

Abstract
Background
Reactivation tuberculosis (TB) arises from those who are latently infected or from those who have been previously treated. The mechanism of reactivation TB in either situation is not well understood. A 13-gene mce1 operon of M. tuberculosis was previously shown to be associated with latent infection in mice, and may also play a role in reactivation.
Methods
We tested mce1 operon Mycobacterium tuberculosis mutants in a Cornell mouse model to examine disease progression and reactivation.
Results
In BALB/c mice, the wild type, mce1 operon mutant, and mce1R (negative transcriptional regulator of the mce1 operon) mutant M. tuberculosis strains were equally susceptible to orally administered isoniazid and pyrazinamide. However, after cessation of the treatment, the mce1R mutant rapidly and progressively proliferated in mouse lungs and spleen, while the other strains remained latent. The reactivation of the mce1R mutant was associated with disease progression in the mouse lungs.
Conclusions
This observation demonstrates that the constitutive expression of the mce1 genes by M. tuberculosis in latent state can cause reactivation TB. The constitutive expression of the mce1 genes in the mce1R mutant may allow this mutant to maintain its lipid metabolism, enabling it to survive long term and proliferate inside granulomas.
Keywords: Reactivation tuberculosis, mce1 operon, Mycobacterium tuberculosis, Cornell mouse model
INTRODUCTION
A large proportion of approximately 8 million new cases of tuberculosis (TB) arise from those who have latent TB infection (LTBI) [1]. The lifetime risk of reactivation TB in those with LTBI with no underlying medical conditions is 2-23%, but the risk increases to 5 to 15% annually in persons coinfected with HIV [2, 3]. Also, many of those who develop reactivation TB have a history of previous treatment for TB, often incomplete [4]. Efforts to prevent reactivation TB are hampered by our limited understanding of the mechanism of LTBI and progression to active disease.
One major obstacle to studying LTBI and reactivation TB has been the lack of an ideal animal model. Two murine models of latent M. tuberculosis infection are recognized. While neither of these models can be said to truly represent the human latent or reactivation TB, they have provided useful information [5-8]. In the first model, sometimes referred to as the low-dose model, mice are infected aerogenically with 5-10 colony forming units (cfu) of M. tuberculosis. The pulmonary bacterial burden stabilizes at around ~3 to 4 log10 cfu, which is maintained for about 15-18 months [5, 8]. After this point, the infection reactivates and the mice die. Flynn et al. have shown using this model that reactive nitrogen intermediates as well as TNF-α play a role in preventing reactivation disease [7, 9]. There are variations of this model that use different aerogenic inoculum doses to accelerate the disease outcome [10].
The other widely used mouse model to study latent M. tuberculosis infection is the Cornell model [11-14]. Scanga et al. reported on the utility of several versions of the Cornell model and concluded that it is highly dependent upon the parameters used to establish latency and that each version has certain limitations [8]. The Cornell model has the advantage of achieving very low or undetectable numbers of bacilli and maintaining those low levels for many weeks; however, it has the disadvantage of artificially inducing latency with antibiotics [8]. This model may be more akin to those human patients who remain latently infected after completing treatment for active disease.
The genome of M. tuberculosis contains four related copies of a cluster of genes called mce operons (mce1-4) [15]. A phylogenomics analysis of the operons shows them to encode possible ATP-binding cassette (ABC) transporters [16]. We previously reported that the disruption of the mce1 renders the M. tuberculosis mutant hypervirulent in BALB/c mice and unable to stimulate a Th1-type immune response [17]. The mce1 operon genes are negatively regulated by Mce1R in intracellular M. tuberculosis [18]. An M. tuberculosis mutant disrupted in mce1R constitutively expresses the mce1 genes in vivo and causes accelerated immunopathologic response in the infected animal [19].
Based on above observations, we used the Cornell murine model of latent infection to examine the outcome of latent infection with the mce1 operon and mce1R mutants. Surprisingly, each of these mutants caused an unexpected course of infection in this model, and provided new insights into the mechanism of reactivation disease that had not been previously considered.
MATERIALS AND METHODS
Animals
Eight-to 10-week-old female BALB/c mice (Charles River, Rockland, Mass.) were maintained and studied in a Biosafety Level-3 (BL3) animal facility at the University of California, Berkeley.
