implied that the interaction between selenium and irinotecan involved more than a direct cytotoxic effect on the tumor cells. In as much as previous work in vivo, our laboratory had demonstrated an antiangiogenic and tumor vasculature maturation effect of MSC treatment alone [2], and an effect of MSC combined with irinotecan on proangiogenic molecular markers including hypoxia-inducible factor 1 alpha (HIF-1α) [3]. Therefore, an investigation of the specific effects of MSC on hypoxia-related protein HIF-1α appeared warranted.
A daily supplementation of 200 μg of selenium was reported to reduce the recurrence of various types of cancers [4, 5] but disappointingly, recent results from a prevention trial using low doses of selenomethionine (SLM) (200 μg/d) alone and in combination with vitamin E (400 IU/d) did not prevent prostate cancer [6]. The anti-tumor activity of selenium in combination therapy has been reported by our group [1]. Organoselenium compound, selenomethionine, has been used in the preclinical model as a suicide prodrug that converted into active metabolite methylselenol using methioninase gene therapy [7]. The molecular mechanism(s) of cytotoxic concentrations of selenium have been studied under normoxic conditions and were shown to modulate multiple pathways including apoptosis [8]. To our knowledge, there are no studies on the effect of selenium on cancer cells cultured under hypoxic conditions. Studies carried out in our laboratory indicate that apoptosis is not the mechanism of action of MSC in combination with irinotecan in our model [3]. Therefore, low oxygen hypoxic conditions were utilized in vitro as an alternate approach to mimic the hypoxic tumor microenvironment in xenografts to determine whether the therapeutic efficacy observed in vivo can be achieved in vitro against hypoxic cells.
HIF-1α is a transcription factor that is stabilized under hypoxic conditions and activates several genes, thus enabling cells to adapt and survive under low oxygen hypoxic conditions [9, 10]. Furthermore, anticancer drug resistance has been implicated with the high expression of HIF-1α [11, 12]. HIF-1α regulation is mediated by several factors including reactive oxygen species (ROS). The tumor microenvironment is often in redox imbalance, with accumulation of ROS [13]. ROS results in stabilization of HIF-1α through inhibition of prolylhydroxylases (PHDs) that hydroxylate proline molecules of HIF-1α, leading to ubiquitylation by von Hippel–Lindau protein (VHL) and degradation by proteosomes [14]. PHDs belong to a super family of iron-and 2-oxoglutarate-dependent dioxygenases. PHD2 is important for the hydroxylation of HIF-1α [15, 16]. Since the ROS is involved in HIF-1α stability through inhibition of PHDs, studies were carried out to investigate the antioxidant property of MSC in the downregulation of ROS that stabilizes PHDs, leading to the degradation of HIF-1α under hypoxic conditions.
The present study was initiated to investigate the critical role of HIF-1α in chemotherapy response and potential mechanism of HIF-1α regulation by MSC. The proposed hypothesis is that downregulation of ROS, stabilization of PHD2 and 3 by MSC results in HIF-1α inhibition. To this end, hypoxic tumor cells, expressing HIF-1α levels similar to those in xenografts, and tumor cells with HIF-1α KD were developed to evaluate and compare the effects of HIF-1α inhibition by shRNA and MSC on the antitumor response to irinotecan against FaDu xenografts.
Materials and methods
Animals
Eight- to 12-week-old female Foxn1nu nude mice (nu/nu, body weight 20–25 g) were obtained from Harlan Sprague–Dawley Inc. (Indianapolis, IN) and kept five mice per cage with water and food ad libitum according to an institutionally approved animal protocol.
Drugs
MSC was purchased from Sigma (St. Louis, MO) and dissolved in sterile saline at a concentration of 1 mg/ml. Irinotecan (Camptosar) was purchased from Pharmacia & Upjohn Co., division of Pfizer (New York, NY) as a solution of 20 mg/ml in 5-ml vials. SN-38 (7-ethyl-10-hydroxycamptothecin) was supplied from Pharmacia Corporation (Kalamazoo, MI). Methylselenic acid (MSA) was purchased from PharmaSe Inc., (Lubbock, TX).
Establishment of tumor xenografts
The human HNSCC FaDu was purchased from American Type Culture Collection (Rockville, MD, USA). The xenografts were initially established by injecting subcutaneous (s.c.) 106 cultured cells into nude mice, and the tumors were passed several generations by transplanting ~50-mg non-necrotic tumor (2–3 pieces) via a trocar when tumor reached to 1 g. FaDu cells (1 × 106 in 0.1 ml) specifically knocking down HIF-1α by shRNA or the negative control (scrambled shRNA) with no effect on HIF-1α were implanted s.c. in the nude mice and passed several generations with documentation of stable KD of HIF-1α. Tumor growth was measured using Vernier calipers [1]. All the studies were conducted in accordance with protocols approved by the Animal Care and Use Committee at Roswell Park Cancer Institute, an AAALAC accredited facility.
Drug doses and schedule
Irinotecan was administered by i.v. push once weekly for 4 weeks (on day 0, 7, 14, and 21) at the maximum tolerated dose (MTD) (100 mg/kg/week × 4) 7 days after the tumor transplantation when the tumors reached ~200–250 mg. MSC (0.2 mg/day/mouse, MTD) was administered orally (p.o.) daily for 28 days (from day –7 to 21) with the first dose started 7 days before irinotecan and at the same time of tumor transplantation. The treatment was concluded on day 21. For in vivo experiments, 5 mice were used for each experimental group with bilateral tumors. All experiments were performed at least twice. In a separate experiment to determine the effect of MSC alone on HIF-1α and its transcriptionally regulated genes (VEGF and CAIX) expression, tumors were harvested from the mice (N = 6) treated with MSC for 14 days from the day of tumor transplantation and untreated controls (N = 6).
For the in vitro studies, SN38 and MSA, the active metabolites of irinotecan and MSC, respectively, were used. To assess cytotoxicity, tumor cells were exposed to MSA for 24 h and to SN38 for 2 h. For combination studies, cells were exposed to MSA for 24 h and to SN38 during the last 2 h.
Antitumor activity
Antitumor activity was determined as described previously [1] in the tumors of HIF-1α KD, their scrambled negative control, and the parental FaDu tumors. Tumor responses were assessed daily during treatment and at least twice weekly thereafter. Tumor response was expressed as partial tumor response (PR) when tumor weight was reduced by at least 50% of initial tumor size, and complete tumor response (CR) was defined when no tumor was palpated at that initial site of tumor. Animals with CR were maintained for 3 months after the treatment was concluded: they were considered to be cured if there was no detectable tumor at the site of transplant, and the lack of tumor cells was confirmed histologically. All animals were killed when the tumor weight reached 2 g according to the Institute’s approved animal protocols.