Bacterial strains and growth conditions
M. tuberculosis H37Rv wild type (ATCC 25618), M. tuberculosis H37Rv mce1 operon mutant, M. tuberculosis H37Rv mce1R mutant, complemented M. tuberculosis H37Rv mce1R mutant, and Erdman wild type were grown in Middlebrook 7H9 broth supplemented with 10% ADC (albumin-dextrose-catalase, Becton Dickinson), 0.2% glycerol and 0.05% Tween 80 or on Middlebrook 7H11 agar containing OADC (oleic acid–albumin-dextrose-catalase supplement, Becton Dickinson), 0.5% glycerol and the antifungal agent cycloheximide (100 mg/ml) (Sigma-Aldrich). The two mutant strains (mce1 operon mutant and mce1R mutant) were generated from M. tuberculosis H37Rv background by homologous recombination with a two-step counter-selection strategy as described in detail previously [17, 18, 20].
Infection and antibiotic treatment of mice
In the initial set of experiments, mice were infected via tail vein with approximately 5 × 105 cfu mce1 operon and mce1R mutant bacilli following the traditional Cornell model. After 4 weeks of infection, mice were given isoniazid (INH, 100 μg/ml) and pyrazinamide (PZA, 15 mg/ml) in drinking water ad libitum for 4 weeks. After a period of 4 weeks, some of the mice were treated with dexamethasone 0.08 mg/day intraperitoneally 6 days a week for 20 weeks. In the second set of experiments, mice were infected via tail vein with a lower dose (5 × 102 cfu per animal). They were otherwise treated similarly to those of the first set of experiments. In both experiments, mice were followed for clinical symptoms, organ cfu determination, and histology after cessation of the antibiotics.
When we observed an unexpected response with the mce1R mutant in the initial set of experiments, we studied this mutant further with the aerosol infection route. Mice were infected with approximately 100 to 200 cfu of the mce1R mutant, complemented mce1R mutant, or wild type H37Rv per mouse with an aerosol generation device (Inhalation Exposure System; Glas-Col, Terre Haute, IN). The inoculum doses in each group were assessed as described below by harvesting the right lung of four mice (from each group of mice) 24 h post-infection. At 4 weeks post-infection, the mice were initiated on treatment with INH (100 μg/ml) and PZA (15 mg/ml) delivered ad libitum in drinking water. The duration of treatment was 4 and 8 weeks and the mice were followed without cortisone or dexamethasone administration for clinical symptoms, organ cfu determination, and histology after cessation of the antibiotics.
Microbiologic and histopathologic examination
At designated time points, four or five animals from each group were sacrificed and lungs and spleen were harvested and examined grossly, histologically, and microbiologically. The number of tubercle bacilli in the corresponding organ homogenates in PBS–Tween 80 (0.05%) was assessed by plating part of the homogenate on 7H11 agar (Difco) plates. The cfu counts were enumerated 3 to 4 weeks later. Data are presented as the mean value ± SD of four or five mice per group and the experiment was performed twice with similar results. Differences between the mean of cfu counts obtained from each treatment group were analyzed by Student’s t test and were considered significant at P < 0.05.
Tissue samples for histopathologic studies were prepared as previously described [21, 22]. Briefly, left lung was fixed in 10% buffered formalin and sectioned. Histopathology slides were made commercially (Histology Consultation Service, Everson, WA, USA), and lung sections were either stained with haematoxylin and eosin (H&E) or by the Ziehl-Neelsen method for acid-fast bacilli. Lung sections from four mice per each group per time point were analyzed by a veterinary pathologist specializing in mouse pathology, blinded to the sources of the specimens. The lung sections were evaluated according to the following set of criteria: 1) percentage of lung parenchymal lesion; 2) severity of organization of the lesions (in order of severity-- diffuse, coalescing nodular, nodular, alveolar septal thickening); 3) type of macrophages present (foamy, epithelioid); 4) lymphocyte distribution (mild, moderate, marked, severe); 5) areas of necrosis (focal, multiple focal, diffuse, coalescing), 6) involvement of airways (small to large numbers of inflammatory cells); and 7) the number of neutrophils present.
RESULTS
High dose tail vein infection of mice treated for 4 weeks
At 5 × 105 dose, all mice infected with the mce1R mutant rapidly progressed to death, and hence, this dose could not be used with this mutant in the traditional Cornell model. Detailed results of the BALB/c infection with the mce1R mutant are published elsewhere [20]. However, with the wild type and mce1 operon mutant M. tuberculosis, bacilli were recovered from organs during the course of dexamethasone treatment as follows: one of 3 mice infected with the mutant sacrificed at 12 weeks of treatment with dexamethasone (or after 24 weeks of tail vein infection) showed 470 cfu from the spleen and 17 cfu from the lung homogenate. No cfu’s were recovered from any other mice sacrificed at 4, 8 or 20 weeks of treatment with dexamethasone. Each of 2 of 3 mice infected with the wild type, sacrificed at 20 weeks, had 17 cfu from the spleen and no cfu from lungs. No cfu’s were recovered from earlier time points (4, 8, and 12 weeks).
Low-dose tail-vein infection of mice treated for 4 weeks
Because of the high pathogenicity of the mce1R mutant at 5 × 105 inoculum dose, we repeated the tail-vein infection with a lower dose. Surprisingly, at 5 × 102 dose, a mean of about 104 mce1R mutant bacilli were recovered from lungs of 3 mice sacrificed after 4 weeks after the antibiotics were stopped and before the dexamethasone was initiated (Fig. 1). The number of bacilli continued to increase until it reached about 106 bacilli per lung after 12 weeks of dexamethasone treatment. No wild type or mce1 operon mutants were recovered from lungs at any time point in mice given dexamethasone, but after 4 weeks of cessation of the drugs, we did see recovery of both in mice not given dexamethasone (Fig. 1B).
Figure 1.
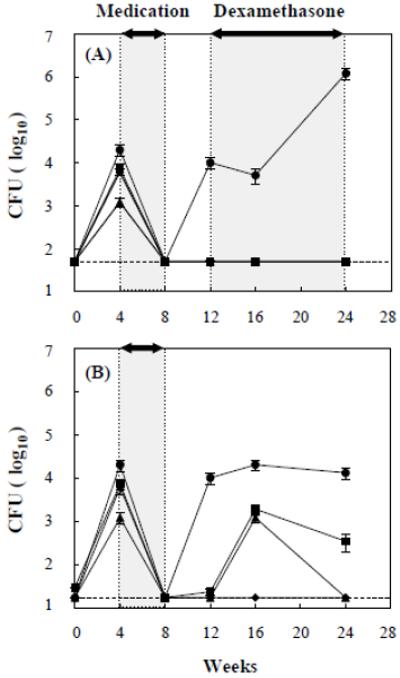
Tail-vein infection with low-dose (5 × 102) Erdman wild type (◆), M. tuberculosis H37Rv wild type (■), mce1 operon mutant (▲), and mce1R mutant (●). Antibiotics were given for 4 weeks in each group. Mice in panel A were treated with dexamethasone, while those in panel B were not. The cfu recovery from lungs is shown as the mean value ± SD of three mice per group at the indicated periods of infection. Dotted line (----) indicates a limit of detection.
Aerosol infection in mice treated for 4 weeks
The rapid reactivation of the mce1R mutant after cessation of the antibiotics was unexpected. We thus studied mce1R in more detail, using the aerosol infection model, comparing two different durations of antibiotic administration (4 and 8 weeks). BALB/c mice were infected with M. tuberculosis H37Rv wild type, mce1R mutant, and complemented mce1R strain via the inhalation route. At 24h post-infection, four mice were sacrificed from each infected group for cfu recovery in the mouse lungs and spleen. The initial inoculum doses were approximately 1.3 - 1.4 × 102 for each mouse per group (Fig. 2). At 4 weeks post-infection, no significant difference in bacterial cfu recovery from lungs (~1.0 - 2.5 × 106) was observed in mice infected with any of the M. tuberculosis stains (Fig. 2A). The number of cfu from spleen of mice infected with the mce1R mutant (9.5 ± 3.16 × 104) was similar to that observed in spleen of wild type H37Rv (1.3 ± 0.4 × 104) or complemented mce1R strain (4.3 ± 1.4 × 104) (Fig. 2B).
Figure 2.