Tumor measurement
Two axis (mm) of tumor (L, longest axis; W, shortest axis) were measured with the aid of a Vernier caliper. Tumor weight (mg) was estimated as a formula: tumor weight = ½ (L × W2). Tumor measurements were taken daily for the first 10 days and at least 3 times a week for the first 4 weeks of post-therapy and twice a week thereafter.
Cell culture and hypoxia growth studies
FaDu cells were maintained as monolayer cultures in RPMI 1640 medium supplemented with 10% fetal bovine serum. The cells were regularly tested and found to be free of mycoplasma by mycoplasma Plus PCR primer set (Stratagene, La Jolla, CA). Hypoxic conditions were applied to cells by changing to medium equilibrated with an atmosphere containing 0.5% O2 and placing the culture flasks into a hypoxia chamber (IN VIVO2 400, Ruskinn Technology Ltd., Cincinnati, OH) maintained at 0.5% O2. Cells were treated with MSA ± SN38 and further incubated for 24 h in the hypoxia chamber at 37°C.
Silencing of HIF-1α
Stable HIF-1α KD FaDu cells were generated using pSi-lencer 2.1-U6-hygro plasmid (Ambion) expressing HIF-1α shRNA controlled via the U6 promoter according to the manufacturer’s protocols. The HIF-1α shRNA- plasmid was kindly provided by Jensen, RL, University of Utah, Salt Lake City, UT [17]. The siRNA 1589 sequence (UUCAAGUUGGAAUUGGUAG) was used to create HIF-1α KD, along with the scrambled negative control, pSil2.1_hygro-Neg (AAUUAGCGUAGAUGUAAUGUG). Into a 6-well cell culture plate was seeded 1.5 × 105 cells and incubated overnight. Cells were transfected with 10 nmol shRNA plasmid using Lipofectamine RNAiMax (Invitrogen, Carlsbad, CA), and transfectants were selected with 300 μg/ml hygromycin in RPMI 1640. Several clones were selected and tested for their HIF-1α KD levels.
In vitro growth inhibition of parental, KD FaDu tumor cells by SN38 ± MSA
Four to five thousand cells were seeded in the 96-well plates and incubated overnight at 37°C under normoxic culture conditions. The concentrations of MSA that caused ~10% cell growth inhibition (IC10 = 1 μM) and SN38 that caused ~50% growth inhibition (IC50 = 0.1 μM) under normoxic conditions were used for cytotoxic studies. Cells were exposed to MSA for 24 h and SN38 for 2 h under normoxic and hypoxic conditions. For combination studies, cellular exposure to SN38 occurred during the last 2 h of 24 h exposure to MSA. Cells were washed three times, and fresh medium was added and further incubated for 24–48 h under normoxic and hypoxic conditions. Cell growth inhibition was determined by using the sulforhodamine (SRB) assay as described previously [18]. Briefly, cells were fixed with 10% trichloroacetic acid, washed, and stained with SRB. After removing the unbound dye, optical density was measured at 570 nm using a plate reader (model EL340; Bio Tek Instruments, Winooski, VT). Percent growth inhibition was calculated considering growth of untreated control cells as 100%. HIF-1α KD FaDu cells were tested for growth after SN38 treatment under normoxia and hypoxia conditions. HIF-1α shRNA-scramblednegativecells were used as control for determining the HIF-1α KD effects with cytotoxic agents.
Quantification of HIF-1α mRNA by RT–PCR
Cells were collected from the flask after treatment under normoxic and hypoxic conditions. To minimize the effect of exposure of the hypoxic cells to air during processing, all the procedures were performed in the cold except the trypsinization for the detachment of cells at 37°C for 2–3 min. For determining HIF-1α RNA levels, cDNA was prepared from the cells using the cells-to-CT Kit (Applied Biosystems, Austin, TX). For isolation of cDNA according to the manufacturer’s protocols, 0.5 × 106 cells were used. Briefly, cells were lysed with lysis buffer and used as the source for mRNA. The cDNA synthesizing master mix was prepared with the proportion of reagents, and cell lysate was added and reverse transcribed using the PCR machine. The cDNA was used for performing RT–PCR as per the procedure described [19].
Protein extraction and Western blot analysis
Tumors were collected and snap-frozen in liquid nitrogen, chopped into small pieces, and homogenized with a Polytron homogenizer using the lysis buffer (50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 0.5% SDS, 0.5% sodium deoxycholate, 0.5% Nonidet P40) with a protease inhibitor cocktail (Roche, Indianapolis, IN). The homogenate was centrifuged, and the supernatant was used for the protein measurement. To isolate protein, cells were washed with cold PBS and scrapped into cold PBS, keeping the flask on ice to avoid degradation of HIF-1α protein. Cells were pelleted by centrifugation at 4°C and further processed for the isolation of protein using the lysis buffer. Western blotting was performed by loading and separating 40 μg of protein in 4–20% gradient SDS–PAGE, followed by transfer to nitrocellulose membranes. Primary antibodies for HIF-1α (Upstate, Lake Placid, NY and R & D Systems, Minneapolis, MN) and VEGF (Santa Cruz Biotechnology Inc, Santa Cruz, CA) PHD2 and 3 (Abcam Inc, Cambridge, MA) were used with 1:500 dilution and incubated for 1 h, washed with phosphate buffer saline with Tween-20 (PBST) milk for 1 h, and further incubated with HRP-conjugated donkey anti-rabbit (Santa Cruz Biotechnology Inc., Santa Cruz, CA) secondary antibody for 1 h. Lumi-Light PLUS Western Blotting Kit (Roch, Indianapolis, IN) was used for detecting the proteins.
Measurement of ROS
Total ROS was measured using 5-(and 6-)carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA), C400 (Molecular Probes Inc., Molecular probes Invitrogen, Carlsbad, CA) according to manufacturer’s procedure. The released carboxy dichlorofluorescein (carboxy-DCF) after oxidation of carboxy-H2DCFDA was measured using a microplate spectrofluorometer. Briefly, cells at 50–60% confluence were incubated with 10 μM Carboxy-H2DCFDA for 30 min in 96-well plates at 37°C under normoxic conditions. After washing with PBS, cells were treated with and without MSA and incubated under hypoxic and normoxic conditions. Fluorescence was measured with excitation 480 nm and emission 530 nm. Data obtained without MSA treatment were used as normal values.
Immunohistochemistry
Specimens were fixed in 10% neutral-buffered formalin overnight and processed traditionally for paraffin blocks, and then conventional H & E staining was performed for histopathological evaluation. Immunohistochemical staining was carried out on 4- to 5-μm sections on charged slides after conventional deparaffinization and quenching of endogenous peroxidase with 3% H2O2. Casein (0.03%) in PBS-T (PBS with 500 μl/l Tween-20) for 30 min at room temperature was used for non-specific binding in each immunoassay. All incubations were done at room temperature unless otherwise specified. All immunohistochemical procedures were carried out using an automated immunostainer (Dako, Carpinteria, CA). Nuclear counter-staining was done with hematoxylin before being dehydrated, cleared, and cover slipped. All washing was done with PBS-T, and chromogen DAB (Dako) was used for 5 min for visualization. An isotype-matched negative control was used on a duplicate slide in place of the primary antibody at the same concentration as the primary antibody. All histopathological and immunohistological analysis, counting, and interpretation were done by a board certified and experienced pathologist (Károly Tóth, MD, PhD).