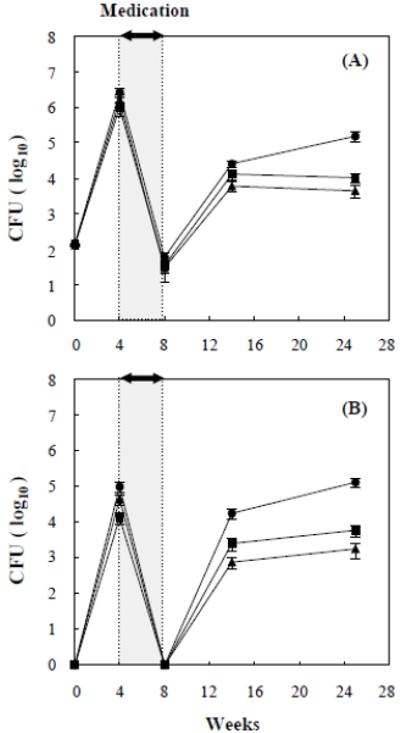
Aerosol infection with wild type M. tuberculosis H37Rv (■), mce1R mutant (●), and complemented mce1R mutant (▲). Infected BALB/c mice were given antibiotics for 4 weeks in each group after 4 weeks of infection. The cfu recovery from lungs (A) and spleens (B) is shown as the mean value ± SD of four or five mice per group at the indicated periods of infection.
After 4 weeks of infection, mice were started on treatment with INH and PZA. The 4-week course of INH-PZA resulted in undetectable numbers of viable bacilli in spleen and in low numbers (~3.0 - 6.0 × 10) of bacilli from lungs (Fig. 2A, B). At 6 weeks after the 4-week antibiotic course (14 weeks post-infection), no significant difference in bacterial cfu recovery from lungs was observed in mice infected with wild type H37Rv (1.3 ± 0.4 × 104), complemented mce1R strain (6.0 ± 1.8 × 103), or mce1R mutant (2.6 ± 0.5 × 104) (Fig. 2A). But, the cfu recovery from spleen of mice infected with the mce1R mutant (1.7 ± 0.5 × 104) became progressively greater than that of mice infected with H37Rv wild type (2.4 ± 0.9 × 103) (P < 0.005) or complemented mce1R strain (7.5 ± 2.5 × 102) (P < 0.001) (Fig. 2B).
Most importantly, by 25 weeks post-infection, the number of cfu from lungs and spleen of mice infected with the mce1R mutant continued to increase to 1.5 ± 0.4 × 105 and 1.3 ± 0.3 × 105, respectively, whereas the cfu recovery from organs of mice infected with H37Rv wild type and complemented mce1R strain remained unchanged around 1.0 ± 0.3 × 104 and 4.4 ± 1.6 × 103 in lungs, and 1.7 ± 0.7 × 103 and 5.8 ± 1.5 × 103 in spleen, respectively.
Lung histopathology in aerosol-infected mice treated for 4 weeks
Histopathologic examination of lungs of mice was performed prospectively in all mice aerosol-infected with wild type M. tuberculosis H37Rv, mce1R mutant, and complemented mce1R strain. First, mice were sacrificed after 4 weeks of infection just before the antibiotics were started. A large proportion of the lung parenchyma (25-75%) in all of the mice contained multifocal and coalescing nodular lesions comprised of a mixed population of foamy and epithelioid macrophages (Fig. 3, row 1). The histology of these lung sections was, for the most part, indistinguishable. There was no obvious change in the proportion of lung parenchymal lesions after treatment for 4 weeks in any of the infected mice. One of the lung sections from a mouse infected with the mce1R mutant showed small numbers of degenerate neutrophils in the parenchyma, and mild intralumenal degenerative neutrophils.
Figure 3.
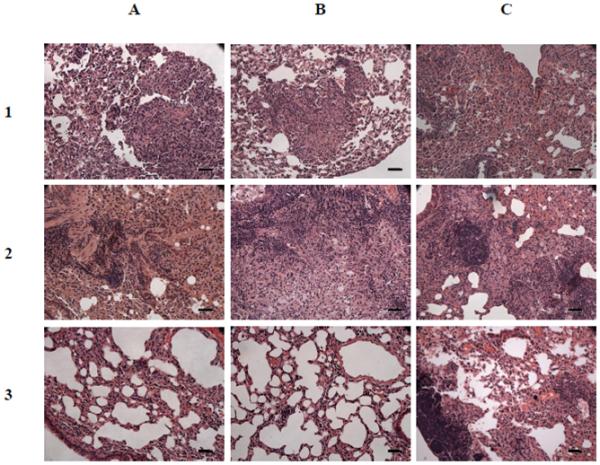
Histology (H&E stain) of lung sections of mice infected for 4 weeks before treatment was initiated (row 1), at the end of 8 weeks of treatment (row 2), and 20 weeks after end of an 8-week treatment (row 3). Mice were infected with H37Rv (column A), complemented mce1R mutant strain (column B), and mce1R mutant strain (column C) of M. tuberculosis via the inhalation route. Magnification, × 200; scale bar, 40 μm.