Detection of HIF-1α-targeted gene carbonic anhydrase IX (CAIX)
The HIF-1α transcriptionally regulated gene CAIX was used to detect hypoxic tumor cells [20]. Antigen retrieval was done with citrate buffer (pH 6.0) in a microwave for 10 min. The primary antibody M75 (20 μg/ml, gift from Dr. Pastorek, Institute of Virology, Slovak Academy of Sciences, Slovak Republick) was used for 90 min at room temperature. The Elite Universal Kit (Vector labs, Burlingame, CA) was used as the detection system as published earlier [21]. Semiquantitative assessment of the total proportion of multiple CAIX tumor cell foci was estimated after examining the entire section of each tumor and was expressed as a percentage of the whole viable tumor in the section. Tumors were cut and embedded to get the biggest cross section for examination. Since in most cases strong staining (cytoplasmic membrane) was present, further categorization of the intensity was not considered.
HIF-1α detection
For detecting HIF-1α in human tissues, we have developed and optimized a multilayer procedure implementing several improvements to increase sensitivity and decrease non-specific staining. Human squamous cell carcinoma xenograft (A253) was used as a known positive control, because this tumor contains well-differentiated areas without microvessels, and tumor cells in these fields are strongly positive for the hypoxia marker CAIX [21]. Therefore, tumor cell nuclei in these areas are expected to be positive for hypoxia and associated with high levels of HIF-1α. Normal human liver was used as a known negative control. Antigen retrieval was done with Target Retrieval Solution (TRS—Dako, Carpinteria CA) in a pressure cooker (Cell Marque Inc., Rocklin, CA) for 3 min according to the manufacturer’s protocol. Endogenous biotin was blocked with a biotin blocking kit (Dako). The primary monoclonal antibody, mouse anti-human anti-HIF-1α (H1 alpha 67) from Novus Biologicals (Littleton, CO) was applied at 0.4 μg/ml concentration overnight at 4°C. This anti-HIF-1α antibody was diluted in 2.5% goat serum in PBS-T (phosphate-buffered saline with 0.05% Tween-20). Incubation with goat versus mouse biotinylated secondary antibody (Jackson ImmunoResearch Labs, West Grove, PA) for 15 min was followed by Elite ABC reagent (Vector Labs) for 20 min. The amplification reagent from the Catalyzed Signal Amplification (CSA) System from Dako was then used at 1:35 dilution (PBS-T) on slides for 10 min. Streptavidin conjugated to horseradish peroxidase (Zymed/Invitrogen, San Francisco, CA) was used for 20 min as the last layer. The chromogen DAB (Dako) was used for 1 min to visualize the immunoreaction. All washes between the various steps were done with a PBS-T rinse. This was followed by 0.03% casein (PBS-T) for 5 min with a blow step before the next reagent was applied in order to considerably reduce background. In place of the primary antibody, an isotype match (mouse IgG2b also at 0.4 μg/ml) was placed on a duplicate slide as a negative control. Semiquantitative analysis of HIF-1α positivity was assessed by determining the microscopic fields positive for HIF-1α nuclear staining in the entire immunostained section of the tumor including perinecrotic areas. Strong specific nuclear (not cytoplasmic) immunostaining was present uniformly; therefore, classification of staining intensity was not necessary.
Vascular Endothelial Growth Factor-A (VEGF-A)
Vascular Endothelial Growth Factor-A was detected by the method published earlier [21] with the improvement of using the antigen retrieval with TRS in microwave for 10 min before immunostaining.
Detection of intratumoral microvessels using CD34
CD34 was used to detect endothelial cells and thus to visualize and count intratumoral microvessels. Antigen retrieval was not required for this antibody. Rat anti-mouse CD34 primary antibody (Abcam, Cambridge, MA) was applied to slides at 2 μg/ml for 1 h. It was followed by a biotinylated secondary antibody for 30 min and by the streptavidin–horseradish peroxidase complex (Zymed/Invitrogen) for 30 min. Rat IgG2a (2 μg/ml) as an isotype-matched control was used on a duplicate slide in place of the primary antibody as a negative control. All CD34 positive intratumoral microvessels were counted at 400× in each microscope field on the viable parts of the entire section without any selection. Single CD34-positive endothelial cells without any lumen formation were not counted. Microvessel density changes were expressed as a percentage of controls.
Statistical analysis
Statistical analysis was performed with the calculated mean values ± SD using GraphPad Prism software (La Jolla, CA) with unpaired two-tailed Student’s t test, and significance was set a P ≤ 0.05.
Results
MSC treatment and HIF-1α KD resulted in similar tumor growth inhibition, antiangiogenic activity, and inhibition of HIF-1α
Tumor growth inhibition was correlated with tumor microvessel density in MSC-treated parental and in HIF-1α KD tumors. HIF-1α KD FaDu cells were generated by transfecting HIF-1α shRNA, and stable KD cells were selected. The inhibition of HIF-1α mRNA was confirmed by RT–PCR in the stable KD cells (data not shown). The data in Fig. 1a indicate that growth of parental FaDu tumor xenografts treated with MTD (0.2 mg/mouse/day) of MSC, and untreated HIF-1α KD xenografts were similarly inhibited compared with untreated control (scrambled shRNA) tumors. The data in Fig. 1 also indicate that CD34 staining (Fig. 1b) and microvessel density (Fig. 1c) were similarly decreased in the parental tumor treated with MSC and untreated HIF-1α KD tumors. The results shown in Fig. 1d reveal that HIF-1α protein inhibition by MSC with parental tumors and HIF-1α KD tumors were similarly affected. Collectively, the data demonstrate that the anti-angiogenic, antitumor activity and HIF-1α inhibition of parental tumors treated with MSC and HIF-1α KD tumors were similar. These data support the hypothesis that HIF-1α is a critical target of MSC in vivo.
Fig. 1.

Effects of MSC (0.2 mg/mouse/day) and HIF-1α KD on FaDu tumor growth vasculature and HIF-1α protein in xenografts. HIF-1α KD results in growth inhibition of FaDu tumors, and the inhibition of HIF-1α was comparable to MSC-treated parental FaDu tumor, correlated with decreased microvessel density. a Tumor growth of FaDu control (scrambled), MSC-treated parental FaDu, and HIF-1α KD xenografts. b Representative Photomicrographs (200×) demonstrate the effects of MSC and HIF-1α KD on CD34 endothelial cell marker for angiogenesis evaluation. Arrows showing the microvessels stained with CD 34. c Effect of MSC and HIF-1α KD on microvessel density. The data are representative of 6–10 tumors of each group. d HIF-1α protein levels in FaDu xenografts with MSC treatments and HIF-1α KD. Scrambled shRNA-negative control tumors were used to compare HIF-1α KD effect
MSC decreases HIF-1α and its transcriptionally regulated genes VEGF and CAIX in FaDu xenografts
The data in Fig. 2 represent the levels of HIF-1α, VEGF, and CAIX expression in control, MSC treated, and KD FaDu tumor xenografts. Representative photomicrographs (Fig. 2a) of HIF-1α immunostaining show several tumor cell nuclei with HIF-1α expression (arrows) around a small necrotic area (N) in the control tumor (left upper panel). In contrast, the MSC-treated tumor does not contain HIF-1α-positive tumor cell nuclei around the necrosis (left middle panel) and no detectable HIF-1α expression in HIF-1α KD tumors (left bottom panel). Among the untreated tumor, 5 out of 6 contained microscopic HIF-1α-positive areas, when compared to 1 out of 6 MSC-treated tumors, and there was no tumor with HIF-1α expression among HIF-1α KD tumors (Fig. 2b). Similar to the HIF-1α expression, the HIF-1α transcriptionally regulated genes VEGF and CAIX expression was decreased with MSC treatment. Representative photomicrographs (Fig. 2a) of VEGF and CAIX immunostaining illustrates that both VEGF and CAIX positive, hypoxic (brown) cell fraction in the control (middle and right upper panel) was remarkably decreased after MSC treatment (middle and right middle panel) and was decreased or not detectable in HIF-1α KD tumors (middle and right bottom panel). Figure 2c shows that the average fraction of VEGF-positive tumor cells present in the untreated control (~20%) significantly decreased to about 10% found in MSC-treated and HIF-1α KD tumors. There was an average of 30% hypoxic fraction in the untreated control tumors compared with an average of 10% after MSC treatment, and CAIX-positive cells were not present in HIF-1α KD tumors as shown in Fig. 2d. In support of this finding, while the percentage of positive cells for CAIX was increased after irinotecan (100 mg/kg) treatment alone in FaDu xenografts, these cells were eliminated by pre-treatment with MSC followed by irinotecan (data not shown). HIF-1α KD resulted in inhibition of HIF-1α, VEGF, and CAIX expression, and the MSC treatment similarly reduced HIF-1α and its transcriptionally regulated genes VEGF and CAIX. These data support the hypothesis that HIF-1α is a critical target of MSC.
Fig. 2.

Immunohistochemical evaluation of the effects of MSC and HIF-1α KD on HIF-1α and its transcriptionally regulated genes VEGF and CAIX. Semiquantitative assessment of hypoxic tumor cells fraction indicates that MSC treatment results in significant reduction in hypoxic tumor cells and correspondingly inhibited the expression of the associated HIF-1α protein. a Representative photomicrographs (×200) demonstrate the perinecrotic numerous HIF-1α-positive tumor cell nuclei (arrows) are seen in the untreated tumor (left upper panel) compared to the MSC-treated (left middle panel) and the HIF-1α KD tumors (left lower panel) without HIF-1α positive, hypoxic tumor cells even around (arrow) the necrosis (N = necrosis). VEGF was expressed predominantly at the periphery of the tumors with much less or no expression at the center part (middle panels). Several VEGF-positive tumor cells (brown) are seen in the control (middle upper panel) when compared to MSC-treated (middle center panel) and to the HIF-1 KD tumors (middle lower panel) containing much less VEGF-positive tumors. A large number of CAIX-positive (brown) tumor cells in FaDu tumor xenograft control (right upper panel) when compared to MSC-treated and HIF-1α KD tumors containing much less (right middle panel) and no CAIX-positive hypoxic tumor cells (right lower panel), respectively. b Graphic presentation of HIF-1α-positive tumors in control, MSC-treated parental, and untreated HIF-1α KD, FaDu xenografts. c and d Quantitation of tumor cell fraction based on VEGF (c) and CAIX (d) immunostaining indicates that MSC treatment and HIF-1α KD resulted in significant reduction in VEGF- and CAIX-positive tumor cells
Regulation of HIF-1α, VEGF, and PHDs by MSA
Data in Fig. 3a demonstrate that inhibition of HIF-1α protein by MSA is time-dependent, evident of effective inhibition at 24-h exposure of hypoxic cells to MSA. The data in Fig. 3b indicate that inhibition of HIF-1α by MSA corresponded with inhibition of HIF-1α transcriptionally regulated gene VEGF, a proangiogenic marker. The data in Fig. 3c indicate that treatment with MSA of normoxic and hypoxic cells did not result in inhibition of HIF-1α mRNA. Thus, MSC does not appear to inhibit the HIF-1α synthesis.
Fig. 3.
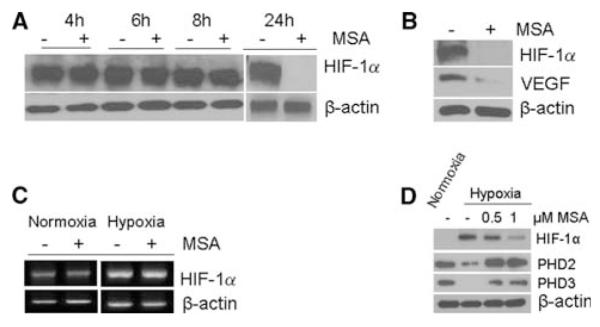
Effects of selenium treatment on the expression of HIF-1α, PHDs, and HIF-1α transcriptionally regulated gene VEGF. Selenium treatment results in inhibition of HIF-1α, VEGF and stabilizes PHDs. FaDu cells were treated with 0.5 and 1 μM MSA under normoxia and hypoxia. Cells were harvested, processed for protein and RNA extraction. a MSA effect on HIF-1α synthesis or accumulation. FaDu cells treated with and without MSA and exposed to hypoxia for different time intervals from 4 to 24 h. HIF-1α protein levels were determined by Western blot. b HIF-1α and VEGF expression in hypoxic cells treated with and without MSA (1 μM) for 24 h, and protein was determined by Western blot. c RT–PCR analysis of HIF-1α mRNA in FaDu cells with and without MSA under hypoxia and normoxia d Downregulation of PHD2 and 3 expression in FaDu cells under hypoxia and MSA treatment restored the normal levels. Hypoxic cells treated with 0.5 and 1 μM MSA for 24 h. PHD2 and 3 levels were determined by Western blot analysis. β-Actin is used as loading control
To characterize a possible regulator of HIF-1α by PHDs, the effects of MSA on the expression levels of PHD2 and PHD3 were evaluated and compared with the effects on HIF-1α in cells growing under normoxic (20% O2) and hypoxic (0.5% O2) conditions. The results outlined in Fig. 3d demonstrate that while PHD2 and PHD3 are expressed in normoxic cells, low levels of PHD2 and no detectable levels of PHD3 were observed in cells grown under hypoxic conditions. Treatment of normoxic cells with MSA did not alter the expression levels of the PHDs (data not shown). Under hypoxia, treatment with MSA resulted in a dose-dependent inhibition of HIF-1α. In contrast, PHD2 and PHD3 were upregulated in hypoxic cells treated with MSA. Collectively, these results demonstrated that HIF-1α is a target of MSA, and its inhibition could be mediated through the upregulation of PHD2 and PHD3 under hypoxic conditions.