Mice treated for 4 weeks were examined 17 weeks after cessation of the treatment (25 weeks after the initial infection) (Fig. 4). The lung parenchymal involvement in mice infected with H37Rv and the complemented mutant was < 25-50%, while that of mice infected with the mce1R mutant was < 25%. In all mice, the distribution of the lung lesions was multifocal and nodular, containing a mixed population of foamy and epithelioid macrophages. No significant difference in the number of acid-fast bacilli was observed from the lung sections of mice infected with wild type H37Rv, complemented mce1R strain, or mce1R mutant (data not shown).
Figure 4.
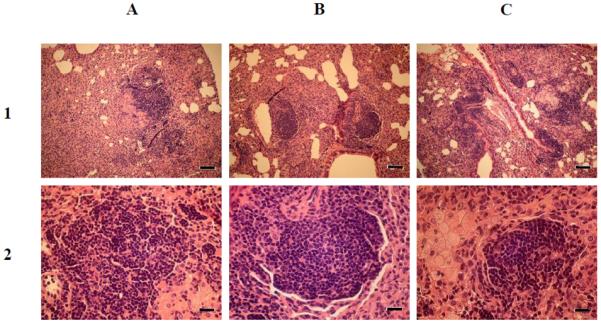
Histopathology of lung sections from BALB/c mice infected with wild type M. tuberculosis H37Rv (A), complemented mce1R mutant (B), and mce1R mutant (C). The mice were infected via the inhalation route and medicated for 4 weeks as described in Fig. 2. Lung sections from four mice per each group were harvested at 17 weeks post-medication (25 weeks post-infection) and stained with H&E. Lung tissues in all groups show a granulomatous interstitial pneumonia. Magnifications, × 100 (row 1) and × 400 (row 2); scale bars, 80 μm (row 1) and 20 μm (row 2).
Aerosol infection in mice treated for 8 weeks
Because the wild type and complemented mutant could still be recovered from mice treated for 4 weeks, we extended the treatment to 8 weeks. The bacterial cfu recoveries at time points before the 8-week medication course were similar to those of the 4-week medication course.
The 8-week course of INH-PZA resulted in undetectable numbers of viable bacilli in lungs and spleens in all infected groups at the end of the course (Fig. 5). At 6 weeks after the 8-week treatment course, (18 weeks post-infection), 1.3 ± 1.0 × 10 cfu’s from lungs and 2.0 ± 1.8 × 10 cfu from spleen of mice infected with the mce1R mutant were recovered, whereas no cfu’s were recovered from any of the organs of mice infected with H37Rv wild type or complemented mce1R strain (Fig. 5A, B).
Figure 5.
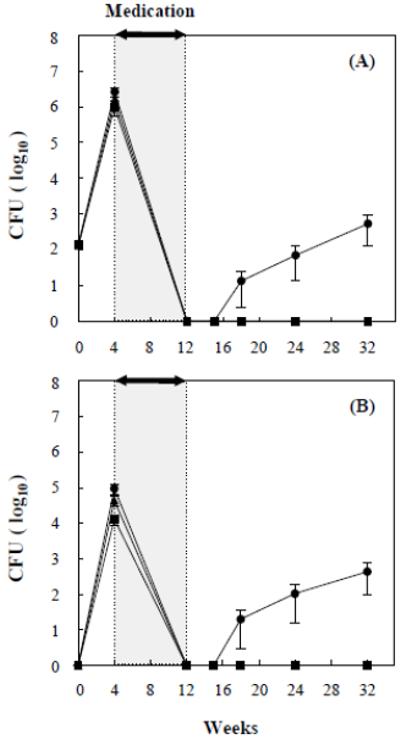
Modified Cornell model of infection with wild type M. tuberculosis H37Rv (■), mce1R mutant (●), and complemented mce1R mutant (▲). BALB/c mice were infected via the inhalation route and antibiotics were given for 8 weeks in each group after 4 weeks of infection. The cfu recovery from lungs (A) and spleens (B) is shown as the mean value ± SD of four or five mice per group at the indicated periods of infection.