MSC downregulated the hypoxia-induced reactive oxygen species (ROS)
Studies were carried out to determine the relationship between HIF-1α and ROS under conditions of hypoxia and whether ROS regulation by MSA results in upregulation of PHDs. Data outlined in Fig. 4 demonstrate that hypoxia induces ROS, similar results to the upregulation of HIF-1α as outlined in Fig. 3a, and treatment with MSA resulted in downregulation of ROS to a level comparable with normoxic controls treated with and without MSA. This is a new and novel finding, which suggests the possibility that inhibition of ROS by MSA resulting in stabilization of PHDs is an important pathway in the regulation of HIF-1α.
Fig. 4.

Effect of MSA on ROS under hypoxia and normoxia. MSA downregulates the hypoxia-induced ROS. FaDu cells were treated with and without MSA (1 μM) under normoxia and hypoxia. The ROS levels in terms of fluorescence intensity were measured with plate reader. The intensity of fluorescence was normalized with control
Hypoxic cells are resistant to SN38, and HIF-1α inhibition resulted in reversal of resistance
The effect of MSA and SN38 alone and in combination on the growth of FaDu parental tumor cells under normoxic and hypoxic conditions was evaluated, and the results are shown in Fig. 5. While 1 μM MSA was relatively non-toxic against normoxic cells, this concentration produced 50% inhibition of tumor cells grown under hypoxic conditions. In contrast, normoxic cells were sensitive to SN38 with an IC50 of 0.1 μM, and this concentration resulted in ~30% growth inhibition under hypoxic conditions. MSA in combination with SN38 treatment further enhanced the cytotoxic effects under hypoxic conditions resulted in 90% growth inhibition. In contrast, the combination did not enhance cytotoxic effects under normoxic conditions. Similar to MSA treatment, HIF-1α KD resulted in enhanced cytotoxic effects of SN38 under hypoxic conditions but not under normoxic conditions. Thus, the hypoxic cells overexpressing HIF-1α are resistant to SN38, and treatment with MSA resulted in sensitization and reversal of resistance to SN38.
Fig. 5.

Effect of MSA and SN-38 alone and in combination on the growth of FaDu tumor cells in normoxic and hypoxic conditions in vitro. Hypoxic cells are highly sensitive to MSA and resistant to SN38 compared to normoxic cells. HIF-1α KD and MSA treatment sensitizes the cells to SN38 treatment under hypoxia. MSA reverses SN38 resistance in hypoxic tumors. Normoxic and hypoxic parental FaDu and HIF-1α KD cells were treated with 1 μM of MSA for 24 h and 0.1 μM (IC50) of SN38 for 2 h alone and in sequential combination with cellular exposure to SN38 during the last 2 h of 24 h pre-treatment with MSA. Cells were washed, and fresh medium was added and further incubated for 24–48 h for assessment of tumor growth inhibition in vitro. Scrambled shRNA FaDu cells were used as negative control for HIF-1α KD FaDu cells. The experiments were repeated at least three times
HIF-1α knockdown resulted in enhanced therapeutic efficacy of irinotecan against FaDu xenografts
To demonstrate that inhibition of HIF-1α correlates with enhanced therapeutic efficacy of irinotecan, the antitumor activity of irinotecan in combination with MSC against parental FaDu tumor xenografts and irinotecan alone against KD tumor xenografts was evaluated. The results summarized in Fig. 6 support the hypothesis that effective and sustained inhibition of HIF-1α by MSC is necessary, but not sufficient for optimal therapeutic outcome. However, the inhibition of HIF-1α sensitizes tumor cells to the subsequent treatment with the anticancer drug irinotecan. This inhibition of HIF-1α is necessary and sufficient for the effective treatment outcome in combination with chemotherapy. The data in Fig. 6 are an outline of the growth of individual mice bearing parental or KD FaDu tumor cells treated as follows: Saline—control and KD tumors; MSC (0.2 mg/mouse/day × 28 days)—parental and KD tumors; Irinotecan (100 mg/kg/week × 4)—parental and KD tumors; MSC + irinotecan—parental tumors. The data presented in Fig. 6 B and E demonstrate that the growth of MSC-treated parental tumors and KD tumors was equally significantly inhibited by ~30% compared to the untreated control (Fig. 6a). Mice bearing tumors were killed at different time points when the tumor reached around 2 g, according to the institutional guidelines. Treatment of parental tumors with MTD dose of irinotecan resulted in significant growth inhibition of FaDu tumor with 50% of treated mice having long-term survival (90 days post-termination of therapy) (Fig. 6c). In contrast, treatment of parental tumors with MSC + irinotecan and KD tumors with irinotecan alone resulted in highly effective treatment achieving 100% and 90% of mice having long-term survival, respectively (Fig. 6d, g).
Fig. 6.

Antitumor activity of irinotecan against parental and HIF-1α KD FaDu xenografts. Therapeutic efficacy between MSC and irinotecan against parental FaDu tumor xenografts overexpressing HIF-1α is comparable to HIF-1α KD xenografts treated with irinotecan alone, a direct implication of HIF-1α as a target of MSC. The left panel represents antitumor activity of oral MSC (0.2 mg/mouse/d × 28) (b), irinotecan (100 mg/kg/week × 4, i.v.) (c) alone and in combination (d) against parental FaDu xenografts. Right panel represents the antitumor activity of MSC (f) and irinotecan alone (g) against HIF-1α KD FaDu xenografts. Tumor transplantation (~50 mg pieces) was initiated on day –7, and MSC was administered orally from day –7 to 21. Irinotecan was administered i.v. on day 0, 7, 14, and 21 when the treatment was concluded. Kinetics of tumor response in individual tumor was assessed daily during treatment and thrice a week thereafter for up to 120 days. The data are representative of an experiment with five mice (10 tumors) for each group, and the tumor growth curves show the individual tumor weight changes. The calculated percent tumor-free survival cure (h) with parental tumors represents the average of 10 experiments, and the data with HIF-1α KD represent the average of the two separate experiments with 10 tumors for each single experimental group
The data presented in Fig. 6h summarize the experiments and show the percent of long-term survival (cure) of parental FaDu xenograft tumors treated with irinotecan + MSC and FaDu KD xenografts treated with irinotecan alone. Treatment of parental tumors with irinotecan alone resulted in ~32% long-term survival. In contrast, the percentages of long-term survival that are considered cure were increased to 100% and >90% when HIF-1α was inhibited either by MSC or by KD. Collectively, the data support the hypothesis that effective and sustained inhibition of HIF-1α by MSC translates into increased therapeutic efficacy in vivo against tumor cells expressing HIF-1α.