At 24 and 32 weeks post-infection, the number of cfu recovered from mice infected with the mce1R mutant continued to increase from 7.5 ± 5.5 × 10 to 5.3 ± 4.0 × 102 in the lungs, and from 1.1 ± 0.9 × 102 to 4.4 ± 3.8 × 102 in the spleen, whereas the cfu recovery from organs of mice infected with H37Rv wild type and complemented mce1R strain remained undetectable. Interestingly, no mce1R mutant was detected from lungs or spleen of any mice at 3 weeks after cessation of the 8-week antibiotic course, suggesting that during this period, this mutant was indeed in a “latent” state.
Lung histopathology in aerosol-infected mice treated for 8 weeks
At the end of 8 weeks of treatment, the proportion of the parenchymal lung involvement in all mice decreased from the period just before the treatment was started (Fig. 3). In those infected with H37Rv or the complemented mce1R mutant the proportion of parenchymal involvement decreased to less than 25% in most of the lung sections (Fig. 3, row 2). In mice infected with the mce1R mutant, the parenchymal involvement also decreased to 25-50% (Fig. 3, C2). Granulomatous lesions were rare and small in mice infected with the mce1R mutant, and no such lesions were observed in mice infected with the other strains.
Lung sections of the remaining mice were harvested at 20 weeks after cessation of the 8-week treatment (32 weeks post-infection) (Fig. 3, row 3). At this time point, the lung lesions were much milder than those in lungs of mice observed after 17 weeks after 4 weeks of treatment (Fig. 4). The only group with nodular lung lesions was observed in mice infected with mce1R mutant (Fig. 3, C3). Lung sections of mice infected with mce1R mutant showed mild granulomatous pneumonia with multifocal nodular lesions characterized by scattered foci of alveolar macrophages with occasional foamy alveolar macrophages, and large focal aggregates of lymphocytes within the nodule (Fig. 3, C3).
DISCUSSION
Here, we used a variant of the Cornell mouse model of latent TB infection to study latent infection by M. tuberculosis mce1 operon and mce1R mutants. The unexpected observation was the rapid post-treatment proliferation of the mce1R mutant. At similar infection inoculum doses, the mce1R mutant rapidly proliferated in mouse lungs and spleen after the cessation of antibiotics, regardless of whether the mice were infected via tail vein or by the aerosol route. The mce1R mutant as well as the other strains exhibited a similar level of susceptibility to the drugs during the treatment course as evidenced by the low or absent cfu counts at the end of each treatment course (Figs 1, 2, 5). All infected mice initially responded clinically to the treatment as evidenced by progressive resolution of the lung lesions (Fig. 3). However, among mice treated for 8 weeks, only those infected with the mce1R mutant showed recrudescence of the lung lesions after treatment cessation. This recrudescence correlated with increasing number of cfu’s recovered from the lungs (Fig. 5). Thus, the mce1R mutant appears to cause a true post-treatment reactivation TB in mice. The fact that the mce1R is regulated in vivo [18, 19] suggests that this response can occur in a natural course of infection with wild type M. tuberculosis in humans. Indeed, post-treatment reactivation TB in humans is not uncommon in those who do not complete a standard 6-month course of treatment, even with drug-susceptible TB.
We showed that at 3 weeks after cessation of the drugs, the mce1R mutant could not be cultured from lungs (Fig. 5). Thus, this phase of infection was consistent with this organism being in a “latent” state of infection. Here the drugs may have reduced the bacterial count to below the threshold recoverable on the 7H11 agar plate. We note that it is possible that the treatment that resulted in no recovery of the wild type represents complete sterilization and not latent infection. However, at least in one experiment, we did observe later recovery of wild type from a subset of infected mice, as depicted in Fig. 1B.
Six of the mce1 genes (mce1A-F) encode cell wall proteins resembling substrate proteins of ATP-binding cassette (ABC) transporters, while the upstream genes yrbE1A and yrbE1B encode proteins that resemble ABC transporter permeases [16]. ABC transporters use an ATPase to supply energy for translocation of a substrate across a membrane [23]. Dassa and Bouige have suggested that RvO655, an ATPase called Mkl, may serve this function for M. tuberculosis [23]. Joshi et al have shown that RvO655 and mce1 are functionally linked and that the mce1 operon may encode an ABC transporter involved in lipid import [24]. Interestingly, the mce1 operon carries a gene fadD5, which encodes a fatty acyl co-A synthetase, putatively involved in fatty acid catabolism [15]. Dunphy et al recently suggested that FadD5 may be involved in recycling mycolic acids from dying M. tuberculosis inside granulomas [25]. Mce1R belongs to a family of GntR negative transcriptional regulators involved in lipid transport and metabolism in other bacterial organisms [18, 26]. Another member of the mce family operons, mce4, was recently shown to serve as a cholesterol importer necessary for persistent infection in mice [27]. These observations together suggest that the mce operon family is involved in lipid transport across M. tuberculosis cell wall and membrane, possibly for the organism to gain nutrients (carbon as energy source) during its long-term survival inside granulomas.