Discussion
Under hypoxic conditions, HIF-1α transactivates several genes [10] that promote cell survival and reduce the efficacy of the cytotoxic agents [22]. The surrogative tumor hypoxia microenvironment markers such as HIF-1α [23], CAIX [24], VEGF [25], and Glut-1 [26] correlate with low oxygen conditions, and their increased expression has been linked to poor outcome of chemotherapy in various cancers [27]. However, the expression of CAIX, VEGF, and Glut-1 was transcriptionally regulated by HIF-1α under hypoxic conditions [10]. Hypoxic tumors are typically characterized with increased angiogenesis, chaotic vasculature, and these characteristics result in decreased drug delivery [21].
Tumor hypoxia is known to increase resistance to cytotoxic agents [28]. The study by Brown and Cowen [29] documents the inhibition of HIF-1α using a small molecule or genetic approach could overcome drug resistance. There are several small molecule inhibitors of HIF-1α, many of them were identified by the National Cancer Institute (NCI) small molecule library and some of these are in clinical trial as reviewed by Adamski et al. [22]. Of these, geldalamycin (17AAG), topotecan, and PX-478 appear to show promise. There are several natural compounds including barbeine, manassantin B1, manassantin A, psedolaric acid B, bisphenol A, laurenditerpinol, and 4-O-methylsaucernol shown to inhibit HIF-1α but their mechanisms have not been fully investigated [30]. Recent studies [31] reported N-acetylcysteine (NAC) to be a potential inhibitor of HIF-1α associated with antitumor activity. Semenza and his colleagues screened more than 3,000 Food and Drug Administration approved drugs to find HIF-1α inhibitors and found 20 potent inhibitors. Among these 20 inhibitors, digoxin and other cardiac glycosides [32] as well as the anthracycline chemotherapeutic agents doxorubicin, daunorubicin [33] were identified as inhibitors of HIF-1α and demonstrated that HIF-1α inhibition resulted in growth inhibition of tumor. The molecular mechanism of HIF-1α inhibition may be drug-specific. Digoxin and related compounds inhibit HIF-1α synthesis and anthracyclines inhibit HIF-1α by blocking its binding to DNA. These studies support the development of HIF-1α inhibitors in cancer therapy.
The HIF-1α-targeted approach for cancer therapy is promising due to its key role in the adaptation of cells to the hypoxic tumor microenvironment by enhancing the angiogenesis [34] in solid tumors and upregulating genes to increase anaerobic glycolysis that prevent apoptosis [35] and support the survival of tumor cells. Gene-specific inhibition of HIF-1α by siRNA has been shown to reduce cell growth and increase apoptosis in pancreatic cancer cells [36], squamous cell carcinoma [37], gastric cancer [38], colon cancer [39], and gliomas [17].
We have demonstrated that MSC treatment resulted in inhibition of proangiogenic markers including HIF-1α in FaDu xenografts [3] and did not emphasize the significance of HIF-1α or investigate the mechanism of HIF-1α regulation by MSC. This observation provided the basis for focusing on mechanism of MSC targeting on HIF-1α. To further investigate that the inhibition of HIF-1α is the cause for the observed therapeutic synergy with anticancer drug, HIF-1α-stable KD FaDu cells were developed. We are hypothesizing that inhibition of HIF-1α by MSC or by KD should result in similar treatment response to irinotecan. Indeed, implantation of FaDu HIF-1α KD cells resulted in significant growth inhibition similar to the parent line treated with MSC (Fig. 1a) Furthermore, treatment of FaDu HIF-1α KD tumors with irinotecan resulted in complete response rates similar to that of parent tumors treated with the combination of MSC and irinotecan (Fig. 6). Also, the tumor growth inhibition by MSC and KD was correlated with the decrease in microvesssel density, and HIF-1α downregulation indicated that HIF-1α inhibition might be the cause for the decreased microvessel density. These results are consistent with the hypothesis that the effect of MSC on HIF-1α is an important element in the therapeutic efficacy observed.
In this report, we have demonstrated that treatment with small molecule organic selenium (MSC) results in degradation of HIF-1α and enhances the efficacy of irinotecan against hypoxic cells in vitro and xenografts. Furthermore, we have shown that downregulation of ROS, stabilization of PHD2 and PHD3 by selenium as the possible effects responsible for the degradation of HIF-1α. The decrease of ROS is responsible for the HIF-1α inhibition. Hypoxic induction of ROS has been implicated in the inhibition of PHD and accumulation of HIF-1α under hypoxia. Accordingly, MSC being an antioxidant could inhibit HIF-1α accumulation by decreasing the production of ROS. Selenium is in a clinical trial at our institute, demonstrating that supranutritional, high doses of 7,200 μg of selenomethionine per day was well tolerated in cancer patients without toxicity [40]. This high dose was required to achieve a plasma concentration of 20–30 μM: a concentration determined essential for therapeutic efficacy in the in vivo preclinical model in our laboratory (Rustum’s unpublished data). The recently reported randomized, controlled selenium (SLM) and vitamin E cancer prevention trial (SELECT) determined that SLM administration of 200 μg did not decrease the risk of prostrate cancer [6]. The results generated in our laboratory clearly demonstrated that MSC at MTD (0.2 mg/mouse/day) produced significant antitumor response ~30% tumor growth inhibition [2]. Therapeutic efficacy was obtained only when combined with chemotherapy against advanced tumor xenografts [1]. The degree of response is MSC or SLM dose-dependent (Rustum’s unpublished data). This finding is partially consistent with the recent editorial comment by Platz [41], and an article published by Chan et al. [42] indirectly suggest that selenium activity may vary according to the type of selenium used, dose administered, and the levels of antioxidants due to manganese superoxide dismutase (SOD2) genotype.
HIF-1α inhibition by various anticancer compounds is mediated by different mechanism, mainly inhibiting the synthesis of HIF-1α. In contrast to this, we have demonstrated HIF-1α degradation by selenium as shown in Fig. 3a indicating there is no inhibitory effect on HIF-1α synthesis during 8 h of treatment. The degradation was noticed after16–18 h (data not shown), and complete degradation was observed after 24 h, indicating that selenium could be degrading the HIF-1α instead of inhibiting the synthesis. To further investigate the mechanism of HIF-1α degradation, we have determined the MSA effect on PHD2 and 3, the regulators of HIF-1α hydroxylation on proline molecules before proteosome-mediated degradation. The induced expression of PHD 2 and 3 has been reported under hypoxic conditions [43]. In contrast, the degradation of PHDs by E3 ubiquitin ligases Siah1a/2 under hypoxic conditions has also been reported [44, 45]. As shown in the Fig. 3d, MSA treatment resulted in stabilization of PHD2 and 3 that were inhibited under hypoxia. These stabilized PHDs may be involved in HIF-1α degradation.
In our model system, FaDu cells in vitro express HIF-1α only under hypoxic conditions. In normoxia, HIF-1α is not expressed, and MSA has no synergistic interaction with SN38 against different human tumor cell lines including FaDu. However, under hypoxia, FaDu cells are resistant to SN38, more sensitive to selenium (MSA), and display increased sensitivity to SN38 in the presence of MSA (Fig. 5). Furthermore, MSA treatment resulted in degradation of HIF-1α in FaDu cells under hypoxic culture conditions (Fig. 3) and sensitize the cells to SN38 treatment that led to the increased cytotoxic effects of SN38 (Fig. 5). This clearly indicates that SN38 in combination with MSA is effective under hypoxia. In contrast, under normoxic conditions with no detectable HIF-1α, no enhanced cytotoxic effect was observed with the combination (Fig. 5).
Despite several attempts in the past to reproduce the therapeutic efficacy of SN38 combined with MSA under normoxic conditions, we were not able to demonstrate the synergistic effect that we achieved in vivo with several tumor xenografts. Here, we are reporting for the first time that MSA enhanced the cytotoxic effect of SN38 under hypoxic conditions. Others have also reported favorable drug interactions, which occur only under hypoxia [39]. An earlier report of the synergestic activity of MSA in combination with docetaxel in a prostrate cancer cell model [18] could be due to their dependence on HIF-1α for development [46, 47], and prostate cancer cells that grow under normoxic culture conditions express HIF-1α [48]. The contribution of HIF-1α to cell survival was demonstrated by the reduced growth rate of FaDu HIF-1α KD under hypoxic conditions (Fig. 5).
In conclusion, MSC degrades HIF-1α through down-regulation of ROS and stabilization of PHD2 and 3. HIF-1α KD xenografts are highly sensitive to irinotecan in vivo like parental tumor xenografts treated with combination of MSC and irinotecan. This indicates HIF-1α is a critical therapeutic target, and its inhibition by MSC is a critical mechanism of sensitization of tumor cells to subsequent treatment with irinotecan. These findings suggest that MSC could potentially be useful in cancer therapy in combination with anticancer agents to which hypoxic tumors may otherwise be resistant.
Acknowledgments
This study was supported by a grant from National Cancer Institute 1R21CA133682-01A2 (S. Chintala) and a Comprehensive Cancer Center Support grant CA 016056 from National Cancer Institute, Bethesda, Maryland, USA. We thank Dr. Harry Slocum for the review of this manuscript, Dr. Jaromir Pastorek for providing antibody M75 for CAIX studies, and Rebecca Dean for technical support.
Footnotes
Conflict of interest statement None.
References
- 1.Cao S, Durrani FA, Rustum YM. Selective modulation of the therapeutic efficacy of anticancer drugs by selenium containing compounds against human tumor xenografts. Clin Cancer Res. 2004;10:2561–2569. doi: 10.1158/1078-0432.ccr-03-0268. [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharya A, Seshadri M, Oven SD, et al. Tumor vascular maturation and improved drug delivery induced by methylselenocysteine leads to therapeutic synergy with anticancer drugs. Clin Cancer Res. 2008;14:3926–3932. doi: 10.1158/1078-0432.CCR-08-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin MB, Li ZR, Toth K, et al. Potentiation of irinotecan sensitivity by Se-methylselenocysteine in an in vivo tumor model is associated with downregulation of cyclooxygenase-2, inducible nitric oxide synthase, and hypoxia-inducible factor 1alpha expression, resulting in reduced angiogenesis. Oncogene. 2006;25:2509–2519. doi: 10.1038/sj.onc.1209073. [DOI] [PubMed] [Google Scholar]
- 4.Fleet JC. Dietary selenium repletion may reduce cancer incidence in people at high risk who live in areas with low soil selenium. Nutr Rev. 1997;55:277–279. doi: 10.1111/j.1753-4887.1997.tb01617.x. [DOI] [PubMed] [Google Scholar]
- 5.Clark LC, Combs GF, Jr, Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276:1957–1963. [PubMed] [Google Scholar]
- 6.Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miki K, Xu M, Gupta A, et al. Methioninase cancer gene therapy with selenomethionine as suicide prodrug substrate. Cancer Res. 2001;61:6805–6810. [PubMed] [Google Scholar]
- 8.Sinha R, Said TK, Medina D. Organic and inorganic selenium compounds inhibit mouse mammary cell growth in vitro by different cellular pathways. Cancer Lett. 1996;107:277–284. doi: 10.1016/0304-3835(96)04373-x. [DOI] [PubMed] [Google Scholar]
- 9.Patiar S, Harris AL. Role of hypoxia-inducible factor-1alpha as a cancer therapy target. Endocr Relat Cancer. 2006;13(Suppl 1):S61–S75. doi: 10.1677/erc.1.01290. [DOI] [PubMed] [Google Scholar]
- 10.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 11.Liu L, Ning X, Sun L, et al. Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 2008;99:121–128. doi: 10.1111/j.1349-7006.2007.00643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Unruh A, Ressel A, Mohamed HG, et al. The hypoxiainducible factor-1 alpha is a negative factor for tumor therapy. Oncogene. 2003;22:3213–3220. doi: 10.1038/sj.onc.1206385. [DOI] [PubMed] [Google Scholar]
- 13.Chandel NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 14.Callapina M, Zhou J, Schmid T, et al. NO restores HIF-1αlpha hydroxylation during hypoxia: role of reactive oxygen species. Free Radic Biol Med. 2005;39:925–936. doi: 10.1016/j.freeradbiomed.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 15.Berra E, Benizri E, Ginouves A, et al. HIF prolylhydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1αlpha in normoxia. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brahimi-Horn MC, Chiche J, Pouyssegur J. Hypoxia signalling controls metabolic demand. Curr Opin Cell Biol. 2007;19:223–229. doi: 10.1016/j.ceb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Gillespie DL, Whang K, Ragel BT, et al. Silencing of hypoxia inducible factor-1alpha by RNA interference attenuates human glioma cell growth in vivo. Clin Cancer Res. 2007;13:2441–2448. doi: 10.1158/1078-0432.CCR-06-2692. [DOI] [PubMed] [Google Scholar]
- 18.Azrak RG, Frank CL, Ling X, et al. The mechanism of methylselenocysteine and docetaxel synergistic activity in prostate cancer cells. Mol Cancer Ther. 2006;5:2540–2548. doi: 10.1158/1535-7163.MCT-05-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chintala S, Li W, Lamoreux ML, et al. Slc7a11 gene controls production of pheomelanin pigment and proliferation of cultured cells. Proc Natl Acad Sci U S A. 2005;102:10964–10969. doi: 10.1073/pnas.0502856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pastorekova S, Ratcliffe PJ, Pastorek J. Molecular mechanisms of carbonic anhydrase IX-mediated pH regulation under hypoxia. BJU Int. 2008;101(Suppl 4):8–15. doi: 10.1111/j.1464-410X.2008.07642.x. [DOI] [PubMed] [Google Scholar]