Beste et al recently reported that in M. bovis BCG, the mce1 operon may be involved in the bacillus switching to slow growth rate, which supports the idea that the regulation of the mce1 operon by mce1R can affect M. tuberculosis replication in vivo [28]. The mce1 genes in the mce1R mutant are constitutively expressed, while in the wild type, these genes are repressed during the early phase of infection in vivo [18, 19]. The expression of the mce1 genes may allow M. tuberculosis to import and metabolize lipids for its survival during persistent state. When the number of organism is reduced by treatment to a level undetectable by culture, wild type strains not expressing the mce1 genes may become restricted for replication by the host immune response, while the mce1R mutant strains expressing the mce1 genes would continue to take up and metabolize lipids and replicate. We have shown previously that wild type M. tuberculosis expresses mce1 genes after about 8 weeks into infection in mice [19]. We do not know at this time what triggers expression or repression of these genes in vivo. In a course of persistent infection, an infected host may harbor a mixed population of strains expressing and not expressing these genes. The progression to reactivation disease vs remaining in latent state may depend on the balance of these two bacterial populations. Factors that disrupt this balance, such as immunosuppression, old age, large initial infectious inoculum doses, or certain M. tuberculosis strain-related factors could lead to reactivation TB.
Acknowledgements
Financial support: This work was supported by the Korea Research Foundation Grant (KRF-2005-214-C00221) funded by the Korean Government (MOEHRD) and by the National Institute of Health, USA (R21AI063350).
Footnotes
Potential conflicts of interest: No conflict
References
- 1.Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA. 1999;282:677–86. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 2.Parrish NM, Dick JD, Bishai WR. Mechanisms of latency in Mycobacterium tuberculosis. Trends Microbiol. 1998;6:107–12. doi: 10.1016/s0966-842x(98)01216-5. [DOI] [PubMed] [Google Scholar]
- 3.Raviglione MC, Snider DE, Jr., Kochi A. Global epidemiology of tuberculosis. Morbidity and mortality of a worldwide epidemic. JAMA. 1995;273:220–6. [PubMed] [Google Scholar]
- 4.Chiang CY, Riley LW. Exogenous reinfection in tuberculosis. Lancet Infect Dis. 2005;5:629–36. doi: 10.1016/S1473-3099(05)70240-1. [DOI] [PubMed] [Google Scholar]
- 5.Adams LB, Mason CM, Kolls JK, Scollard D, Krahenbuhl JL, Nelson S. Exacerbation of acute and chronic murine tuberculosis by administration of a tumor necrosis factor receptor-expressing adenovirus. J Infect Dis. 1995;171:400–5. doi: 10.1093/infdis/171.2.400. [DOI] [PubMed] [Google Scholar]
- 6.Flynn JL, Scanga CA, Tanaka KE, Chan J. Effects of aminoguanidine on latent murine tuberculosis. J Immunol. 1998;160:1796–803. [PubMed] [Google Scholar]
- 7.Orme IM. A mouse model of the recrudescence of latent tuberculosis in the elderly. Am Rev Respir Dis. 1988;137:716–8. doi: 10.1164/ajrccm/137.3.716. [DOI] [PubMed] [Google Scholar]
- 8.Scanga CA, Mohan VP, Joseph H, Yu K, Chan J, Flynn JL. Reactivation of latent tuberculosis: variations on the Cornell murine model. Infect Immun. 1999;67:4531–8. doi: 10.1128/iai.67.9.4531-4538.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohan VP, Scanga CA, Yu K, et al. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun. 2001;69:1847–55. doi: 10.1128/IAI.69.3.1847-1855.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rhoades ER, Frank AA, Orme IM. Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tuber Lung Dis. 1997;78:57–66. doi: 10.1016/s0962-8479(97)90016-2. [DOI] [PubMed] [Google Scholar]
- 11.McCune RM, Tompsett R. Fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. I. The persistence of drug-susceptible tubercle bacilli despite prolonged antimicrobial therapy. J Exp Med. 1957;104:737–62. doi: 10.1084/jem.104.5.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCune RM, Tompsett R, McDermott W. Fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. II. The conversion of tuberculous infection to the latent state by the administration of pyrazinamide and a companion drug. J Exp Med. 1957;104:763–802. doi: 10.1084/jem.104.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCune RM, Feldmann FM, Lambert HP, McDermott W. Microbial persistence. I. The capacity of tubercle bacilli to survive sterilization in mouse tissues. J Exp Med. 1966;123:445–68. doi: 10.1084/jem.123.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCune RM, Feldmann FM, McDermott W. Microbial persistence. II. Characteristics of the sterile state of tubercle bacilli. J Exp Med. 1966;123:469–86. doi: 10.1084/jem.123.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 16.Casali N, Riley LW. A phylogenomic analysis of the Actinomycetales mce operons. BMC Genomics. 2007;8:60–82. doi: 10.1186/1471-2164-8-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimono N, Morici L, Casali N, Cantrell S, Sidders B, Ehrt S, Riley LW. Hypervirulent mutant of Mycobacterium tuberculosis resulting from disruption of the mce1 operon. Proc Natl Acad Sci USA. 2003;100:15918–23. doi: 10.1073/pnas.2433882100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casali N, White AM, Riley LW. Regulation of the Mycobacterium tuberculosis mce1 operon. J Bacteriol. 2006;188:441–9. doi: 10.1128/JB.188.2.441-449.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uchida Y, Casali N, White A, Morici L, Kendall LV, Riley LW. Accelerated immunopathological response of mice infected with Mycobacterium tuberculosis disrupted in the mce1 operon negative transcriptional regulator. Cell Microbiol. 2007;9:1275–83. doi: 10.1111/j.1462-5822.2006.00870.x. [DOI] [PubMed] [Google Scholar]
- 20.Parish T, Stoker NG. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology. 2000;146:1969–75. doi: 10.1099/00221287-146-8-1969. [DOI] [PubMed] [Google Scholar]
- 21.Senaratne RH, De Silva AD, Williams SJ, et al. 5′-Adenosinephosphosulphate reductase (CysH) protects Mycobacterium tuberculosis against free radicals during chronic infection phase in mice. Mol Microbiol. 2006;59:1744–53. doi: 10.1111/j.1365-2958.2006.05075.x. [DOI] [PubMed] [Google Scholar]
- 22.Lima P, Sidders B, Morici L, Reader R, Senaratne R, Casali N, Riley LW. Enhanced mortality despite control of lung infection in mice aerogenically infected with a Mycobacterium tuberculosis mce1 operon mutant. Microbes Infect. 2007;9:1285–90. doi: 10.1016/j.micinf.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dassa E, Bouige P. The ABC of ABCS: a phylogenetic and functional classification of ABC systems in living organisms. Res Microbiol. 2001;152:211–29. doi: 10.1016/s0923-2508(01)01194-9. [DOI] [PubMed] [Google Scholar]
- 24.Joshi SM, Pandey AK, Capite N, Fortune SM, Rubin EJ, Sassetti CM. Characterization of mycobacterial virulence genes through genetic interaction mapping. Proc Natl Acad Sci USA. 2006;103:11760–5. doi: 10.1073/pnas.0603179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunphy K, Senaratne RH, Mazuzawa M, Kendall LV, Riley LW. Attenuation of Mycobacterium tuberculosis functionally disrupted in a fatty acyl-CoA synthetase gene fadD5. J Infect Dis. doi: 10.1086/651452. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DiRusso CC, Black PN. Bacterial long chain fatty acid transport: gateway to a fatty acid-responsive signaling system. J Biol Chem. 2004;279:49563–6. doi: 10.1074/jbc.R400026200. [DOI] [PubMed] [Google Scholar]
- 27.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci USA. 2008;105:4376–80. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beste DJ, Espasa M, Bonde B, Kierzek AM, Stewart GR, McFadden J. The genetic requirements for fast and slow growth in mycobacteria. PLoS One. 2009;4(4):e5349. doi: 10.1371/journal.pone.0005349. [DOI] [PMC free article] [PubMed] [Google Scholar]