- 21.Bhattacharya A, Toth K, Mazurchuk R, et al. Lack of microvessels in well-differentiated regions of human head and neck squamous cell carcinoma A253 associated with functional magnetic resonance imaging detectable hypoxia, limited drug delivery, and resistance to irinotecan therapy. Clin Cancer Res. 2004;10:8005–8017. doi: 10.1158/1078-0432.CCR-04-1306. [DOI] [PubMed] [Google Scholar]
- 22.Adamski JK, Estlin EJ, Makin GW. The cellular adaptations to hypoxia as novel therapeutic targets in childhood cancer. Cancer Treat Rev. 2008;34:231–246. doi: 10.1016/j.ctrv.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Haugland HK, Vukovic V, Pintilie M, et al. Expression of hypoxia-inducible factor-1alpha in cervical carcinomas: correlation with tumor oxygenation. Int J Radiat Oncol Biol Phys. 2002;53:854–861. doi: 10.1016/s0360-3016(02)02815-8. [DOI] [PubMed] [Google Scholar]
- 24.Loncaster JA, Harris AL, Davidson SE, et al. Carbonic anhydrase (CA IX) expression, a potential new intrinsic marker of hypoxia: correlations with tumor oxygen measurements and prognosis in locally advanced carcinoma of the cervix. Cancer Res. 2001;61:6394–6399. [PubMed] [Google Scholar]
- 25.Cvetkovic D, Movsas B, Dicker AP, et al. Increased hypoxia correlates with increased expression of the angiogenesis marker vascular endothelial growth factor in human prostate cancer. Urology. 2001;57:821–825. doi: 10.1016/s0090-4295(00)01044-x. [DOI] [PubMed] [Google Scholar]
- 26.Airley R, Loncaster J, Davidson S, et al. Glucose transporter glut-1 expression correlates with tumor hypoxia and predicts metastasis-free survival in advanced carcinoma of the cervix. Clin Cancer Res. 2001;7:928–934. [PubMed] [Google Scholar]
- 27.Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26:225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 28.Teicher BA. Hypoxia and drug resistance. Cancer Metastasis Rev. 1994;13:139–168. doi: 10.1007/BF00689633. [DOI] [PubMed] [Google Scholar]
- 29.Brown LM, Cowen RL, Debray C, et al. Reversing hypoxic cell chemoresistance in vitro using genetic and small molecule approaches targeting hypoxia inducible factor-1. Mol Pharmacol. 2006;69:411–418. doi: 10.1124/mol.105.015743. [DOI] [PubMed] [Google Scholar]
- 30.Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today. 2007;12:853–859. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Gao P, Zhang H, Dinavahi R, et al. HIF-dependent anti-tumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230–238. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H, Qian DZ, Tan YS, et al. Digoxin and other cardiac glycosides inhibit HIF-1αlpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105:19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee K, Qian DZ, Rey S, et al. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci USA. 2009;106:2353–2358. doi: 10.1073/pnas.0812801106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Tang N, Wang L, Esko J, et al. Loss of HIF-1αlpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell. 2004;6:485–495. doi: 10.1016/j.ccr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 35.Erler JT, Cawthorne CJ, Williams KJ, et al. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol. 2004;24:2875–2889. doi: 10.1128/MCB.24.7.2875-2889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mizuno T, Nagao M, Yamada Y, et al. Small interfering RNA expression vector targeting hypoxia-inducible factor 1 alpha inhibits tumor growth in hepatobiliary and pancreatic cancers. Cancer Gene Ther. 2006;13:131–140. doi: 10.1038/sj.cgt.7700871. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Q, Zhang ZF, Rao JY, et al. Treatment with siRNA and antisense oligonucleotides targeted to HIF-1αlpha induced apoptosis in human tongue squamous cell carcinomas. Int J Cancer. 2004;111:849–857. doi: 10.1002/ijc.20334. [DOI] [PubMed] [Google Scholar]
- 38.Stoeltzing O, McCarty MF, Wey JS, et al. Role of hypoxiainducible factor 1alpha in gastric cancer cell growth, angiogenesis, and vessel maturation. J Natl Cancer Inst. 2004;96:946–956. doi: 10.1093/jnci/djh168. [DOI] [PubMed] [Google Scholar]
- 39.Pencreach E, Guerin E, Nicolet C, et al. Marked activity of irinotecan and rapamycin combination toward colon cancer cells in vivo and in vitro is mediated through cooperative modulation of the mammalian target of rapamycin/hypoxia-inducible factor-1{alpha} axis. Clin Cancer Res. 2009;15:1297–1307. doi: 10.1158/1078-0432.CCR-08-0889. [DOI] [PubMed] [Google Scholar]
- 40.Fakih MG, Pendyala L, Brady W, et al. A Phase I and pharmacokinetic study of selenomethionine in combination with a fixed dose of irinotecan in solid tumors. Cancer Chemother Pharmacol. 2008;62:499–508. doi: 10.1007/s00280-007-0631-4. [DOI] [PubMed] [Google Scholar]
- 41.Platz EA. Selenium, genetic variation, and prostate cancer risk: epidemiology reflects back on selenium and vitamin E cancer prevention trial. J Clin Oncol. 2009;27:3569–3572. doi: 10.1200/JCO.2009.22.2117. [DOI] [PubMed] [Google Scholar]
- 42.Chan JM, Oh WK, Xie W, et al. Plasma selenium, manganese superoxide dismutase, and intermediate- or high-risk prostate cancer. J Clin Oncol. 2009;27:3577–3583. doi: 10.1200/JCO.2008.18.8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stiehl DP, Wirthner R, Koditz J, et al. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen-sensing system. J Biol Chem. 2006;281:23482–23491. doi: 10.1074/jbc.M601719200. [DOI] [PubMed] [Google Scholar]
- 44.Nakayama K, Frew IJ, Hagensen M, et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell. 2004;117:941–952. doi: 10.1016/j.cell.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 45.Moller A, House CM, Wong CS, et al. Inhibition of Siah ubiquitin ligase function. Oncogene. 2009;28:289–296. doi: 10.1038/onc.2008.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong H, Semenza GL, Simons JW, De Marzo AM. Up-regulation of hypoxia-inducible factor 1alpha is an early event in prostate carcinogenesis. Cancer Detect Prev. 2004;28:88–93. doi: 10.1016/j.cdp.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 47.Kimbro KS, Simons JW. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr Relat Cancer. 2006;13:739–749. doi: 10.1677/erc.1.00728. [DOI] [PubMed] [Google Scholar]
- 48.Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]